

Abstract
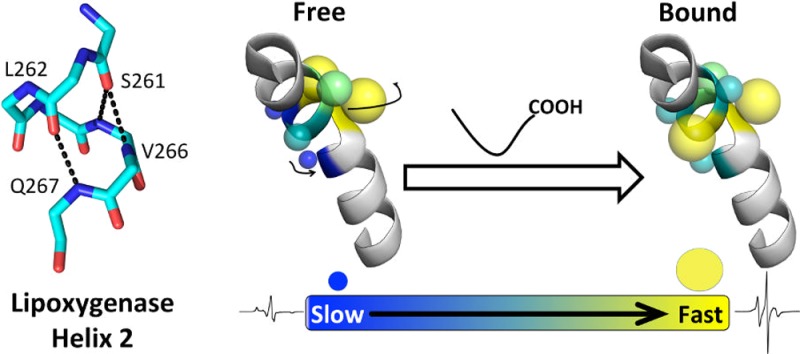
The second helix in lipoxygenases adapts to permit substrate access to the active site, but details of this process are varied and poorly understood. We therefore examined the dynamics of helix 2 in solutions of spin-labeled soybean lipoxygenase-1 and spin relaxation at 60 K of the spin-labels by catalytic iron. Helix 2 in soybean lipoxygenase structures is surface-exposed and contains one turn of π-helix, centrally located. A site-directed spin-label scan of 18 of the 21 helix 2 residues, and electron paramagnetic resonance, showed that the π-helical segment became unusually mobile, on a nanosecond time scale, under conditions favoring substrate binding (pH 9 and lipid addition), while segments before and after had relatively unchanged dynamics. Backbone dynamics of residues in the π-helical segment appeared to be correlated, at pH 9. Samples also were frozen to examine the polarity and proticity of the local environments, the effect of the local environment on intrinsic relaxation, and dipolar relaxation by two symmetries of catalytic iron. The average hyperfine tensor component, Azz, of four π-helix residues decreased by 1.75 G, with an increase in pH from 7 to 9, while it remained unaffected for nearby buried residues. Power saturation data suggested the change in polarity specific to the π-helix altered the intrinsic relaxation rates. Different symmetries of iron contributed to distance-dependent magnetic relaxation. We interpret these data to mean that a π-helix in the second helix of plant lipoxygenases is highly dynamic and is the site where lipid chains penetrate to inner helices that outline the substrate pocket.
Variation in the site of oxidation of polyunsaturated fatty acids by lipoxygenases is important in the physiology of plants and humans because the hydroperoxide products (ROOH) are the starting points in a variety of lipid mediator pathways.1−3 Lipoxygenase structures share a common fold, but the determinants of specificity in the oxidation reaction remain elusive. The current state of understanding is that a helix toward the N-terminus of lipoxygenases (helix 2) contains elements of an entrance to the substrate channel. Indeed, lipid binding near helix 2 has been demonstrated by spectroscopy4 and by crystallography.5,6 There are substantial structural differences in helices 2, but fewer elsewhere, in the cases where there are both ligand-free and ligand-bound crystal structures of lipoxygenases. Here, we examine dynamics of helix 2 in solutions of spin-labeled soybean lipoxygenase-1 (SBL1) in an effort to reveal fluctuations that could facilitate entry of a lipid into the substrate channel. Measurements using electron paramagnetic resonance (EPR) with spin-labels report on lifetimes of motional states that are in the range of ∼0.1–100 ns, times characteristic of backbone and side chain fluctuations of proteins in solution.7
Lipoxygenases require acyl chains with one or more pentadienyl moieties as substrates, as in linoleic acid or arachidonic acid. Substrate preferences may include free or esterified fatty acids. The lipoxygenase catalytic cycle involves both oxidation states of a metal ion (generally iron8 or manganese9) in a proton-coupled electron transfer reaction.10−12 The metal is located centrally in these largely helical proteins of ∼70–100 kDa. Known lipoxygenases are highly specific in product formation, for instance, in leading to 5S-, 8R-, 12S-, or 15S-hydroperoxides of arachidonate. An idea that has guided studies of lipoxygenase specificity for many years is that if the positions of the carboxy (polar) and methyl ends of the acyl chain within the substrate channel were reversed, then a 5S- or a 15S-lipoxygenase product of arachidonic acid could result from minor adjustments of a conserved protein fold.13 This concept apparently is confirmed by the finding that human 5S-lipoxygenase switches to yield 15S-products when it is pseudophosphorylated by a Ser to Asp mutation5 or when the cavity volume is reduced by mutation.14 Unresolved is how substrate acyl chains, in either configuration, enter lipid binding sites of various lipoxygenases. Helices 2, 11, and 12 in several animal lipoxygenases are displaced from native configurations when substrate or inhibitors bind,5,15,16 and a bacterial lipoxygenase has an elongated helix 2 with α2A and α2B inserts forming a lid over a bound phospholipid.6 These observations focus attention particularly on helices 2 across the entire lipoxygenase family.
Lipoxygenases from soybean have served as models for the structure and function of the entire family.17,18 Four soybean isoforms, differing in specificity with respect to converting linoleate lipids to 13S-hydroperoxides (equivalent to 15S-hydroperoxide from arachidonate), have highly similar overall structures, but they differ in the shapes of their putative entrances to the active site channel.19 In the major isoform, SBL1, the substrate channel is lined primarily with hydrophobic side chains of residues in helix 11 (SBL1 residues 535–545). A third helix, helix 21, lies parallel to helix 11 and contributes S747 and L754 to the channel. Access to the binding site appears to be blocked at the surface by L541 and T259 (in helix 2).18 Although an opening to the channel could easily be made by rotation of these side chains, extended rearrangement seems to be more suitable for admitting a long acyl chain. In all four crystal structures of ligand-free soybean isoforms,17−20 helices 2 have one turn of π-helix toward the middle (S261–Q267 in SBL1), covering the seven residues preceding a proline. This region contains two (i + 5, i) π-helical hydrogen bonds [266–261 (also bifurcated H-bond from residue 265) and 267–262]. The result is a bend of ∼27° between the axes of the two ends of the helix.
The importance of helix 2 in SBL1 was highlighted by our recent study that defined the location of the polar end of a bound paramagnetic lysolipid, using site-directed spin labeling and pulsed dipolar EPR.4 Residues in helix 2, the loop preceding it, and helix 11 are close to the headgroup of that lipid. Now, we substituted a spin-labeled cysteine for 18 of 21 residues in the helix to examine the dynamic state of helix 2. Site-directed spin labeling opens a unique window on nanosecond dynamics of macromolecules in solution.7 For residues buried in a structure, EPR spectra of spin-labels reflect primarily overall rotational correlation times. In contrast, spin labeling peripheral helices provides information about side chain packing, backbone dynamics, surface exposure, and changes thereof. The peripheral nature of helix 2 in SBL1 is illustrated in Figure 1. We examined the effects of the nonreactive diamagnetic substrate analogue, lyso-oleoylphosphatidylcholine (LOPC), on each SBL1 mutant. The SBL1 isoform has an activity optimum at pH 8–9 and little activity at pH 6; crystal structures were determined at pH 7.0 and ∼5.6.17,18 Thus, pH-dependent dynamics of the spin-label signals were characterized to connect the enzymology to the structure. These and EPR power saturation experiments show that the π-helical segment of helix 2 is most sensitive to the presence of a substrate analogue. This suggests a model in which an acyl chain gains access to the substrate cavity by extended interactions with helix 2 residues, rather than by entering through small rearrangements of selected side chains that block access to the active site.
Figure 1.

Helix 2 in soybean lipoxygenase-1 (SBL1). The solvent-accessible surface of the large domain in the SBL1 structure (PDB entry 1YGE, residues 170–841), with the N-terminal domain as a cartoon for reference, was rendered with PyMol (Schrödinger). In the large domain, residues 255–275 constitute helix 2 and are illustrated as a cartoon (blue). Iron and associated water in the catalytic center can be seen through the semitransparent surface. The sequences of helices 2 in the four isoforms of soybean lipoxygenases for which structures are available are given below as a WebLogo (redrawn from http://weblogo.berkely.edu). The residue numbering refers to the SBL1 sequence, and the letters below the sequence numbers refer to the SBL1 sequence. The names of other isoforms are lipoxygenase-3 (PDB entry 1RRL), VLXB (PDB entry 2IUJ), and VLXD (PDB entry 2IUK). The lowered letter “I” between residues 273 and 274 indicates the insertion of isoleucine into VLXD at this point.
Experimental Procedures
Materials
The spin-label reagent, (1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl) methanethiosulfonate (MTSL), was from Toronto Chemical. The substrate analogue, 1-oleoyl-2-hydroxy-sn-glycero-3-phosphocholine, “lyso-oleoyl-phosphatidylcholine” (LOPC), was from Avanti Polar Lipids.
Protein Mutagenesis and Preparation for EPR
Proteins with single-cysteine replacements of native side chains in helix 2 were expressed in CodonPlus-RILP(DE-3) cells (Agilent) using a cysteine-free SBL1 construct previously reported.21 The construct appends a (His)6 N-terminal tail. Spin labeling and purification were performed according to the previous report.
EPR samples were prepared by buffer exchange in a centrifugal concentrator. Protein, at 0.15 mM (15 mg/mL) for frozen samples and at least 0.2 mM for solution samples, in 0.02 M tris (pH 7.2), was diluted with an equal part of selected 0.18 M buffer containing 60% (w/v) sucrose, giving final protein concentrations of 0.075 mM (frozen) or 0.1 mM (solution) in 0.1 M buffer and 30% sucrose. The pH values of buffers used for samples were determined at 21 °C. Sample buffers for solution EPR were tris (pH 7.2 or 9.0), and for frozen samples, at room temperature they were bis-tris (pH 6.4) and tricine (pH 8.4) with 30% sucrose. The thermal coefficients of pKa’s for bis-tris and tricine are similar, resulting in more basic pH values at the freezing temperature. Thus, the pH values of frozen samples were estimated to be ∼7 and ∼9, respectively.
Samples were aerobic in both solution and frozen states. Oxygen is a secondary substrate that diffuses to a lipoxygenase substrate radical after the rate-limiting step of the reaction.22 Whether protein radical relaxation by oxygen is significant depends on the radical spin–lattice relaxation rate23 and will be examined separately for the spin-labeled SBL1 cases.
EPR Spectroscopy
Room-temperature (21–22 °C) EPR spectroscopy was performed with an E600 Bruker spectrometer, equipped with an SHQE_R2 high-sensitivity resonator, operating at X-band frequencies (9.375–9.409 GHz) with the quartz dewar of a cryostat in place. Magnetic field values at the sample were calibrated with a DPPH (2,2-diphenyl-1-picrylhydrazyl) standard. Instrument settings for solution samples were 0.1 mT for the modulation amplitude (100 kHz) and 2 mW for the power. For comparative displays, solution spectra were normalized to the same second integral and adjusted to equivalent frequencies by adding a small constant magnetic field value to the recorded values, as needed. Simulations of some spectra were made with EasySpin (http://www.easyspin.org24).
Power saturation experiments were generally conducted at 60 K, using a helium-flow ESR 9/10 cryostat (Oxford Instruments), an ITC503 controller (Oxford), and a CERNOX sensor (Lakeshore Cryotronics) under the sample. Sample volumes of ∼200 μL were contained in 4 mm quartz tubes. The SHQE_R2 resonator was used for most power saturation runs, but selected experiments with a DM2 resonator were conducted at the EPR Center of the Medical College of Wisconsin (Milwaukee, WI). Automated power saturation settings were 4 mT scans (encompassing the center of the spin-label spectrum), a sweep of 0.05 mT/s, a modulation amplitude of 0.2 mT (100 kHz), and a delay between power steps of 10 s. Reported values of power included the separate calibration constants of the different resonators (Bruker Biospin), and saturation of a representative sample occurred at essentially identical powers in both resonators. An additional calibration for the quatrz inserts was not conducted. The extent to which using 100 kHz ωm is bordering (differently among mutants) on violation of the slow passage requirement will be examined in further studies.
The hyperfine value, as 2Azz, of spectra recorded at 60 K and at X-band was measured from spectra recorded under nonsaturating conditions (0.2 μW) in 20 mT sweeps.
Analysis of Power Saturation EPR Data
The peak-to-peak intensity, I, of the derivative signal in the mI = 0 region in EPR spectra of frozen samples was measured. From the Bloch equations, the absorption amplitude, Abs, of a magnetic resonance signal depends on the rotating B1 field and relaxation times, T1 and T2, as in eq 1.
| 1 |
Characteristic values of P50 can be extracted from I versus √P plots, as the power at which the derivative signal intensity has fallen to half the value predicted by the linear region, or fits to the data can be used to obtain an optimized value of P50. The appropriate function for fits of I versus √P depends on whether the signal is homogeneously or inhomogeneously broadened (or between homogeneously and inhomogeneously).25−27 We find, for our samples at 60 K, the inhomogeneous case applies (Figure S1 of the Supporting Information), so that data fits following eq 2 also yield P50. Deviations of fits when P50 is larger arise when dipolar interaction with a fast-relaxing metal is significant28 or when the sample is heterogeneous.
| 2 |
where c is a scaling factor and the square root of the microwave power, √P, is proportional to B1.
Optimized values of P50 were obtained by least-squares fitting data for powers from 0.1 μW to 1.0 mW to eq 2. Experimental log(I/√P) versus log(P) plots deviate from prediction: residuals in fitting the data with eq 2 increased to as much as 12% of P50 in samples with larger values of P50. Although the fits can be improved using a stretched exponent (b/2 instead of 1/2 in eq 2),29 the exponent differed from mutant to mutant and is of questionable significance, so only b = 1 is used in the evaluations presented here.
Enzyme Kinetics
Helix 2 mutants I257R1, T259R1/I265R1, Q267R1, and F274R1 were selected for analysis of enzyme kinetics. Solutions of linoleic acid substrate (4–80 μM) were usually prepared in sodium borate (0.2 M, pH 9.2) with 20 μg/mL Tween 20. When inhibition by LOPC was examined, the buffer substrate was prepared in 0.8% methanol with no Tween 20.4 Equal volumes of enzyme (20 nM) and substrate solutions were rapidly mixed and monitored optically for product formation as described previously.4 The enzymatic rate for each substrate solution was measured five times. Kinetic constants (kcat and Km) were obtained by nonlinear fits to the Michaelis–Menten equation (using KaleidaGraph, Synergy Software). Each effective substrate concentration was measured by completely oxidizing the substrate with a small amount of lipoxygenase.
Results
Side Chain Dynamics of Helix 2 for SBL1 in Solution
Helices 2 in crystal structures of four soybean isoforms17−19,30 have 21 or 22 residues and are divided into two axis directions by a stretch of π-helix (SBL1, S261–Q267). The π-helix precedes a conserved proline (P268 in SBL1). Eighteen of the native amino acids in helix 2 of cysteine-free SBL1 were mutated to cysteine and spin labeled with the MTSL reagent. The resulting spin-labeled side chain was designated R1 by convention.31 The potential mutants not included in this study are V266R1 (had a tendency to precipitate), P268C (did not express well), and A269R1 (not tried). Some data for mutant E256R1 are included here, but the native Glu at this position is unique among isoforms of soybean lipoxygenase and is part of a salt bridge connecting helices 2 and 11.20 This salt bridge contributes to the unusually high pH optimum of SBL1 enzymatic activity.4
Each spin-labeled mutant (0.1 mM) was examined by EPR at ambient temperature and under three solvent conditions, in the order pH 7.2, 9.0, and 9.0 with the lipid LOPC (0.2 mM). This choice was made because of the difference between the optimal pH for SBL1 activity (pH 8–9) and pH values at which crystal structures of SBL1 were obtained (pH ≤7).17,18 The pH-induced changes in the spectra were reversible within the time (1–2 h) required to prepare and measure EPR samples.
Residues in the middle of helix 2, the π-helical region, had the greatest sensitivity of EPR spectra to changes in pH or LOPC addition. The spectra of S261R1–Q267R1 are shown in Figure 2, with separate columns for the three solvent and ligand conditions. Remarkable differences are evident when spectra of samples at pH 7.2 (left column) are compared with those at pH 9.0 (middle column). At pH 7.2, the spectra of residues S261R1–Q267R1 show variation in side chain mobility that repeats in ≥i + 4 residues, consistent with one side of a surface helix interacting closely with neighboring residues in other elements of the structure while the other side is solvent-exposed. The bars on the left of the figure illustrate this point by showing the magnitude of the magnetic field shift of the first low-field maximum in pH 7.2 spectra, relative to that value for the very mobile residue, I265R1. In contrast, spectra at pH 9.0 (middle column) have largely lost evidence of this helical variation. Instead, all of the pH 9.0 spectra of the middle part of helix 2 are quite similar to each other and reflect a degree of motion that is intermediate between the extremes of the spectra of pH 7.2 samples. Most strikingly when the pH is increased from 7.2 to 9.0, side chains of S263R1 and Q267R1 become more mobile while those of S261R1, Q264R1, and I265R1 become less so. Residue I265R1 is particularly mobile at pH 7.2, consistent with the natural side chain, I265, being surface-exposed in the crystal structure. Estimates of rotational times corresponding to the spectra at pH 7.2 could be as varied as ∼45 ns (overall protein rotation, plus an order parameter of 0.75, and hydrated radius of 3.4 nm32) for S263R1 to ∼2.5 ns for I265R1 (assuming isotropic motion). The presence of several coexisting EPR subspectra is most apparent after LOPC is added to samples at pH 9.0 (right column, Figure 2). A highly mobile component increases upon addition of LOPC, as discussed further in the following section.
Figure 2.

Dynamic changes in middle portion of helix 2 in response to pH and lipid. The low-field region (in the box of the full spectrum at the top) of X-band EPR spectra (9.404 GHz) for spin-label residues 261–267 (100 mM) in SBL1 helix 2 is shown. Spectra were recorded at 295 K. The three columns show EPR spectra for samples prepared at pH 7.2 (first column of spectra), pH 9.0 (middle column), and pH 9.0 with addition of lyso-oleoylphosphatidylcholine (LOPC, 200 mM). The bars, left of the spectra, indicate the magnetic field shift of the first maximum in the EPR spectra of the pH 7.2 samples compared to that of I265R1. The dashed line is a visual guide to illustrate the range in magnitude of these shifts in pH 7.2 samples.
The solvent conditions have a much smaller influence on the EPR spectra of spin-labels at the N- and C-terminal portions of helix 2 (Figure 3). Except for I257R1, neither variation of pH nor addition of LOPC substantially influences the degree of immobilization of the spin-labels at the N-terminal end of helix 2 (residues 255R1–260R1), indicating that this end of the helix has a relatively stable structure under the conditions examined. Spectra of mutant I257R1 do change significantly with pH, likely because residue 257 is preceded by a glutamic acid, E256. The C-terminal end of the helix (residues F270R1–D275R1) appears to be generally more mobile than the N-terminal portion (compare the first two columns with the second two in Figure 3). The EPR spectra are multicomponent, with variable amounts of a sharp mobile component for some C-terminal mutants. Some variations that are seen with solvent conditions may be related to the fact that natural residues E271 and D275 are likely ionized. The first and last entries in Figure 3 show EPR data for two residues outside of helix 2. Residue V237R1 (top, first column) is in a loop preceding helix 2 and was found near the polar end of a bound lysolecithin in a previous study.4 Its motional state does not change upon addition of LOPC (Figure 3). However, spin-labeled L541R1 (bottom, fourth column) does respond to addition of LOPC, as might be expected for its role in forming one end of the substrate channel near the surface.18 Note, though, that the close neighbor of L541, T259R1, responds little to addition of LOPC.
Figure 3.

Response of N-terminal and C-terminal regions of helix 2 to changes in pH and addition of lipid. Solution X-band EPR spectra are shown for spin-labeled residues at the N-terminal (residues 255–260) and C-terminal (residues 270–275) ends of SBL1 helix 2. Spectra with solid lines are from samples at pH 9.0. For the effect of pH, dotted lines are results for samples at pH 7.2; for the effect of addition of LOPC, dotted lines are results for LOPC addition at pH 9.0. Other experimental conditions are the same as those given in the legend of Figure 2. Data for two spin-labeled mutants outside of helix 2, V237R1 (top left) and L541R1 (bottom right), are also included.
In contrast to the high variability in side chain motion in helix 2, the room-temperature EPR spectra of several mutants spin-labeled at selected positions in other helices of SBL1, residues L480R1 (helix 9), A569R1 (helix 13), A619R1 (helix 15), and F782R1 (helix 23), studied previously,18 have solution EPR spectra much less sensitive to pH or addition of LOPC, although they do reflect changes in protein rotational correlation time with and without 30% sucrose. These results make global unfolding at higher pH unlikely to be the origin of changes in EPR spectra with pH (Figure 2).
In summary, Figures 2 and 3 show that side chains in the π-helical middle segment of SBL1 helix 2 have unique motional properties compared with those at the ends of the same helix. The change in pH from 7.2 to 9.0 alters the side chain states from ones in which motional freedom is related to the surface or buried nature of the side chain (pH 7.2) to one in which all the π-helix side chains have almost the same motional properties (pH 9.0). We interpret this to mean that backbone fluctuations have increased at pH 9.0 as a result of nanosecond processes that influence all residues in the π-helix almost equally. One likely mechanism for this process might include π- to α-helical transitions, proceeding in reversible steps,33 in which π-hydrogen bonds (i + 5 → i) break and re-form as α-hydrogen bonds (i + 4 → i). We further speculate that the sharp component of the EPR signal, the magnitude of which increases upon substrate binding (Figure 2), may arise from single-residue displacements from the helical structure. In that view, deformations of the π-helix might also include steps variously described as propagating “α-aneurisms” or “loop-outs”.34,35
A sequence of the same length as helix 2 in SBL1 is found in many plant lipoxygenase sequences. Sequence equivalents of SBL1 residues L255, K260, P268, and D275 are uniformly conserved, and the N- and C-terminal regions have a high degree of sequence similarity; however, residues 262–267 (the π-helical region) are more variable. All four structures of soybean lipoxygenases have one π-helical turn in the equivalent position. Sequence alignment suggests that all plant lipoxygenases have a bent helix 2 with a π-helical turn, as well. Animal lipoxygenases have different strategies by which helix 2 may adapt to binding substrate.
Effect of a Lipid Substrate Analogue on Helix 2 of SBL1
Lysolecithins are known substrates of lipoxygenases,36 and a spin-labeled lysolecithin with an oleoyl chain is an inhibitor of SBL1.4 In the study presented here, unlabeled, monounsaturated 1-oleoyl-2-hydroxy-sn-glycero-3-phosphocholine (LOPC) is chosen as a nonreactive substrate analogue for data shown in Figures 2–4. The EPR spectra from R1 mutations in the midsection of helix 2 were examined after addition, at pH 9.0, of 2 equiv of LOPC (Figure 2, right column). An overview of the effect of adding LOPC to the spin-labeled mutants at residues 263–265 and 267 is that the fraction of a mobile component increases after LOPC is added. In separate experiments, the concentration of LOPC was varied from 20 to 1000 μM with mutant Q264R1 (100 μM), and the resulting changes in the spectra suggested a binding constant in the tens of micromolar range. Reversibility in the influence of LOPC on spectra was demonstrated for mutant I265R1 by reversing the effect with repeated centrifugal concentrator exchanges with pH 7.2 buffer, and re-recording of EPR spectra.
Figure 4.
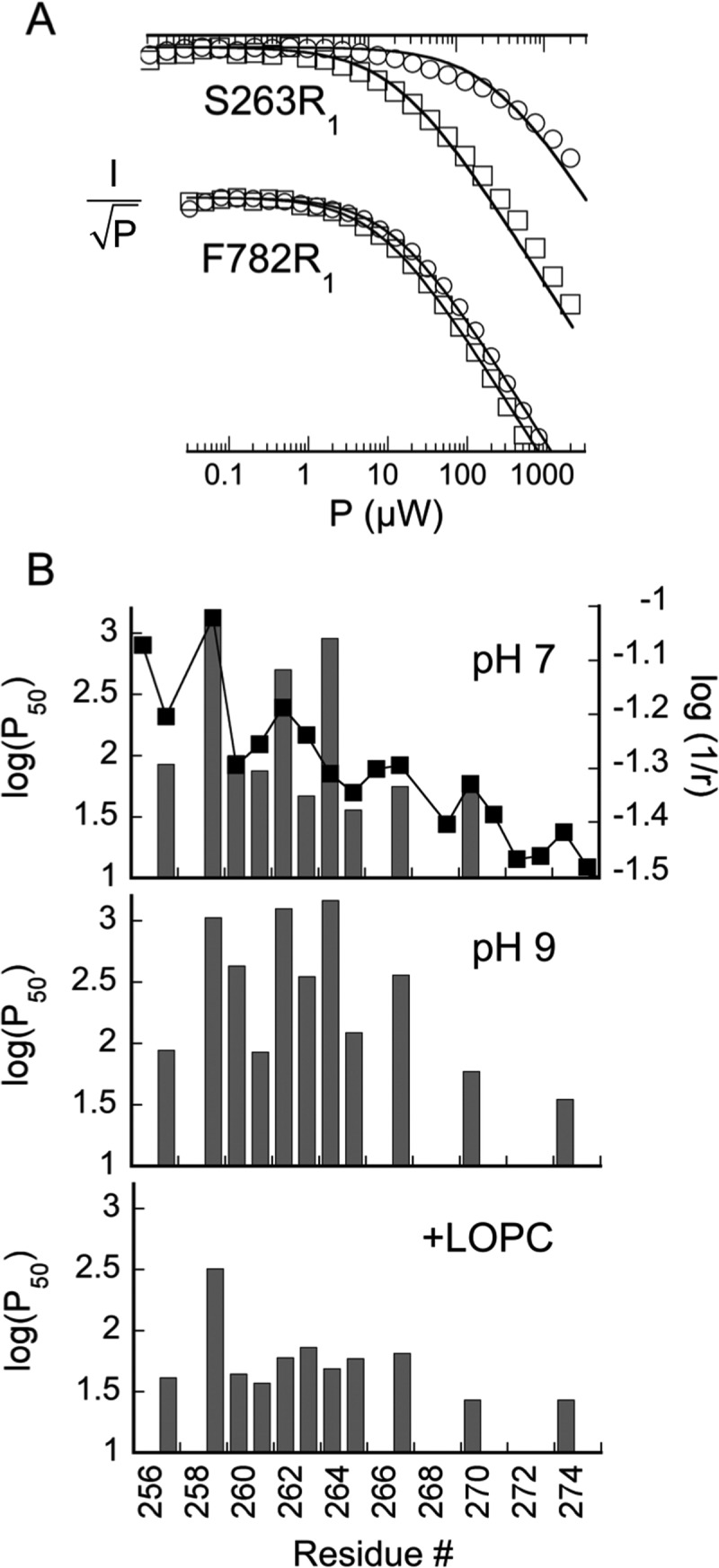
Summary of power saturation data at 60 K for R1 mutants in helix 2. (A) Representative power saturation plots for two spin-labeled mutants near (S263R1) and farther (F782R1) from the expected LOPC binding site. Results from samples at pH 9.0 with LOPC [(□) 75 μM protein and 150 μM lipid] and without [(○) 85 μM protein] are compared. (B) Graphical summary of saturation results for spin-labeled SBL1 helix 2 at pH 7 (top) and at pH 9, without (middle) and with addition of LOPC (bottom). Values of log(P50) (units of microwatts, left ordinate) are compared across the spin-labeled residues in helix 2. For comparison, in the top panel, pH 7 data are compared with log(r)−1 (r units in angstroms, right ordinate), where r is the distance from iron of the final atom of the natural side chain in the 1YGE structure.
Because the affinity of LOPC for SBL1 might vary among the R1 mutants, kinetic data for representative mutants were obtained (Table S1 of the Supporting Information). Km values with the linoleic acid substrate at pH 9.0 varied from 5 to 20 μM, values similar to those of other spin-labeled mutants of SBL1 studied previously.4 The inhibition constant (Ki) for addition of LOPC to S263R1 was determined to be ∼35 μM using a competitive inhibition model.
Influence of Catalytic Iron on Power Saturation of R1 Side Chains
In an effort to determine how adding the substrate analogue LOPC to SBL1 might change the average structure of helix 2, frozen samples were examined at 60 K. EPR spectra of 60 K samples should provide structural information about changes in the polarity of spin-label environments, and also about changes in the distance of spin-labels from iron if there are significant dipolar interactions between these spins. In power saturation comparisons, polarity effects may contribute to the intrinsic relaxation times T1 and T2, while dipolar interactions make a distance-dependent magnetic contribution to relaxation. Although a diamagnetic state suitable for comparison with the high-spin ferrous state of spin-labeled SBL1 presently is not available, the scan of spin-label substitutions across helix 2, and other locations, provides a distance-dependent framework for understanding and separating intrinsic from magnetic components of relaxation rates. The buffers were different from those used for room-temperature EPR, but they were chosen so that the estimated pH values for 60 K samples were comparable to those of the solution samples. All samples contained 30% sucrose.
In proteins, the local polar and/or protic environment of introduced spin-labels is manifest in the correlation of a decreasing gxx and an increasing Azz as the polarity or proticity of the environment increases.37,38 The outer hyperfine separation, 2Azz, was determined for samples of each spin-labeled mutant in helix 2. Values of Azz for frozen samples at pH 7.0 correlate well with differences in the surface exposure of side chains (Table 1). For instance, residues 259, 262, and 270 are not surface-exposed and, when spin-labeled, had Azz values 3.31–3.47 mT, while the other helix 2 residues are surface-exposed and had higher Azz values (3.61–3.77 mT). The switch to pH 9 leads generally to less distinction in, and lower values of, Azz (3.58 mT average), for the side chains that have solvent exposure in the crystal structures obtained at pH ≤7. The change in Azz is evidently a consequence of a lower solvent “protonicity” when the pH is increased, meaning primarily that there was a decrease in the average number of H-bonds between water and nitroxide. After the addition of LOPC, there were very small further changes in the EPR parameters characterizing frozen samples (Azz = 3.60 mT on average). These observations led to an expectation of some variations in the intrinsic relaxation times (T1 and T2) across the helix 2 mutants when the pH of the sample is changed from 7 to 9 (see the discussion below of Figure 4B, middle panel).
Table 1. Comparison of Hyperfine Splittings (Azza) at 60 K for Spin-Labeled SBL1b with Variations in pH and Lipid (LOPC) and Predicted Water Exposurec.
| residue | Azz at pH 7 | Azz at pH 9 | Azz with LOPC | water exposure |
|---|---|---|---|---|
| I257R1 | 3.74 | 3.73 | 3.61 | 58 |
| T259R1 | 3.47 | 3.47 | 3.61 | 6 |
| K260R1 | 3.72 | 3.51 | 3.58 | 57 |
| S261R1 | 3.64 | 3.57 | 3.56 | 20 |
| L262R1 | 3.43 | 3.43 | 3.54 | 0 |
| S263R1 | 3.77 | 3.53 | 3.61 | 20 |
| Q264R1 | 3.75 | 3.55 | 3.64 | 94 |
| I265R1 | 3.74 | 3.56 | 3.60 | 68 |
| Q267R1 | 3.61 | 3.54 | 3.61 | 18 |
| F270R1 | 3.31 | 3.60 | 3.61 | 1 |
| F274R1 | NDd | 3.65 | 3.66 | 10 |
| L541R1 | NDd | 3.54 | 3.66 | 27 |
Values of Azz are given in millitesla.
Residues 257–274 are on helix 2, and residue 541 is on helix 11.
Values of water exposure are given in square angstroms and were determined using the DSSP software package39 with the crystal structure of PDB entry 1YGE.
Measurements of F274R1 and L541R1 at pH 7 were not determined.
Spin-labeled mutants in helix 2 were examined by continuous wave (CW) power saturation at 60 K (Figure 4). A previous study yielded coordinates for the spins of F270R1 and F782R1,4 from which the calculated distances to iron are ∼20.9 and ∼26.5 Å, respectively. Distances from iron of natural helix 2 side chain ultimate atoms are generally ≤22 Å, with the hydroxyl of T259 only 10.5 Å from iron. Representative power saturation plots are compared in Figure 4A for F782R1 and S263R1 under the same sets of conditions (pH ∼9) with LOPC present (squares) or absent (circles). Values of P50 for F782R1 are near the lower limit we detected either with (24 μW) or without (38 μW) lipid. Close interaction of LOPC with residue 782 is not expected, and little effect of its addition was observed (Figure 4A, bottom curve). In contrast, it is more difficult to saturate the spins of S263R1 (in helix 2) when LOPC is absent (P50 of 240 μW) than when it is present (P50 of 74 μW). The lines in Figure 4A are least-squares fits (eq 2) for a single relaxation rate. The deviation of the fit to the S263R1 data is typical of cases in which angle-dependent magnetic relaxation terms are significant29 but can also indicate sample heterogeneity.
The variation in power saturation with residue position in helix 2 is summarized in Figure 4B for the solvent conditions at pH 7.0, pH 9.0 without LOPC, and pH 9.0 with LOPC added (top, middle, and bottom panels, respectively). Here, the power at which the peak-to-peak derivative amplitude falls to half the value expected for a linear response, P50, is the value reported. In the top panel, experimental log(P50) values (left ordinate) are compared with distances, as log(1/r) (right ordinate), between iron and the final atom in the natural side chain, from the crystal structure18 (■). Generally, the saturation results at pH 7.0 are consistent with increasing distance of spin-labeled side chains from iron, progressing from the N- to C-terminal ends of helix 2. Of course, an exact correspondence of nitroxide spin–iron distances with natural side chain–iron distances is not expected. However, the slope, approximately 4–5 for the left ordinate/right ordinate, suggests there is a substantial dipolar component to relaxation under these conditions. (Dipolar relaxation has r–6 dependence.)
The three panels of Figure 4B are quite different from each other in the sequence variation of P50 values and evidently include changes in both intrinsic and magnetic spin relaxation. In the switch to pH 9 (middle panel), compared with pH 7 samples (top panel), the P50 values of some residues are little changed (residues 259, 261, and 270), while major changes are evident in values of P50 for residues 260 and 262–267. With the exception of residue 262, residues with large increases in P50 values at pH 9 are those with Azz values suggesting less H-bonding by water to the nitroxide at the higher pH (Table 1). Measured values of T1 at 60 K generally are smaller when the spin is in nonpolar solvents than in polar and/or protic ones.40 It seems likely then that change in buffer pH from 7 to 9 (reduction in proticity) does lead to decreased values of T1, making it harder to saturate (increased P50 values) exposed residues (at pH 7.0) than buried ones.
The bottom panel of Figure 4B shows uniformly reduced P50 values when LOPC is added to SBL1 at pH 9.0. Known magnetic properties of iron in lipoxygenase may offer an explanation. The structure of the ferrous iron in resting lipoxygenase is similar to that of the iron center in bacterial reaction centers, and adding a spin-label to lipoxygenase may provide an analogue of the quinones in the reaction centers.29,41,42 Previous studies of SBL1 in ferrous states establish that there are two interconvertible symmetries at the iron center, summarized as five-coordinate and six-coordinate, as shown by magnetic circular dichroism.43 Resting ferrous SBL1 is high-spin and has a mixture of five- and six-coordinate states, with zero-field splittings (D) of 10 cm–1 (five-coordinate) and 13 cm–1 (six-coordinate), respectively, and an E/D of ∼0.1 for either geometry. Adding a lipid under nonreacting conditions to ferrous SBL1 changes the population of the two iron species in the five- and six-coordinate mix to mostly six-coordinate,43 resulting in only the D = 13 cm–1 levels contributing to relaxation. For comparison, the zero-field splittings of ferrous iron in bacterial reaction centers are D ∼ 5.2 cm–1 and E/D ∼ 0.25.41 Although the energies of the effective excited state levels of iron in SBL1 are somewhat higher than those in the reaction centers, we suggest that the across-the-board reduction in P50 values when LOPC is added (Figure 4B, bottom panel) results because now only the 13 cm–1 (six-coordinate) excited energy levels are available to contribute to relaxation. Because we find no loss of specific activity for samples after LOPC treatment, it is unlikely that reduced P50 values seen in the bottom panel of Figure 4B result from a loss of iron.
The relaxation properties of spin-labels on helix 2, summarized in Figure 4B, are a combination of effects from distance-dependent dipolar relaxation by iron, the polarity and proticity of the spin-label environment, changes in energy levels of the iron center, and other intrinsic relaxation contributions. Of interest is making a connection between the dramatic changes in side chain motion in solution (Figure 2) and changes in structure of the bend in helix 2. In spite of uncertainties in extracting the distance component from complex relaxation data, responses of π-helical residues 262R1–264R1 to a change in pH (Figure 4B) are dramatic and do suggest that the spin-label side chains have a distribution of locations that is, on average, closer to iron at the higher pH. The r–6 dependence of dipolar relaxation will magnify small shifts toward iron. At pH 9, the structure of the middle of helix 2 is more flexible and R1 side chains may be redistributed away from the surface exposure seen in crystal structures. The addition of the LOPC substrate analogue reverses that trend (bottom panel of Figure 4B). Thus, solution EPR spectra and relaxation studies both provide evidence that the middle of helix 2 senses the presence of bound lipid in SBL1.
Conclusions
In conclusion, the single turn of π-helix in the second helix of SBL1 has changes in dynamics and in relaxation by iron in response to changes in pH and lipid binding. These results speak to two subjects, structural transitions involving π-helices and the mechanism(s) by which the substrate accesses the lipoxygenase active site cavity. These insights result from a site-directed spin-label scan over the full length of helix 2, so that the special nature of the π-helical region (residues S261–Q267) is distinguished from other parts of helix 2.
The minimal π-helix structure has two consecutive (i + 5 → i) H-bonds in a six-amino acid sequence. The insertion of a single amino acid into an α-helix is suggested as the evolutionary origin of π-helices, so that α-helical bulges (“aneurisms”) and loop-outs may be included in the same category.34 An estimated 15% of proteins contain qualifying insertions in α-helices, and these are often associated with functionality.34,44 Indeed, another π-helical segment (in helix 9) in lipoxygenases is well-known for the role of contributing side chains that coordinate the catalytic metal.17,18,44 The four isoforms of soybean lipoxygenase for which crystal structures exist all have π-helical inserts in helix 2, as well, and an associated bend in the helix direction. In SBL1, we found dramatic changes in the spin-label mobility of the helix 2 π-helical residues when the sample pH was increased from 7 to 9, but minimal pH-dependent changes occurred in the neighboring α-helical segments. At the higher pH, spin-labeled residues in the π-helix all had very similar EPR spectra that are characteristic of nanosecond motional averaging. These results suggest that the backbone H-bonds in the π-helices are sufficiently weaker than those in the α-helices that base-catalyzed breaking and re-forming of amide H-bonds occur in nanoseconds rather than microseconds. The site-directed spin-label approach, at conventional EPR frequencies, appears to be ideally suited for studying the dynamics of π-helices in proteins.
An entrance large enough to permit entry of a fatty acid is not evident in the SBL1 crystal structures, although side chain rotation of residues in helices 2 (T259) and 11 (L541) may provide one.18 While only helix 2 residues L255, G258, T259, and K260 contribute to forming the mouth of the substrate channel in SBL1,18 we found that the π-helical part of helix 2, particularly residues 263–267, underwent increases in nanosecond dynamics, at room temperature, associated with substrate binding (Figure 2, right column). The π-helix part of helix 2 lies adjacent to a reverse turn in helix 11, residues 547–551, and helix 11 contributes numerous side chains to the substrate cavity. Our data led to a model in which an acyl chain makes extended initial contact with helix 2, and this contact makes use of the inherent flexibility in the π-helical region to allow an acyl chain to pass between helices 2 and 11 as it enters the substrate cavity. This model might be called a “middle-first” entry mode for substrate, in contrast to historical discussions13 couched in terms of “head-first” and “tail-first” entry through a localized site. The middle-first entry would permit different orientations of the chain within the cavity and hence different stereochemical outcomes. Factors favoring docking of the polar end of lipid substrates, by their bulk or by charge, at the mouth of the cavity likely determine the actual stereochemical specificity in product formation.
Structural studies of other lipoxygenases have provided end points that suggest rearrangement of a helix 2 when substrate or inhibitors are present.5,6,15,16 Implicit in a rearrangement is motion. Our study sheds light on the details of helix 2 dynamics in SBL1. At the pH optimum of the enzymatic reaction (pH 9), the middle section of helix 2 has robust nanosecond backbone fluctuations (Figure 2). At lower pH, the middle portion has a defined helical structure, corresponding to the substrate-free crystal structures, with side chain motion reflecting surface exposure. The observed motion at pH 9 is accentuated when a lipid is present. We interpret these results to mean that residues, including 261–267, in SBL1 are the point of entry and exit of the binding of the acyl chain to SBL1.
Acknowledgments
We thank Professor Brian Bennett and Candice Klug (National Biomedical EPR Center of the Medical College of Wisconsin) for valuable discussion and assistance in data acquisition using the DM2 X-band resonator. We also thank Xianfang Wu and Fayi Wu for discussion and assistance with cloning and mutagenesis.
Glossary
Abbreviations
- CW
continuous wave
- DPPH
2,2-diphenyl-1-picrylhydrazyl
- EPR
electron paramagnetic resonance
- LOPC
1-oleoyl-2-hydroxy-sn-glycero-3-phosphocholine
- MTSL
1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methylmethanethiosulfonate
- PDB
Protein Data Bank
- P50
microwave power at which the signal amplitude is half the unsaturated value
- ROOH
lipid hydroperoxide products of lipoxygenase
- SBL1
soybean lipoxygenase-1
- T1 and T2
spin–lattice and spin–spin relaxation times, respectively.
Supporting Information Available
Representative X-band EPR spectra of S263R1 at 60 K and a simulation (Figure S1) and steady state enzyme kinetic constants of spin-labeled mutants of SBL1 (Table S1). This material is available free of charge via the Internet at http://pubs.acs.org.
Supported by National Institutes of Health Grant GM06528 to B.J.G. and, in part, by National Institutes of Health Grant EB001980 that supports the National Biomedical EPR Center of the Medical College of Wisconsin.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Newcomer M. E.; Gilbert N. C. (2010) Location, location, location: Compartmentalization of early events in leukotriene biosynthesis. J. Biol. Chem. 285, 25109–25114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan C. N.; Petasis N. A. (2011) Resolvins and protectins in inflammation resolution. Chem. Rev. 111, 5922–5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liavonchanka A.; Feussner N. (2006) Lipoxygenases: Occurrence, functions and catalysis. J. Plant Physiol. 163, 348–357. [DOI] [PubMed] [Google Scholar]
- Gaffney B. J.; Bradshaw M. D.; Frausto S. D.; Wu F. Y.; Freed J. H.; Borbat P. (2012) Locating a Lipid at the Portal to the Lipoxygenase Active Site. Biophys. J. 103, 2134–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert N. C.; Rui Z.; Neau D. B.; Waight M. T.; Bartlett S. G.; Boeglin W. E.; Brash A. R.; Newcomer M. E. (2012) Conversion of human 5-lipoxygenase to a 15-lipoxygenase by a point mutation to mimic phosphorylation at serine-663. FASEB J. 26, 3222–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garreta A.; Val-Moraes S. P.; Garcia-Fernandez Q.; Busquets M.; Juan C.; Oliver A.; Ortiz A.; Gaffney B. J.; Fita I.; Manresa A.; Carpena X. (2013) Structure and interaction with phospholipids of a prokaryotic lipoxygenase from Pseudomonas aeruginosa. FASEB J. 27, 4811–4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbell W. L.; Lopez C. J.; Altenbach C.; Yang Z. (2013) Technological advances in site-directed spin labeling of proteins. Curr. Opin. Struct. Biol. 23, 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney B. J. (1996) Lipoxygenases: Structural Principles and Spectroscopy. In Annual Reviews of Biophysics and Biomolecular Structure (Stroud R. M., Ed.) pp 431–459, Annual Reviews, Palo Alto, CA. [DOI] [PubMed] [Google Scholar]
- Su C.; Sahlin M.; Oliw E. H. (2002) Manganese lipoxygenase has a mononuclear redox center. Adv. Exp. Med. Biol. 507, 171–176. [DOI] [PubMed] [Google Scholar]
- Hu S.; Sharma S. C.; Scouras A. D.; Soudackov A. V.; Carr C. A.; Hammes-Schiffer S.; Alber T.; Klinman J. P. (2014) Extremely elevated room-temperature kinetic isotope effects quantify the critical role of barrier width in enzymatic C-H activation. J. Am. Chem. Soc. 136, 8157–8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr C. A. M.; Klinman J. P. (2014) Hydrogen Tunneling in a Prokaryotic Lipoxygenase. Biochemistry 53, 2212–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell E. A. C.; Jamil R.; Gauld J. W. (2013) Gaining insight into the chemistry of lipoxygenases: A computational investigation into the catalytic mechanism of (8R)-lipoxygenase. JBIC, J. Biol. Inorg. Chem. 18, 343–355. [DOI] [PubMed] [Google Scholar]
- Gardner H. W. (1989) Soybean lipoxygenase-1 enzymically forms both (9S)- and (13S)-hydroperoxides from linoleic acid by a pH-dependent mechanism. Biochim. Biophys. Acta 1001, 274–281. [DOI] [PubMed] [Google Scholar]
- Hofheinz K.; Kakularam K. R.; Adel S.; Anton M.; Polymarasetty A.; Reddanna P.; Kuhn H.; Horn T. (2013) Conversion of pro-inflammatory murine Alox5 into an anti-inflammatory 15S-lipoxygenating enzyme by multiple mutations of sequence determinants. Arch. Biochem. Biophys. 530, 40–47. [DOI] [PubMed] [Google Scholar]
- Choi J.; Chon J. K.; Kim S.; Shin W. (2008) Conformational flexibility in mammalian 15S-lipoxygenase: Reinterpretation of the crystallographic data. Proteins 70, 1023–1032. [DOI] [PubMed] [Google Scholar]
- Xu S.; Mueser T. C.; Marnett L. J.; Funk M. O. (2012) Crystal Structure of 12-Lipoxygenase Catalytic-Domain-Inhibitor Complex Identifies a Substrate-Binding Channel for Catalysis. Structure 20, 1490–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyington J. C.; Gaffney B. J.; Amzel L. M. (1993) The 3-Dimensional Structure of an Arachidonic-Acid 15-Lipoxygenase. Science 260, 1482–1486. [DOI] [PubMed] [Google Scholar]
- Minor W.; Steczko J.; Stec B.; Otwinowski Z.; Bolin J. T.; Walter R.; Axelrod B. (1996) Crystal structure of soybean lipoxygenase L-1 at 1.4 Å resolution. Biochemistry 35, 10687–10701. [DOI] [PubMed] [Google Scholar]
- Youn B.; Sellhorn G. E.; Mirchel R. J.; Gaffney B. J.; Grimes H. D.; Kang C. (2006) Crystal structures of vegetative soybean lipoxygenase VLX-B and VLX-D, and comparisons with seed isoforms LOX-1 and LOX-3. Proteins 65, 1008–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrzypczak-Jankun E.; Amzel L. M.; Kroa B. A.; Funk M. O. Jr. (1997) Structure of soybean lipoxygenase L3 and a comparison with its L1 isoenzyme. Proteins 29, 15–31. [PubMed] [Google Scholar]
- Coffa G.; Imber A. N.; Maguire B. C.; Laxmikanthan G.; Schneider C.; Gaffney B. J.; Brash A. R. (2005) On the relationships of substrate orientation, hydrogen abstraction, and product stereochemistry in single and double dioxygenations by soybean lipoxygenase-1 and its Ala542Gly mutant. J. Biol. Chem. 280, 38756–38766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp M. J.; Klinman J. P. (2003) Kinetic studies of oxygen reactivity in soybean lipoxygenase-1. Biochemistry 42, 11466–11475. [DOI] [PubMed] [Google Scholar]
- Galli C.; MacArthur R.; AbuSoud H. M.; Clark P.; Stuehr D. J.; Brudvig G. W. (1996) EPR spectroscopic characterization of neuronal NO synthase. Biochemistry 35, 2804–2810. [DOI] [PubMed] [Google Scholar]
- Stoll S.; Schweiger A. (2006) EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 178, 42–55. [DOI] [PubMed] [Google Scholar]
- Haas D. A.; Mailer C.; Robinson B. H. (1993) Using Nitroxide Spin Labels: How to Obtain T(1e) from Continuous Wave Electron-Paramagnetic Resonance-Spectra at All Rotational Rates. Biophys. J. 64, 594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenbach C.; Greenhalgh D. A.; Khorana H. G.; Hubbell W. L. (1994) A Collision Gradient-Method to Determine the Immersion Depth of Nitroxides in Lipid Bilayers: Application to Spin-Labeled Mutants of Bacteriorhodopsin. Proc. Natl. Acad. Sci. U.S.A. 91, 1667–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug C. S.; Eaton S. S.; Eaton G. R.; Feix J. B. (1998) Ligand-induced conformational change in the ferric enterobactin receptor FepA as studied by site-directed spin labeling and time-domain ESR. Biochemistry 37, 9016–9023. [DOI] [PubMed] [Google Scholar]
- Hirsh D. J.; Beck W. F.; Innes J. B.; Brudvig G. W. (1992) Using Saturation Recovery EPR to Measure Distances in Proteins: Applications to Photosystem-II. Biochemistry 31, 532–541. [DOI] [PubMed] [Google Scholar]
- Lakshmi K. V., and Brudvig G. W. (2000) Electron Paramagnetic Resonance Distance Measurements in Photosynthetic Reaction Centers. In Distance Measurements in Biological Systems by EPR (Berliner L. W., Eaton S. S., and Eaton G. R., Eds.) Kluwer, New York. [Google Scholar]
- Skrzypczak-Jankun E.; Borbulevych O. Y.; Zavodszky M. I.; Baranski M. R.; Padmanabhan K.; Petricek V.; Jankun J. (2006) Effect of crystal freezing and small-molecule binding on internal cavity size in a large protein: X-ray and docking studies of lipoxygenase at ambient and low temperature at 2.0 angstrom resolution. Acta Crystallogr D 62, 766–775. [DOI] [PubMed] [Google Scholar]
- Langen R.; Oh K. J.; Cascio D.; Hubbell W. L. (2000) Crystal structures of spin labeled T4 lysozyme mutants: Implications for the interpretation of EPR spectra in terms of structure. Biochemistry 39, 8396–8405. [DOI] [PubMed] [Google Scholar]
- Dainese E.; Sabatucci A.; van Zadelhoff G.; Angelucci C. B.; Vachette P.; Veldink G. A.; Agro A. F.; Maccarrone M. (2005) Structural stability of soybean lipoxygenase-1 in solution as probed by small angle X-ray scattering. J. Mol. Biol. 349, 143–152. [DOI] [PubMed] [Google Scholar]
- Armen R.; Alonso D. O. V.; Daggett V. (2003) The role of α-, 3(10)-, and π-helix in helix → coil transitions. Protein Sci. 12, 1145–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooley R. B.; Arp D. J.; Karplus P. A. (2010) Evolutionary origin of a secondary structure: π-Helices as cryptic but widespread insertional variations of α-helices that enhance protein functionality. J. Mol. Biol. 404, 232–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe L. J.; Sondek J.; Shortle D.; Lattman E. E. (1993) The α-Aneurysm: A Structural Motif Revealed in an Insertion Mutant of Staphylococcal Nuclease. Proc. Natl. Acad. Sci. U.S.A. 90, 3275–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L. S.; Kim M. R.; Sok D. E. (2006) Linoleoyl lysophosphatidylcholine is an efficient substrate for soybean lipoxygenase-1. Arch. Biochem. Biophys. 455, 119–126. [DOI] [PubMed] [Google Scholar]
- Plato M.; Steinhoff H. J.; Wegener C.; Torring J. T.; Savitsky A.; Mobius K. (2002) Molecular orbital study of polarity and hydrogen bonding effects on the g and hyperfine tensors of site directed NO spin labelled bacteriorhodopsin. Mol. Phys. 100, 3711–3721. [Google Scholar]
- Bordignon E.; Brutlach H.; Urban L.; Hideg K.; Savitsky A.; Schnegg A.; Gast P.; Engelhard M.; Groenen E. J. J.; Mobius K.; Steinhoff H. J. (2010) Heterogeneity in the Nitroxide Micro-Environment: Polarity and Proticity Effects in Spin-Labeled Proteins Studied by Multi-Frequency EPR. Appl. Magn. Reson. 37, 391–403. [Google Scholar]
- Kabsch W.; Sander C. (1983) Dictionary of Protein Secondary Structure: Pattern-Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Bowler B. E.; Eaton G. R.; Eaton S. S. (1999) Electron spin lattice relaxation rates for S=1/2 molecular species in glassy matrices or magnetically dilute solids at temperatures between 10 and 300 K. J. Magn. Reson. 139, 165–174. [DOI] [PubMed] [Google Scholar]
- Calvo R.; Isaacson R. A.; Abresch E. C.; Okamura M. Y.; Feher G. (2002) Spin-lattice relaxation of coupled metal-radical spin-dimers in proteins: Application to Fe2+-cofactor (Q(A), Q(B), φ) dimers in reaction centers from photosynthetic bacteria. Biophys. J. 83, 2440–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsh D. J.; Brudvig G. W. (1993) Long-Range Electron-Spin Spin Interactions in the Bacterial Photosynthetic Reaction-Center. J. Phys. Chem. 97, 13216–13222. [Google Scholar]
- Pavlosky M. A.; Zhang Y.; Westre T. E.; Gan Q. F.; Pavel E. G.; Campochiaro C.; Hedman B.; Hodgson K. O.; Solomon E. I. (1995) Near-Infrared Circular-Dichroism, Magnetic Circular-Dichroism, and X-ray-Absorption Spectral Comparison of the Nonheme Ferrous Active-Sites of Plant and Mammalian 15-Lipoxygenases. J. Am. Chem. Soc. 117, 4316–4327. [Google Scholar]
- Fodje M. N.; Al-Karadaghi S. (2002) Occurrence, conformational features and amino acid propensities for the π-helix. Protein Eng. 15, 353–358. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.