

Abstract
Rett syndrome is a neurodevelopmental disorder caused by Mecp2 gene mutations. In RTT patients and Mecp2-null (Mecp2−/Y) mice, norepinephrine (NE) content drops significantly, which may play a role in breathing arrhythmia, sleep disorders and sudden death in RTT. However, the underlying mechanisms for the NE defect are not fully understood. The NE defect may result from decreased NE biosynthesis, loss of catecholaminergic neurons or both. Although deficiency in tyrosine hydroxylase (TH) has been demonstrated, it is possible that dopamine β-hydroxylase (DBH), the critical enzyme converting dopamine to NE, is also affected. To test these possibilities, we studied DBH expressions at mRNA and protein levels in pontine catecholaminergic neurons of Mecp2−/Y mice identified with breathing abnormalities. In comparison to the WT, Mecp2−/Y mice at 2 months of age showed ~50% decrease in the expressions of DBH and TH, at both protein and mRNA levels in the locus coeruleus (LC) region. Consistently, DBH and TH immunoreactivity was markedly decreased in LC neurons of Mecp2−/Y mice. No evidence was found for selective deficiency in TH- or DBH-containing neurons in Mecp2−/Y mice, as almost all TH-positive cells expressed DBH. By counting TH-immunoreactive cells in the LC, we found that the Mecp2−/Y mice lost only ~5% of the catecholaminergic neurons as compared to wild-type, although their LC volume shrank by ~15%. These results strongly suggest that the NE defect in Mecp2−/Y mice is likely to result from deficient expression of not only TH but also DBH without significant loss of catecholaminergic neurons in the LC.
Keywords: Rett syndrome, catercholaminergic neurons, norepinephrine, dopamine β-hydroxylase, tyrosine hydroxylase
Introduction
Rett syndrome (RTT) is a severe neurodevelopmental disorder affecting 1 in 10,000 female births [1,2]. RTT patients undergo an abrupt loss of acquired language and purposeful movement at 6-18 months of age. Subsequently, mental and growth retardation become significant, accompanying with autistic syndromes, cardiac arrhythmia, respiratory irregularities, sleep disorders and epilepsy [2]. About 26% percent of RTT patients die suddenly and unexpectedly [3], likely attributable to abnormalities in brainstem autonomic regulations of the cardio-respiratory systems [4-6].
Mutations in a single gene encoding methyl-CpG-binding protein 2 (MeCP2) on the X chromosome are the major causes of RTT. Mutations or deletion of the Mecp2 gene in mice recapitulate major phenotypes of RTT [7,8]. Studies on RTT patients and mouse models have shown defects in monoamine systems [9-11]. Of them, the norepinephrine (NE) system appears to be the primary site affected by MeCP2 deficiency, which is supported by several lines of evidence. In the brainstem [9,12] and whole-brain preparations [10], the NE content is lower in Mecp2-null mice than the WT. There is a significant reduction in the NE metabolite 3-methoxy-4-hydroxyphenylethylene (MHPG) in the cerebrospinal fluid of RTT patients [13]. The drops in NE content and NE metabolites appear earlier or more severe than those of other neurotransmitter systems [9-11]. The application of exogenous NE or the NE uptake inhibitor desipramine improves respiratory rhythm. Interestingly, desipramine increases the number of TH-positive neurons in the medulla, and extends the lifespan of Mecp2-null mice [9,14,15].
However, it is unclear what mechanisms underlie the defects in the noradrenalinergic system. The NE defect can be a result of a loss of catecholaminergic neurons, lack of expression of NE biosynthetic enzymes, or both. Indeed, the reduced expression of tyrosine hydroxylase (TH), a rate-limiting enzyme for biosynthesis of catecholamines (dopamine, NE and epinephrine), has been observed in the medulla and pons of Mecp2 null mice [9,11,12,16]. Another, perhaps more critical, enzyme for NE biosynthesis is dopamine β-hydroxylase (DBH) that converts dopamine to NE and determines the final product of catecholamine metabolism to be dopamine or NE. Defects in DBH may have impacts on the NE biosynthesis, TH activity and the development of breathing disorders in RTT patients. To test the hypothesis that DBH is defective in Mecp2-null mice, we undertook these studies.
Methods
Animals breeding and genotyping
Mecp2tm1–1Bird mice on a C57BL/6 background, developed by Dr. Adrian Bird (Wellcome Trust Centre for Cell Biology, Institute of Cell and Molecular Biology, University of Edinburgh, Edinburgh, UK), were obtained from The Jackson Laboratory (Bar Harbor, ME) and used as a model for RTT [7]. Owing to the variable phenotypes in Mecp2−/+ females [9,15], all experiments were performed in hemizygous Mecp2 null males (Mecp2−/Y) and WT C57BL/6 males. The Mecp2−/Y mice were purchased from the Jackson Laboratory or generated by cross-breeding heterozygous Mecp2tm1–1Bird knock-out females with C57BL/6 males. All procedures were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and approved by the Georgia State University Institutional Animal Care and Use Committee.
Plethysmograph recording
Conscious mice of 8-10 weeks old without anesthesia were kept in a plethysmograph chamber (~ 40 ml). Another chamber with the same volume was used as a reference chamber that was connected to the same gas source. The chambers were constantly ventilated with air in a flow rate of 50 ml/min, and the animal was allowed to stay in the chamber for 10 min for adapting the chamber environments before recording, followed by a recording for 30 minutes at room temperature. The recorded waveforms in our system resembled that published on the website (www.buxco.com) of Buxco Electronics Inc.
Western blot
Male C57BL/6 and Mecp2−/Y mice (8-10 weeks old) were anesthetized by inhalation of lethal dose halothane (Halocarbon Laboratories, River Edge, NJ), and decapitated. The pons or LC region was dissected free or micropunched. Proteins were obtained with Tissue Extraction Reagent I (Invitrogen) and concentrations determined by the Bradford method. For immuneblotting, primary antibodies against DBH (Immunostar, 1:200) and TH (Sigma, 1:8000) were used. The signal of β-actin (Sigma, 1:4000) was used as loading control. The blotting signals were detected with a chemiluminescent detection system (Pierce). The photograph was processed with Photoshop 7.0 software. To quantify the immunobloting signals, all signals from TH and DBH were normalized to β-actin.
Reverse transcription-PCR and real-time quantitative PCR
The total RNAs were extracted from the pons or LC areas using RNeasy Mini kit (Qiagen). A half microgram of total RNAs was used to synthesize first-strand cDNAs with random hexamer primers. A housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was amplified as an internal control. The presence of TH(GenBank: NM009377), DBH (GenBank: NM138942), MeCP2 (GenBank: NM010788) and GAPDH (GenBank: NM008084) cDNAs was detected with specific primers (detailed primer sequences available upon request). A 905bp segment of TH, 958bp of DBH and 889bp of MeCP2 were amplified.
The cDNAs obtained with reverse transcription reaction were quantified with real-time PCR on an ABI PRISM® 7500 (Applied Biosystems, Warrington, UK) device with Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen). Data was analyzed with GeneAmp 5700 SDS software in 2−ΔΔCT method [17]. Threshold cycles (Ct) were automatically set, and quantitative analysis of TH and DBH expression in Mecp2−/Y mice relative to WT controls was calculated after normalizing to the levels of reference GAPDH gene.
Immunocytochemistry
C56BL/6 and Mecp2−/Y mice at 8 weeks of age were anesthetized as described above, and fixed by transcardial perdusion with 4% (w/v) paraformaldehyde. The brain was removed, and the transverse pontine sections (30 or 40 μm) were cut from brainstem on a cryostat (Leica, Wetzlar, Germany). Diaminobenzidine (DAB) immunocyto-chemistry was performed to detect single immunostaining of TH or DBH. Briefly, the floating sections were incubated with primary antibodies against TH (Sigma, 1:4000) and DBH (Immunostar, 1:2000), respectively, followed by biotin conjugated secondary antibodies (1:1000). The sections were finally exposed to horseradish peroxidase (HRP)-conjugated streptavidin, and reacted with DAB. The DAB reaction was visualized using a Zeiss Axioscope 200 microscope. The immunoreactive intensity and neuronal morphology were compared in 40 μm sections. Meanwhile, the counting of TH immuno-positive neurons was performed in all sections (40μm) or in one out of every other section (30μm). Cell counting was based on the presence of nucleus (lack of staining) in TH immunoreactive neurons. The area of LC was measured in one of every other section with TH immunostaining using ImageJ software (NIH). The LC area of the missing section in between was estimated as an average of its adjacent sections. Subsequently, the LC volume was obtained by adding all areas in these section and multiplication by section thickness (30μm). Double labelings of TH and DBH were performed in 40μm sections. The TH and DBH expression were detected with a Texas red-conjugated and a FITC-conjugated secondary antibody (both from Jackson ImmunoResearch, 1:500), respectively. The fluorescence images were taken with a LSM 510 Zeiss confocal microscope (Jena, Germany).
Statistics analysis
Student’s t test was used for quantitative PCR, western blot and LC volume. The cell number was analyzed with Mann-Whitney test.
Results
Confirmation of breathing abnormalities in Mecp2−/Y mice
In this study, the lack of Mecp2 was first verified in all mutant mice with genotyping protocol provided by Jackson Laboratory. Then, all mice underwent functional tests to detect abnormal autonomic function. For such a purpose, we examined breathing activity using a plethysmograph. Consistent with previous reports [9,18,19], Mecp2−/Y mice displayed alternating periods of fast and slow respiratory frequency and variable inspiratory amplitude with frequent hyperpnea and apnea, which was markedly different from the WT mice of the same age (Suppl Fig. 1).
Decreases in protein expression of DBH and TH in the pons and LC of Mecp2−/Ymice
Since the majority of noradrenergic neurons are located in pons, we first examined the pontine expressions of DBH with Western blot. For a comparison purpose, parallel studies were carried out on TH as well. Expressions of both enzymes were detected with the expected molecular weight (72kDa for DBH and 60kDa for TH, respectively) in each sample. The band intensity of both proteins was clearly lower in Mecp2−/Y mice than in the WT (Fig. 1A). Quantitative analysis was performed by measuring photo-intensity of each band and normalizing it to β-actin in the same sample. The results showed that Mecp2−/Y mice had deficits in pontine DBH and TH proteins by 50.4±8.8% (n=4 animals for each group, P<0.01), and 55.9±12.2% (n=6, P<0.01), respectively (Fig. 1B).
Figure 1. Down-regulation of DBH and TH protein levels in Mecp2−/Y mice.
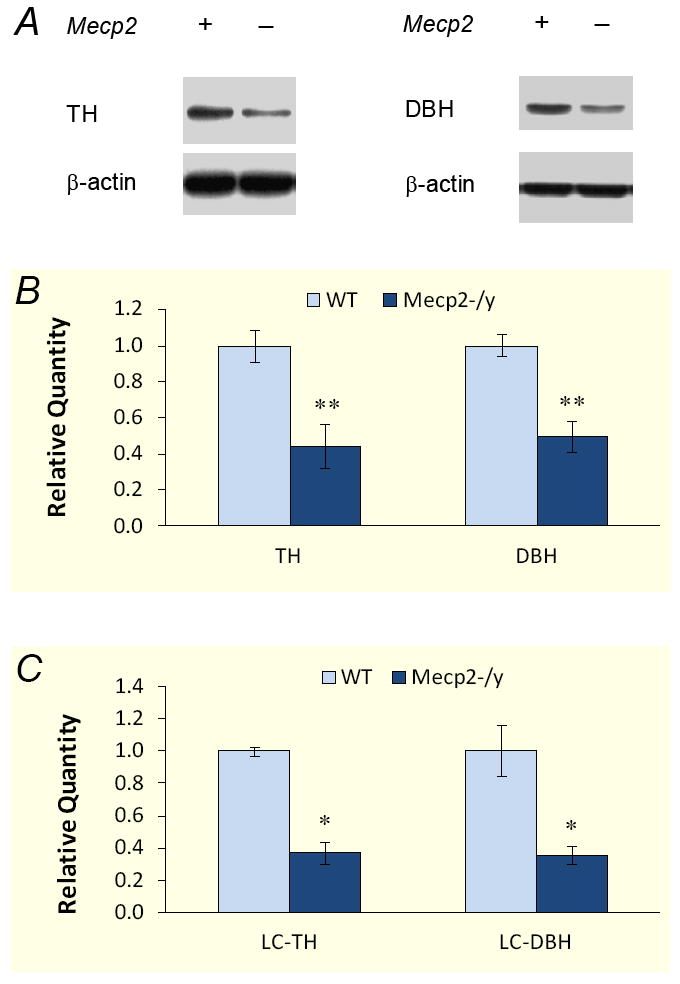
A. Representative western blot for DBH and TH proteins. Pontine extracts were obtained from WT and Mecp2−/Y mice. The intensity of TH (60kDa) and DBH (72kDa) are clearly lower in the Mecp2−/Y mice than the WT. β-Actin (43kDa) was used as loading control. B. Quantitative analysis of Western blot. The presented data was obtained from 6 animals for TH and 4 animals for DBH analysis. The quantity of target proteins was normalized to β-actin. Pontine TH and DBH proteins reduced about 50% in the Mecp2−/Y mice (**, P<0.01). C. Similarly, both TH and DBH decreased by over 60% in LC region of Mecp2−/Y animals (*, P<0.05, n=4 for each group).
The expression of two critical enzymes were further investigated in the micropunches of LC tissues where the A6 neurons are located that are known to produce more than 50% NE in the central nervous system. Consistently, both DBH and TH were reduced by 64.4%±5.6% (n=4, P<0.05) and 62.9%±6.7% (n=4, P<0.05), respectively, in the LC tissue of Mecp2−/Y mice in comparison to WT (Fig. 1C), indicating that the NE biosynthesis in the LC is impaired.
Down-regulation of DBH and TH transcripts in the pons and LC of Mecp2−/Y mice
To understand whether the lower expression of DBH also involves transcriptional mechanisms as TH does, we studied the levels of DBH and TH transcripts. In regular RT-PCR, a single band of DBH and TH in the expected sizes was detected in the pontine tissue of both groups of animals. The photointensity of both DBH and TH products, especially the former, was obviously lower in the Mecp2−/Y mice than the WT (Fig. 2A). Quantitative analysis of the mRNA levels was performed using the 2−ΔΔCT real-time PCR method [17]. In comparison to the WT, the expression of pontine DBH was significantly reduced in Mecp2 null animals by 41.9±4.9% (n=12 samples from 4 animals for each group, P<0.001, unpaired t test). The TH transcripts were reduced to a similar degree by 44.1±7.6% (n=12, P<0.001; Fig. 2B) in pons of Mecp2 null animals. Consistently, both DBH and TH transcripts were dropped by 57.2%±7.4% (n=24, P<0.001) and 57.5%±6.6% (n=24, P<0.001) in LC micropunches, respectively (Fig. 2C), indicating that transcription of these two critical enzymes for NE biosynthesis are affected by Mecp2 knockout.
Figure 2. Reduction in DBH and TH transcripts in Mecp2 null mice.
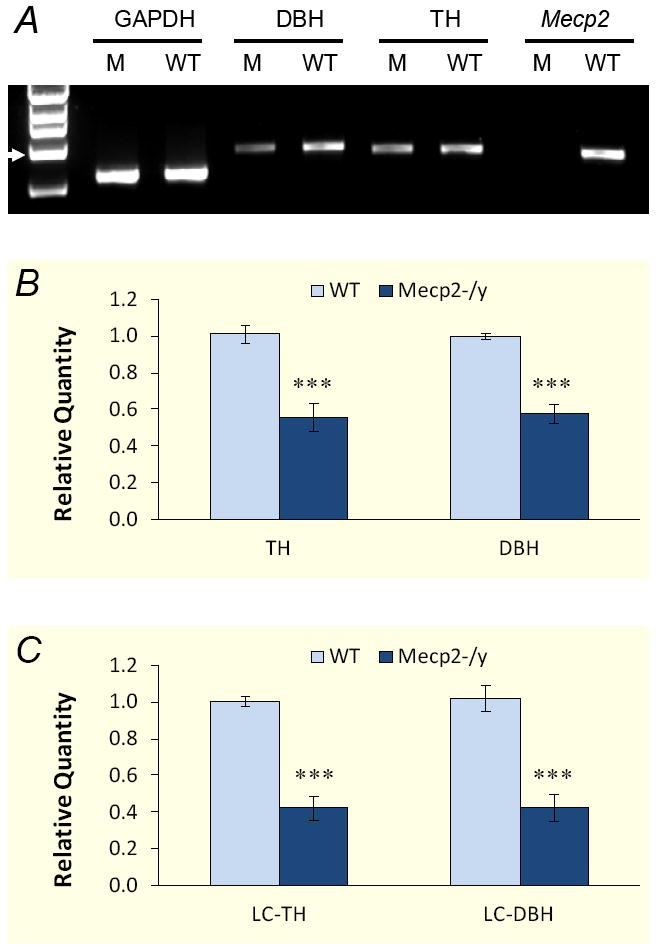
A. RT-PCR analysis showed decreased expression of DBH and TH transcripts in Mecp2 null mutants. Total RNAs (0.5μg each sample) were used for reverse transcription to synthesis the first strand of cDNA. The cDNAs were amplified with DBH- and TH-specific primers. PCR products were tested with gel electrophoresis. Arrow head indicates 1 kb. B. Decreased expression of DBH and TH was confirmed by quantitative real-time PCR. Each bar represents data obtained from 4 animals. The expression of both DBH and TH decreased by nearly 50% in Mecp2−/Y animals (***, P<0.001). C. The expression of both DBH and TH transcripts dropped nearly 60% in LC region of Mecp2−/Y animals (***, P<0.001).
Immunocytochemical evidence for the defects of DBH and TH expression in locus coeruleus
The dramatic decrease in DBH and TH proteins and mRNAs could be a result of a loss of catecholaminergic neurons or ill expressions of DBH and TH. To address these, we performed immunocytochemical studies on the LC (A6) catecholaminergic neurons in transverse pontine sections. The immunoreactivity for both DBH and TH, especially the former, was lower in the Mecp2−/Y mice than in the WT (Fig. 3A-D). Although individual neurons were well recognized in both groups of animals, the WT tissue showed neurons with the primary dendrites and sometimes the secondary and tertiary dendrites revealed. In comparison, only soma was seen in the Mecp2−/Y mouse pontine sections, consistent with the deficient mRNA and protein levels in Mecp2−/Y mice.
Figure 3. DBH and TH immunoreactivity in locus coeruleus (LC).
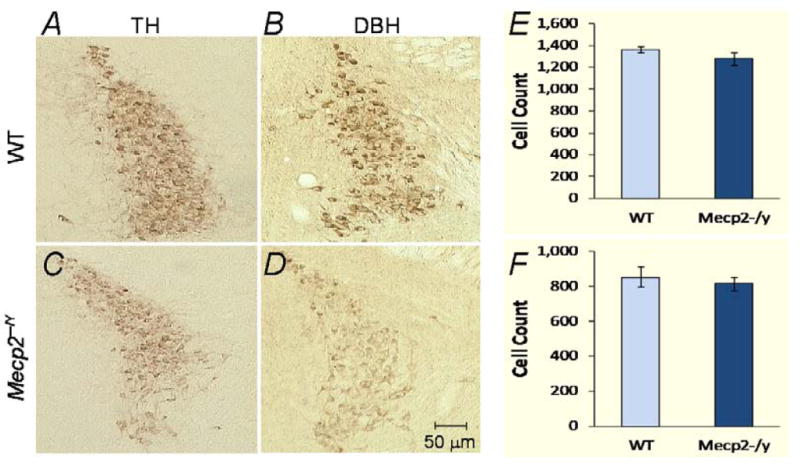
The expression of DBH and TH was examined in LC. A-D. The HRP - DAB method was used to visualize the immunoreactivity in transverse pontine sections. A marked reduction of both TH and DBH immunoreactivities is seen in the Mecp2−/Y mice. In addition, positive immunostains were only located in soma of Mecp2−/Y neurons, but also seen in proximal and secondary dendrites of the WT. E,F. TH-immunoreactive neurons were counted in transverse pontine sections stained with the HRP-DAB method. A cell count was considered when the nucleus (devoid of TH staining) was seen. Mecp2−/Y mice showed a 4.5% reduction in TH-positive LC neurons by a counting in alternative sections (30μm thick) from 3 pairs of mice (E), and a 6.2% reduction by counting all cells in every tissue section (40μm thick) from 2 pairs of mice (F). No significant difference (P>0.05, Mann-Whitney test) was found in either or combined analysis (5.4%). Data are shown as means ± S.D.
The decrease in DBH and TH expressions may have impact on neurotransmitter production leading to a switch of NE-ergic neurons to dopaminergic ones. Thus, we further studied the LC catecholaminergic neurons to determine whether there is a selective loss of DBH- or TH- immunoreactive cells using double immunestainings with FITC (DBH) and Texas red (TH). We found that almost all DBH-positive cells were also positively labeled with TH (Fig. 4A), suggesting that there may not be a change in neurotransmitter phenotype of LC neurons in Mecp2−/Y mice.
Figure 4. Lack of selective loss of catecholaminergic neurons in LC of Mecp2−/Y mice.
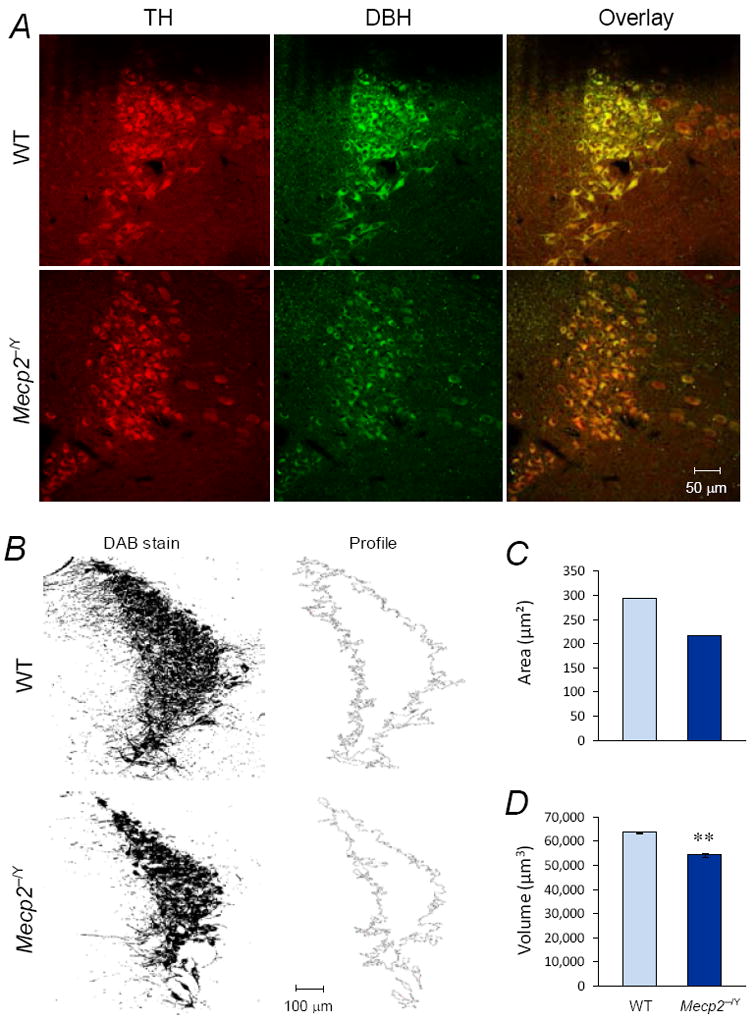
A. Double labelings of TH (red) and DBH (green) were conducted in LC neurons with immunofluorescence method under an identical condition. Two transverse pontine sections presented were obtained from a WT (upper) and Mecp2−/Y mice (lower), respectively. Although the Mecp2−/Y mouse showed weaker staining of TH and DBH, almost all TH-positive cells expressed DBH in both WT and Mecp2−/Y mice. No evident was found for selective expression of either TH or DBH in these mice. B. To measure the LC volume, the occupancy areas of TH-immunoreactive cells were detected (left), and extracted from the background (right). C. The LC area of representative sections in B was shown as means ± S.D. D. In comparison to WT, Mecp2−/Y mouse showed smaller LC volume (**, P<0.01).
TH-positive neurons in the LC were counted in five pairs of WT and Mecp2−/Y mice with DAB method. A cell count was considered when the nucleus of the cell was clearly seen. In 2 pairs of mice, TH-positive neurons were counted in all 40μm brainstem sections along the entire LC column. In the other 3 pairs, TH-positive neurons were counted in alternative 30μm brainstem sections. Both methods showed that the number of LC TH-positive neurons was ~5% less in Mecp2−/Y mice than in the WT (Fig. 3E,F). This seems to be an underestimate, as some of the weakly TH-immunoreactive cells might have been missed in Mecp2−/Y mice.
Realizing that the overall areas of the LC were smaller in Mecp2−/Y mice, we measured the LC volume in a series of transverse sections. We found that the total volume of the LC was ~15% less in Mecp2−/Y mice (Fig. 4B-D). Considering 20-30% less in the body weight of the Mecp2−/Y mice, the shrinkage in the LC nuclei seems moderate. Taken together, these results suggest that there is no major loss of LC catecholaminergic neurons in the Mecp2−/Y mice.
Discussion
This is the first demonstration of the defect in DBH, the rate-limiting enzyme specific for NE production, in Mecp2−/Y mice. The evidence was found by systematic studies of the DBH expression at the mRNA, protein and immunocytochemical levels and by rigorous comparison with the TH defect and the DBH expression in WT mice. We have also examined two potential mechanisms underlying the NE defect in Mecp2−/Y mice, i.e., the expression of rate-limiting enzymes for NE biosynthesis and total number of catecholaminergic neurons in the LC, as defect in each could lead to NE deficiency.
Previous studies have shown that NE content drops by nearly 50% in the whole brain [10] and the medulla oblongata of Mecp2−/Y mice of 2 months old [9]. As a major source of central NE, pontine NE-ergic neurons produce more than 70% NE in central nervous system and are very likely targeted by Mecp2 knockout. Although no significant change in pons is indicated in early study, a recent study states a clear reduction of pontine NE content [12]. The latter is also supported by our data and others’ [11,12,16], showing that the expression of TH is declined in the LC.
TH is the first and common rate-limiting enzyme for biosynthesis of catecholamines (DA, NE and epinephrine), and has been widely used as a loose indicator for NE-ergic neurons in previous studies. However, DBH is the critical enzyme controlling NE production and determines the neurotransmitter phenotype of catecholaminergic neurons. Given that the expression of TH and DBH could be differentially regulated during the development of catecholamine systems [20,21], DBH appears to be a direct and more specific indicator for NE-ergic neurons. A deficiency in DBH may lead to accumulation of dopamine that could potentially serve as a feedback inhibitor reducing TH activity/expression. Indeed, we have observed a major decrease in DBH expression in LC neurons, comparable to the defective TH expression shown in the present study as well as by others [11,12,16]. The decrease in DBH/TH protein expressions is likely to occur at the transcriptional level, as our RT-PCR and quantitative PCR also showed a similar decrease in the mRNAs of these enzymes. MeCP2-binding sites have been found in the TH promoter [11,22], and similar studies are necessary to demonstrate whether DBH is also subject to the direct regulation by MeCP2. In addition, indirect mechanisms may be also involved, such as suppression of BDNF in Mecp2−/Y mice [23,24].
Since a loss of DBH may cause a poor NE production and potentially could switch the NE-ergic neurons to DA-ergic in Mecp2−/Y mice, we have investigated this possibility using immunofluresence. However, we did not find selective loss of DBH-containing neurons in the LC. The reduction of both DBH and TH to a similar degree thus may help retain the neurochemical phenotypes of LC neurons. Therefore, the decreased expressions of these NE-biosynthetic enzymes are likely to play a role in the reduction in NE content found in RTT patients and Mecp2−/Y mice.
In contrast to the major suppression of DBH and TH expression, the Mecp2-knockout does not seem to compromise survival of LC catecholaminergic neurons. Our results have shown that TH and DBH were co-localized in most LC neurons, and the number of LC TH-immunoreactive neurons is only about 5% less in 2-month-old Mecp2−/Y mice than in the WT. Such a small reduction in cell numbers, however, is inconsistent with observation by Roux et al. [16] showing that Mecp2−/Y mice lose about 20%TH-positive neurons in LC. This discrepancy could be caused by the employment of different antibodies and immunostaining methods, as the weaker immnostaining may not have allowed a counting of every catecholaminergic cells in the LC. Unlike its effect on the LC neurons, the Mecp2-knockout causes a drastic loss of TH-immunoreactive neurons in the medulla oblongata [9], suggesting that different mechanisms may exist for the regulation of these brainstem neurons. With respect to LC neurons, the lack of NE content is more likely to be related to the deficient expression of DBH/TH in the LC than a loss of catecholaminergic neurons.
In addition to the two mechanisms that we have examined, the NE defect may be produced by an elevated activity of NE degradation enzymes. However, the decrease in NE metabolite concentration in human subjects does not seem to suggest an enhanced NE degradation in RTT [13].
The existence of most of catecholaminergic neurons in the Mecp2−/Y mice is an encouraging finding, as therapeutical modalities may be formulated by targeting on mechanisms endogenous in these cells such as NE uptake and enzymes for NE biosynthesis and degradation. Indeed, the NE uptake inhibitor desipramine has been recently shown to improve respiratory rhythm activity, increase the number of TH-immunoreactive neurons, and extend lifespan of Mecp2-null mice [14,15]. Since there is a dramatic loss of monoaminergic neurons in the medulla [9], and since the action sites of desipramine are still unclear [14,15], our results showing that most LC neurons survive the Mecp2 knockout appear to provide a cellular basis of the potential desipramine targets. Concerning the population mass of LC neurons, it is likely that substantial norepinephrinergic neurons remain available for participating in the modulation of NE content in the central nervous system of the Mecp2−/Y mice and perhaps RTT patients as well.
In conclusion, we have demonstrated DBH deficiency in Mecp2−/Y mice in terms of its transcripts, protein expressions and immunoreactivity, providing the first evidence for the defect of this NE-specific rate limiting enzyme. Furthermore, we found the existence of most, if not all, NE-ergic neurons in the locus coeruleus, which is encouraging as therapeutical intervention may be formulated by targeting at these neurons.
Supplementary Material
Acknowledgments
This work was supported by the NIH (HD060959), the American Heart Association (09GRNT2010037) and the International Rett Syndrome Foundation (IRSF).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Francke U. Mechanisms of disease: neurogenetics of MeCP2 deficiency. Nat Clin Pract Neurol. 2006;2:212–221. doi: 10.1038/ncpneuro0148. [DOI] [PubMed] [Google Scholar]
- 2.Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Kerr AM, Armstrong DD, Prescott RJ, Doyle D, Kearney DL. Rett syndrome: analysis of deaths in the British survey. Eur Child Adolesc Psychiatry. 1997;6(Suppl 1):71–74. [PubMed] [Google Scholar]
- 4.Julu PO, Kerr AM, Hansen S, Apartopoulos F, Jamal GA. Immaturity of medullary cardiorespiratory neurones leading to inappropriate autonomic reactions as a likely cause of sudden death in Rett’s syndrome. Arch Dis Child. 1997;77:464–465. doi: 10.1136/adc.77.5.463c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guideri F, Acampa M, Hayek G, Zappella M, Di Perri T. Reduced heart rate variability in patients affected with Rett syndrome. A possible explanation for sudden death. Neuropediatrics. 1999;30:146–148. doi: 10.1055/s-2007-973480. [DOI] [PubMed] [Google Scholar]
- 6.Weese-Mayer DE, Lieske SP, Boothby CM, Kenny AS, Bennett HL, et al. Autonomic nervous system dysregulation: breathing and heart rate perturbation during wakefulness in young girls with Rett syndrome. Pediatr Res. 2006;60:443–449. doi: 10.1203/01.pdr.0000238302.84552.d0. [DOI] [PubMed] [Google Scholar]
- 7.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 8.Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, et al. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- 9.Viemari JC, Roux JC, Tryba AK, Saywell V, Burnet H, et al. Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. J Neurosci. 2005;25:11521–11530. doi: 10.1523/JNEUROSCI.4373-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ide S, Itoh M, Goto Y. Defect in normal developmental increase of the brain biogenic amine concentrations in the mecp2-null mouse. Neurosci Lett. 2005;386:14–17. doi: 10.1016/j.neulet.2005.05.056. [DOI] [PubMed] [Google Scholar]
- 11.Samaco RC, Mandel-Brehm C, Chao HT, Ward CS, Fyffe-Maricich SL, et al. Loss of MeCP2 in aminergic neurons causes cell-autonomous defects in neurotransmitter synthesis and specific behavioral abnormalities. Proc Natl Acad Sci U S A. 2009;106:21966–21971. doi: 10.1073/pnas.0912257106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taneja P, Ogier M, Brooks-Harris G, Schmid DA, Katz DM, et al. Pathophysiology of locus ceruleus neurons in a mouse model of Rett syndrome. J Neurosci. 2009;29:12187–12195. doi: 10.1523/JNEUROSCI.3156-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zoghbi HY, Percy AK, Glaze DG, Butler IJ, Riccardi VM. Reduction of biogenic amine levels in the Rett syndrome. N Engl J Med. 1985;313:921–924. doi: 10.1056/NEJM198510103131504. [DOI] [PubMed] [Google Scholar]
- 14.Roux JC, Dura E, Moncla A, Mancini J, Villard L. Treatment with desipramine improves breathing and survival in a mouse model for Rett syndrome. Eur J Neurosci. 2007;25:1915–1922. doi: 10.1111/j.1460-9568.2007.05466.x. [DOI] [PubMed] [Google Scholar]
- 15.Zanella S, Mebarek S, Lajard AM, Picard N, Dutschmann M, et al. Oral treatment with desipramine improves breathing and life span in Rett syndrome mouse model. Respir Physiol Neurobiol. 2008;160:116–121. doi: 10.1016/j.resp.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 16.Roux JC, Panayotis N, Dura E, Villard L. Progressive noradrenergic deficits in the locus coeruleus of Mecp2 deficient mice. J Neurosci Res. 2009 doi: 10.1002/jnr.22312. In press. [DOI] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Stettner GM, Huppke P, Brendel C, Richter DW, Gartner J, et al. Breathing dysfunctions associated with impaired control of postinspiratory activity in Mecp2-/y knockout mice. J Physiol. 2007;579:863–876. doi: 10.1113/jphysiol.2006.119966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bissonnette JM, Knopp SJ. Effect of inspired oxygen on periodic breathing in methy-CpG-binding protein 2 (Mecp2) deficient mice. J Appl Physiol. 2008;104:198–204. doi: 10.1152/japplphysiol.00843.2007. [DOI] [PubMed] [Google Scholar]
- 20.Badoyannis HC, Sharma SC, Sabban EL. The differential effects of cell density and NGF on the expression of tyrosine hydroxylase and dopamine beta-hydroxylase in PC12 cells. Brain Res Mol Brain Res. 1991;11:79–87. doi: 10.1016/0169-328x(91)90024-r. [DOI] [PubMed] [Google Scholar]
- 21.Hirsch MR, Tiveron MC, Guillemot F, Brunet JF, Goridis C. Control of noradrenergic differentiation and Phox2a expression by MASH1 in the central and peripheral nervous system. Development. 1998;125:599–608. doi: 10.1242/dev.125.4.599. [DOI] [PubMed] [Google Scholar]
- 22.Yasui DH, Peddada S, Bieda MC, Vallero RO, Hogart A, et al. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc Natl Acad Sci U S A. 2007;104:19416–19421. doi: 10.1073/pnas.0707442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen WG, Chang Q, Lin Y, Meissner A, West AE, et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- 24.Guo H, Hellard DT, Huang L, Katz DM. Development of pontine noradrenergic A5 neurons requires brain-derived neurotrophic factor. Eur J Neurosci. 2005;21:2019–2023. doi: 10.1111/j.1460-9568.2005.04016.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.