

Abstract
Vascular ATP-sensitive K+ channels are inhibited by multiple vasoconstricting hormones via the PKC pathway. However, the molecular substrates for PKC phosphorylation remain unknown. To identify the PKC sites, Kir6.1/SUR2B and Kir6.2/SUR2B were expressed in HEK293 cells. Following channel activation by pinacidil, the catalytic fragment of PKC inhibited the Kir6.1/SUR2B currents but not the Kir6.2/SUR2B. Phorbol 12-myristate 13-acetate (PMA, a PKC activator) had similar effects. Using Kir6.1–Kir6.2 chimeras two critical protein domains for the PKC-dependent channel inhibition were identified. The proximal N-terminus of Kir6.1 was needed, but there was no PKC phosphorylation site in this region. The distal C-terminus of Kir6.1 was crucial where there are several consensus PKC sites. Mutation of Ser354, Ser379, Ser385, Ser391 or Ser397 to non-phosphorylatable alanine reduced PKC inhibition moderately but significantly. Combined mutations of these residues had greater effects. The channel inhibition was almost completely abolished when five of them were jointly mutated. In-vitro phosphorylation assay showed that 4 of the serine residues were necessary for the PKC-dependent 32P incorporation into the distal C-terminal peptides. Thus, a motif containing 4 phosphorylation repeats is identified in the Kir6.1 subunit underlying the PKC-dependent inhibition of the Kir6.1/SUR2B channel. The presence of the phosphorylation motif in the Kir6.1 but not in its close relative Kir6.2 suggests that the vascular KATP channel may have undergone evolutionary optimization allowing it to be regulated by a variety of vasoconstricting hormones and neurotransmitters.
Introduction
ATP sensitive K+ (KATP) channels play an important role in vascular tone regulations (1–3). Such a function attributes to the channel regulation by a variety of vasodilating and vasoconstricting hormones and neurotransmitters (3–7). Therefore, the understanding of molecular basis for the channel regulation has a major impact on the design of therapeutical modalities by targeting at specific molecular substrates of the channel. It is known that the major isoform of KATP channels in vascular smooth muscles (VSM) is composed of Kir6.1 and SUR2B (8–12). Genetic disruption of Kir6.1 or SUR2 indeed results in a phenotype of Prinzmetal angina with a high rate of sudden death (13,14), consistent with the importance of the channel in vascular regulations.
Experimental evidence suggests that vasoconstrictors act on the vascular KATP channel through the PKC signaling system. Our previous studies have shown that the Kir6.1/SUR2B channel and its counterpart in VMS cells are inhibited by vasopressin, and the channel inhibition can be abolished by specific PKC blockers (6). Similar observations have been made by other groups with endothelin (15), muscarinic M3 receptor agonist (16) and angiotensin II (17). The effect of angiotensin II on vascular KATP channel requires translocation of PKCε to plasma membranes (18,19). By comparing effects of acetylcholine M3-receptor on Kir6.1/SUR2B and Kir6.2/SUR2B channels, Quinn et al. (16) suggest that the Kir6.1/SUR2B channel inhibition is mediated via a direct effect of PKC rather the change in phosphatidyl-4,5-bisphosphate concentrations. They also show evidence for Kir6.1 phosphorylation using in-vitro biochemical assay (16). Consistently, purified PKC inhibits the cloned Kir6.1/SUR2B and VMS-endogenous KATP channels in inside-out patches where cytosolic soluble components are absent (4,20). Although these previous studies have significantly improved our understanding of the vascular KATP channel regulation by vasoconstrictors, the molecular substrate of PKC remain unknown. Therefore, we performed these studies to identify the critical protein domain and amino acid residues for the PKC phosphorylation.
Materials and Methods
Molecular Biology
Rat Kir6.1 (GenBank #D42145), mouse Kir6.2 (GenBank #D50581) and mouse SUR2B (GenBank #D86038) cDNAs were used in the present study. The cDNAs were cloned in a eukaryotic expression vector pcNDA3.1 and used for mammalian cell expression (5,6). Kir6.1-Kir6.2 chimeras were produced by overlap extension using PCR (pfu DNA polymerase, Stratagene, La Jolla, CA) (21). Site-specific mutations were made using a site-directed mutagenesis kit (Stratagene). The orientation of the constructs and correct mutations were confirmed with DNA sequencing. The constructs were expressed in HEK293 (American type culture collection, Rockville, MD) as detailed in our previous reports (5,6).
Patch clamp experiments were performed at room temperature as described previously (5,6). In brief, the bath solution for whole-cell recording contained (in mmol/L): 10 KCl, 135 potassium gluconate, 5 EGTA, 5 glucose, and 10 HEPES (pH = 7.4). The pipette solution contained 10 KCl, 133 potassium gluconate, 5 EGTA, 5 glucose, 1 K2ATP, 0.5 NaADP and 10 HEPES (pH = 7.4), in which the free Mg2+ concentration was adjusted to 1mmol/L using MgCl2. PMA and other chemicals were purchased from Sigma if not otherwise stated. Chemicals were prepared in high concentration stork solution in double distilled H2O or DMSO, and were diluted in bath solution to experimental concentrations immediately before usage. Glibenclamide, pinacidil and PMA were applied to cells using a perfusion system. PKC inhibitory peptide 19–31(PKCi, 10μmol/L, Calbiochem, La Jolla, CA) was applied to the pipette solution. To avoid ATP degradation, all ATP-containing solutions were made immediately before experiments and used for no longer than 4hrs. Inside-out patches were performed with symmetric high K+ in bath solution and pipette: 10 KCl, 135 potassium gluconate, 5 EGTA, 5 glucose, and 10 HEPES (pH = 7.4), with [Mg2+] adjusted to 1mmol/L using MgCl2. After giga-seal was formed, the patch was excised and intracellular side was exposed to bath solution. Constant single voltage of −60mV were applied to the patch from holding potential of 0mV. The effect of PMA was expressed as % current inhibition: i.e., current amplitude inhibited by PMA exposure divided by the difference of the maximum channel activation by pinacidil from the maximum inhibition by glibenclamide.
Western blot
MBP-Kir6.1Cs with or without mutants were generated with the distal C-terminus segment (residues 346–420) of Kir6.1 fused to C-terminus of Maltose binding protein (MBP) using same methods detailed in our previous report (5). The constructs were then transformed into protease-deficient E.coli BL21, in which MBP-fusion peptides were induced with 0.3mmol/L isopropylthiogalactoside for 2 hours. The MBP-fusion peptides were purified using amylose resin according to the procedure by the provider (New England Biolabs). The peptides were run on in 10% SDS-page gel and transferred to PVDF membrane (Bio-Rad, Hercules, CA). Western blot was carried out using rabbit primary antibody against Kir6.1 C-terminus (KRNSMRRNNSMRRSN, corresponding to amino acid residues 382–396 of rat Kir6.1, Sigma, 1:2000) and secondary goat anti-rabbit IgG conjugated with alkaline phosphotase. The protein bands were depicted with bromochloroindolyl phosphate (BCIP) and nitro blue tetrazolium (NBT).
In vitro phosphorylation
The fusion peptides were treated with the catalytic fragment of PKC (cPKC, Biomol, Plymouth, PA) in following condition: ~5μg fusion peptides in 5μL modified elution buffer (200 NaCl, 30 tris-HCl, 6 EDTA, 10 maltose, pH 7.4), 5μL of 5X reaction buffer (125 tris-HCl, 0.1 EGTA, pH 7.5), 5μL Mg-ATP solution (20 MOPS, 25 β-glycerophosphate, 5 EGTA, 1 Na3VO4, 1 dithiothreitol, 75 MgCl2, 0.5 ATP and pH 7.2,), 10ng cPKC in 10μL H2O and 1μL of 5μCi/μl of 32P-γ-labeled ATP (Perkin-Elmer, San Francisco, CA). After 60min of reaction, 5μl of 5X protein loading buffer were added to each sample to terminate the reaction. The samples were subjected to electrophoresis in 10% SDS-page gel. The gel was colored with Coomassie blue and photographed. The gel was then fixed and dried. Autoradiography was taken using a Fuji BAS 2500 Imaging Plate. The in vitro phosphorylation experiment was repeated twice.
Data were presented as means ± s.e. Differences in means were tested with the ANOVA or Student t test and were accepted as significant if P ≤ 0.05.
Results
PKC-dependent inhibition of the Kir6.1/SUR2B channel
Whole-cell currents were recorded with high K+ in both bath and pipette solutions. The Kir6.1/SUR2B currents remained small in 8–10 min of baseline recording, and were strongly activated by 10μmol/L pinacidil and inhibited by 10μmol/L glibenclamide (Fig 1A, B). At maximum activation, application of 100nmol/L PMA strongly inhibited the Kir6.1/SUR2B currents (77.5±3.7%, n=19), which was blocked by PKCi, a specific PKC inhibitor (Fig 1C, F). The inactive phorbol ester, 4α-phorbol-12,13-didecanoate (4α-PDD), did not affect Kir6.1/SUR2B currents (Fig 1F).
Figure 1.

Kir6.1/SUR2B and Kir6.2/SUR2B channels expressed in HEK293 cells. A. Whole-cell currents were recorded from a cell transfected with Kir6.1/SUR2B. Symmetric concentrations of K+ (145mmol/L) were applied to the pipette and bath solutions. The cell was held at 0mV, and pulse voltages from −120 to 80mV with a 20mV increment were applied. The current amplitude increased in response to pinacidil (Pin, 10μmol/L). The pinacidil-induced currents were strongly inhibited by PMA (100nmol/L) and completely inhibited by glibenclamide (Glib, 10μmol/L). B. The time course for the Kir6.1/SUR2B channel modulation. Whole-cell currents were recorded with a holding potential at 0mV and command pulses of −80mV in every 3 seconds. After whole-cell configuration was formed, the cell was perfused with extracellular solution for a ~2 min baseline recording. The currents were strongly activated by pinacidil (10μmol/L), and the maximum activation was reached in 3–4 min of the exposure. The currents were inhibited by PMA (100nmol/L) in around 5min and further inhibited by glibenclamide. The lower panel shows individual currents produced by a single command pulse. C. Kir6.1/SUR2B currents were recorded with PKCi (10μmol/L) in the pipette solution. PKCi almost completely blocked Kir6.1/SUR2B currents inhibition by PMA. D. E. Kir6.2/SUR2B currents were recorded from transfected HEK293 cells with the same treatment as for the Kir6.1/SUR2B. PMA did not affect the pinacidil-activated Kir6.2/SUR2B currents. F. Summary of PMA effects on Kir6.1/SUR2B and Kir6.2/SUR2B currents activated by pinacidil. PMA inhibited Kir6.1/SUR2B currents strongly (77.5±3.7%, n=19). Non-active PMA analog, 4α-phorbol 12, 13-didecanoate (4α-PDD, 100nmol/L), had little effects on the Kir6.1/SUR2B currents (13.3±8.2%, n=4). In the presence of PKCi, the PMA effect was blocked (7.1±6.0%, n=4). PMA had no inhibitory effects on Kir6.2/SUR2B currents (−0.7±0.7%, n=5).
In inside-out patches, channel activity was low in the absence of nucleotides. The Kir6.1/SUR2B channel was activated in the presence of MgADP/ATP, which was further augmented with pinacidil application (Fig 2A). When the catalytic fragment of PKC (cPKC) was applied to the internal patch membranes, the Kir6.1/SUR2B currents were markedly inhibited (61.1±3.8%, n=6, Fig 2A, C). These results thus indicate that the Kir6.1/SUR2B channel is inhibited by PKC independently of cytosolic soluble components, consistent with previous reports (20). In contrast, PMA had no effects on Kir6.2/SUR2B channel in whole-cell recordings (Fig 1 D–F), neither the cPKC in inside-out patches (Fig 2B, C), suggesting that the SUR2B subunit is not critical.
Figure 2.
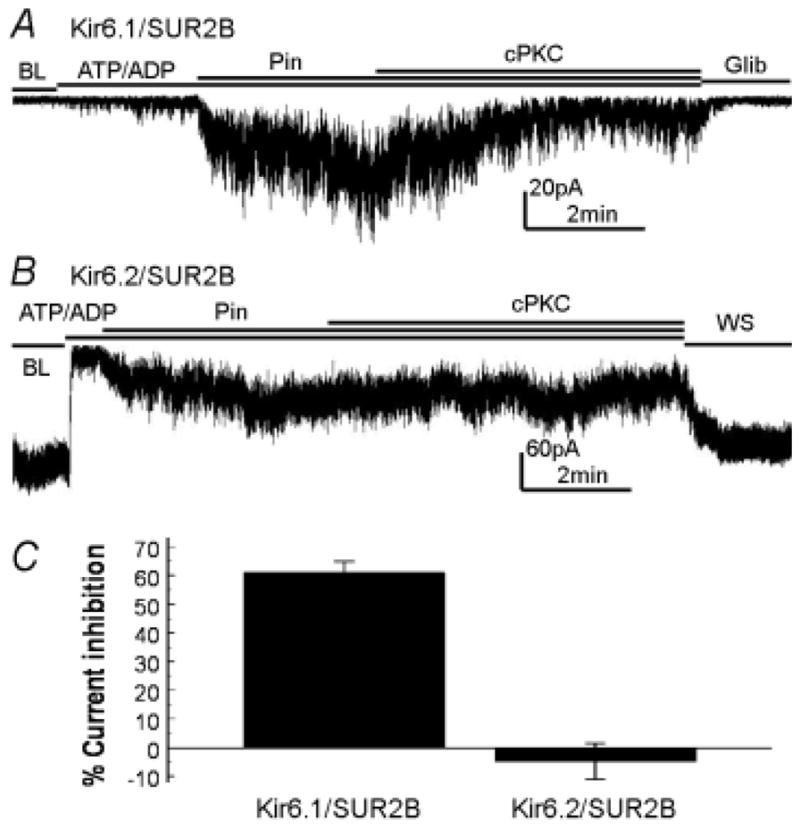
Effects of the recombinant catalytic fragment of PKC on Kir6.1/SUR2B and Kir6.2/SUR2B currents in inside-out patches. A. At baseline (BL) without nucleotides, the Kir6.1/SUR2B channels were closed. Application of 1mmol/L ATP / 0.5mmol/L ADP to the internal patch membrane slightly activated the currents, which were strongly activated by pinacidil (Pin, 10μmol/L). Application of cPKC inhibited the Kir6.1/SUR2B currents. B. Without nucleotides, the Kir6.2/SR2B channel was open. The currents were inhibited by 1mmol/L ATP / 0.5mmol/L ADP and activated by pinacidil. Unlike Kir6.1/SUR2B, cPKC has no effects on Kir6.2/SUR2B currents under same experimental condition. C. Summary of cPKC effects on Kir6.1/SUR2B and Kir6.2/SUR2B currents that were prior activated by pinacidil. The Kir6.1/SUR2B currents were significantly inhibited by cPKC (61.1±3.8%, n=6), while the Kir6.2/SURB currents were not affected (−4.9±6.3%, n=3).
Critical protein domains for the PKC-dependent channel inhibition
The differential PKC sensitivity of the Kir6.1/SUR2B from Kir6.2/SUR2B suggests that critical protein domains for PKC regulation are located on the Kir subunit. Thereby, we constructed Kir6.1-Kir6.2 chimeras and expressed them with SUR2B in HEK293 cells. The Kir6.1 and Kir6.2 were divided into three regions: the N-terminus, the core sequence containing two transmembrane domains and the pore loop, and the C-terminus (Fig. 3). Six chimeras were thus obtained, all of which showed functional channels and were sensitive to pinacidil and glibenclamide. When the N-terminus of Kir6.1 was replaced with that of Kir6.2 (named as 211), the channel inhibition by PMA was greatly diminished (Fig 3A, D). Similar result was observed in the 112 whose C-terminus was from Kir6.2 (Fig 3B, D). Consistent with this, replacement of either N-terminus or C-terminus of Kir6.2 (122 and 221 respectively) did not confer the PMA sensitivity to the chimerical channels (Fig. 3D). When both N- and C-termini of Kir6.1 were constructed, the 121 channel responded to PMA just like Kir6.1 (Fig 3C, D). Therefore, both N- and C-termini are required for the PKC-dependent channel inhibition.
Figure 3.

Responses of Kir6.1-Kir6.2 chimeras to PMA. All chimerical channels were expressed with SUR2B. A. When N-terminus of Kir6.1 was replaced with that of Kir6.2, PMA failed to inhibit the 211 channel. B. When the C-terminus of Kir6.1 was replaced with that of Kir6.2, the 112 channel lost response to PMA. C. Construction of both Kir6.1 N- and C-termini in the Kir6.2 core sequence resulted in PMA sensitivity as wt Kir6.1/SUR2B. D. Summary of PMA inhibition on chimeras. Kir6.1 N-terminus, core and C-terminus refer to residues 1–71, residues 72–186, and residues 187–424, respectively. Kir6.2 N-terminus, core and C-terminus refer to residues 1–70, residues 71–176, and residues 177–390.
We further divided the N-terminus into two segments and the C-terminus into three. These chimeras showed similar responses to pinacidil and glibenclamide as the wt channels. When the distal N-terminus was replaced, the Kir6.1-21_111 channel responded to PMA to the same extend (71.1±4.2%, n=5, Fig. 4A) as Kir6.1. Replacement of the proximal N-terminus, however, significantly decreased PMA effects, although such an effect was somehow incomplete (36.7±7.8%, n= 5, Fig. 4B). A serine residue (Ser40) was found in the proximal N-terminus of Kir6.1 but not in Kir6.2. When the Ser40 was mutated to the corresponding residue (Lys39) in Kir6.2, the Kir6.1-S40K channel was still inhibited as the wt Kir6.1 (Fig 5E). Since no PKC site was found in the N-terminus, how this protein domain is involved in the PKC action remains to be understood. When the proximal C-terminus was replaced, the Kir6.1-11_211 responded to PMA to the same degree as the wt channel (Fig 4C). With the middle or distal segment swapped, the Kir6.1-11_121 and Kir6.1-11_112 channel response to PMA was significantly diminished (Fig 4D, E). The PMA inhibition was further decreased in a chimera that both middle and distal segments were from Kir6.2 (Kir6.1-11_122, Fig 4F).
Figure 4.

Dissection of critical protein domains for the PKC-dependent channel inhibition. The N-terminus was further divided into two segments at residue 35 in Kir6.1 and residue 34 in Kir6.2. The C-terminus was divided into 3 segments at residue 275 and 363 in Kir6.1 as well as residue 265 and 354 in Kir6.2. A. Kir6.1-21_111, the distal N-terminus of Kir6.1 replaced with that of Kir6.2, was inhibited by PMA as much as the wt Kir6.1/SUR2B channel (P>0.05, n=5). B. The PMA effect was disrupted in Kir6.1-12_111, where the proximal N-terminus of Kir6.1 was replaced (P<0.001, n=5). C. Replacement of proximal C-terminus of Kir6.1 did not affect PMA sensitivity (P>0.05, n=4). D.–F. Replacement the middle segment of the C-terminus, the distal C-terminus or both disrupted the channel inhibition to various degrees (P<0.05; P<0.01; and P<0.001 respectively). G.–I. Construction of necessary protein segments in Kir6.2 core. G. The distal C-terminus conferred partial PMA effects to Kir6.2-core channel (P<0.001 compared to both Kir6.2 and Kir6.1, n=5). H. When the middle and distal segments were constructed, the channel displayed PMA inhibition with no significant difference from the wt Kir6.1 channel (P>0.05, n=4). I. In Kir6.2-21_221′ in which the distal C-segment was elongated from residue 364 to 346 in Kir6.1, PMA fully inhibited the channel (P>0.05, n=5). J. Summary of the effect of N- and C-terminal segments on the channel inhibition. Data were obtained from 4–6 cells in each construct. The amino acid sequences of important distal C-terminus (Kir6.1-346-424 and corresponding sequence in Kir6.2) were shown in low panel with putative PKC phosphorylation sites highlighted.
Figure 5.

Mutagenesis analysis of potential PKC phosphorylation sites. A. Mutation Ser403 of Kir6.1 to alanine did not affect the PMA inhibition. B. Mutation of Ser385 decreased PMA effects. C, D. When 4 and 5 potential PKC sites were mutated to alanine simultaneously, the channel inhibition was largely eliminated. E. Summary of the mutations. 3A, Kir6.1-S385A/Ser391A/S397A; 4A, Kir6.1-S379A/S385A/Ser391A/S397A; 5A, Kir6.1-S354A/S379A/S385A/Ser391A/S397A. Data were obtained from 4–9 patches. White bars, P>0.05; Black bars, P<0.05.
We then used chimeras with the Kir6.2 core to search for the necessary protein domain in the C-terminus. The Kir6.2-21_211 showed full PMA sensitivity (69.9±6.7%, n=4, Fig 4H), and the Kir6.2-21_221 was partially inhibited by PMA (33.7±3.1%, n=5, Fig 4G). With a slight extension of the distal segment, the Kir6.2-21_221′ channel showed full PMA response (78.8±2.5%, n=5, Fig 4I), suggesting that the distal segment contains the essential C-terminus elements for PKC phosphorylation.
Phosphorylation of a stretch of serine repeats in the C terminus by PKC
There are multiple consensus PKC sequences in the distal C-terminus from residue 346 to the end (residue 424). Particularly remarkable are the SXRRXN/SXRKXN repeats at residues 379, 385, 391 and 397. We thus performed alanine mutational screening. Mutation of any individuals produced moderate but significant reduction of the PKC-dependent inhibition (Fig. 5B, E). Mutation of Ser354 in the same protein domain showed a similar effect (Fig. 5E). A combined mutation of 3 or 4 (Ser379, Ser385, Ser391, Ser397) had greater effects (Kir6.1-3A, Kir6.1-4A, Fig. 5C, E). When all of these 5 serine residues are jointly mutated (Kir6.1-5A), the PKC-dependent channel inhibition was almost completely eliminated (13.3±4.5%, n=5, note that there were ~10% desensitizations contributing to the remaining effect) (Fig. 5D, E).
To confirm that these serine residues are indeed phosphorylated, we generated several C-terminal peptides of Kir6.1 (residues 346–424) with and without mutations on the serine residues. After extraction and purification, the peptides were subjected to SDS-page separation, western blot, and in-vitro phosphorylation. In Coomassie blue stain and Western blot, a 52kd band was detected which was consistent with the peptide size (Fig 6B), while there was another 49kd band indicating a degraded protein fragment. In vitro phosphorylation experiment showed strong 32P incorporation in both bands of the wt peptide (Fig. 6 C), consistent with our functional assays. The strength of 32P incorporation was substantially reduced in the Kir6.1-3A peptide. The Kir6.1-4A and Kir6.1-5A peptides were only weakly phosphorylated. Comparing the phosphorylation patterns, we found that Ser354 was not phosphorylated (See analysis in Figure 6 legend). The 32P incorporation to the Kir6.1-4A and Kir6.1-5A peptides was almost completely eliminated in comparison to the wt, consistent with our mutational analysis in this region (Ser403A, Ser404A and Thr414A, Fig 5E), suggesting that residues other than the Ser379, Ser385, Ser391, Ser397 have, if any, very little effect on the PKC-dependent channel inhibition.
Figure 6.

In vitro phosphorylation on MBP-fusion proteins. A. Four MBP-fusion proteins were constructed by linking Kir6.1 distal C fragment (residues 346–424) to MBP with or without mutations on critical serine residues. The sequence underlined is the antigen for Western detection. B. All the constructs showed two bands with Western blot. The band around 52kd represented the intact fusion proteins (arrow), while the band of ~49kd was a degraded protein fragment (arrowhead). These bands were weekly detected in 3A, 4A and 5A by Western because mutations were made in antigen region. Low panel showed protein input colored with Coomassie blue. C. In vitro phosphorylation showed strong 32P incorporation in both 52kd and 49kd bands in wt. The 32P incorporation was marked reduced in 3A. The 4A and 5A were only weekly phosphorylated in the 52kd band. By comparing 3A and 4A, the phosphorylation of 49kd band on 3A must occur on Ser379. The 49kd of 4A was not phosphorylated also suggested that Ser354 was not phosphorylated. In addition, phosphorylation on the 52kd of 4A and 5A suggested there are unidentified phosphorylation site(s) located C-terminal to Ser397, but they were not functionally important according to mutational analysis in Figure 5E.
Discussion
The present studies show evidence for the molecular basis underlying the Kir6.1/SUR2B channel inhibition by PKC. The channel inhibition relies on a short motif consisting of 4 phosphorylation repeats in the Kir6.1 subunit. Graded channel inhibitions are produced by phosphorylation of different numbers of these serine residues. The presence of the phosphorylation motif in the Kir6.1 but not in its close relative Kir6.2 suggests that the vascular KATP channel may have undergone evolutionary optimization allowing it to be regulated by a variety of vasoconstrictors.
PKC acts on several isoforms of KATP channels. Previous studies indicate that PKC activation leads to inhibition of the vascular Kir6.1/SUR2B channel, and this effect is likely to be mediated by direct phosphorylation of the channel protein (16,20). PKC activates the pancreatic isoform (Kir6.2/SUR1) and striated muscular isoform (Kir6.2/SUR2A) of KATP channels (22). Expressed with SUR2B, the Kir6.2 channel is activated by PKC or shows no response, depending on recording conditions (16,20). A critical PKC phosphorylation site (Thr180) has been found in Kir6.2 mediating the fast channel activation (22). It is unclear however whether a corresponding site in Kir6.1 (Thr190) plays a similar role, as mutation of the residue leads to non-functional channel (5,20). PKC also causes the Kir6.2/SUR1 and Kir6.2/SUR2A channel internalization (23). Since the effect of PKC on the Kir6.1/SUR2B (inhibition) is clearly contrast to Kir6.2-containing channels (activation or no effect), the Kir subunit is likely to be targeted by PKC regulation. The differential responses of the Kir6.1 and Kir6.2 to PKC allow chimerical dissections of critical protein domains. Indeed, our studies on the Kir6.1-Kir6.2 chimeras have shown that proximal N-terminus and distal C-terminus of Kir6.1 are critical for the PKC-dependent channel inhibition.
The proximal N-terminus has only 7 residues different between Kir6.1 and Kir6.2 in their primary sequences, none of which can be phosphorylated. The lack of PKC phosphorylation sites in the protein domain suggests that it is likely to be involved in the channel gating or interaction of protein domains. Previous studies have indeed shown that the proximal N-terminus is important for Kir6.2 channel gating by multiple channel regulators such as ATP and phosphatidyl-4,5-bisphosphate (24–27). A notable difference between Kir6.1 and Kir6.2 channels is that in the absence of nucleotide the Kir6.2 channels are spontaneously open while the Kir6.1 channels are completely closed (28), a phenomenon that is also explained as fast rundown of the Kir6.1 channels (29). Using Kir6.1-Kir6.2 chimeras, Kondo et al. found that the both N- and C-termini are important for this difference while N-terminus seems more critical, in which 5 residues in proximal N-terminus are found to play a major role in the difference (28).
The distal C-terminus (residues 346–424) of Kir6.1 is critical for the PKC-dependent channel inhibition. This protein domain is a direct target of PKC as multiple PKC phosphorylation sites are found in this narrow region. Our results indicate that there are at least four phosphorylation sites in this region, i.e., residues 379, 385, 391 and 397. Our patch clamp study suggests Ser354 is another, a result that is not supported by our in vitro phosphorylation assay. Therefore it is unclear whether Ser354 can be phosphorylated in vivo. Of them Ser397 in the human Kir6.1 has been previously reported to be phosphorylated by an unknown kinase in a global phosphorylation screening study (30). None of the rest has been studied previously. Interestingly, the effects of phosphorylation at these residues are additive or cumulative. Mutation of each individual reduces PKC effects by ~20%. Combined mutations of multiple residues have greater effects. The channel inhibition is almost completely abolished with mutations of all five (including Ser354). Multiple phosphorylation sites often results in sequential phosphorylation, which does not seem to be the case in the Kir6.1/SUR2B, as our results suggest none of the residues shows a dominant effect over others. Of the 5 putative phosphorylation sites, 4 serine residues appear in a clear pattern of repeats (SXRRXN/SXRKXN). To our knowledge, this is the first demonstration regarding such a phosphorylation motif in ion channels.
The distal C-termini of inward rectifier K+ channels play a critical role in channel trafficking and thus control the number of functional channels on cell membrane (31–34). There is an ER retention sequence (RKR, residues 381–383 in Kir6.1 and 369–371 in Kir6.2) (35), which warrants the Kir6 subunit targeting membrane together with the SUR subunit. A dileucine motif (Leu355, Leu356) in Kir6.2 is involved in channel endocytosis (23). Our results indicate that the distal C-terminus of Kir6.1 is also involved in channel activity control (gating). In the narrow C-terminal region where last 4 PKC sites are found, there are 9 alkaline residues including the ‘RKR’ ER retention signal, making this region extremely positively charged. The positive charges appear to favor the open state in the presence of pinacidil or other channel openers. These charges are largely neutralized following phosphorylation of the serine repeats, which may attenuate the channel opening.
It is noteworthy that although we used PMA and pinacidil to optimize the experimental condition, similar PKC-dependent channel inhibition has been shown to play a role in the channel modulation by vasopressin in our previous studies (6). Consistently, endothelin (15), muscarinic M3 receptor agonist (16) and angiotensin II (17) also inhibit the vascular KATP channels in a PKC-dependent manner. Since some of these previous studies were performed in mesenteric arteries and dissociated VSM cells, the channel modulation mechanism demonstrated in the present study is of physiological significance.
Why does the PKC phosphorylation motif exist in Kir6.1 but not in Kir6.2? The phosphorylation motif render 16 phosphorylation sites in a KATP channel with four Kir6.1 subunits. This as well as the cumulative nature of each phosphorylation may allow the channel to be elaborately modulated according to the levels of PKC activation, which is consistent with functional needs of the vascular KATP channel targeted by a variety of vasoconstricting hormones and neurotransmitters. The presence of the phosphorylation motif in the Kir6.1 but not in its close relative Kir6.2 suggests that the vascular KATP channel may have undergone evolutionary optimization serving for the vascular regulation under various physiological and pathophysiological conditions.
Acknowledgments
We thank Dr. Susumu Seino and Dr. Yoshihisa Kurachi for the gifts of Kir6.1 and SUR2B cDNAs. We also thank Dr. Xiaozhou Zhang and Mr. Binh T. Ha for their help in western blot and protein purification respectively. This work is supported by the NIH (HL067890).
Reference List
- 1.Jackson WF. Microcirculation. 2005;12:113–127. doi: 10.1080/10739680590896072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Minami K, Miki T, Kadowaki T, Seino S. Diabetes. 2004;53:S176–S180. doi: 10.2337/diabetes.53.suppl_3.s176. [DOI] [PubMed] [Google Scholar]
- 3.Quayle JM, Nelson MT, Standen NB. Physiological Reviews. 1997;77:1165–1232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- 4.Cole WC, Malcolm T, Walsh MP, Light PE. Circulation Research. 2000;87:112–117. doi: 10.1161/01.res.87.2.112. [DOI] [PubMed] [Google Scholar]
- 5.Shi Y, Wu Z, Cui N, Shi W, Yang Y, Zhang X, Rojas A, Ha BT, Jiang C. American Journal of Physiology. 2007;293:R1205–1214. doi: 10.1152/ajpregu.00337.2007.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi W, Cui N, Shi Y, Zhang X, Yang Y, Jiang C. American Journal of Physiology. 2007;293:R191–199. doi: 10.1152/ajpregu.00047.2007. [DOI] [PubMed] [Google Scholar]
- 7.Standen NB, Quayle JM, Davies NW, Brayden JE, Huang Y, Nelson MT. Science. 1989;245:177–180. doi: 10.1126/science.2501869. [DOI] [PubMed] [Google Scholar]
- 8.Li L, Wu J, Jiang C. Journal of Membrane Biology. 2003;196:61–69. doi: 10.1007/s00232-003-0625-z. [DOI] [PubMed] [Google Scholar]
- 9.Zhang H, Bolton TB. British Journal of Pharmacology. 1995;114:662–672. doi: 10.1111/j.1476-5381.1995.tb17190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morrissey A, Rosner E, Lanning J, Parachuru L, Dhar CP, Han S, Lopez G, Tong X, Yoshida H, Nakamura TY, Artman M, Giblin JP, Tinker A, Coetzee WA. BMC Physiol. 2005;5:1. doi: 10.1186/1472-6793-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamada M, Isomoto S, Matsumoto S, Kondo C, Shindo T, Horio Y, Kurachi Y. Journal of Physiology-London. 1997;499:715–720. doi: 10.1113/jphysiol.1997.sp021963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cui Y, Tran S, Tinker A, Clapp LH. American Journal of Respiratory Cell and Molecular Biology. 2002;26:135–143. doi: 10.1165/ajrcmb.26.1.4622. [DOI] [PubMed] [Google Scholar]
- 13.Miki T, Suzuki M, Shibasaki T, Uemura H, Sato T, Yamaguchi K, Koseki H, Iwanaga T, Nakaya H, Seino S. Nature Medicine. 2002;8:466–472. doi: 10.1038/nm0502-466. [DOI] [PubMed] [Google Scholar]
- 14.Chutkow WA, Pu JL, Wheeler MT, Wada T, Makielski JC, Burant CF, McNally EM. Journal of Clinical Investigation. 2002;110:203–208. doi: 10.1172/JCI15672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park WS, Ko EA, Han J, Kim N, Earm YE. Journal of Cardiovascular Pharmacology. 2005;45:99–108. doi: 10.1097/01.fjc.0000150442.49051.f7. [DOI] [PubMed] [Google Scholar]
- 16.Quinn KV, Cui Y, Giblin JP, Clapp LH, Tinker A. Circulation Research. 2003;93:646–655. doi: 10.1161/01.RES.0000095247.81449.8E. [DOI] [PubMed] [Google Scholar]
- 17.Kubo M, Quayle JM, Standen NB. Journal of Physiology-London. 1997;503:489–496. doi: 10.1111/j.1469-7793.1997.489bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayabuchi Y, Davies NW, Standen NB. Journal of Physiology-London. 2001;530:193–205. doi: 10.1111/j.1469-7793.2001.0193l.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sampson LJ, Davies LM, Barrett-Jolley R, Standen NB, Dart C. Cardiovascular Research. 2007;76:61–70. doi: 10.1016/j.cardiores.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 20.Thorneloe KS, Maruyama Y, Malcolm AT, Light PE, Walsh MP, Cole WC. Journal of Physiology-London. 2002;541:65–80. doi: 10.1113/jphysiol.2002.018101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piao HL, Cui NR, Xu HX, Mao JZ, Rojas A, Wang RP, Abdulkadir L, Li L, Wu JP, Jiang C. Journal of Biological Chemistry. 2001;276:36673–36680. doi: 10.1074/jbc.M106123200. [DOI] [PubMed] [Google Scholar]
- 22.Light PE, Bladen C, Winkfein RJ, Walsh MP, French RJ. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:9058–9063. doi: 10.1073/pnas.160068997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu KL, Huang CS, Jan YN, Jan LY. Neuron. 2003;38:417–432. doi: 10.1016/s0896-6273(03)00256-3. [DOI] [PubMed] [Google Scholar]
- 24.Schulze D, Krauter T, Fritzenschaft H, Soom M, Baukrowitz T. Journal of Biological Chemistry. 2003;278:10500–10505. doi: 10.1074/jbc.M208413200. [DOI] [PubMed] [Google Scholar]
- 25.Cukras CA, Jeliazkova I, Nichols CG. Journal of General Physiology. 2002;120:437–446. doi: 10.1085/jgp.20028621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Proks P, Gribble FM, Adhikari R, Tucker SJ, Ashcroft FM. Journal of Physiology-London. 1999;514:19–25. doi: 10.1111/j.1469-7793.1999.019af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nichols CG. Nature. 2006;440:470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 28.Kondo C, Repunte VP, Satoh E, Yamada M, Horio Y, Matsuzawa Y, Pott L, Kurachi Y. Receptors & Channels. 1998;6:129–140. [PubMed] [Google Scholar]
- 29.Babenko AP, Bryan J. Journal of Biological Chemistry. 2001;276:49083–49092. doi: 10.1074/jbc.M108763200. [DOI] [PubMed] [Google Scholar]
- 30.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 31.Pearson WL, Skatchkov SN, Eaton MJ, Nichols CG. Journal of Membrane Biology. 2006;213:187–193. doi: 10.1007/s00232-006-0058-6. [DOI] [PubMed] [Google Scholar]
- 32.Ma D, Zerangue N, Raab-Graham K, Fried SR, Jan YN, Jan LY. Neuron. 2002;33:715–729. doi: 10.1016/s0896-6273(02)00614-1. [DOI] [PubMed] [Google Scholar]
- 33.Yoo D, Flagg TP, Olsen O, Raghuram V, Foskett JK, Welling PA. Journal of Biological Chemistry. 2004;279:6863–6873. doi: 10.1074/jbc.M311599200. [DOI] [PubMed] [Google Scholar]
- 34.Grishin A, Li H, Levitan ES, Zaks-Makhina E. Journal of Biological Chemistry. 2006;281:30104–30111. doi: 10.1074/jbc.M602439200. [DOI] [PubMed] [Google Scholar]
- 35.Zerangue N, Schwappach B, Jan YN, Jan LY. Neuron. 1999;22:537–548. doi: 10.1016/s0896-6273(00)80708-4. [DOI] [PubMed] [Google Scholar]