

Abstract
Traumatic brain injury (TBI) afflicts up to 2 million people annually in the United States and is the primary cause of death and disability in young adults and children. Previous TBI studies have focused predominantly on the morphological, biochemical, and functional alterations of gray matter structures, such as the hippocampus. However, little attention has been given to the brain ventricular system, despite the fact that altered ventricular function is known to occur in brain pathologies. In the present study, we investigated anatomical and functional alterations to mouse ventricular cilia that result from mild TBI. We demonstrate that TBI causes a dramatic decrease in cilia. Further, using a particle tracking technique, we demonstrate that cerebrospinal fluid flow is diminished, thus potentially negatively affecting waste and nutrient exchange. Interestingly, injury-induced ventricular system pathology resolves completely by 30 days after injury as ependymal cell ciliogenesis restores cilia density to uninjured levels in the affected lateral ventricle.
Key words: : β-tubulin, basal body, hydrocephalus, lateral fluid percussion, particle tracking
Introduction
Traumatic brain injury (TBI) is the leading cause of death in children and young adults ranging from 15 to 30 years of age in the United States. TBI occurs at an alarming frequency of once every 23 seconds, resulting in 1.7 million incidences annually, including approximately 52,000 deaths.1,2 Many who survive the initial traumatic incident endure continuing difficulties with higher cognitive and motor functions. The majority of TBI research to date has focused on alterations in neuronal function and circuitry that are believed to underlie the cognitive impairment associated with TBI. However, a critical component of central nervous system (CNS) homeostasis involving the ventricular system and cerebrospinal fluid (CSF) flow has been largely neglected in TBI research. This knowledge gap is surprising because dysfunction in this system contributes to cognitive difficulties and TBI can lead to hydrocephalus.3,4 The brain's ventricular system is lined by ciliated ependymal cells, which contribute to the normal CSF flow, and ciliary defects have yielded hydrocephalus in both animal models5–10 as well as human disease.11–15 Because hydrocephalus is occasionally observed in TBI patients,3,4 we explored whether concussive brain injury alters cilia function and CSF flow.
The CNS ventricular system is comprised of interconnected fluid spaces that subserve four important functions. First and foremost, this system provides intracranial buoyancy, acting to reduce the apparent mass of the brain by suspension in CSF.16 Second, when the cranium suffers a jolt or impact, the CSF and the ventricular system serve as a shock absorber, thus diminishing the potential physical injury of the brain tissue. Third, continuous CSF one-way flow through the ventricular system generates a constant mechanism for removal of metabolic waste. Fourth, the CSF serves to transport hormones and critical nutrients to other areas of the brain. This interchange allows for homeostatic regulation of the distribution of neuroendocrine and other essential factors, to which slight changes can cause dramatic (cognitive) effects or damage to the nervous system.
The ventricular system is comprised of paired symmetric lateral ventricles, which communicate with the mid-line third ventricle though the interventricular foramina (foramina of Monro). The third ventricle terminates in the cerebral aqueduct (aqueduct of Sylvius), which, in turn, drains into the fourth ventricle. Because of the small volume of the third and fourth ventricles, CSF flows at a relatively fast rate, circulating in a one-way predictable path. The unidirectionality of CSF flow is ideal for clearing potentially harmful metabolic waste and carrying chemical information “downstream.” The ependymal cells lining the ventricles interface between CSF and brain parenchyma and are crucial for delivery of nutrients and waste removal, and, as mentioned above, all of these cells are ciliated. CSF within the ventricular system functions as a means of nonsynaptic volume transmission, providing a route of communication for signals mediating sleep,17–19 circadian rhythms,20 seasonal reproduction,21 and sexual behavior.22
The importance of the ependymal cell in normal ventricular development and maintaining patency of the interventricular foramina has recently been emphasized in several genetically modified mouse models, in which dysfunctional cilia result in hydrocephalus.5–10 In addition to the important role functional ependymal cilia play in ventricular development, inhibition of ciliary beating by intraventricular injection of G-protein-coupled receptor alpha i2 (Gαi2) small interfering RNA in mature mice resulted in ipsilateral hydrocephlus, emphasizing the importance of cilia function in postdevelopment ventricular homeostasis.10 Additionally, the continuous beating by ependymal cilia contributes to CSF flow and underlies the critical nutrient exchange and waste removal between the parenchyma and CSF. By constantly moving the CSF immediately adjacent to the ependymal layer, beating cilia enhance metabolic exchange between ependymal cells and CSF.15 Therefore, diminished cilia beating and ependymal cell function caused by brain injury could have far-reaching pathological ramifications.
Methods
Lateral fluid percussion injury
Experiments were performed on 5- to 7-week-old, 20- to 25-g C57BL/6J male mice (The Jackson Laboratory, Bar Harbor, ME). All experimental procedures and protocols for animal studies were approved by the Children's Hospital of Philadelphia's Institutional Animal Care and Use Committee (Philadelphia, PA) in accord with the international guidelines on the ethical use of animals. Experiments were designed to minimize the number of animals required. Animals used were cared for, handled, and medicated to minimize suffering (National Research Council, National Academy Press, Washington, DC, 1996). Lateral fluid perscussion injury (LFPI) was performed as previously reported.23–26 In brief, on day 1, animals were anesthetized with a ketamine (10 mg/100 g body weight; JHP Pharmaceuticals, Parsippany, NJ) and xylazine (1 mg/100 g body weight; Phoenix Pharmaceutical, Phoenix, AZ) mixture, and a parietal craniectomy on the right side was performed using a 3-mm trephine. A Luer-loc needle hub (3-mm inner diameter) was secured above the skull opening with cyanoacrylate adhesive and dental acrylic. On day 2, the hub was filled with saline and connected by high-pressure tubing to the LFPI device (Department of Biomedical Engineering, Virginia Commonwealth University, Richmond, VA). The injury was induced by a brief pulse of saline onto the intact dura, which delivers a pressure between 1.8 and 2.1 atmospheres, generating a mild-to-moderate brain injury.23,26 Sham animals received all of the above-mentioned procedures except the fluid pulse. Animals were warmed with an electric heating pad until they could move freely, before they were returned to their cages and fed with regular diet and tap water.
Immunofluorescent staining
Thirty minutes, 18 h, 7 days, or 30 days after LFPI, injured and respective time-matched sham animals were anesthetized with 5% chloral hydrate and perfused with saline, followed by 4% paraformaldehyde (PFA; Sigma-Aldrich, St. Louis, MO). Brains were postfixed for 2 h at room temperature (RT) and 50-μm-thick frontal (also known as coronal) sections were cut with a VT1000S vibratome (Leica Microsystems Inc., Buffalo Grove, IL). All brain sections were±1 mm from bregma. This was ensured by grossly verifying the presence of the corpus callosum and the absence of the hippocampus in each slice. For cilia staining, sections were incubated with a mouse monoclonal antibody (Ab) against type IV ß-tubulin (1:1000 in phosphate-buffered saline; AbCam, Cambridge, MA) before visualization with Alexa Fluor® 488–conjugated goat anti-mouse immunoglobulin (IgG; 1:200; Molecular Probes, Grand Island, NY). Basal body staining was conducted by incubating sections with a rabbit polyclonal Ab against γ-tubulin (1:500; AbCam). These sections were subsequently incubated with biotinylated goat anti-rabbit IgG (1:250; Vector Laboratories Inc., Burlingame, CA) and then with streptavidin/cyanine 3 (1:250; Sigma-Aldrich) for visualization. Primary Ab incubation was applied for 90 minutes at RT and continued overnight at 4°C, and secondary Abs were subsequently applied for 90 minutes at RT.
Confocal images were acquired with the Olympus Fluoview 1000 System (Olympus America, Center Valley, PA), with the Z-step kept at 0.5 μm. Consistent confocal settings (laser intensity, confocal aperture, photomultiplier tube, gain, offset, and resolution) were optimized and remained unchanged during the imaging of slices from both sham and injured animals for each time point. The staining pattern was confirmed in three LFPI and time-matched sham animals at each individual time point. From each mouse, 3–5 sections through the lateral ventricle at the rostral levels on the ipsilateral side were assessed.
Ependymal cell loss assay
Seven days after injury or sham procedure, 3 LFPI mice and 3 sham mice were processed for this assay. Fixed frontal sections (within the rostrocaudal range±1 mm relative to bregma) through the lateral ventricles were stained with Hoechst (Molecular Probes), a nuclear dye to stain all cells in the tissue. Confocal images of nuclei of the ependymal cells lining the ipsilateral lateral ventricle were compared between injured and sham mice. In healthy animals, ependymal cells cover the brain ventricles evenly (Supplementary Fig. 1) (see online supplementary material at http://www.liebertpub.com). In case of ependymal cell loss, obvious gaps between stained ependymal nuclei can be observed along the banks of the ventricles in the sections.
Pre-embedding immunogold staining
Seven days after injury, 3 LFPI mice and 3 sham-operated controls were perfused with a mixture of 4% PFA and 0.5% glutaraldehyde (GTA; Sigma-Aldrich). Brains were removed and postfixed in the aforementioned fixative for 3 h at RT. After incubation with the γ-tubulin Ab for basal body staining as described above, 50-μm-thick vibratome sections (±1 mm relative to bregma) were incubated with Ultra-Small gold-conjugated goat anti-rabbit IgG (1:100 in acetylated bovine serum albumin; Aurion, Wageningen, The Netherlands) for 6 h at RT. They were post-fixed with 2% GTA for 30 min. Immunogold-stained sections were silver-enhanced with the R-Gent SE-EM kit (Aurion) for 45 min at RT and then saved in enhancement condition solution (Aurion) for transmission electron microscopic (TEM) processing.
Transmission electron microscopy
The right hemisphere of 3 immunogold-stained sections (for basal body staining) and 3 unstained sections (for cilia “9+2 microtubule arrangement”) from each mouse were cropped and treated with 0.1 M of cacodylate buffer twice, 1% tannic acid, and then 1% NaSO4, each for 10 min. They were subsequently washed in deionized water (DW) twice and then dehydrated with a graded ethanol series (50–100%). After the last 100% ethanol treatment, slices were placed in 100% acetonitrile (2×10 min each) followed by serial infiltration with epoxy resin (Embed 812): acetonitrile/epoxy (2:1); acetonitrile/epoxy (1:1); and then acetonitrile/epoxy (1:2), 30 min each. Lateral ventricles were trimmed out and well oriented in Beem Capsules (2–3 right ventricle slices from 1 mouse in each capsule), embedded in 100% epoxy, and heat-incubated for 24 h. All chemicals for TEM processing were purchased from Electron Microscopic Services (Hatfield, PA).
Trimmed epoxy blocks were set on a 2050 Reichert Jung Supercut microtome (Leica Microsystems Inc.). Slices (1-μm-thick) were cut perpendicular to the plane of the 50-μm vibratome slices. As such, each “thick” slice is a 50-μm-wide belt. They were collected in drops of DW on heated slide glasses (60°C). To identify area of interest (ventricular cilia), dried slices were stained with 1% methylene blue for 13 sec. Excess dye was washed away with tap water, and the samples were differentiated with 95% ethanol for 15 sec. When cilia were identified under the microscope, the epoxy block was set on a Richert Ultra Cut S microtome (Leica Microsystems). Ultra-thin slices were cut at 80 nm and floated on DW. They were mounted on 250 mesh nickel grids (Electron Microscopic Services). Grids were then dried thoroughly on filter paper and saved in grid cassettes for at least 24 h before contrast staining. Dried sample grids were stained with 1% uranyl acetate in ethanol and then 0.1% lead citrate in DW, 30 min each. Once again, grids were dried on filter paper before observation under a Hitachi 7000 Electron Microscope (Hitachi High Technologies America, Pleasanton, CA).
Motile cilia are composed of nine peripheral doublet microtubles surrounding a central pair of single microtubules, a 9+2 axoneme. Radial spokes project from the central pair and interact with the peripheral doublets, lending to the stability of the axonemal structure, whereas the dynein arms generate the motion of the cilia by creating a sliding motion between the adjacent peripheral doublets.27,28 Transverse sections of cilia were observed to test whether LFPI affects the 9+2 microtubule arrangement normal structure of cilia. Immunogold-stained basal body transverse and longitudinal sections were also observed by electron microscope (EM). These images were taken using a Hitachi EM scope with an attached CCD camera (Hitachi Technologies America). Cilia and basal body morphology were then compared between LFPI and sham animals.
Scanning electron microscopy
Seven days after injury or sham procedure, 3 LFPI mice and 3 sham mice were sacrificed and their brains were processed as in the protocol for immunogold staining. Vibratome sections at 300 μm (±1 mm relative to bregma) were dehydrated in a progression of increasing ethanol concentrations, up to 100% ethanol, as described previously.29 Specimens were then critical-point dried in CO2, mounted on scanning electron microscopy (SEM) stubs, and sputter coated with gold palladium to a depth of 12 nm. The surface of brain slices was then examined with an AMR-1400 SEM (Phillips, Amsterdam, The Netherlands) at an accelerating voltage of 20 kV. Representative photomicrographs were taken at various angles to effectively display the specimen so that any errors in assessment resulting from the tilt of the specimen or other artifacts are minimized.
Magnetic resonance imaging
Three mice were used for ventricular volume analysis with repeated magnetic resonance imaging (MRI) imaging. They were all imaged on the same day of injury, before injury procedure, and this measurement was used as a within-subjects control. After this scan, LFPI was induced as described above. All mice were imaged again at 18 h, 7 days, and 4 weeks after LFPI. Whole-brain MRI images were obtained with a ClinScan system (7 T horizontal bore Bruker Magnet; Bruker Biospin, Ettlingen, Germany; and Siemens syngo software; Siemens Healthcare Global AG, Erlangen, Germany) using the linear body coil and a mouse brain 2×2 surface coil array. Mice were anesthetized with isofluorane and their vital signs (heart rate, respiratory rate and body temperature) monitored throughout the scan. A three-dimensional data set of the whole-mouse brain (in plane field of view of 25×21 mm, 192 data points, and 48 slices of 160-μm thickness resulting in voxels with dimensions of 130×130×160 microns) was acquired using a turbo spin echo technique with a turbo factor of 31. The repetition rate was 1500 ms, whereas the echo time (TE) of 3.12 ms corresponded to an effective TE of 66 ms. The bandwidth was 592 Hz/Px and five excitations were averaged. A magnetization restore pulse and the generalized autocalibrating partially parallel acquisitions technique with an acceleration factor of 2 were used to speed up the acquisition. MRI images were opened and volumes of the brain ventricles on both sides were analyzed with ImageJ software (National Institutes of Health, Bethesda, MD).
Particle tracking with fluorescent beads
Three LFPI mice and 3 sham controls were sacrificed at 18 h, 7 days, or 30 days after LFPI or sham procedure. Brains were removed and rinsed in cold CO2/O2-balanced sucrose cutting media. Live frontal slices of 250-μm thickness were cut through the lateral ventricle (±1 mm relative to bregma) with a VT 1200S vibratome (Leica Microsystems GmbH, Wetzlar, Germany) and collected in CO2/O2-balanced artificial CSF (aCSF). Latex beads with yellow-green fluorescence (2μm in diameter; Sigma-Aldrich) were diluted in aCSF and added to cover glass-bottomed dishes that held single slices before video recording under the Olympus FV 1000 confocal system (Olympus America). A total of 450 frames of images were taken in 30 sec using the XYT “round-trip” module, which reaches the highest imaging speed of the system (15 frames/sec). The resolution was automatically set at 256×256 pixels and 6×zoom. The 10×optical lens was used for video recording of both green fluorescent and differential interference contrast channels. This way, beating cilia could be identified while simultaneously imaging moving fluorescent beads. At least 3–6 slices were used from each mouse and 5 beads were tracked in each slice. Videos were opened with the MetaMorph software program (Molecular Devices, LLC, Sunnyvale, CA), and the tracks of single beads were traced for at least 15 frames (1 sec). To reduce experimental variability, only beads moving along the main stream of CSF at the focus of beating cilia that could be followed for at least 15 frames of recording were used. Velocity was calculated and compared between sham and LFPI groups at each time point.
Statistical analysis
Because of the nested nature of the data and modest sample size, a linear mixed-effect model was used to determine statistical significance when comparing different treatment groups, with α=0.05. Each dot in the group data plot in Figure 6 is a mean from multiple beads from a single brain slice. IBM SPSS (version 21; IBM, Armonk, NY) software was used to run the linear mixed-effect analysis.
FIG. 6.

(A) Bead velocity at different time points after lateral fluid perscussion injury (LFPI) and their time-matched sham controls. Fluorescent beads were added to live brain slices containing the lateral ventricles. Time-lapse movies were taken with a high-speed imaging protocol under Olympus Fluoview 1000 (Olympus America, Center Valley, PA). MetaMorph software (Molecular Devices, LLC, Sunnyvale, CA) was used to track single beads and were tracked for at least 1 sec. Each plotted point is an average value of multiple beads from a single brain slice. Each symbol (triangle, circle, and square) signifies slices derived from the same animal. Colored lines indicate the means for each animal; the black line indicates the group mean. (B) Trace (green) of moving beads tracked with the MetaMorph program (Molecular Devices). Each bead was traced for 15 frames (1 sec). The longer trace corresponds to a greater velocity of the moving beads. Orange indicates the end point of the moving bead. The red number “1” indicates single tracking. Color image is available online at www.liebertpub.com/neu
Results
Concussive brain injury leads to rapid and protracted loss of ventricular ependymal cilia without overt ependymal cell loss
The ventricular system of the brain is lined with ciliated ependymal cells (Fig. 1). One week after nonpenetrating concussive brain injury, the ipsilateral lateral ventricular surface was nearly devoid of cilia (Fig. 2). The loss of cilia was evident within 30 min of injury and persisted for at least 1 week (Fig. 3). Brain slices prepared from animals 30 days after LFPI demonstrated that the ipsilateral lateral ventricle was repopulated with cilia, resulting in a ventricle that morphologically appeared no different from ventricles in brain slices prepared from age-matched sham animals (Fig. 3D,D′). Further, no cilia loss was evident in the contralateral lateral ventricle; therefore, all subsequent experiments were conducted solely on the ipsilateral lateral ventricle. We then checked whether LFPI could induce ependymal cell loss. Hoechst staining showed no overt loss of ependymal cell nuclei in the ipsilateral lateral ventricle (Supplementary Fig. 1B) (see online supplementary material at http://www.liebertpub.com) of injured mice, compared to its contralateral side (Supplementary Fig. 1A) (see online supplementary material at http://www.liebertpub.com) or the ipsilateral lateral ventricle of sham mice (Supplementary Fig. 1C) (see online supplementary material at http://www.liebertpub.com). Because no gross cell loss was evident, no further analysis was undertaken because there would need to have been large-scale cell death to account for the considerable loss of cilia.
FIG. 1.

Brain cilia lining healthy ventricle. (A) Photomicrograph montage showing immunofluorescent stained cilia (arrows) with type IV β-tubuline. Brain slice from a naïve mouse at the rostrocaudal level precisely at bregma (anterior/posterior, 0 mm), relevant to Fig. 31 of Paxinos and Franklin.59 3V, third ventricle; ac, anterior commissure; cc, corpus callosum; cg, cingulum; CPu, caudate putamen; ic, internal capsule; LV, lateral ventricle; st, stria terminalis. (B) Scanning electron microscope (SEM) image showing cilia on the lateral wall of the lateral ventricle from a sham mouse. (C) Higher magnification of the ventricular cilia under SEM. Scale bar is 500 μm in (A), 100 μm in (B), and 10 μm in (C). Color image is available online at www.liebertpub.com/neu
FIG. 2.
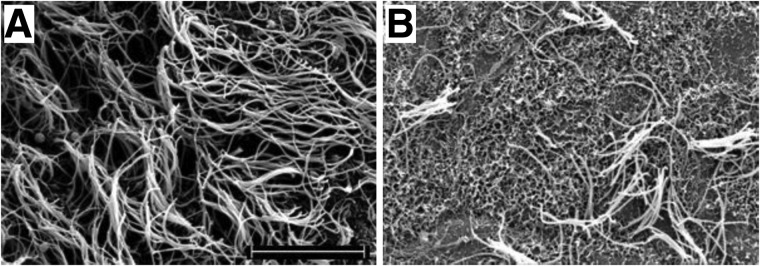
Scanning electron microscope images of lateral ventricular cilia. Samples from slices derived from mice 7 days after sham (A) or LFPI (B) procedure. Scale bar is 10 μm.
FIG. 3.

Lateral fluid perscussion injury results in overt decrease in density of lateral ventricular cilia ipsilateral to injury. Immunofluorescent staining against type IV β-tubulin in slices from time-matched sham (top row) and injured (bottom row) mice. Time points from left to right (A–D) are 30 min, 18 h, and 7 and 30 days, respectively. LV, lateral ventricle. Scale bar is 5 μm. Color image is available online at www.liebertpub.com/neu
Anatomy, physiology, and ultrastructure of surviving cilia
Basal bodies anchor the axonemal scaffold proteins and are interconnected by a series of cytoskeletal fibers both within and between ependymal cells subserving a potential mechanism to allow coordinated ciliary beating.30 Immunostaining for β-tubulin demonstrates a paucity of organized basal bodies in the ependymal layer of animals 7 days postinjury, compared to the ordered array observed in tissue from age-matched sham animals (Fig. 4A,B). Interestingly, basal body ultrastructure examined in transverse as well as longitudinal sections demonstrated no injury-induced alterations (Fig. 4C–F). The paucity of well-demarcated basal bodies in the TBI specimen is consistent with the significant level of ciliary loss noted by SEM and immunofluorescence (Figs. 2 and 3). Additionally, TEM demonstrated no overt differences in post-TBI surviving cilia because the 9+2 microtubule arrangement was intact and undisturbed (Fig. 5).
FIG. 4.

Photomicrographs showing basal bodies of lateral ventricular cilia in brain slices from sham (right column) and time-matched injured mice (left column). (A and B) Immunofluorescent staining. Each cluster of red dots represents all basal bodies of one individual ependymal cell. (C–F) Transmission electron micrscope images showing ultrastructure of basal bodies identified with immunogold-silver staining. Only a small fraction of one ependymal cell with basal bodies is shown. Note the asymmetrical gold staining (i.e., the staining occurs only on one side of each basal body). Each cluster of gold particles represents a single basal body at the electron microscope level. (C and D) Sagittal sections of cilia and basal bodies. (E and F) Transverse sections of cilia and basal bodies. Scale bars are 5μm in (A & B) and 100 nm in (C and F). Color image is available online at www.liebertpub.com/neu
FIG. 5.

Transmission electron micrscope images showing ultrastructure of ventricular cilia 7 days after sham procedure (A), compared to age-matched injured cilia (B). The 9+2 microtubule arrangement is shown at higher magnification of cilia in transverse sections (see insets). m, mitochondria; mv, microvilli. In order to demonstrate the ultrastructure of the cilia, it was necessary to locate comparable intact ependymal cells. Because of the high magnification, only a part of an individual ependymal cell in an injured and sham brain slice is shown. Scale bar is 500 nM.
Lateral fluid percussion injury does not lead to hydrocephalous
TBI has been reported to occasionally lead to hydrocephalous.3,4 Further, it has been recently demonstrated using transgenic (Tg) technology that inhibition of ventricular ciliary beating leads to hydrocephalus in rodents.5,6,9,31,32 Additionally, deficits in ventricular ciliary beating also significantly enhance the potential development of hydrocephalus and ventriculomegaly in humans.9,33 Therefore, to determine whether LFPI caused hydrocephalus, animals were subjected to sequential MRIs initiated before injury and followed 4 weeks after injury. The volume of the ipsilateral lateral ventricle was calculated using ImageJ software (National Institutes of Health). Interestingly, despite the loss of ependymal cilia, volume in the ipsilateral lateral ventricle in injured animals was not significantly different at any time point from the preinjury volume measured in those same animals (data not shown). A Friedman's test comparing the preinjury volume to the volumes at 1, 2, and 4 weeks post-LFPI demonstrated no significant differences (Friedman's test statistic=3.000; p=0.445). The average volumes (±standard deviation) in microliters were 3.58±1.07 (preinjury), 3.97±1.59 (1 week post-LFPI), 4.58±1.38 (2 weeks post-LFPI), and 4.096±0.56 (4 weeks post-LFPI; n=5 animals). These data suggest that substantial, but transient, loss of cilia in one ventricle postdevelopment is probably insufficient to cause hydrocephalus (Supplementary Fig. 2) (see online supplementary material at http://www.liebertpub.com).
Cerebrospinal fluid surface velocity is diminished after brain injury
Whereas LFPI does not appear to cause hydrocephalus, surface CSF flow is also important for the continued delivery of nutrients and removal of catabolic waste from the parynchema.34 Further, it also serves as a functional conduit for endocrine transport.35 To determine whether surface CSF velocity is altered after TBI, fluorescent latex beads were applied to the ipsilateral lateral ventricle of slices from sham and injured animals and tracked for the duration that they remained in the original focal plane of the beating cilia. Bead velocity measured in slices from sham animals was significantly faster, compared to bead velocity obtained in sliced from injured animals, 18 h and 7 days after LFPI (Fig. 6). Because the number of slices for each animal varied, it was necessary to use a linear mixed-model analysis. Significant differences were found in the bead velocities in slices generated from of sham versus brain injured mice at 18 h postinjury (F(1,2.131)=30.701; p=0.027) and 7 days postinjury (F(1,3.545)=26.762; p=0.009). Not surprisingly, bead velocity in slices from animals 30 days postinjury returned to values not significantly different from slices from time-matched sham animals (F(1, 28.000)=1.243; p=0.274), consistent with observations of the repopulation of cilia in the ipsilateral lateral ventricle 30 days after LFPI (Fig. 3).
Discussion
To the best of our knowledge, this is the first analysis of the effects of TBI on ventricular cilia. We have demonstrated that LFPI, a model of closed-head injury, leads to a rapid, protracted, but reversible, shearing of motile cilia lining the ipsilateral lateral ventricle with reduction of CSF flow velocity over the ependymal surface of that specific ventricle.
Within 30 min of LFPI, large swathes of cilia were sheared from the ependymal cells lining the ipsilateral lateral ventricle. We hypothesize that the unilateral delivery of a single brief pressure wave transient to the exposed dura creates a fluid pressure wave in the CSF, decimating the most exposed and thus vulnerable cilia lining the ventricle. It has previously been shown that the mechanical pressure wave preferentially affects larger, loosely packed cells, compared to smaller, more tightly packed cells.36 This observation stems from seismology; large and unsupported objects are more prone to breakage, compared to smaller objects that simply rise and fall during the traveling pressure waves.37 A similar phenomenon may be occurring in the present situation; longer cilia are more susceptible to shearing because they are subject to more of the physical stress during the fluid wave. We have demonstrated that fewer cilia result in slower CSF velocity. We cannot rule out that surviving cilia may be immature and therefore shorter and more resilient to physical sheer stress. No alterations in the ultrastructure of surviving cilia were observed because the 9+2 microtubule arrangement of motile cilia was conserved and appeared undisturbed; thus, gross ultrastructural permeation does not explain the resilience of the surviving cilia. Whereas transmission EM demonstrated that, at the ultrastructural level, the basal bodies are intact 7 days postinjury, immunofluorescence for γ-tubulin demonstrated an overall lack of organization within each cell. However, in this animal model of nonpenetrating head trauma, the loss of cilia was completely restored by 30 days after injury.
Cilia dysfunction induced by Tg technology,5,6,9 antisense RNA injection,10 or pharmacologically38 all cause hydrocephalus or, to a lesser extent, ventricular dilatation. Because of the existing correlation between cilia dysfunction and alterations in ventricular size, we hypothesized that the large-scale loss of cilia after the fluid pulse may also cause an increase in ipsilateral lateral ventricle size. However, serial MRIs taken before and followed for 2 weeks after brain injury demonstrated no alteration in ventricular size or volume. We believe that the LFPI used in this study did not result in a significant change in ventricular volume because the deciliation was restricted to the ipsilateral lateral ventricle, was incomplete, and fully reversible. Though reported as possible sequela of TBI, hydrocephalus is not a consistent finding in TBI patients.3,4 Further, a lack of ventricular enlargement is not surprising, because the precipitating mechanism(s) underlying the development of hydrocephalus are unimportant. Rather, it is a series of ependymal cell alterations that lead to hydrocephalus. That is, initially, cilia are lost, followed by a disappearance of the microvilli,39 possibly resulting from extensive stretching of the ependymal layer.40 These changes culminate in the death of the ependymal cells. We hypothesize that it is ependymal cell death that leads to hydrocephalus. Because cilia loss in our model was not accompanied by ependymal cell loss (Supplementary Fig. 1) (see online supplementary material at http://www.liebertpub.com), cellular viability may underlie and mediate the robust ciliogenesis restoring ipsilateral lateral ventricle function and preventing hydrocephalus.
Although the exact function of the ependymal cilia is not known, it is thought that cilia contribute to CSF circulation,6,41 thereby allowing for robust exchange between nutrient delivery and waste removal.34 The most profound consequence of injury-induced shearing of ipsilateral lateral ventricular cilia that we demonstrate is the significant and protracted decrease in CSF flow reflected in the velocity of exogenously applied fluorescent beads. The velocity of these beads was drastically reduced at 18 h after injury with restoration of CSF velocity toward contralateral or sham injured values, but still significantly reduced at 7 days postinjury. Because ciliogenesis repopulates the damaged ventricular surface, the bead velocity, and, by proxy, the CSF flow rate, returns to normal as well at 30 days after injury. Though the clinical ramifications of this finding are not directly known, several sequelae can be extrapolated. First and foremost is compromised diffusion and waste removal, thus diminishing the clearance of harmful metabolites (e.g., amyloid).42 The inability to robustly clear normal amyloid depositions may underlie the proposed linkage between brain injury and the development of Alzheimer's disease.43–46 Second, because CSF supplies micronutrients and peptides to neuronal networks, diminished CSF flow may contribute and enhance pathologies, as has been suggested with various bipolar disorders and schizophrenia.47–49 This may be especially true of blast injury and repetitive injury that leads to chronic traumatic encephalopathy,50 a tau-protein–linked neurodegenerative disease.51 Further, CSF is a conduit for inflammatory cytokines and chemokines to be rapidly delivered to the entire CNS. Thus, decreased CSF velocity may blunt an inflammatory response to CNS microbial infection, as is often encountered in TBI. Additionally, respiratory cilia have recently been demonstrated to detect microbes and assist in the clearance.52 Thus, if ependymal cilia subserve the same function, deciliation would contribute to CNS infection post-TBI.
Several endocrine glands are situated along various sites of the ventricular system and collectively comprise the circumventricular organ system. This system uses CSF as a transport conduit for various hormones and peptides. Therefore, diminished CSF flow would negatively affect the function of this system.53
Recently, motile cilia have received substantial attention as cellular sensors. Respiratory cilia have been demonstrated to exhibit both mechanosensitivity54 and chemosensitivity to bitter tastants,55 quorum-sensing molecules,52 and alterations in pH.56 Brain ependymal cilia have recently been demonstrated to detect alterations in viscosity57 and thus may modulate CSF composition. Thus, cilia denudation may have more-profound effects on CNS homeostasis than merely depressed CSF flow and amalgamation.
It is important to stress that the model used for these studies reflects mild TBI, and that more periventricular damage may be observed in a severe TBI model. Even so, we demonstrate a profound, yet reversible, loss of ependymal cilia with a resultant decrease in CSF velocity in the ipsilateral lateral ventricle. Though additional studies are necessary to further determine the contribution of ependymal cilia to CSF homeostasis and overall CNS function, recent reports have demonstrated that motile cilia in the respiratory, reproductive, and ventricular system can exhibit sensory function for detection of alterations in fluid composition as well as presence of microbial factors.52,55,57,58 Thus, loss of cilia function after TBI may have serious implications in short- and long-term CNS function.
Supplementary Material
Acknowledgments
The authors thank Michael Schell for critiquing an earlier draft of the manuscript and Kathleen Joy Propert for statistical guidance. This study was supported in part by National Institutes of Health grants (R01HD059288 and R01NS069629; to A.S.C.).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Faul M., Xu L., Wald M.M., and Coronado V.G. (2010). Traumatic brain injury in the United States: emergency department visits, hospitalizations and deaths 2002– 2006. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control: Atlanta, GA [Google Scholar]
- 2.Rutland-Brown W., Langlois J.A., Thomas K.E., and Xi Y.L. (2006). Incidence of traumatic brain injury in the United States, 2003. J. Head Trauma Rehabil. 21, 544–548 [DOI] [PubMed] [Google Scholar]
- 3.Beyerl B., and Black P.M. (1984). Posttraumatic hydrocephalus. Neurosurgery 15, 257–261 [DOI] [PubMed] [Google Scholar]
- 4.Guyot L.L., and Michael D.B. (2000). Post-traumatic hydrocephalus. Neurol. Res. 22, 25–28 [DOI] [PubMed] [Google Scholar]
- 5.Lechtreck K.F., Delmotte P., Robinson M.L., Sanderson M.J., and Witman G.B. (2008). Mutations in Hydin impair ciliary motility in mice. J. Cell Biol. 180, 633–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banizs B., Pike M.M., Millican C.L., Ferguson W.B., Komlosi P., Sheetz J., Bell P.D., Schwiebert E.M., and Yoder B.K. (2005). Dysfunctional cilia lead to altered ependyma and choroid plexus function, and result in the formation of hydrocephalus. Development 132, 5329–5339 [DOI] [PubMed] [Google Scholar]
- 7.Kosaki K., Ikeda K., Miyakoshi K., Ueno M., Kosaki R., Takahashi D., Tanaka M., Torikata C., Yoshimura Y., and Takahashi T. (2004). Absent inner dynein arms in a fetus with familial hydrocephalus-situs abnormality. Am. J. Med. Genet. A 129A, 308–311 [DOI] [PubMed] [Google Scholar]
- 8.Takaki E., Fujimoto M., Nakahari T., Yonemura S., Miyata Y., Hayashida N., Yamamoto K., Vallee R.B., Mikuriya T., Sugahara K., Yamashita H., Inouye S., and Nakai A. (2007). Heat shock transcription factor 1 is required for maintenance of ciliary beating in mice. J. Biol. Chem. 282, 37285–37292 [DOI] [PubMed] [Google Scholar]
- 9.Ibañez-Tallon I., Pagenstecher A., Fliegauf M., Olbrich H., Kispert A., Ketelsen U.P., North A., Heintz N., and Omran H. (2004). Dysfunction of axonemal dynein heavy chain Mdnah5 inhibits ependymal flow and reveals a novel mechanism for hydrocephalus formation. Hum. Mol. Genet. 13, 2133–2141 [DOI] [PubMed] [Google Scholar]
- 10.Mönkkönen K.S., Hakumäki J.M., Hirst R.A., Miettinen R.A., O'Callaghan C., Männistö P.T., and Laitinen J.T. (2007). Intracerebroventricular antisense knockdown of G alpha i2 results in ciliary stasis and ventricular dilatation in the rat. BMC Neurosci. 8, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bush A., Chodhari R., Collins N., Copeland F., Hall P., Harcourt J., Hariri M., Hogg C., Lucas J., Mitchison H.M., O'Callaghan C., and Phillips G. (2007). Primary ciliary dyskinesia: current state of the art. Arch. Dis. Child 92, 1136–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Badano J.L., Mitsuma N., Beales P.L., and Katsanis N. (2006). The ciliopathies: an emerging class of human genetic disorders. Annu. Rev. Genomics Hum. Genet. 7, 125–148 [DOI] [PubMed] [Google Scholar]
- 13.al-Shroof M., Karnik A.M., Karnik A.A., Longshore J., Sliman N.A., and Khan F.A. (2001). Ciliary dyskinesia associated with hydrocephalus and mental retardation in a Jordanian family. Mayo Clin. Proc. 76, 1219–1224 [DOI] [PubMed] [Google Scholar]
- 14.Reichler I.M., Hoerauf A., Guscetti F., Gardelle O., Stoffel M.H., Jentsch B., Walt H., and Arnold S. (2001). Primary ciliary dyskinesia with situs inversus totalis, hydrocephalus internus and cardiac malformations in a dog. J. Small Anim. Pract. 42, 345–348 [DOI] [PubMed] [Google Scholar]
- 15.Roth Y., Kimhi Y., Edery H., Aharonson E., and Priel Z. (1985). Ciliary motility in brain ventricular system and trachea of hamsters. Brain Res. 330, 291–297 [DOI] [PubMed] [Google Scholar]
- 16.Noback C., Strominger N.L., Demarest R.J., and Ruggiero D.A. (2005). The Human Nervous System: Structure and Function. 6th ed. Humana Press: Totowa, NJ [Google Scholar]
- 17.Opp M.R., and Krueger J.M. (1991). Interleukin 1-receptor antagonist blocks interleukin 1-induced sleep and fever. Am. J. Physiol. 260, R453–R457 [DOI] [PubMed] [Google Scholar]
- 18.Opp M.R., Obal F., Jr., and Krueger J.M. (1991). Interleukin 1 alters rat sleep: temporal and dose-related effects. Am. J. Physiol. 260, R52–R58 [DOI] [PubMed] [Google Scholar]
- 19.Krueger J.M., Fang J., Taishi P., Chen Z., Kushikata T., and Gardi J. (1998). Sleep. A physiologic role for IL-1 beta and TNF-alpha. Ann. N. Y. Acad. Sci. 856, 148–159 [DOI] [PubMed] [Google Scholar]
- 20.Silver R., LeSauter J., Tresco P.A., and Lehman M.N. (1996). A diffusible coupling signal from the transplanted suprachiasmatic nucleus controlling circadian locomotor rhythms. Nature 382, 810–813 [DOI] [PubMed] [Google Scholar]
- 21.Skinner D.C., and Malpaux B. (1999). High melatonin concentrations in third ventricular cerebrospinal fluid are not due to Galen vein blood recirculating through the choroid plexus. Endocrinology 140, 4399–4405 [DOI] [PubMed] [Google Scholar]
- 22.Lehman M., and Silver R. (2000). CSF signaling in physiology and behavior. Prog. Brain Res. 125, 415–433 [DOI] [PubMed] [Google Scholar]
- 23.Witgen B.M., Lifshitz J., Smith M.L., Schwarzbach E., Liang S.L., Grady M.S., and Cohen A,S. (2005). Regional hippocampal alteration associated with cognitive deficit following experimental brain injury: a systems, network and cellular evaluation. Neuroscience 133, 1–15 [DOI] [PubMed] [Google Scholar]
- 24.Schwarzbach E., Bonislawski D.P., Xiong G., and Cohen A.S. (2006). Mechanisms underlying the inability to induce area CA1 LTP in the mouse after traumatic brain injury. Hippocampus 16, 541–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonislawski D.P., Schwarzbach E.P., and Cohen A.S. (2007). Brain injury impairs dentate gyrus inhibitory efficacy. Neurobiol. Dis. 25, 163–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cole J,T, Mitala C.M., Kundu S., Verma A., Elkind J.A., Nissim I., and Cohen A,S. (2010). Dietary branched chain amino acids ameliorate injury-induced cognitive impairment. Proc. Natl. Acad. Sci. U. S. A. 107, 366–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Antunes M.B., Gudis D.A., and Cohen N.A. (2009). Epithelium, cilia, and mucus: their importance in chronic rhinosinusitis. Immunol. Allergy Clin. North Am. 29, 631–643 [DOI] [PubMed] [Google Scholar]
- 28.Gudis D.A., and Cohen N.A. (2010). Cilia dysfunction. Otolaryngol. Clin. North Am. 43, 461–472, vii [DOI] [PubMed] [Google Scholar]
- 29.Antunes M.B., Woodworth B.A., Bhargave G., Xiong G., Aguilar J.L., Ratner A.J., Kreindler J.L., Rubenstein R.C., and Cohen N.A. (2007). Murine nasal septa for respiratory epithelial air-liquid interface cultures. Biotechniques 43, 195–196, 198, 200 passim. [DOI] [PubMed] [Google Scholar]
- 30.Gonzalez Santander R., Martinez Cuadrado G., Rubio Saez M., Bujan Varela J., and Larana Sole A. (1984). Ultrastructural basis of the coordination of ciliary movement. Contractile function of the interconnecting cross-striated roots in ependymal epithelium. Acta Anat. (Basel) 118, 82–90 [PubMed] [Google Scholar]
- 31.Sapiro R., Kostetskii I., Olds-Clarke P., Gerton G.L., Radice G.L., and Strauss J.F., III (2002). Male infertility, impaired sperm motility, and hydrocephalus in mice deficient in sperm-associated antigen 6. Mol. Cell Biol. 22, 6298–6305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Z., Tang W., Zhou R., Shen X., Wei Z., Patel A.M., Povlishock J.T., Bennett J., and Strauss J.F., III., (2007). Accelerated mortality from hydrocephalus and pneumonia in mice with a combined deficiency of SPAG6 and SPAG16L reveals a functional interrelationship between the two central apparatus proteins. Cell Motil. Cytoskeleton 64, 360–376 [DOI] [PubMed] [Google Scholar]
- 33.Afzelius B.A. (2004). Cilia-related diseases. J. Pathol. 204, 470–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stadlbauer A., Salomonowitz E., van der Riet W., Buchfelder M., and Ganslandt O. (2010). Insight into the patterns of cerebrospinal fluid flow in the human ventricular system using MR velocity mapping. Neuroimage 51, 42–52 [DOI] [PubMed] [Google Scholar]
- 35.Feng C.Y., Wiggins L.M., and von Bartheld C.S. (2011). The locus ceruleus responds to signaling molecules obtained from the CSF by transfer through tanycytes. J Neurosci 31, 9147–9158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toth Z., Hollrigel G.S., Gorcs T., and Soltesz I. (2007). Instantaneous perturbation of dentate interneuronal networks by a pressure wave-transient delivered to the neocortex. J. Neurosci. 17, 8106–8117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bolt B. (1993). Earthquakes. 3rd ed. W.H. Freeman: New York [Google Scholar]
- 38.Nakamura Y., and Sato K. (1993). Role of disturbance of ependymal ciliary movement in development of hydrocephalus in rats. Childs Nerv. Syst. 9, 65–71 [DOI] [PubMed] [Google Scholar]
- 39.Bannister C.M., and Chapman S.A. (1980). Ventricular ependyma of normal and hydrocephalic subjects: a scanning electronmicroscopic study. Dev. Med. Child Neurol. 22, 725–735 [DOI] [PubMed] [Google Scholar]
- 40.Kiefer M., Eymann R., von Tiling S., Müller A., Steudel W.I., and Booz K.H. (1998). The ependyma in chronic hydrocephalus. Childs Nerv. Syst. 14, 263–270 [DOI] [PubMed] [Google Scholar]
- 41.Tissir F., Qu Y., Montcouquiol M., Zhou L., Komatsu K., Shi D., Fujimori T., Labeau J., Tyteca D., Courtoy P., Poumay Y., Uemura T., and Goffinet A.M. (2010). Lack of cadherins Celsr2 and Celsr3 impairs ependymal ciliogenesis, leading to fatal hydrocephalus. Nat. Neurosci. 13, 700–707 [DOI] [PubMed] [Google Scholar]
- 42.Rubenstein E. (1998). Relationship of senescence of cerebrospinal fluid circulatory system to dementias of the aged. Lancet 351, 283–285 [DOI] [PubMed] [Google Scholar]
- 43.Jellinger K.A., Paulus W., Wrocklage C., and Litvan I. (2001). Traumatic brain injury as a risk factor for Alzheimer disease. Comparison of two retrospective autopsy cohorts with evaluation of ApoE genotype. BMC Neurol. 1, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith D.H., Uryu K., Saatman K.E., Trojanowski J.Q., and McIntosh T.K. (2003). Protein accumulation in traumatic brain injury. Neuromolecular Med 4, 59–72 [DOI] [PubMed] [Google Scholar]
- 45.van den Heuvel C., Thornton E., and Vink R. (2007). Traumatic brain injury and Alzheimer's disease: a review. Prog. Brain Res. 161, 303–316 [DOI] [PubMed] [Google Scholar]
- 46.Johnson V.E., Stewart W., and Smith D.H. (2010). Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer's disease? Nat. Rev. Neurosci. 11, 361–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rothermundt M., Falkai P., Ponath G., Abel S., Bürkle H., Diedrich M., Hetzel G., Peters M., Siegmund A., Pedersen A., Maier W., Schramm J., Suslow T., Ohrmann P., and Arolt V. (2004). Glial cell dysfunction in schizophrenia indicated by increased S100B in the CSF. Mol. Psychiatry 9, 897–899 [DOI] [PubMed] [Google Scholar]
- 48.Linderholm K.R., Skogh E., Olsson S.K., Dahl M.L., Holtze M., Engberg G., Samuelsson M., and Erhardt S. (2012). Increased levels of kynurenine and kynurenic acid in the CSF of patients with schizophrenia. Schizophr. Bull. 38, 426–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Regenold W.T., Phatak P., Marano C.M., Sassan A., Conley R.R., and Kling M.A. (2009). Elevated cerebrospinal fluid lactate concentrations in patients with bipolar disorder and schizophrenia: implications for the mitochondrial dysfunction hypothesis. Biol. Psychiatry 65, 489–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith D.H., Johnson V.E., and Stewart W. (2013). Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat. Rev. Neurol. 9, 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goldstein L.E., Fisher A.M., Tagge C.A., Zhang X.L., Velisek L., Sullivan J.A., Upreti C., Kracht J.M., Ericsson M., Wojnarowicz M.W., Goletiani C.J., Maglakelidze G.M., Casey N., Moncaster J.A., Minaeva O., Moir R.D., Nowinski C.J., Stern R.A., Cantu R.C., Geiling J., Blusztajn J.K., Wolozin B.L., Ikezu T., Stein T.D., Budson A.E., Kowall N.W., Chargin D., Sharon A., Saman S., Hall G.F., Moss W.C., Cleveland R.O., Tanzi R.E., Stanton P.K., and McKee A.C. (2012). Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med 4, 134ra160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee R.J., Xiong G., Kofonow J.M., Chen B., Lysenko A., Jiang P., Abraham V., Doghramji L., Adappa N.D., Palmer J.N., Kennedy D.W., Beauchamp G.K., Doulias P.T., Ischiropoulos H., Kreindler J.L., Reed D.R., and Cohen N.A. (2012). T2R38 taste receptor polymorphisms underlie susceptibility to upper respiratory infection. J. Clin. Invest. 122, 4145–4159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tanriverdi F., De Bellis A., Ulutabanca H., Bizzarro A., Sinisi A.A., Bellastella G., Paglionico V.A., Mora L.D., Selcuklu A., Unluhizarci K., Casanueva F.F., and Kelestimur F. (2013). Five years prospective investigation of anterior pituitary function after traumatic brain injury: is hypopituitarism long-term after head trauma associated with autoimmunity? J. Neurotrauma 30, 1426–1433 [DOI] [PubMed] [Google Scholar]
- 54.Winters S.L., Davis C.W., and Boucher R.C. (2007). Mechanosensitivity of mouse tracheal ciliary beat frequency: roles for Ca2+, purinergic signaling, tonicity, and viscosity. Am. J. Physiol. Lung Cell Mol. Physiol. 292, L614–L624 [DOI] [PubMed] [Google Scholar]
- 55.Shah A.S., Ben-Shahar Y., Moninger T.O., Kline J.N., and Welsh M.J. (2009). Motile cilia of human airway epithelia are chemosensory. Science 325, 1131–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sutto Z., Conner G.E., and Salathe M. (2004). Regulation of human airway ciliary beat frequency by intracellular pH. J. Physiol. 560, 519–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rios M., Hermoso M., Sanchez T.M., Croxatto H.B., and Villalon M.J. (2007). Effect of oestradiol and progesterone on the instant and directional velocity of microsphere movements in the rat oviduct: gap junctions mediate the kinetic effect of oestradiol. Reprod. Fertil. Dev.19, 634–640 [DOI] [PubMed] [Google Scholar]
- 58.Andrade Y.N., Fernandes J., Vázquez E., Fernández-Fernández J.M., Arniges M., Sánchez T.M., Villalón M., and Valverde M.A. (2005). TRPV4 channel is involved in the coupling of fluid viscosity changes to epithelial ciliary activity. J. Cell Biol. 168, 869–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Paxinos G., and Franklin K.B.J. (2001). The Mouse Brain in Stereotaxic Coordinates. 2nd ed. Academic Press: Hong Kong [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.