

Background: Bacteria evade host defense peptides by degradation.
Results: Actin protects the LL-37 antimicrobial peptide and enables its bactericidal action in the presence of bacterial proteases.
Conclusion: Sites of infection and necrosis are likely to contain high amounts of actin that will protect LL-37 from degradation.
Significance: Components released from lysed cells can be used to enhance local immunity.
Keywords: Actin, Antimicrobial Peptide (AMP), Host Defense, Protease, Transglutaminase
Abstract
Host defense peptides play an important host-protective role by their microcidal action, immunomodulatory functions, and tissue repair activities. Proteolysis is a common strategy of pathogens used to neutralize host defense peptides. Here, we show that actin, the most abundant structural protein in eukaryotes, binds the LL-37 host defense peptide, protects it from degradation by the proteases of Pseudomonas aeruginosa and Porphyromonas gingivalis, and enables its antimicrobial activity despite the presence of the proteases. Co-localization of LL-37 with extracellular actin was observed in necrotized regions of samples from oral lesions. Competition assays, cross-linking experiments, limited proteolysis, and mass spectrometry revealed that LL-37 binds by specific hydrophobic interactions to the His-40–Lys-50 segment of actin, located in the DNase I binding loop. The integrity of the binding site of both LL-37 and actin is a prerequisite to the binding. Our results demonstrate that actin, presumably released by dead cells and abundant in infected sites, might be utilized by the immune system to enhance spatio-temporal immunity in an attempt to arrest infection and control inflammation.
Introduction
Antimicrobial peptides are components of the innate immunity that mediate a broad range of antimicrobial activities and that are being explored as an alternative for classic antibiotic agents (1–4). Although highly diverse, antimicrobial peptides share the features of a net positive charge and the ability to adopt an amphipathic structure in solution. The bacterial targets of peptides include the outer and inner membranes and cytoplasmic components (5–7). Antimicrobial peptides also function as immunomodulators that alter gene expression and host chemokine and cytokine production to elicit or inhibit a proinflammatory response (4, 8, 9) and to promote wound healing mechanisms (10, 11). Due to the recognition of their immunoregulatory functions, antimicrobial peptides are often referred to as “host defense peptides” and “alarmins.”
LL-37 is the only human member of the cathelicidin antimicrobial peptide family (12, 13). It is processed from the human cathelicidin antimicrobial peptide 18-kDa (hCAP18) precursor expressed by neutrophils, subpopulations of lymphocytes and monocytes, and by epithelia (14). LL-37 has been shown to exert a broad spectrum of antimicrobial activities (1, 7, 15) and to express multiple immunoregulatory and wound healing activities (16–18). The murine cathelicidin-related antimicrobial peptide is the mouse homologue of LL-37, and the only cathelicidin in mice. Mice lacking the murine cathelicidin-related antimicrobial peptide were shown to be susceptible to necrotic skin infection caused by Group A Streptococcus (19) and to infection by Escherichia coli (20). Deficiency in salivary LL-37 in patients with morbus Kostmann (21) or with Papillon-Lefevre syndrome (22) has been correlated with severe periodontal disease.
Susceptibility to proteases is an obvious limitation of antimicrobial peptides (23–25). Many bacterial pathogens, including oral ones, express and secrete a wide variation of virulence-associated proteases. Although some oral antimicrobial peptides (cystatins) show some resistance to bacterial cleavage (and low protease inhibitory activity) (26), we (1, 15, 31, 32) and others (27–30) have shown the capacity of pathogens such as Pseudomonas aeruginosa and the oral Porphyromonas gingivalis to degrade the peptides directed against them.
P. gingivalis is a highly proteolytic oral anaerobe that is associated with periodontal disease (33, 34). Others (27) and we (31) showed that P. gingivalis can inactivate LL-37 by cleavage. Surprisingly, P. gingivalis was unable to cleave LL-37 in the presence of saliva (32). Saliva does not inhibit the P. gingivalis Arg-gingipain cysteine protease that we identified as the protease that cleaves LL-37 (32), but rather it protects LL-37 from degradation in a dose-dependent manner (32). This protection was specific, because saliva did not prevent P. gingivalis degradation of a control protein (32). Importantly, the salivary protection of LL-37 enabled the antimicrobial activity of LL-37 in the presence of proteases (32). In this study, we sought to identify salivary components that enable the antimicrobial activity of LL-37 despite the presence of pathogenic proteases.
Actin is the most abundant protein in the eukaryotic cell. It is a dynamic structural protein with multiple functions, among which are cytoskeleton formation, cell division, motility, adhesion, and signaling (35). Actin exists as monomer (globular, G-actin) or polymer (actin filament, F-actin). These actin species transform into one another by different factors. G-actin is polymerized into F-actin by an increase in monovalent or divalent cation concentration and by specific positively charged proteins and peptides. F-actin filaments may form bundles via actin-bundling proteins (36), which cross-link two filaments by attaching their two discrete actin-binding sites to two actin monomers located on separate filaments. Actin filaments are also bundled by polycations, including polycationic proteins or peptides and polyamines. These bundling factors act by nonspecific electrostatic interactions, which eliminate repulsion between negatively charged actin filaments (37). LL-37, which is a polycationic polypeptide, also acts as an actin-bundling factor (38, 39). The LL-37-induced actin bundles contribute to the accumulation of sputum (40), which is a primary cause of bacterial infections and death in patients with cystic fibrosis (41). We showed previously that in addition to electrostatic interactions, site-specific hydrophobic interactions between actin and LL-37 also have a significant role in bundling, especially at physiological ionic strength, where the bundling effect of polycations decreases. This is due to the relatively high concentration of monovalent cations that mask the electrostatic interactions between the polycations and actin (41).
Here, we found that actin protects LL-37 against microbial proteases and that this protection enables the antimicrobial activity of this peptide in the presence of the proteases. We further studied the structural aspects of LL-37-actin interactions using cross-linking, limited proteolysis, competition with actin-binding proteins, and mass spectrometry. We found that LL-37 is tightly bound to the His-40–Lys-50 segment (subdomain 2) of actin by specific hydrophobic interactions and that the integrity of the amino acid sequence of this actin segment and of LL-37 is a prerequisite to the tight binding.
EXPERIMENTAL PROCEDURES
Materials
Deoxyribonuclease I (DNase I), ATP, ADP, dithiothreitol (DTT), and subtilisin were purchased from Sigma. The peptides used in this study are presented in Table 1; LL-37 (LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES), scrambled LL-37 (sLL-37) (GLKLRFEFSKIKGEFLKTPEVRFRDIKLKDNRISVQR), Q22A-LL-37 (Q22A), alanine substitution of glutamine in position 22 of LL-37 (LLGDFFRKSKEKIGKEFKRIVARIKDFLRNLVPRTES), I20S/I24S-LL-37 (I20S/I24S), serine substitution of isoleucines in positions 20 and 24 (LLGDFFRKSKEKIGKEFKRSVQRSKDFLRNLVPRTE, and tetramethylrhodamine-labeled LL-37 (r-LL-37) were purchased from Genemed Synthesis, Inc. (San Antonio, TX). The peptides were purified by HPLC (greater than 90% purity) and were determined by mass spectrometry. Yeast cofilin and bacterial transglutaminase (TGase)3 were generous gifts of Prof. Emil Reisler, UCLA, and Prof. György Hegyi, Eötvös Loránd University, Budapest, Hungary, respectively. rhCAP18, the His-tagged recombinant cathelicidin domain of hCAP18 (lacking LL-37 at its C terminus), a kind gift of Prof. R. L. Gallo University of California at San Diego (42), was expressed in E. coli BL21(DE3) and purified under denaturing conditions using Ni-NTA columns as described before (32).
TABLE 1.
Peptides used in this study
| Peptide | MICa | k dissociation with actin | Sequence |
|---|---|---|---|
| μm | |||
| LL-37 | 1.1 | 1.86 × 10−7 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES |
| Scrambled LL-37 | ∼40 | 1.38 × 10−4 | LLGDVFGRSFEKIGVEFRKIVQDIRDFRLNLKPRTES |
| Q22A | 1.1 | Not measured | LLGDFFRKSKEKIGKEFKRIVARIKDFLRNLVPRTES |
| I20S/I24S | 11 | Not measured | LLGDFFRKSKEKIGKEFKRSVQRSKDFLRNLVPRTES |
a Data were measured with B. subtilis. MIC means minimal inhibitory concentration.
Saliva
Saliva collection was described before (32). Briefly, whole saliva was collected from five healthy donors and clarified by centrifugation at 1,500 × g for 10 min followed by filtration (0.2 μm; Whatman Schleicher & Schuell). Saliva samples were pooled and kept in aliquots at −20 °C.
Granulation Tissue
Granulation tissues located peri-apical to a lower right first molar were collected from lesions diagnosed with peri-apical x-rays of otherwise healthy donors after obtaining informed consent and in accordance with a protocol that was approved by the institutional ethical committee. The tooth was extracted using a combination of an elevator followed by dental forceps. The lesion, which was located in the socket, was removed using a dental curette. The specimens were sent to the laboratory in a formaldehyde solution. Macroscopic findings revealed soft granulation tissue.
Bacterial Strains and Growth Conditions
Bacillus subtilis PY79 (a kind gift of Prof. S. Ben-Yehuda, Hebrew University, Jerusalem, Israel) and P. aeruginosa PAO1 (from our laboratory stock) were grown in LB Broth (Difco) at 37 °C under aerobic conditions. P. gingivalis ATCC 33277 (from our laboratory stock) was grown in Wilkins Chalgren medium II, (Oxoid, UK) in anaerobic jars (Oxoid) at 37 °C. For supernatant collection, overnight cultures of P. aeruginosa and 4-day cultures of P. gingivalis were centrifuged at 20,000 × g for 10 min, and the supernatant was collected and transferred through a 0.2-μm filter (BD Biosciences). Bacterial purity was determined by phase contrast microscopy.
Pulldown of Salivary Factors That Bind rhCAP18
Ni-NTA-agarose beads (110 μl, Qiagen, Germany) were washed three times in PBS (2 min, 3,000 rpm at 4 °C) and incubated with 300 μg of rhCAP18 in 500 μl of PBS (or with PBS without rhCAP18 for background control) for 15 min at 4 °C. The beads were washed three times as described above and incubated with 1 ml of clarified saliva for 1 h at 4 °C. The beads were washed as follows: once with 5 mm imidazole, 0.3 m NaCl in PBS; once with PBS containing 5 mm imidazole, 0.5 m NaCl; once with PBS containing 5 mm imidazole, 1 m NaCl; once with 30 mm imidazole in PBS; and once with 50 mm imidazole in PBS followed by elution with PBS containing 500 mm imidazole.
Immunofluorescence
The granulation tissues were fixed overnight at 4 °C in 4% paraformaldehyde in PBS, cryopreserved overnight at 4 °C in 30% sucrose, embedded in OCT (Tissue-Tek), and cryo-sectioned. Sections (15 μm thick) were post fixed in ice-cold methanol (4 h, 4 °C), and processed for immunofluorescence as follows. Sections were washed three times in 1× PBS and blocked in blocking buffer (5% fetal bovine serum, 0.1% Triton X-100 in PBS) for 1 h at room temperature. Blocking was followed by incubation with 1:100 rabbit anti LL-37 (32) and 1:100 goat anti-actin (Santa Cruz Biotechnology) overnight at 4 °C. The next day, the sections were washed three times in PBS, incubated with fluorescently conjugated secondary antibody (Cy-2 anti-rabbit and Cy-3 anti-goat, Jackson ImmunoResearch) in blocking buffer for 1 h at room temperature, washed three times in PBS, dipped in Hoechst (1:10,000) to stain nuclei, and mounted with Fluoromount G (Southern Biotech). Images were taken using Nikon C2 confocal laser scanning microscope.
Preparation of Actin
CaATP-G-actin was prepared from dried acetone powder derived from the back and leg muscles of rabbits by the method of Spudich and Watt (43) that without gel filtration yields highly homogeneous actin in a purity greater than 90%. CaATP-G-actin was stored in a buffer containing 5 mm Tris-HCl, 0.2 mm CaCl2, 0.2 mm ATP, 0.5 mm β-mercaptoethanol, pH 8.0 (CaATP-G-buffer). MgATP-G-actin was obtained by incubating CaATP-G-actin with 0.2 mm EGTA and 0.1 mm MgCl2 at room temperature for 5 min. MgATP-G-actin was diluted for further treatments in MgATP-G-buffer containing 5 mm MOPS, 0.1 mm MgCl2, 0.2 mm EGTA, 0.2 mm ATP, and 0.5 mm DTT, pH 7.4. MgF-actin was polymerized from MgATP-G-actin or CaATP-G-actin by a 30-min incubation with 2 mm MgCl2 at room temperature. The concentration of unlabeled rabbit skeletal muscle α-G-actin was determined spectrophotometrically using the extinction coefficients E2901% = 11.5 cm−1. (The optical density of actin was measured in the presence of 0.5 m NaOH, which shifts the maximum of absorbance from 280 to 290 nm). Molecular masses of skeletal actin, yeast cofilin, DNase I, and LL-37 were assumed to be 42, 15.9, 31.3, and 4.5 kDa, respectively.
SDS-PAGE
Samples were separated on 12% SDS gels and visualized by Coomassie Blue unless stated otherwise. Representative gel pictures are presented.
Bundling of F-actin Filaments
Following the addition of native or modified LL-37 to MgF-actin, samples were centrifuged at 20,800 × g at 4 °C for 8 min (in an Eppendorf centrifuge). The supernatants were separated by 12% SDS-PAGE and analyzed by densitometry using the TINA 2.07d software. Presented data are mean ± S.D. of three independent experiments.
Western Immunodetection
Reaction mixtures were loaded on a 17% SDS-PAGE, and LL-37 was detected by Western immunodetection using rabbit antisera generated against the synthetic LL-37 peptide as described before (32).
Protection Assay
LL-37 or Q22A (1.1 μm in PBS) were incubated with or without 3 μm F-actin or BSA as control for 10 min at 37 °C. 5 μl of the protease-containing supernatant of P. aeruginosa diluted 1:100 or of P. gingivalis diluted 1:10 were added (or not) to the 50-μl reaction mixture and incubated for 30 min at 37 °C. The reaction mixtures were added to the wells of 96-well plates (Nunc, Denmark) containing 150 μl of B. subtilis PY79 cells (at mid-logarithmic growth) and diluted 1:5,000 in LB (to complete a 200 μl final volume). For measuring the protection of the bacterial growth inhibition activity of LL-37, the plates were transferred to a GENIOS Microplate Reader (TECAN, Austria), and absorbance's at A595 nm were measured during incubation at 37 °C every 17 min (after automated mixing/aeration for 500 s) to generate growth curves. Percent growth inhibition of each treatment was compared with growth at late logarithmic growth phase of untreated bacteria (0% growth inhibition). To measure the protection of the bactericidal activity of LL-37, mixtures of LL-37, actin, proteases, and bacteria were prepared in microplates as described above and incubated with shaking for 60 min at 37 °C, and then serial dilutions were plated on LB agar overnight at 30 °C. Colonies were counted and display as CFU/ml.
Effect of Actin on the Arg-Gingipain Activity of P. gingivalis
The effect of actin on the trypsin-like activity of the P. gingivalis Arg-gingipains was determined using Nα-benzoyl-l-arginine 4-nitroanilide hydrochloride (BAPNA) (Sigma), an arginine protease chromogenic substrate. BAPNA solution was prepared by dissolving 1 mg of BAPNA in 200 μl of dimethyl sulfoxide and adding 1.8 ml of BAPNA buffer (0.2 m Tris-HCl, pH 8.0, 0.1 m NaCl, 0.05 m CaCl2, and 0.05 m l-cysteine). Aliquots of 100 μl of BAPNA solution were added to wells of 96-well plates containing 20 μl of P. gingivalis culture supernatant (or BAPNA buffer when indicated) and 80 μl of 3 μm F-actin, 3 μm BSA, protease inhibitor mixture (all in PBS) or PBS. Plates were incubated at 37 °C for 30 min, and absorbance was measured at 405 nm using a GENIOS Microplate Reader (Tecan, Austria).
Effect of Actin on LL-37 Binding to B. subtilis
Tetramethylrhodamine-labeled LL-37 (1.1 μm) was incubated with or without F-actin (3 μm) in a total volume of 180 μl for 10 min at 37 °C. 20 μl of B. subtilis grown overnight (∼1–2 × 107) were added, and the mixture was incubated for 10 min at room temperature. Labeling of B. subtilis with the tetramethylrhodamine LL-37 was measured by flow cytometry using the Eclipse analyzer (Icyte) and analyzed using FlowJo 7.6.5 software (Treestar).
In-gel Proteolysis and Mass Spectrometry Analysis
Protein bands excised from SDS gels were reduced with 2.8 mm DTT (60 °C for 30 min), modified with 8.8 mm iodoacetamide in 100 mm ammonium bicarbonate (in the dark, at room temperature for 30 min), and digested in 10% acetonitrile and 10 mm ammonium bicarbonate with modified trypsin or Glu C (Promega) overnight at 37 °C. The resulting tryptic peptides were resolved by reverse phase chromatography on 0.075 × 200-mm fused silica capillaries (J & W Scientific) packed with reprosil reversed phase material (Dr. Maisch GmbH, Germany). The peptides were eluted with linear 65-min gradients of 5–45% and 15 min at 95% acetonitrile with 0.1% formic acid in water at flow rates of 0.25 μl/min. Mass spectrometry was performed by an ion-trap mass spectrometer (OrbitrapXL, Thermo) in a positive mode using repetitively a full MS scan followed by collision-induced dissociation of the seven most dominant ions selected from the first MS scan. The mass spectrometry data were analyzed using the Protein Discoverer 1.3 (Thermo) using the Sequest search engine, searching against the human section of the Uniprot database. The data were filtered according to accuracy and 1 or 5% false discovery rate.
TGase Cross-linking
Unless stated otherwise actin, LL-37 and TGase were mixed together simultaneously and incubated at room temperature for 90 min. The reaction was stopped by the addition of protein sample buffer and incubation in boiling water for 5 min. Samples were subjected to 12% SDS-PAGE, visualized by Coomassie Blue, Western immunodetection, or rhodamine fluorescence and evaluated by densitometry. For measurement of B. subtilis growth inhibition by LL-37 cross-linked products, the cross-linked samples were incubated with B. subtilis for kinetic measurements as mentioned above in the protection assay.
Statistics
Unless specified, all presented data are mean ± S.D. of three independent experiments performed in triplicate. All presented SDS gels and Western immunodetection blots are representative of three independent experiments. Student's t test and one-way analysis of variance were used for calculation of p value.
RESULTS
Actin Protects LL-37 from Degradation by the Proteases of P. gingivalis and P. aeruginosa and Enables Its Anti-microbial Activity Despite the Presence of the Proteases
As described before, saliva was found to protect LL-37 and rhCAP18, the recombinant cathelicidin domain of the LL-37 precursor hCAP18, from degradation by P. gingivalis (32). Attempts to pull down salivary factors that bind LL-37 using anti-LL-37 antibodies were unsuccessful, possibly due to steric hindrance by the available antibodies. We therefore used the His-tagged rhCAP18 as bait to pull down salivary components that bind rhCAP18 and that possibly protects LL-37 from degradation by bacterial proteases. Mass spectrometry analysis of the SDS-PAGE band of the salivary proteins that bound rhCAP18 (Fig. 1A) identified actin with high confidence (Fig. 1B). Furthermore, LL-37 was found to co-localize with extracellular actin in necrotized regions of samples of granular tissues taken from human oral lesions (Fig. 1C). Actin was previously shown to interact with LL-37 (39, 40) suggesting that it might mask the sites cleaved by the microbial proteases. Indeed, actin was found to prevent proteolysis of LL-37 by the proteases of P. gingivalis and P. aeruginosa (Fig. 2, A and B, respectively). Addition of actin slightly reduced the antimicrobial activity of LL-37 (Fig. 2D) but not in a statistically significant manner. Moreover, actin enabled the microbial growth inhibition and bactericidal activities of LL-37 in the presence of each of these proteases (Fig. 2, C and D and E and F, respectively). Similar actin protection of LL-37 was found against subtilisin, the protease of B. subtilis (data not shown). As can be seen in Fig. 3A, actin did not inhibit the R-gingipain protease that cleaves LL-37 (32). In addition, BSA used as control did not protect LL-37 from degradation (Fig. 3B) demonstrating that the prevention of LL-37 digestion is specific and mediated by LL-37's interaction with actin, rather than by protease inhibition or competition of actin with LL-37 as substrate for the protease.
FIGURE 1.

Actin binds rhCAP18 and LL-37. Coomassie Blue-stained SDS-PAGE (A) and mass spectrometry analysis (B) of salivary factors (open arrow) that were pulled down (see “Experimental Procedures”) with Ni-NTA-agarose beads coated (+) or not (−) with rhCAP18 (closed arrow) used as bait. C, co-localization of LL-37 (red) with extracellular actin (green). Confocal microscopy imaging of immunofluorescence (see under “Experimental Procedures”) was performed on a sample of granular tissue collected from a human oral lesion. The merged channel contains nuclei staining with Hoechst (blue). Right panel shows the inset indicated in the merged image. Arrows point to co-localization of actin and LL-37.
FIGURE 2.

F-actin protects and enables the antibacterial activity of LL-37 in the presence of bacterial proteases. A and B, Western immunodetection and densitometry analysis (mean ± S.D. of two independent experiments) of 3 μm LL-37 incubated with 5 μl of culture supernatant (SN) of P. gingivalis (P.g) (A) or P. aeruginosa (P.a) (B), in the absence or presence of 9 μm F-actin. C and D, growth inhibition of B. subtilis by 1.1 μm LL-37 treated or not with P. gingivalis (P.g) (C) or P. aeruginosa (P.a) (D) supernatant (SN), in the presence or absence of 3 μm F-actin. E and F, killing of B. subtilis (displayed as colony-forming units, CFU) by 1.1 μm LL-37 treated or not with P. gingivalis (P.g) (E) or P. aeruginosa (P.a) (F) supernatant (SN) in the presence or absence of 3 μm F-actin. * represents p < 0.05 in Student's t test, ** represents p < 0.01 in one-way analysis of variance.
FIGURE 3.
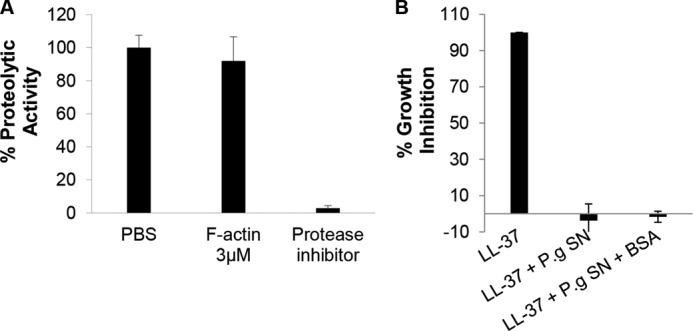
Prevention of LL-37 proteolysis by microbial proteases is mediated by protection rather than protease inhibition. A, actin does not inhibit the proteolytic activity of R-gingipain of P. gingivalis (P.g). Protease activity (quantified using BAPNA as an R-gingipain substrate, see “Experimental Procedures”) of P. gingivalis conditioned medium co-incubated with or without 3 μm F-actin or with 1 μl of protease inhibitor mixture used as control. B, unlike actin, BSA (see Fig. 2C) does not protect the antimicrobial activity of LL-37 from the proteases of P. gingivalis. Presented data are mean ± S.D. of two independent experiments performed in triplicate. SN, supernatant.
Binding of tetramethylrhodamine-labeled LL-37 to B. subtilis cells was maintained in the presence of actin (Fig. 4), demonstrating that the actin-LL-37 interactions do not hamper the ability of LL-37 to bind and act on the target microbial cell wall and supporting our results shown in Fig. 2 that actin maintains the antimicrobial activity of LL-37 while protecting it from restriction by the microbial protease.
FIGURE 4.

Binding of LL-37 to the bacterial membrane is maintained in the presence of F-actin. FACS histogram (representative experiment) (A) and analysis of three independent experiments (B) of B. subtilis PY79 labeling when incubated with 1.1 μm tetramethylrhodamine-labeled LL-37, in the presence (blue dashed in A) or absence (red in A) of 3 μm F-actin, are shown. Gray histogram in A represents the background of B. subtilis without LL-37.
LL-37 Is Cross-linked to Monomeric (G) and Polymeric (F) Actin by TGase, and the Reaction Is Not Sensitive to Ionic Strength
TGase catalyzes isopeptide bond formation in the reaction, R-CONH2 + R′-NH2 → R-CONH-R′ + NH3, in which R-CONH2, the acceptor, is a glutamine residue, and R′-NH2, the donor, is an alkylamine. In protein-protein interactions, the alkylamine is a lysyl residue, and the TGase reaction produces both intermolecular and intramolecular cross-links (44, 45). Transglutaminase treatment of actin has been used to study the structure and function of both monomeric (45) and polymeric (46) actin.
As can be seen in Fig. 5A by both Coomassie Blue staining and Western immunodetection, LL-37 is cross-linked to G-actin by TGase. LL-37 is also cross-linked to F-actin (Figs. 5B and 6A). The cross-linking reaction indicates a close interaction of the cationic peptide with actin. The main product of the reaction with both G- and F-actin is an actin-LL-37 cross-linked heterodimer, although actin-dimer-LL-37 and actin oligomers-LL-37 also appear as minor products with G-actin (Fig. 5, A and B) but not with F-actin (Fig. 5B). Similar amounts of the actin-LL-37 cross-linked product are formed when using CaATP- and MgATP-G-actin. However, less than half of the actin-LL-37 cross-linked product is formed with F-actin (Fig. 5B).
FIGURE 5.

Cross-linking of LL-37 to actin by TGase. A, SDS-PAGE of cross-linking of 10 μm LL-37 to 8 μm CaATP-G-actin by 0.2 mg/ml TGase. Lane a, actin only; lane b, actin cross-linked to LL-37 (see “Experimental Procedures”) visualized by Coomassie Blue (left) and by Western immunodetection (right, lane b*). B, effect of NaCl concentration on 0.2 mg/ml TGase-induced cross-link formation between 4 μm CaATP-G or Mg-F-actin and 9 μm LL-37 (mean ± S.D. of three independent experiments). C, actin-LL-37 cross-link formation is preferred over LL-37 dimerization. 66 μm LL-37 were incubated with (lanes a and c) and without (lane b) 0.4 mg/ml TGase in the absence (lane b) and in the presence (lane c) of 60 μm CaATP-G-actin at room temperature for 2 h (lanes a and b), Coomassie blue stain; lane a*, lane b*, and lane c*, Western immunodetection. D, antimicrobial activity of LL-37 is lost upon formation of LL-37-LL-37 or LL-37-actin covalent cross-links. 1.1 μm LL-37 was incubated with or without 1.1 μm F-actin and with 0.4 mg/ml TGase at room temperature for 2 h. Following incubation samples were added to B. subtilis cells in a 96-well plate, and antimicrobial activity was measured as described under “Experimental Procedures.”
FIGURE 6.

Pretreatment of LL-37 (but not of actin) with TGase inhibits actin-LL-37 cross-linking. A, 8 μm LL-37 was reacted with 0.2 mg/ml TGase at room temperature for 2 h and then cross-linked with 4 μm CaATP-G-actin, MgATP-G-actin, or Mg-F-actin for an additional 2 h. Lane a, actin alone; lanes b, e, and h, actin and TGase; lanes c, f, and j, LL-37 and TGase added simultaneously to actin and incubated for 2 h; lanes d, g, and i, LL-37 was preincubated for 2 h at room temperature with TGase before adding it to actin. B, effect of LL-37 pretreatment with TGase on the LL-37-actin cross-link formation. 82 μm LL-37 were treated with 1.8 mg/ml TGase at room temperature for 2 h and then 9 μm treated or untreated LL-37 and 0.2 mg/ml TGase was added to 4 μm actin and incubated for an additional 2 h (mean ± S.D. of three independent experiments). C, effect of actin pretreatment with TGase on the LL-37-actin cross-link formation. 8 μm actin was incubated with 0.2 mg/ml TGase on ice for 24 h and then 10 μm LL-37 was added, and the incubation was continued for an additional 24 h. Lanes a–d, CaATP-G-actin; lanes e and f, MgF-actin. Lane a, untreated actin; lane b, TGase-pretreated actin, no LL-37; lanes c and e, TGase pretreated actin with LL-37; lanes d and f, untreated actin with LL-37 and TGase added simultaneously; M, molecular weight marker.
The amount of G-actin-LL-37 cross-linked product was not influenced by the increase in NaCl concentration during the cross-linking process. In F-actin, the quantity of the cross-linked product was also the same at low ionic strength and in 100 mm NaCl and was only slightly decreased in the presence of 200 mm NaCl (Fig. 5B). The relative insensitivity of the cross-linking to the increase in ionic strength, which masks electrostatic interactions between the positively charged LL-37 and the negatively charged actin, indicates that hydrophobic interactions also mediate binding of LL-37 to actin.
Cross-linking of LL-37 to Actin Competes with LL-37 to LL-37 Cross-linking and Both Cross-linking Reactions Inhibit the Antimicrobial Activity of the Peptide
TGase treatment of LL-37 in the absence of actin leads to the formation of covalently cross-linked LL-37 dimers (Fig. 5C, lane a). The production of LL-37 cross-linked dimers is inhibited upon the simultaneous addition of actin, LL-37, and TGase (Fig. 5C, lane c*). This indicates that actin-LL-37 cross-link formation is preferred over LL-37 dimerization. Thus, the TGase-catalyzed reaction between actin and LL-37 successfully competes with the reaction of dimerization between two LL-37 molecules. We also found that both LL-37 to LL-37 and LL-37 to actin cross-link formations strongly inhibit the antimicrobial activity of the peptide (Fig. 5D) possibly because the strong covalent binding of LL-37 to actin or between two LL-37 molecules prevents (by steric hindrance) the flexibility needed for the proper interaction of the peptide with the bacterial membrane.
Pretreatment of LL-37 (but Not of Actin) with TGase Inhibits Actin-LL-37 Cross-linking
Pretreatment of LL-37 with TGase, which leads to covalently cross-linked dimer formation, was found to strongly diminish the amount of the LL-37-actin cross-link formed compared with the amount generated when actin, LL-37, and TGase are added simultaneously (Fig. 6A). The amount of the actin-LL-37 cross-linked product generated with TGase-pretreated LL-37 is only about 20% that formed in the reaction with nontreated LL-37 in the case of CaATP- and MgATP-G-actin and 50% with MgF-actin (Fig. 6B). This extensive inhibition may indicate that the single glutamine of LL-37 (Gln-22) has a major role in both the LL-37-LL-37 and LL-37-actin cross-linking reaction. In the LL-37 dimer, either the Gln-22 of one of the LL-37 molecules is linked to a lysine of the partner LL-37 or the glutamines of both LL-37 peptides react with lysines in their counterpart. Gln-22 may not react with actin in the case of TGase-pretreated LL-37 possibly because either two glutamine-lysine cross-links connect the two LL-37s in the dimer (leaving no available glutamine on LL-37) or because there is only one cross-link, but the remaining single free glutamine of the dimer is not available for the reaction. Another possibility is that the glutamine of LL-37 is involved in an intramolecular cross-link in the peptide itself, and therefore, it cannot be cross-linked with actin. This latter possibility is suggested by the finding that the extent of inhibition of the actin-LL-37 cross-linking with pretreated LL-37 (Fig. 6B) is greater than the extent of LL-37 dimerization (see Fig. 5C, lane a, Coomassie Blue); therefore, LL-37 dimerization cannot be the sole reason of the inhibition. The effect of TGase pretreatment of actin on the actin-LL-37 cross-link formation was also studied; however, in this case no inhibition was found (Fig. 6C, lane c compared with d and lane e compared with f). This result indicates that Gln-41, which is considered to be the most reactive glutamine of actin in various TGase-catalyzed reactions (45–47), has no significant part in the LL-37-actin cross-link formation. This is because TGase catalyzes intramolecular cross-link formation (Fig. 9B, lanes b and c) between Gln-41 and Lys-50 (45); therefore, Gln-41 in TGase pretreated actin is not available for the cross-linking.
FIGURE 9.

Analysis of actin's LL-37-binding site by subtilisin digestion. A, cross-linking 6 μm r-LL-37 to 4 μm CaATP-G-actin protects actin's D-loop from subtilisin digestion. TGase reaction was stopped by 1 mm cysteamine; 30 min digestion with 1 μg/ml subtilisin was stopped by 1 mm PMSF; incubations were performed at room temperature. Samples were separated on SDS-PAGE and visualized by Coomassie Blue staining (lanes a–d) or by tetramethylrhodamine fluorescence (lanes c* and d*). Lane a, actin alone; lane b, actin and TGase; lanes c and c*, actin, r-LL-37, and TGase (added simultaneously); lanes d and d*, actin, r-LL-37, and TGase added simultaneously and followed by subtilisin digestion. Illustration on the left shows cleavage of CaATP-G-actin between Met-47 and Gly-48 by subtilisin (small black arrow) and covalent binding between Gln-41 and Lys-50 by TGase (two- headed arrow). B, subtilisin cleavage of the D-loop prevents LL-37-actin cross-linking. Other than using 9 μm LL-37, the same conditions as in A were applied. Lane a, actin; lane b, subtilisin-digested actin; lane c, subtilisin-digested actin treated by TGase; lane d, subtilisin-digested actin treated by TGase and then incubated with LL-37 for an additional 90 min; lane e, actin incubated with LL-37 and TGase (added simultaneously) for 90 min. C, LL-37 bundling of 4 μm MgF-actin digested or undigested by subtilisin in the presence or absence of 100 mm NaCl. 50 μm CaATP-G-actin were digested by subtilisin (at a 168:1 w/w/ratio) for 30 min, stopped by 1 mm PMSF and polymerized to MgF-actin by 2 mm MgCl2 (subtilisin digested F-actin). To 4 μm subtilisin, digested F-actin LL-37 was added in increasing concentrations, and bundling was carried out as described under “Experimental Procedures.”
Gln-22 to Ala Substitution, Replacement of Two Hydrophobic Residues and Scrambling of the LL-37 Sequence Weaken the Actin-LL-37 Interaction
Modified LL-37 peptides (Table 1), Q22A (Q-22 is substituted by Ala), I20S/I24S (hydrophobic Ile-20 and Ile-24 are substituted with Ser) and s-LL-37 (scrambled LL-37) were also used to study LL-37-actin interactions (Fig. 7). As with LL-37, Q22A generated actin-peptide and actin dimer-peptide cross-linked bands in SDS-PAGE. However, the extent of cross-linking obtained in the TGase reaction between Q22A and G- or F-actin is much less than the cross-linked product of the unmodified LL-37 and actin (Fig. 7A). Cross-linking of F-actin with Q22A was also more sensitive to increased ionic strength than that with of LL-37 (100 mm compared with 200 mm NaCl, Fig. 7A), which indicates that the binding of the modified peptide to actin is weaker than that of the unmodified one. Q22A was cross-linked also to TGase pretreated actin, similar to the unmodified LL-37 (Fig. 6C). Pretreatment of actin did not influence the extent of cross-linking with Q22A (data not shown).
FIGURE 7.

Replacing LL-37's Gln-22 with alanine and Ile-20 and Ile-24 with serine, and scrambling reduces binding of LL-37 to actin. A, cross-linking of 4 μm actin with 9 μm Q22A or LL-37 at low ionic strength and in the presence of 100 and 200 mm NaCl. Upper panel, SDS-PAGE. Lower panel, densitometry analysis. Lanes a–d, CaATP-G-actin; lanes e–j, MgF-actin. Lane a, G-actin alone; lane b, G-actin and TGase, no LL-37; lane c, G-actin, LL-37, and TGase; lane d, G-actin, Q22A, and TGase; lane e, F-actin, LL-37, and TGase; lane f, F-actin, Q22A, and TGase; lane g, F-actin, LL-37, TGase, and 100 mm NaCl; lane h, F-actin, Q22A, TGase, and 100 mm NaCl; lane i, F-actin, LL-37, TGase, and 200 mm NaCl; lane j, F-actin, Q22A, TGase, and 200 mm NaCl. B, bundling of 4 μm MgF-actin by 2–12 μm LL-37 and Q22A at low ionic strength and in the presence of 100 mm NaCl. C, bundling of 4 μm MgF-actin by 3–20 μm I20S/I24S and LL-37 at low ionic strength and in the presence of 100 mm NaCl. For bundling procedure see “Experimental Procedures.” D, F-actin protects the antimicrobial activity of Q22A from proteolysis. Antimicrobial activity of LL-37 and of Q22A and their protection assay was performed as described under “Experimental Procedures.” SN, supernatant. E, cross-linking of 4 μm CaATP-G-actin and MgF-actin with 9 μm LL-37 or s-LL37 in the presence of increasing concentrations of NaCl. s-LL-37 or LL-37 was incubated simultaneously with 4 μm actin and 0.2 mg/ml TGase. Upper panel, SDS-PAGE, Lane a, actin only; lane b, actin, LL-37, TGase; lane c, actin, LL-37, TGase, 100 mm NaCl; lane d, actin, LL-37, TGase, 200 mm NaCl; actin, s-LL-37, TGase; lane e, actin, s-LL-37, TGase; lane f, actin, s-LL-37, TGase, 100 mm NaCl; lane g, actin, s-LL-37, TGase, 200 mm NaCl. Lower panel, quantitative assessment of LL-37-actin cross-link obtained from densitometry of SDS-PAGE.
The ability of Q22A to bundle F-actin filaments was significantly reduced compared with its unmodified counterpart (Fig. 7B). This is more conspicuous in 100 mm NaCl than at low ionic strength. In 100 mm NaCl, the electrostatic interactions between the positively charged peptide and the negatively charged actin are partially masked, and the short range hydrophobic interactions have a greater role in the bundling process. Thus, the finding that the actin bundling ability of Q22A is reduced more in 100 mm NaCl than at low ionic strength compared with the bundling ability of the unmodified LL-37 indicates that Gln-22 of LL-37 has an important role in the hydrophobic interaction between LL-37 and actin.
To further study the possible hydrophobic interactions between actin and LL-37, we replaced two hydrophobic residues in LL-37, Ile-20, and Ile-24 with serine residues. The F-actin bundling effect of the modified peptide, I20S/I24S, was compared with that of LL-37 at low ionic strength and in the presence of 100 mm NaCl (Fig. 7C). The bundling ability of I20S/I24S decreased relative to that of LL-37 already at low ionic strength, where ∼60% bundling obtained with 5 μm LL-37 but only with 9 μm I20S/I24S. This difference is much more conspicuous in the presence of 100 mm NaCl, where no significant bundling was found even with 20 μm I20S/I24S. These results strongly support our findings that the hydrophobic interactions between actin and LL-37 are significant.
Although I20S/I24S demonstrated reduced antimicrobial activity compared with LL-37 (a 10-fold increase in the minimal inhibitory concentration, see Table 1), the antimicrobial activity of Q22A is similar to that of the unmodified LL-37 (Table 1). This demonstrates that Gln-22 is not important for the antimicrobial activity of LL-37. Next, we tested how the Gln-22 to Ala substitution affects the ability of actin to protect the peptide against bacterial proteases (Fig. 7D). Although binding of Q22A to actin is weaker than that of LL-37, actin enabled the antimicrobial activity of the modified peptide in the presence of P. aeruginosa supernatant proteases (Fig. 7D). This protection demonstrates that relatively weaker interactions (Fig. 7B) between actin and Q22A are sufficient for the protection of Q22A from proteolytic degradation.
Scrambling the LL-37 sequence (s-LL-37) significantly reduced cross-linking to both G- and F-actin compared with the unmodified peptide (Fig. 7E). As we have shown recently (39), the bundling of actin filaments by s-LL-37, especially in the presence of 100 mm or higher concentrations of NaCl, is significantly lower compared with unmodified LL-37. This again points to the importance of the specific LL-37 sequence in the formation of strong hydrophobic bonds at the actin LL-37-actin interface.
DNase I and Cofilin Inhibit the Cross-linking of LL-37 to Actin
Competition assays with DNase I and cofilin were used to locate the LL-37-binding site in actin. DNase I depolymerizes F-actin and forms a tight complex with the actin monomer by binding mainly to subdomain 2 of actin, which includes the DNase I binding loop (D-loop) (46). Cofilin has an extended binding site on actin, which includes the D-loop that has a secondary role in cofilin binding (48). Recently, we found that both these proteins dissociate LL-37-induced F-actin bundles and that the dissociating effect of DNase I is much stronger than that of cofilin (39). On the basis of these results, we assumed that the DNase-binding site of actin has an important role in LL-37 binding. To study this possibility, we cross-linked LL-37 or tetramethylrhodamine-labeled LL-37 to CaATP-G-actin in the presence of DNase I or cofilin (Fig. 8A). Both proteins inhibited the cross-linking; however, the inhibitory effect of DNase I was much stronger than that of cofilin. These results further confirm that the DNase I-binding site of actin is involved in binding LL-37. We also studied the competition between LL-37 and cofilin for actin binding by examining the effect of increasing concentrations of LL-37 on cofilin binding to actin (Fig. 8B). This is possible because TGase also cross-links cofilin to actin (48) and the band of actin-cofilin cross-link on SDS-PAGE is higher than that of actin-LL-37 cross-link (Fig. 8B, gel panel). Increasing concentrations of LL-37 decreased the amount of cofilin cross-linked to CaATP-G-actin; however, the density of the cofilin-actin band decreased only by ∼40% at the maximal LL-37 concentration used in this experiment (Fig. 8B, lower panel). These results indicate that cofilin and LL-37 partially share their respective binding site on actin.
FIGURE 8.

DNase I and cofilin inhibit cross-linking of LL-37 to actin. A, representative gel (upper panel) and quantitative assessment (lower panel) of CaATP-G-actin (4 μm) cross-linked (“Experimental Procedures”) with 9 μm r-LL-37 or LL-37 by 0.2 mg/ml TGase in the presence and absence of 6 μm DNase I or 12 μm cofilin. All the constituents were incubated simultaneously at room temperature for 90 min. Lane a, actin alone; lane b, actin, TGase; lane c, actin, LL-37, TGase; lane d, actin, r-LL-37, TGase; lane e, actin, r-LL-37, cofilin, TGase; lane f, actin, r-LL-37, DNase I, TGase; *, rhodamine fluorescence. Samples were treated as in Fig. 6B. Evaluation is based on densitometry of SDS-PAGE. B, competition between 8 μm cofilin and 0–10 μm LL-37 for cross-linking to 4 μm CaATP-G-actin by TGase. LL-37 and cofilin were added simultaneously to CaATP-G-actin. For cross-linking procedure, see “Experimental Procedures.” Upper panel, SDS-PAGE. Lane a, actin only; lane b, actin, cofilin, TGase; lane c, actin, cofilin, TGase, 2 μm LL-37; lane d, actin, cofilin, TGase, 4 μm LL-37; lane e, actin, cofilin, TGase, 7 μm LL-37; lane f, actin, cofilin, TGase, 10 μm LL-37. Lower panel, quantitative evaluation of cross-linking of cofilin with actin from densitometry of SDS-PAGE.
LL-37 Binds to the His-40–Lys-50 Segment (Subdomain 2) of Actin
Subtilisin (Fig. 9) digestion, thrombin digestion (Fig. 10), and mass spectroscopy (Fig. 11) were used next to pinpoint the LL-37-binding site on actin. Subtilisin at low concentrations is known to cleave G-actin between Met-47 and Gly-48 in the D-loop to form N-and C-terminal fragments (49). Under nondenaturing conditions, the resulting two fragments remain attached to each other by noncovalent forces and can be reconnected by TGase treatment that covalently cross-links them between Gln-41 and Lys-50 (Fig. 9B, lanes b and c) (45). The CaATP-G-actin-tetramethylrhodamine labeled LL-37 (rLL-37) cross-linked product was found to be resistant to subtilisin digestion (Fig. 9A, gel panel). This indicates that LL-37 that is cross-linked to actin protects actin from subtilisin cleavage. However, LL-37 cannot be cross-linked to subtilisin-digested G-actin even after covalently reconnecting the two fragments by the TGase reaction (Fig. 9B, lanes c and d). These results indicate that the integrity of the D-loop is an absolute prerequisite to the cross-linking of LL-37 to actin.
FIGURE 10.
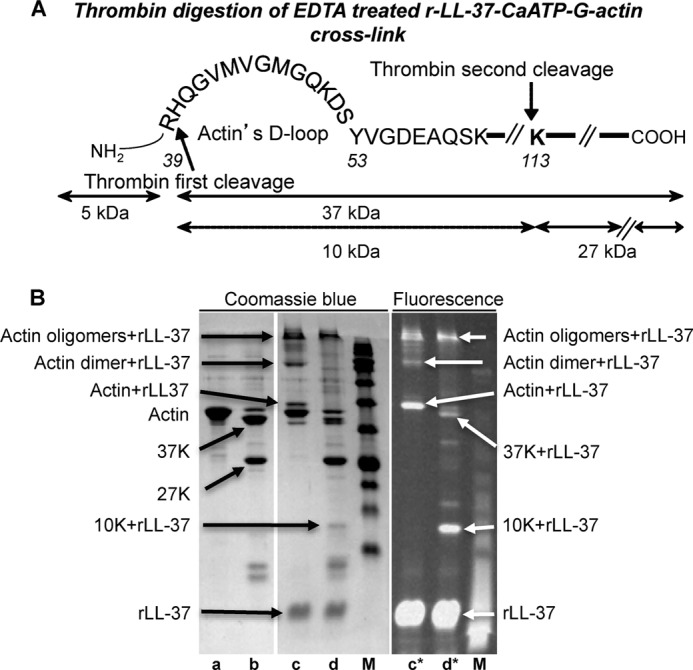
LL-37 binds the actin His-40–Lys-113 10-kDa segment. A, illustration of thrombin digestion of EDTA-treated CaATP-G-actin. B, thrombin digestion of EDTA-treated r-LL-37-CaATP-G-actin cross-link. Lane a, 8 μm CaATP-G-actin was incubated with 1.5 mm EDTA for 21 h; lane b, same as lane a, but 30 μg/ml thrombin was also added together with EDTA, and proteolysis was stopped by 1 mm PMSF; lane c, 8 μm CaATP-G-actin cross-linked with 16 μm r-LL-37 by 0.3 mg of TGase for 2 h; the reaction was stopped by 1 mm cysteamine, and 1.5 mm EDTA was added followed by incubation for 21 h; lane d, same as lane c, but 30 μg/ml thrombin was also added together with EDTA, and proteolysis was stopped as in lane b; M, molecular weight marker. All incubations were performed at room temperature. Samples were separated on SDS-PAGE and visualized by Coomassie Blue stain (left panel) or by fluorescence (right panel, lanes c* and d*).
FIGURE 11.

Identification of the residues involved in cross-linking between actin and LL-37 by mass spectrometry. A, model of the F-actin molecule (green) and the D-loop (gray sticks) using the PyMOL software. Enlarged illustration shows the location of Gln-49 in the D-loop. B and C, relative abundance of the D-loop sequences in actin and in LL-37-actin cross-linked bands. B, His-40–Lys-50 peptide is practically missing from the digest of the actin-LL-37 cross-linked heterodimer, which indicates its involvement in the cross-link formation. C, quantity of VMVGMGQK peptide in the His-40–Lys-50 sequence, containing Gln-49, is greatly reduced in the digest of actin-LL-37 cross-linked heterodimer relative to its quantity in the actin digest. D and E, mass spectrometry analysis and relative abundance of selected peptides in the digest of LL-37-actin or Q22A-actin cross-linked heterodimers. D, mass spectrometry relative abundance analysis show that Gln-22 and five lysines (Lys-8, Lys-10, Lys-12, and to a lesser extent Lys-15 and Lys-18) in LL-37 can participate in the formation of LL-37-actin cross-link. E, similar analysis of the Q22A-actin cross-linked digest indicates the participation of the same lysine residues in the cross-linking reaction. The residues marked in red could participate in the cross-link reaction. A change in font size indicates the significance of the residues in the cross-linking according to the MS calculation of peak area ratio between LL-37 or Q22A and actin-LL-37 or Q22A cross-link.
The ability of LL-37 to bundle F-actin that was polymerized from subtilisin-cleaved and TGase-treated CaATP-G-actin was also studied (Fig. 9C). The ability of the above F-actin species to form bundles upon addition of LL-37 was significantly reduced especially in the 4–9 μm LL-37 range. The reduction in the bundling capability of this actin was more conspicuous in the presence of 100 mm NaCl, which mostly masks the electrostatic interactions, and therefore, the hydrophobic interactions are dominant. These results indicate that the integrity of the D-loop is important not just for the actin-LL-37 cross-link formation but also for the specific hydrophobic interaction between LL-37 and actin.
Thrombin digestion was also used for identification of the LL-37-binding site on actin (Fig. 10, A and B). After the removal of the divalent cation strongly bound to actin by EDTA (50), thrombin cleaves G-actin first at Arg-39 to generate a 5-kDa N-terminal and a 37-kDa C-terminal fragment. Next, the 37-kDa peptide is further cleaved at Lys-113 to a 27-kDa C-terminal and a 10-kDa fragment. This 10-kDa fragment contains the His-40–Lys-113 actin sequence (Fig. 10A) (51). After thrombin digestion of the EDTA-treated tetramethylrhodamine-labeled LL-37 (rLL-37) cross-linked CaATP-G-actin product, we found that rLL-37 is bound to the 10-kDa fragment (Fig. 10B, fluorescent panel, lane d*). This result indicates that the His-40–Lys-113 actin segment, which includes the D-loop, contains the LL-37-binding site of actin.
Mass spectroscopy was used next to localize the sites involved in cross-linking between actin and LL-37. LL-37-G-actin and Q22A-G-actin cross-linked heterodimers, untreated actin, LL-37 and Q22A were digested (see “Experimental Procedures”), and the resulting peptides were analyzed and compared by mass spectroscopy. A peptide, consisting of the His-40–Lys-50 segment of actin, located in subfragment 2 (Fig. 11A) (52) was found at a much higher concentration in the digest of noncross-linked actin than in the digest of LL-37-G-actin cross-linked heterodimer (Fig. 11B). This peptide contains Gln-41 and Gln-49 (Fig. 11A). Our finding that blocking Gln-41 does not affect the actin-LL-37 cross-link formation (Fig. 6C) excludes the participation of Gln-41 in the cross-link, which points to Gln-49 as the Gln donor. Moreover, a peptide consisting of the 43–50-residue actin segment VMVGMGQK containing only Gln-49 as a single Gln was found in the digest of the noncross-linked actin but not in the digest of LL-37-G-actin cross-linked heterodimer (Fig. 11C). This explicitly identifies Gln-49 as a Gln donor on actin in the TGase-catalyzed cross-linking. Similarly, Gln-49 was identified as a Gln donor on actin from the analysis of actin and Q22A-G-actin cross-linked heterodimer digests (data not shown). Comparing the LL-37 and LL-37-G-actin cross-linked heterodimer digests, we found that the 19–23-residue segment of LL-37, RIVQR, containing Gln-22, is found only in the digest of LL-37 but not in that of the heterodimer (Fig. 11D). This finding identifies Gln-22 as the Gln donor on LL-37. This result supports our finding that in the TGase reaction, the Gln donor can be either on actin or on LL-37 (Fig. 6, A and B). We attempted to also identify the Lys acceptors in the cross-linking reaction. When Gln-22 on LL-37 is the Gln donor, then Lys-50 on actin should be the major acceptor because this is the only Lys in the His-40–Lys-50 segment, which was found in the digest of noncross-linked actin, but practically not in the digest of LL-37-G-actin cross-linked heterodimer (Fig. 11B). Identifying the Lys acceptors on LL-37 is more complex. This is because there are several segments in the LL-37 digest that are missing or are present only in small quantities in the digest of LL-37-actin or Q22A-actin cross-linked heterodimers. These segments include L1–Lys-8, L1–Glu-11, Glu-11–Lys-18 (LLGDFFRK, LLGDFFRK SKE, EKIGKEFK respectively), containing Lys-8, Lys-8, Lys-10, Lys-12, Lys-15, and Lys-18 residues, respectively (Fig. 11, D and E).
In summary we identified six possible TGase-induced cross-linking sites between actin and LL-37. (a) When the Gln donor is on actin (Gln-49), the Lys acceptors on LL-37 could be as follows: 1) Lys-8; 2) Lys-10 (this is the most probable lysine acceptor in LL-37); 3) Lys-12; 4) Lys-15; or 5) Lys-18. However, Lys-15 and Lys-18 are less significant lysine acceptors because the IGKEFK peptide, containing the two Lys residues, was found almost in the same quantity in the Q22A and the Q22A-actin cross-linked digest (Fig. 11E). (b), when the Gln donor is on LL-37 (Gln-22), the Lys acceptor on actin is Lys-50. All the cross-linking sites identified connect the His-40–Lys-50 segment of actin with the N-terminal part of LL-37 (Fig. 12). The finding that there are three significant Lys acceptors on LL-37 in the N-terminal part of the peptide points to considerable dynamic flexibility in the LL-37 and/or G-actin D-loop structure. The results of mass spectrometry support the findings of proteolysis and DNase I competition and together indicate that the main binding site of LL-37 on actin is the His-40–Lys-50 segment, located in the D-loop on the subdomain 2 of actin.
FIGURE 12.
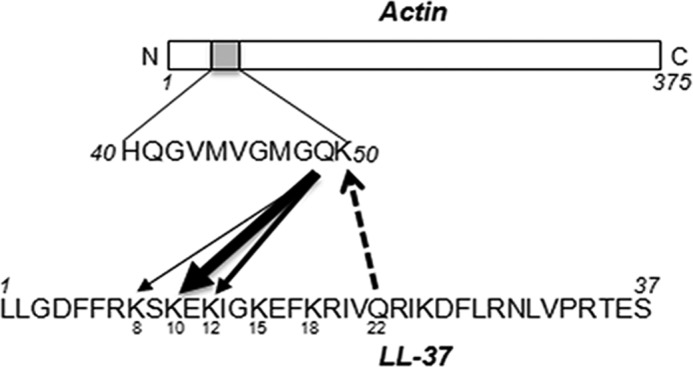
Schematic model of the significant residues that participate in cross-linking of LL-37 and actin by TGase. Arrow thickness signifies the residue's involvement in the cross-linking.
DISCUSSION
Proteolysis is a common strategy employed by pathogens to neutralize the protective actions of host defense peptides (24, 28, 29, 53). One counter-strategy employed by the host is to inhibit the proteases of the invading pathogens (15, 42). Here, we show that extracellular actin from necrotized cells (likely to be abundant in infected sites) binds and is co-localized with LL-37 (Fig. 1C). Another possible source for actin to bind LL-37 may be neutrophils that were recruited to the site of infection and were induced to generate neutrophil extracellular traps for the arrest and extracellular elimination of invading bacteria. Actin was previously described among the neutrophil extracellular trap components (54), and it is likely that it is released to the extracellular surroundings upon breakdown of the neutrophils during the neutrophil extracellular trap release.
We also show that binding to actin protects LL-37 from restriction by the proteases of the two tested bacterial pathogens and enables the microcidal activity of LL-37 despite the presence of active proteases. The mechanism for protecting LL-37 by actin requires precision. Weak binding of actin will result in the digestion of the host defense peptide by the proteases. However, excessively strong binding might hamper the ability of the host defense peptide to bind and kill the bacteria, as seen with LL-37 cross-linked to actin (Fig. 5D). Moreover, while bound to actin, the peptide's microbial binding domain(s) has to be exposed to enable the peptides' microcidal activity.
The strong competition between LL-37 and DNase I for actin binding both in cross-linking (Fig. 8) and bundling experiments (39) indicates that LL-37 binds to the DNase I binding loop (D-loop) of actin (52). This conclusion was supported by the results of subtilisin cleavage of actin, which showed that the integrity of the D-loop is a prerequisite to the tight binding of LL-37 to actin (Fig. 9B). Further support came from thrombin digestion (Fig. 10, A and B) of the tetramethylrhodamine-labeled LL-37-actin cross-linked product, which indicated that LL-37 binds to the His-40–Lys-113 segment of actin. Finally, mass spectrometry localized the binding site to the His-40–Lys-50 segment (Fig. 11) in the D-loop of actin.
The binding between the negatively charged actin and the cationic LL-37 involves both electrostatic and hydrophobic interactions. The presence of hydrophobic interactions is indicated by the finding that in 100 mm NaCl, where the electrostatic interactions are mostly masked and actin filament bundles induced by other cationic proteins and peptides are dissolved (55), LL-37 still bundles actin filaments (Fig. 7B) (39). Substitution of two LL-37 hydrophobic residues, Ile-20 and Ile-24 to Ser, only moderately decreases the F-actin bundling ability of LL-37 at low ionic strength but dramatically in the presence of 100 mm NaCl. This finding strongly supports the existence of significant hydrophobic interactions between actin and LL-37. Surface plasmon resonance measurements (39) demonstrated that the binding between actin and LL-37 is tight (Kd = 1.86·10−7 m−1) and id based on hydrophobic interactions. In addition, the relative insensitivity of LL-37-actin cross-linking to the increase of ionic strength (Fig. 5B) also points to strong hydrophobic interactions. The prerequisite for the tight binding is a closely fitting actin-LL-37 interface, thus the need for the integrity of the binding site on both actin and LL-37.
Decreased binding was observed upon limited subtilisin digestion of G-actin, which cleaves the sequence of the D-loop between Met-47 and Gly-48, as indicated by the reduced bundling of actin filaments in 100 mm NaCl (Fig. 9C) and the complete elimination of actin-LL-37 cross-link formation (Fig. 9B) with subtilisin-digested actin. Altering the LL-37 sequence also affects binding between the two constituents. Scrambling of the LL-37 sequence strongly reduces its binding affinity to actin according to surface plasmon resonance and increased ionic strength sensitivity of bundling (39), and it decreases its ability to cross-link with actin (Fig. 7E). The substitution of Gln-22 in the LL-37 sequence with alanine resulted in inhibitory effects on cross-linking and bundling (Fig. 7, A and B). Interestingly although this substitution reduces the binding affinity of LL-37 to actin, it does not affect its antimicrobial activity (Table 1), and despite the weaker interactions, actin still protects the antibacterial action of Q22A in the presence of the bacterial protease (Fig. 7D). Thus, the presence of Gln-22 in the LL-37 sequence is important for the correct LL-37-actin interaction but not for the antimicrobial activity of the peptide that relies on its binding to the bacterial surface.
TGase cross-linking between proteins is based on a formation of a glutamine-lysyl cross-link (44). Both actin and LL-37 contain glutamine residues, which may participate in the cross-linking reaction. Incubation of LL-37 with TGase leads to LL-37 dimer formation in which the availability of glutamine residue for actin cross-linking is strongly diminished. Cross-link formation between this TGase pretreated LL-37 and actin is reduced but not eliminated (Fig. 6), which points to a role of the sole LL-37 glutamine (Gln-22) in the cross-linking.
Cross-link formation, even though reduced, also takes place between the Q22A LL-37 derivative, which lacks glutamine, and actin. The cross-linking between Q22A and actin indicates that actin glutamines participate in the TGase reaction. Among the two glutamines in the His-40–Lys-50 actin sequence (LL-37-binding site) Gln-41 seemed to be the best candidate for this role because this residue participates in the intrastrand cross-linking in the presence of N-(4-azido-2-nitrophenyl) putrescine between F-actin protomers (46), in intramolecular actin cross-linking to Lys-50 (45), and in the actin cofilin cross-link in both G- and F-actin (48). This residue was also labeled with the fluorescent dansyl ethylenediamine (47) and tetramethylrhodamine cadaverine (56) probes on G-actin by TGase. However, the involvement of Gln-41 in the actin-LL-37 cross-link was excluded by the finding that the pretreatment of actin by TGase, which leads to intramolecular cross-link formation between Gln-41 and Lys-50 (45), does not inhibit the extent of the actin-LL-37 cross-linking (Fig. 6C). Because LL-37 binds to the His-40–Lys-50 segment of actin, Gln-49 appears to be involved in the actin-LL-37 cross-link formation. This conclusion was strongly supported by mass spectrometry, which explicitly identified Gln-49 as the most significant Gln donor on actin in the cross-linking reaction (Fig. 11C). LL-37-actin cross-linked products are formed with both G- and F-actin; however, the yield with F-actin is only half that with G-actin. This is probably due to the fact that the structure of the D-loop in the His-40–Lys-50 segment of F-actin is less flexible than in G-actin; therefore, it is less available for the TGase reaction, which requires structural flexibility of the reactants. Another difference in the TGase reaction between G- and F-actin is that in the reaction with the former, but not with the latter, actin-dimer-LL-37 and actin-higher oligomer-LL-37 cross-linked products also appear in addition to the actin monomer-LL-37 cross-link. The appearance of actin-dimer LL-37 and actin-higher oligomer LL-37 cross-linked products can be explained by our finding that glutamine residues from both LL-37 (Gln-22) and actin (Gln-49) participate in the TGase reaction. Thus, an actin-dimer-LL-37 cross-linked heterotrimer can be produced by cross-linking the Gln-49 residue of an actin monomer to an LL-37 lysine in the first step, and then Gln-22 of the same LL-37 molecule reacts with a lysine (Lys-50) of a second G-actin in the second step. This process can be further continued in the formation of the actin higher oligomer LL-37 cross-linked products. An alternative option for the formation of cross-linked heterotrimer is the reaction of the Gln-49 of actin with an LL-37 lysine in the first step and the reaction of another lysine from the same peptide molecule with Gln-49 of a second actin monomer in the next step. This option is the only possibility for Q22A-actin cross-linked product formation, because Q22A lacks the glutamine residue. Heterotrimer cannot form in the case of F-actin because a single LL-37 molecule cannot react with the two actins because of the steric hindrance caused by the distance between the His-40–Lys-50 segments of the actin protomers in the F-actin structure.
As stated above, the hydrophobic interactions play an important role in the binding of LL-37 to actin. Hydrophobic interactions may also have some role in the mediation of the antimicrobial action of LL-37 as seen in the increase in the minimal inhibitory concentration of I20S/I24S compared with the parent LL-37 (Table 1). However, the positive charge of host defense peptides and of LL-37 in particular (57) was shown to be critical for their selectivity toward microbial membranes that are heavily populated with negatively charged zwitterionic phospholipids on their outermost leaflet (58–60).
We therefore suggest that in the aqueous intercellular environment at physiological ionic strength, the LL-37 hydrophobic domains interact with actin, thus protecting LL-37 from bacterial proteolysis. The positively charged domains of the peptide face the intercellular hydrophilic environment and are exposed and available for attachment to the microbe's surfaces. Under our tested conditions, actin did not reduce the antibacterial activity of LL-37 in a statistically significant manner. However, actin was shown previously to reduce the antimicrobial activity of LL-37 (38).
In conclusion, our results suggest that at a possible cost of a small reduction in the bactericidal activity of LL-37, actin enables the antibacterial activity of LL-37 despite the presence of virulence-associated bacterial proteases.
Acknowledgments
We are very grateful to the Smoler Proteomics Center at the Technion, Haifa, Israel, for technical assistance and analysis of mass spectrometry experiments.
This work was supported by Israel Science Foundation Grant 208/10.
- TGase
- transglutaminase
- Ni-NTA
- nickel-nitrilotriacetic acid
- BAPNA
- Nα-benzoyl-l-arginine 4-nitroanilide hydrochloride
- rhCAP18
- recombinant human CAP18
- r-LL-37
- tetramethylrhodamine-labeled LL-37
- s
- scrambled.
REFERENCES
- 1. Altman H., Steinberg D., Porat Y., Mor A., Fridman D., Friedman M., Bachrach G. (2006) In vitro assessment of antimicrobial peptides as potential agents against several oral bacteria. J. Antimicrob. Chemother. 58, 198–201 [DOI] [PubMed] [Google Scholar]
- 2. Hilpert K., Volkmer-Engert R., Walter T., Hancock R. E. (2005) High-throughput generation of small antibacterial peptides with improved activity. Nat. Biotechnol. 23, 1008–1012 [DOI] [PubMed] [Google Scholar]
- 3. Radzishevsky I. S., Rotem S., Bourdetsky D., Navon-Venezia S., Carmeli Y., Mor A. (2007) Improved antimicrobial peptides based on acyl-lysine oligomers. Nat. Biotechnol. 25, 657–659 [DOI] [PubMed] [Google Scholar]
- 4. Hancock R. E., Nijnik A., Philpott D. J. (2012) Modulating immunity as a therapy for bacterial infections. Nat. Rev. Microbiol. 10, 243–254 [DOI] [PubMed] [Google Scholar]
- 5. Brogden K. A. (2005) Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3, 238–250 [DOI] [PubMed] [Google Scholar]
- 6. Hancock R. E., Sahl H. G. (2006) Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24, 1551–1557 [DOI] [PubMed] [Google Scholar]
- 7. Sochacki K. A., Barns K. J., Bucki R., Weisshaar J. C. (2011) Real-time attack on single Escherichia coli cells by the human antimicrobial peptide LL-37. Proc. Natl. Acad. Sci. U.S.A. 108, E77–E81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Scott M. G., Dullaghan E., Mookherjee N., Glavas N., Waldbrook M., Thompson A., Wang A., Lee K., Doria S., Hamill P., Yu J. J., Li Y., Donini O., Guarna M. M., Finlay B. B., North J. R., Hancock R. E. (2007) An anti-infective peptide that selectively modulates the innate immune response. Nat. Biotechnol. 25, 465–472 [DOI] [PubMed] [Google Scholar]
- 9. Yang D., Biragyn A., Hoover D. M., Lubkowski J., Oppenheim J. J. (2004) Multiple roles of antimicrobial defensins, cathelicidins, and eosinophil-derived neurotoxin in host defense. Annu. Rev. Immunol. 22, 181–215 [DOI] [PubMed] [Google Scholar]
- 10. Hirsch T., Spielmann M., Zuhaili B., Fossum M., Metzig M., Koehler T., Steinau H. U., Yao F., Onderdonk A. B., Steinstraesser L., Eriksson E. (2009) Human β-defensin-3 promotes wound healing in infected diabetic wounds. J. Gene Med. 11, 220–228 [DOI] [PubMed] [Google Scholar]
- 11. Carretero M., Escámez M. J., García M., Duarte B., Holguín A., Retamosa L., Jorcano J. L., Río M. D., Larcher F. (2008) In vitro and in vivo wound healing-promoting activities of human cathelicidin LL-37. J. Invest. Dermatol. 128, 223–236 [DOI] [PubMed] [Google Scholar]
- 12. Cowland J. B., Johnsen A. H., Borregaard N. (1995) hCAP-18, a cathelin/pro-bactenecin-like protein of human neutrophil specific granules. FEBS Lett. 368, 173–176 [DOI] [PubMed] [Google Scholar]
- 13. Lehrer R. I., Ganz T. (2002) Cathelicidins: a family of endogenous antimicrobial peptides. Curr. Opin. Hematol. 9, 18–22 [DOI] [PubMed] [Google Scholar]
- 14. Bals R., Wang X., Zasloff M., Wilson J. M. (1998) The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc. Natl. Acad. Sci. U.S.A. 95, 9541–9546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rosen G., Sela M. N., Bachrach G. (2012) The antibacterial activity of LL-37 against Treponema denticola is dentilisin protease-independent and facilitated by the major outer sheath protein virulence factor. Infect. Immun. 80, 1107–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mookherjee N., Lippert D. N., Hamill P., Falsafi R., Nijnik A., Kindrachuk J., Pistolic J., Gardy J., Miri P., Naseer M., Foster L. J., Hancock R. E. (2009) Intracellular receptor for human host defense peptide LL-37 in monocytes. J. Immunol. 183, 2688–2696 [DOI] [PubMed] [Google Scholar]
- 17. Morioka Y., Yamasaki K., Leung D., Gallo R. L. (2008) Cathelicidin antimicrobial peptides inhibit hyaluronan-induced cytokine release and modulate chronic allergic dermatitis. J. Immunol. 181, 3915–3922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang G., Epand R. F., Mishra B., Lushnikova T., Thomas V. C., Bayles K. W., Epand R. M. (2012) Decoding the functional roles of cationic side chains of the major antimicrobial region of human cathelicidin LL-37. Antimicrob. Agents Chemother. 56, 845–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nizet V., Ohtake T., Lauth X., Trowbridge J., Rudisill J., Dorschner R. A., Pestonjamasp V., Piraino J., Huttner K., Gallo R. L. (2001) Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature 414, 454–457 [DOI] [PubMed] [Google Scholar]
- 20. Chromek M., Slamová Z., Bergman P., Kovács L., Podracká L., Ehrén I., Hökfelt T., Gudmundsson G. H., Gallo R. L., Agerberth B., Brauner A. (2006) The antimicrobial peptide cathelicidin protects the urinary tract against invasive bacterial infection. Nat. Med. 12, 636–641 [DOI] [PubMed] [Google Scholar]
- 21. Pütsep K., Carlsson G., Boman H. G., Andersson M. (2002) Deficiency of antibacterial peptides in patients with morbus Kostmann: an observation study. Lancet 360, 1144–1149 [DOI] [PubMed] [Google Scholar]
- 22. de Haar S. F., Hiemstra P. S., van Steenbergen M. T., Everts V., Beertsen W. (2006) Role of polymorphonuclear leukocyte-derived serine proteinases in defense against Actinobacillus actinomycetemcomitans. Infect. Immun. 74, 5284–5291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Devine D. A., Hancock R. E. (2002) Cationic peptides: distribution and mechanisms of resistance. Curr. Pharm. Des. 8, 703–714 [DOI] [PubMed] [Google Scholar]
- 24. Thomassin J. L., Brannon J. R., Gibbs B. F., Gruenheid S., Le Moual H. (2012) OmpT outer membrane proteases of enterohemorrhagic and enteropathogenic Escherichia coli contribute differently to the degradation of human LL-37. Infect. Immun. 80, 483–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hollands A., Gonzalez D., Leire E., Donald C., Gallo R. L., Sanderson-Smith M., Dorrestein P. C., Nizet V. (2012) A bacterial pathogen co-opts host plasmin to resist killing by cathelicidin antimicrobial peptides. J. Biol. Chem. 287, 40891–40897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baron A. C., Gansky S. A., Ryder M. I., Featherstone J. D. (1999) Cysteine protease inhibitory activity and levels of salivary cystatins in whole saliva of periodontally diseased patients. J. Periodontal Res. 34, 437–444 [DOI] [PubMed] [Google Scholar]
- 27. Devine D. A., Marsh P. D., Percival R. S., Rangarajan M., Curtis M. A. (1999) Modulation of antibacterial peptide activity by products of Porphyromonas gingivalis and Prevotella spp. Microbiology 145, 965–971 [DOI] [PubMed] [Google Scholar]
- 28. Schmidtchen A., Frick I. M., Andersson E., Tapper H., Björck L. (2002) Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol. Microbiol. 46, 157–168 [DOI] [PubMed] [Google Scholar]
- 29. Sieprawska-Lupa M., Mydel P., Krawczyk K., Wójcik K., Puklo M., Lupa B., Suder P., Silberring J., Reed M., Pohl J., Shafer W., McAleese F., Foster T., Travis J., Potempa J. (2004) Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob. Agents Chemother. 48, 4673–4679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Puklo M., Guentsch A., Hiemstra P. S., Eick S., Potempa J. (2008) Analysis of neutrophil-derived antimicrobial peptides in gingival crevicular fluid suggests importance of cathelicidin LL-37 in the innate immune response against periodontogenic bacteria. Oral Microbiol. Immunol. 23, 328–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bachrach G., Altman H., Kolenbrander P. E., Chalmers N. I., Gabai-Gutner M., Mor A., Friedman M., Steinberg D. (2008) Resistance of Porphyromonas gingivalis ATCC 33277 to direct killing by antimicrobial peptides is protease independent. Antimicrob. Agents Chemother. 52, 638–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gutner M., Chaushu S., Balter D., Bachrach G. (2009) Saliva enables the antimicrobial activity of LL-37 in the presence of proteases of Porphyromonas gingivalis. Infect. Immun. 77, 5558–5563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hajishengallis G., Liang S., Payne M. A., Hashim A., Jotwani R., Eskan M. A., McIntosh M. L., Alsam A., Kirkwood K. L., Lambris J. D., Darveau R. P., Curtis M. A. (2011) Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10, 497–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Socransky S. S., Haffajee A. D., Cugini M. A., Smith C., Kent R. L., Jr. (1998) Microbial complexes in subgingival plaque. J. Clin. Periodontol. 25, 134–144 [DOI] [PubMed] [Google Scholar]
- 35. Rottner K., Stradal T. E. (2011) Actin dynamics and turnover in cell motility. Curr. Opin. Cell Biol. 23, 569–578 [DOI] [PubMed] [Google Scholar]
- 36. Puius Y. A., Mahoney N. M., Almo S. C. (1998) The modular structure of actin-regulatory proteins. Curr. Opin. Cell Biol. 10, 23–34 [DOI] [PubMed] [Google Scholar]
- 37. Tang J. X., Janmey P. A. (1996) The polyelectrolyte nature of F-actin and the mechanism of actin bundle formation. J. Biol. Chem. 271, 8556–8563 [DOI] [PubMed] [Google Scholar]
- 38. Weiner D. J., Bucki R., Janmey P. A. (2003) The antimicrobial activity of the cathelicidin LL37 is inhibited by F-actin bundles and restored by gelsolin. Am. J. Respir. Cell Mol. Biol. 28, 738–745 [DOI] [PubMed] [Google Scholar]
- 39. Sol A., Blotnick E., Bachrach G., Muhlrad A. (2012) LL-37 induces polymerization and bundling of actin and affects actin structure. PLoS One 7, e50078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bucki R., Byfield F. J., Janmey P. A. (2007) Release of the antimicrobial peptide LL-37 from DNA/F-actin bundles in cystic fibrosis sputum. Eur. Respir. J. 29, 624–632 [DOI] [PubMed] [Google Scholar]
- 41. Welsh M. J., Smith A. E. (1995) Cystic fibrosis. Sci. Am. 273, 52–59 [DOI] [PubMed] [Google Scholar]
- 42. Zaiou M., Nizet V., Gallo R. L. (2003) Antimicrobial and protease inhibitory functions of the human cathelicidin (hCAP18/LL-37) prosequence. J. Invest. Dermatol. 120, 810–816 [DOI] [PubMed] [Google Scholar]
- 43. Spudich J. A., Watt S. (1971) The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 246, 4866–4871 [PubMed] [Google Scholar]
- 44. Folk J. E., Chung S. I. (1985) Transglutaminases. Methods Enzymol. 113, 358–375 [DOI] [PubMed] [Google Scholar]
- 45. Eli-Berchoer L., Hegyi G., Patthy A., Reisler E., Muhlrad A. (2000) Effect of intramolecular cross-linking between glutamine-41 and lysine-50 on actin structure and function. J. Muscle Res. Cell Motil. 21, 405–414 [DOI] [PubMed] [Google Scholar]
- 46. Hegyi G., Mák M., Kim E., Elzinga M., Muhlrad A., Reisler E. (1998) Intrastrand cross-linked actin between Gln-41 and Cys-374. I. Mapping of sites cross-linked in F-actin by N-(4-azido-2-nitrophenyl) putrescine. Biochemistry 37, 17784–17792 [DOI] [PubMed] [Google Scholar]
- 47. Kim E., Motoki M., Seguro K., Muhlrad A., Reisler E. (1995) Conformational changes in subdomain 2 of G-actin: fluorescence probing by dansyl ethylenediamine attached to Gln-41. Biophys. J. 69, 2024–2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Benchaar S. A., Xie Y., Phillips M., Loo R. R., Galkin V. E., Orlova A., Thevis M., Muhlrad A., Almo S. C., Loo J. A., Egelman E. H., Reisler E. (2007) Mapping the interaction of cofilin with subdomain 2 on actin. Biochemistry 46, 225–233 [DOI] [PubMed] [Google Scholar]
- 49. Schwyter D., Phillips M., Reisler E. (1989) Subtilisin-cleaved actin: polymerization and interaction with myosin subfragment 1. Biochemistry 28, 5889–5895 [DOI] [PubMed] [Google Scholar]
- 50. Muszbek L., Laki K. (1974) Cleavage of actin by thrombin. Proc. Natl. Acad. Sci. U.S.A. 71, 2208–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Muszbek L., Gladner J. A., Laki K. (1975) The fragmentation of actin by thrombin. Isolation and characterization of the split products. Arch. Biochem. Biophys. 167, 99–103 [DOI] [PubMed] [Google Scholar]
- 52. Kabsch W., Mannherz H. G., Suck D., Pai E. F., Holmes K. C. (1990) Atomic structure of the actin:DNase I complex. Nature 347, 37–44 [DOI] [PubMed] [Google Scholar]
- 53. Koziel J., Karim A. Y., Przybyszewska K., Ksiazek M., Rapala-Kozik M., Nguyen K. A., Potempa J. (2010) Proteolytic inactivation of LL-37 by karilysin, a novel virulence mechanism of Tannerella forsythia. J. Innate Immun. 2, 288–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Urban C. F., Ermert D., Schmid M., Abu-Abed U., Goosmann C., Nacken W., Brinkmann V., Jungblut P. R., Zychlinsky A. (2009) Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 5, e1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Muhlrad A., Grintsevich E. E., Reisler E. (2011) Polycation induced actin bundles. Biophys. Chem. 155, 45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Muhlrad A., Kudryashov D., Michael Peyser Y., Bobkov A. A., Almo S. C., Reisler E. (2004) Cofilin induced conformational changes in F-actin expose subdomain 2 to proteolysis. J. Mol. Biol. 342, 1559–1567 [DOI] [PubMed] [Google Scholar]
- 57. Sol A., Ginesin O., Chaushu S., Karra L., Coppenhagen-Glazer S., Ginsburg I., Bachrach G. (2013) LL-37 opsonizes and inhibits biofilm formation of Aggregatibacter actinomycetemcomitans at sub-bactericidal concentrations. Infect. Immun. 81, 3577–3585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zasloff M. (2002) Antimicrobial peptides of multicellular organisms. Nature 415, 389–395 [DOI] [PubMed] [Google Scholar]
- 59. Shai Y. (1999) Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta 1462, 55–70 [DOI] [PubMed] [Google Scholar]
- 60. Matsuzaki K. (1999) Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta 1462, 1–10 [DOI] [PubMed] [Google Scholar]