

Abstract

Experiments and atomistic simulations of polypeptides have revealed structural intermediates that promote or inhibit conformational transitions to the native state during folding. We invoke a concept of “kinetic frustration” to quantify the prevalence and impact of these behaviors on folding rates within a large set of atomistic simulation data for 10 fast-folding proteins, where each protein’s conformational space is represented as a Markov state model of conformational transitions. Our graph theoretic approach addresses what conformational features correlate with folding inhibition and therefore permits comparison among features within a single protein network and also more generally between proteins. Nonnative contacts and nonnative secondary structure formation can thus be quantitatively implicated in inhibiting folding for several of the tested peptides.
Introduction
Theoretical and computational modeling has provided many insights into the remarkable ability of proteins to rapidly fold from unstructured coils into their native, functional conformations.1,2 Especially for small structured proteins, entire folding processes can be investigated via atomistic, equilibrium molecular dynamics (MD) simulations, where the ability to sample multiple folding events (with μs simulations) with a transferable force field is an important milestone in algorithm development and hardware parallelization.3−7 When multiple folding events are observed, the underlying kinetics and conformational features that promote structural transitions and the eventual attainment of the native state can be statistically compared. Such studies reveal important characteristics of the underlying free energy landscape (FEL), the high-dimensional surface of hills and valleys that govern the likelihood of structural transitions and the occupancy probabilities of energetically coherent states, called conformational substates.8,9 For structured proteins, the FEL has been conceptualized as a funnel with a low-energy native ensemble at its global minimum, where near-native intermediates are kinetic neighbors, and a nonnative ensemble composed of freely interconverting conformers at some further reaction distance.10,11 While the majority of protein functions are accomplished via the native ensemble, quantifying the structural and kinetic characteristics of the nonnative ensemble can aid calibration of coarse-grained polypeptide models12−14 and improve our understanding of folding initiation pathways,15,16 protein misfolding,17 protein aggregation,18,19 and synergistic folding (i.e., folding in tandem with a binding partner).20
Although nonnative ensembles recapitulate several properties of idealized random-coil models,21,22 they have also been shown to deviate from polymeric predictions in important ways. Substantial secondary structure can accrue in the nonnative ensemble,6,23,24 and these nucleation locations have been implicated as consistent waypoints in folding pathways.25,26 Lindorff–Larsen et al.5 likewise showed that for transition pathways specifically, secondary structure accumulates before native contacts are formed, a temporal bias that is inconsistent with an idealized nonnative ensemble. From the kinetic perspective, another surprise is that the nonnative ensemble can be modeled as a hub-like transition map,27 where interchange between unfolded peptide geometries is mediated preferentially via the native (hub) ensemble instead of by direct routes28 (see ref (29) for a contrasting interpretation). The minimally frustrated model of protein folding harmonizes some of these observations by recognizing that folding is energetically downhill and will thus avoid the enthalpic frustration of nonnative structure formation.30−33 Analogously, we can ask whether the nonnative ensemble is minimally frustrated in a kinetic sense. Does folding proceed sequentially34 from unfolded to folded substates or are there off-pathway kinetic inhibitors populating the FEL?
Computational studies have invoked Cartesian, angular, topological, subspace-projection, or other structural descriptors to identify nonnative conformational states,25,35−37 but assessing their impact on folding rates requires additional analysis. To quantify the kinetic contribution to folding of specific conformational substates, we present here a methodology that (1) permits comparisons of kinetic inhibition across multiple folding events and between multiple proteins and (2) can query any proposed structural parameters that may impact folding kinetics. Instead of only specifying the existence of kinetic traps, hubs, or preferential pathways in MD trajectories, we quantify the overall kinetic burden, or kinetic frustration, that structural deformations (secondary structure, tertiary structure, standard RMSD-to-native, or others) effect in protein simulations. In the rest of this introduction we give an overview of our model’s assumptions, justification for a topological definition of kinetic frustration, and primary results.
We invoke a kinetic modeling of MD simulation data where a simulation trajectory and its conformers are represented as (1) sets of clusters, or conformational substates, whose kinetically indistinguishable members share conformational features, and (2) transitions, which capture the observed jumps between substates. Such a network of substates (nodes) and edges, when constructed with an appropriate lag-time between sampled trajectory snapshots and clustering criteria, satisfies the properties of a Markov State Model (MSM).38,39 These models are guided by the motivation to equate conformational transitions with probability flow, enabling multistep transition pathways to be associated with a probability and expected duration even if the path itself was never observed within contiguous trajectory frames. By representing a protein’s FEL as an evolving finite markov chain, MSMs permit computation of the stationary distribution, the unique set of substate probabilities that is stable over time. It is then possible to calculate the expected time for any substate to transition to the assigned native ensemble, that is, the mean first passage time (MFPT) or transit time.40 MFPT values express temporal expectations for random walks along the weighted edges of the conformational network.41,42 They are robust43,44 and can be compared to diffusional models of folding45,46 and nanosecond laser T-jump experiments.47 Whereas MFPTs necessarily are a function of two specified end points, our concern is only with those transition paths that terminate at the native ensemble, a convention implicit throughout this study and indicated by the subscript of MFPT values, τnat. Can these values tell us which substates are responsible for accelerating or hindering folding? Not directly, but that exact information is revealed when substates are theoretically removed from the transition network and the change in τnat values among the rest of the nonnative ensemble compared. Kinetic frustration, quantified in frustration scores, f̅nat, captures these changes and quantifies the degree to which a particular conformational substate state inhibits or facilitates transitions to the native state. The terms inhibit and facilitate summarize a substate’s topological neighborhood with respect to the native ensemble: a substate that facilitates folding is highly connected to native or native-like substates, whereas a folding inhibitor promotes transitions to non-native regions of the FEL.
MFPTs and frustration scores, f̅nat, are therefore related but, importantly, distill different information. τnat values reflect expected transit times given a network structure and designated native ensemble, whereas frustration scores quantify the impact on the network given the substate of interest. Nodes that have equal transit times need not share frustration scores, for example (see Figures 1 and 2B). Moreover, because a demarcated native state is inherent to their definition, frustration scores go beyond a quantification of local topology, such as average neighbor connectivity,27 which treats equally links leading toward or away from the native state (for a given nonnative conformational substate). That is, frustration scores are sensitive to the native state location, whereas local connectivities are not (cf. Supporting Information (SI) Figure S6), an advantage that warrants the more computationally demanding node by node perturbation approach presented here. Frustration scores are thus individually computed for all nodes in the nonnative ensemble by observing changes in τnat values when each is removed from the network model (Figure 1),48 a process akin to the eigenvalue estimation problem in matrix perturbation theory.49 Observations obtained by altering the transition network in this way provide a quantitative framework for understanding its unperturbed behavior. Specifically, each frustration score f̅nat i can be understood as the mean percentage change in all transit times from all possible paths as a result of node i, a kinetic interpretation lacking from many local topology metrics. Substates with f̅nat > 0 are labeled facilitators since folding rates would decrease (i.e., τnat increase) in their absence; states with f̅nat < 0 are inhibitors, or kinetic traps, in that folding rates would increase (τnat decrease) if they were to be removed from the conformational landscape. We thus invoke the concept of kinetic frustration because MFPT values alone cannot elucidate these causal relationships.
Figure 1.

Computing frustration scores, f̅nat, for a model transition network. (A) Each conformational substate in the nonnative ensemble (1...knn) is colored according to frustration scores, f̅nat; substate diameters indicate the stationary probability. The native ensemble is represented as a single green substate. Transition probabilities are shown along observed transitions where values above the transition path always denote left → right transitions and values beneath the arrow refer to moving right → left. (B) Computed MFPTs, τnat values, for each nonnative substate to reach the native ensemble. (C) Procedure for computing f̅nat. Each nonnative substate is removed from the transition matrix (states i = 1...4, top to bottom) and transit times for all remaining knn – 1 substates are compared with unperturbed values (left panels: black bars, unperturbed; red, gray, or blue bars, perturbed). Relative changes in transit times (wedges, right panels) are averaged over remaining substates to yield f̅nat (dashed lines). These frustration scores are then depicted by the color scale on the original intact network (A). Substates 1 and 2 have identical MFPTs, whereas f̅nat values indicate substate 2 is a facilitator and increases folding rates from all other substates by an average of 25%, while substate 1 is kinetically neutral. Substate 4 is a kinetic trap, slowing all transit times by 30% on average in the unperturbed network.
Figure 2.

Network representations of substates and transitions. (A) Nodes indicate conformational substates determined with RMSD clustering;64 node diameters are proportional to substate probability. The native ensemble, green, was determined by modularity optimization.65 White diamonds indicate the substate containing the conformation closest to the experimental structure. Black diamonds indicate the substate containing the native conformation (see Methods). Frustration scores, f̅nat, are denoted by the color spectrum, centered at f̅nat = 0. Positive scores, blue, indicate substates that facilitate transition to the native ensemble; negative scores, red, indicate kinetic traps. (B) Comparison of frustration scores, f̅nat, and transit times, τnat (μs), for nonnative substates in part A. Color values correspond to frustration scores as in part A.
Within our simulation data set of 10 fast-folding proteins, substantial kinetic traps were observed for four proteins (TRP, BBL, PB, and HMDM) whereas kinetic inhibition was chiefly absent in the nonnative ensembles of WW, PG, and A3D. The largest frustration scores (i.e., most extreme facilitators) were observed in the WW and PG simulations, but none of the 10 globular proteins examined (Table 1) presented a single facilitator, an ultimate gatekeeper substate, to the native ensemble. As shown in Figure 2, kinetic traps were unequally distributed throughout the nonnative ensembles. The transition networks, or transition maps, did display unique topological features, and we were able to ask to what relative degree secondary structure, tertiary structure, and nonnativeness (standard RMSD-to-native) are associated with positive or negative kinetic frustration. We chose these structural parameters because of their broad interpretability and popularity for monitoring folding progress50,51 but emphasize that the approach is compatible with any geometric or energetic feature that can be computed for all trajectory frames.
Table 1. Simulation Dataa.
| protein | Nres | ttotal (μs) | k | PDB | Temp (K) | Nf | Nu | rnc (Å) |
|---|---|---|---|---|---|---|---|---|
| trp-cage (TRP) | 20 | 208 | 417 | 2JOF | 290 | 12 | 12 | 1.5 |
| BBA | 28 | 325 | 999–1 | 1FME | 325 | 14 | 14 | 2.6 |
| villin headpiece (VHP) | 35 | 125 | 251 | 2F4K | 360 | 34 | 34 | 1.3 |
| WW-domain (WW) | 35 | 1137 | 2274 | 2F21 (4–39) | 360 | 12 | 11 | 1.4 |
| BBL | 47 | 429 | 860 | 2WXC | 298 | 12 | 11 | 4.8 |
| protein B (PB) | 47 | 104 | 2310 | 1PRB (7–53) | 340 | 19 | 19 | 3.4 |
| homeodomain (HMDM) | 52 | 327 | 654 | 2P6J | 360 | 27 | 28 | 3.7 |
| protein G (PG) | 56 | 1155 | 2310–2 | 1MI0 (10–65) | 350 | 12 | 13 | 1.2 |
| alpha 3D (A3D) | 73 | 707 | 1414 | 2A3D | 370 | 12 | 12 | 2.9 |
| lambda repressor (LAMDA) | 80 | 643 | 1293–1 | 1LMB (6–85) | 350 | 10 | 12 | 1.9 |
A summary of the proteins and simulations studied, adapted from Lindorff-Larsen et al.5 Data columns indicate sequence length (Nres), total aggregated simulation duration (ttotal), number of conformational substates (and any substates excised during transition matrix MLE) (k), Protein Data Bank accession code (residue indices), simulation temperature, number of folding events (Nf), number of unfolding events (Nf), and the native-ensemble RMSD cutoff (rnc). All figures and tables order proteins according to increasing sequence length.
Folding is a conformationally heterogeneous process,52 but the recognized prevalence of preferred folding routes53 and transition pathways54 highlights the need for tools linking specific nonnative substates to folding kinetics. Quantifying these relationships is a legitimate aim in its own right, but our findings relate to the wider problem of predicting the kinetic impact of direct perturbations to protein systems. Mutations, small molecule ligands, or solvent conditions that modulate the populations of conformational substates can influence folding rates or folding routes,55−57 and quantifying any such changes therefore has applications to pathway inhibition, aggregation-based diseases, and protein engineering.58−61
Methods
MD Simulations
We applied our analysis to 10 proteins within a large simulation data set generated by D. E. Shaw Research as reported in Lindorff-Larsen et al.5 and analyzed further elsewhere.29,33,62,63 Aggregate simulations of the 10 proteins comprise 5.1 ms of total sampling where each protein undergoes at least 10 folding and unfolding events (Table 1); details for the simulations and folding event classification are contained in the original reference. Clustering and all subsequent analysis was performed on the Cα coordinates. Snapshots were recorded every 200 ps. In this study, multiple simulations for the same protein, if present, were concatenated.
Determining Conformational Substates
To identify conformational substates for each protein, we performed hierarchical clustering with MSMBuilder2.64 Trajectories were first subsampled to obtain snapshots every 50 ns based on implied time scale plots (SI Figure S1), then clustered into substates using root mean squared distance (RMSD) and Ward’s algorithm.66 The number of substates, k (see Table 1), is a heuristic user parameter that was selected to be approximately equal to (simulation frames)/10.67 This parameter has been shown to have little dependence on peptide length, Nres.68 The k values chosen here correspond closely to those in ref (29). The transition probability matrix P was then approximated using the MSMBuilder2 maximum likelihood estimation (MLE) routine, and substates not included in the estimated matrix (i.e., those separate from the primary connected component) were excised from subsequent analysis. Connected singletons (substates with a single member) were retained, however, and constituted 0% of total conformers for BBA, BBL, PB, and TRP and 0.8–10% for A3D, HMDM, LAMDA, PG, VHP, and WW. Distributions of cluster sizes (number of member conformers) and widths (defined as mean pairwise RMSD of any two substate members) are given in the Supporting Information (Figures S4 and S5).
Defining the Native Ensemble
Our network folding model requires a demarcated native state to function as a kinetic end point, that is, a theoretical absorbing state40 where folding is defined as complete. Selecting the largest conformational substate,5 the substate closest to the PDB-deposited coordinates, or a hard RMSD threshold is too restrictive, excluding many substates with ‘native-like’ properties and artificially increasing theoretical τnat values.69 Instead, we chose to designate a native ensemble, or a set of conformational substates that interconvert more frequently with each other than with outside substates. Such a graph property is captured by an algorithm called modularity optimization,65 and is particularly suited for this classification task in that it reflects and adapts to the actual network topology, unlike an RMSD threshold. Modularity optimization proceeds by initially designating each substate as its own ensemble and then iteratively combining them until only highly intraconnected ensembles remain, at which point modularity is maximized. For a transition network, modularity is defined as
where cij is the number of transitions between substates i and j, ki is the total number of transitions to substate i, kj is the number of transitions to substate j, 2m is the total transition count in the network, and δ(si,sj) = 1 when substates i and j reside in the same ensemble s and 0 otherwise (elsewhere lnn or ln denote total edges among nodes in the nonnative or native ensembles, respectively). Other formulations for optimizing modularity are possible, including normalized cut and conductance criteria.70 Maximizing W yields multiple ensembles for each of 10 analyzed transition networks, but only one per network will be defined as the native ensemble. Within each candidate ensemble, five random conformers were sampled from every constituent substate, and the aggregate number of conformers within a cutoff rnc to the PDB-deposited native structure were counted and compared with similar counts from the remaining candidate ensembles. The ensemble with the most conformers under the cutoff was designated to be the native ensemble, and all its substates, not only those under the cutoff, were included (all substates not in the native ensemble were defined to be in the nonnative ensemble). The cutoff itself, rnc, was determined by identifying the RMSD value such that 5% of total trajectory frames were within rnc Å from the PDB-deposited crystal structure (Table 1). For 8 of 10 proteins, 100% of the frames with RMSD < rnc were found in the native ensemble; the same values were 82% for BBA and 93% for BBL. Substates containing the snapshot nearest the experimental native structure were always contained in the assigned native ensemble (Figure 2, white diamonds). The number of substates assigned to the native and nonnative ensembles was kn and knn, respectively (Table 1). Although the nonnative ensembles were partitioned variously during iterations of modularity optimization, the constituent substates of the (eventually defined) native ensemble were in fact identically preserved through all iterations for all proteins. The algorithm converged in seconds for all networks. Modularity optimization operates exclusively on transitions counts; however, we observed that secondary structure also separated cleanly between the native and nonnative ensembles as a result of this classification (SI Figure S2). The identified native ensembles are shown in green in the network representations (Figure 2). For purposes of computing RMSD, a native conformation was defined to be the trajectory snapshot nearest the theoretical mean structure of the entire native ensemble (Figure 2, black diamonds, and Figure 6, tube representations).
Figure 6.

Ensemble representation of kinetic traps compared with native structure. (A) Representative structures of kinetic traps, shown in topological context in SI Figure S8. (B) Comparison of substate widths (intrasubstate pairwise RMSD). Red, substates classified as kinetic traps; gray, all nonnative substates; green, all native substates.
Defining Q and Secondary Structure
The proportion of native contacts present in any trajectory conformer, Q, is a useful reaction coordinate for monitoring folding progress71,72 or modeling energy barriers.73 It is a degenerate quantity in that many distinct conformers could map to an identical Q value.74,75 We defined native contacts as those residue pairs whose separation (Cα–Cα) was less than 10 Å for at least 65% of the conformers within the native ensemble (SI Figure S3). Native contacts separated by fewer than 7 amino acids in the primary structure were excluded. We denote the percentage of native contacts as Qn(t) and the percentage of nonnative contacts as Qnn(t) for some time t.
We quantified the presence of secondary structure in each trajectory frame using Protein Secondary Element Assignment (P-SEA), which labels every residue as in either an unstructured coil, α helix, β sheet, or ‘other’ configuration76 (SI Figure S2). An ‘ideal’ sequence of native secondary structure assignment was defined as the residue-wise assignment most common within the native ensemble and termed the structure sequence. The presence of native secondary structure throughout the simulation was quantified by dividing the number of native-like P-SEA assignments by the total number of β sheet and α helix assignments within the structure sequence. This value is denoted Hn(t). Nonnative secondary structure, which captures the percentage of α and β secondary structure assignment that is unlike that found in the structure sequence, is denoted Hnn(t).
Mean First Passage Times
Having determined the set
of substates defining the native state, we next derived the expected
mean first passage time of each nonnative substate to the native ensemble
as put forward in ref (48) (alternative algorithms for computing transit times are given in
Torchala et al.77). First, we estimated
the symmetric transition probability matrix P from
the clustering results using the MSMBuilder2 MLE method to guarantee
detailed balance.78 This matrix carries
jump probabilities for the embedded discrete Markov chain,40 but can also be expressed as a rate matrix 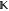 that
approximates the continuous time transition
rates.39
that
approximates the continuous time transition
rates.39
For each nonnative substate i, we modify 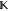 to
have zero transition rates to all substates
previously connected to i. We then compute the formal
matrix exponentiation e(
to
have zero transition rates to all substates
previously connected to i. We then compute the formal
matrix exponentiation e(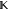 0it) for geometrically
spaced t values (t = 50(1.2r), r ∈ [0,1,...40].
That is, t ∼ 50 ns...74 μs). The fraction
of trajectories, starting at (nonnative) state i,
that will arrive at the native ensemble N before
time t is then given by
0it) for geometrically
spaced t values (t = 50(1.2r), r ∈ [0,1,...40].
That is, t ∼ 50 ns...74 μs). The fraction
of trajectories, starting at (nonnative) state i,
that will arrive at the native ensemble N before
time t is then given by
where j indexes substates in the native ensemble. This fraction consistently converged for all substates (i.e., miniPiN = 0.9964 at tmax out of all proteins). The mean first passage time (see SI Figure S7) is then given by
Frustration Scores
We then ask how these mean first passage times to the native ensemble, or transit times, change in response to network perturbation, that is, the removal of a substate in the nonnative ensemble. To that end, we remove a substate i in the nonnative ensemble from the network and then observe the percentage change between unperturbed (τjN) and perturbed (τjN*) transit times, in both cases for all nonnative substates m ∈ [1...knn ≠ i] (see Figure 1). That is,
where the bar thus indicates the average percentage change in transit time over all knn–1 substates in the nonnative ensemble, and the multiplicative factor allows frustration scores to be interpreted as percentages. Substates in the native ensemble are never removed throughout the procedure. Any isolates resulting from removing node i were discarded when computing f̅nat i, but this was rare ((isolates)/(knn) < 0.01 for all proteins except LAMDA, (isolates)/(knn) < 0.021). Frustration scores f̅nat quantify the kinetic impact, positive or negative, for each nonnative substate i, expressed as percentages in Figure 3 and Table 2. States with positive frustration scores are termed facilitators, those with negative frustration scores are termed inhibitors or kinetic traps. All analysis subsequent to clustering was performed in Matlab.79 Due to the matrix exponentiation, complexity of f̅nat computation is ∼O(N3), and runtimes were between 4 min (PB, knn = 167) and 20h (PG, knn = 2248) on a 12-core cluster.
Figure 3.

Frustration scores. Nonnative substate f̅nat values are shown for all 10 proteins. Because the substates of the native ensemble cannot be associated with f̅nat values, the number of data points corresponds to the number of substates in the nonnative ensemble, knn (Table 1). Values less than zero indicate a kinetic trap, those above zero indicate a substate that facilitates transition to the native ensemble. Central red marks indicate the median; box edges are the 25th and 75th percentiles. See SI Figure S7 for a comparison with frustration scores of phantom networks.
Table 2. Transition Network Summary Statisticsa.
| min f̅nat | max f̅nat | median substrate size | median substrate width (Å) | median neighbors per substate | knn(kn) | transition matrix density (%) | W | (lnn→n)/lnn (%) | |
|---|---|---|---|---|---|---|---|---|---|
| TRP | –1.8 | 0.1 | 7 (11.5) | 3.4 (2.6) | 14 (12) | 387 (30) | 3.6 (22.3) | 0.30 | 5.74 |
| BBA | –0.7 | 2.2 | 5 (12) | 4.3 (2.9) | 8 (16) | 905 (93) | 1.0 (16.2) | 0.42 | 3.89 |
| VHP | –1.6 | 0.6 | 7 (9.5) | 5.3 (4.6) | 13 (16) | 207(44) | 6.1 (19.3) | 0.23 | 10.23 |
| WW | –0.6 | 3.5 | 2 (68) | 4.7 (1.7) | 4 (86) | 2067 (207) | 0.2 (39.2) | 0.46 | 0.68 |
| BBL | –1.6 | 1.1 | 7 (15) | 6.2 (4.8) | 11 (18) | 758 (102) | 1.5 (12.7) | 0.42 | 8.36 |
| PB | –4.2 | 1.4 | 5 (21) | 7.0 (4.7) | 10 (20) | 167 (41) | 5.8 (39.7) | 0.34 | 6.61 |
| HMDM | –1.7 | 1.1 | 5 (18) | 5.6 (4.0) | 8 (29) | 517 (137) | 1.8 (17.8) | 0.44 | 20.86 |
| PG | –0.7 | 3.3 | 4 (78) | 5.5 (2.5) | 6 (27) | 2248 (62) | 0.4 (37.5) | 0.66 | 0.50 |
| A3D | –0.6 | 2.2 | 4 (88.5) | 8.2 (3.6) | 8 (40) | 1346 (68) | 0.6 (48.5) | 0.45 | 0.51 |
| LAMDA | –1.2 | 2.0 | 6 (18.5) | 6.0 (4.6) | 5 (18) | 1181 (112) | 0.6 (14.7) | 0.68 | 2.25 |
Details of transition networks in Figure 2. Columns 1–7: parenthetical values denote properties of the native ensemble, all others to the nonnative ensemble. The range of frustration scores is given in the first two columns. Median substate size refers to the number of trajectory snapshots clustered into each conformational substate. Substate width refers to average intra-substate pairwise RMSD. The number of substates classified by modularity optimization as being in the nonnative (native) ensemble is denoted by knn(kn). Maximum modularity value, W, for the modularity optimization algorithm utilized (see Methods) is also given. The last column shows the ratio between (1) total transition edges connecting nonnative and native ensembles (lnn→n) and (2) the total number of edges in the nonnative ensemble (lnn), expressed as a percentage.
Network Representations
Substate transition matrices
are usually very sparse, especially in the nonnative ensemble (Table 2). Most transitions are forbidden due to the involved
steric clashes, backbone geometry restrictions, and repulsive electrostatics.
Graph-based visualizations of conformation space thus have interpretive
value in conveying only the transitions that do take place as well
as the relative sizes of conformational states.80,81 We used Gephi82 to represent each protein’s
transition network (Figure 2). Network layouts
were optimized using the Force Atlas algorithm, first allowing and
then penalizing node overlap, in both cases with an internode repulsion
strength of 200. Edge weights were scaled according to the transition
matrix, specifically 1000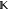 , but
are not differentiated visually in
the figure. The repulsion force acts between all nodes, whereas node
attraction is relative to connecting edge weight, so unconnected nodes
feel zero direct attraction. Node diameters reflect their relative
populations, but the smallest node is shown no smaller than 1/30 the size of the largest node for clarity. Network
radial orientation was rooted with the native ensemble facing east.
, but
are not differentiated visually in
the figure. The repulsion force acts between all nodes, whereas node
attraction is relative to connecting edge weight, so unconnected nodes
feel zero direct attraction. Node diameters reflect their relative
populations, but the smallest node is shown no smaller than 1/30 the size of the largest node for clarity. Network
radial orientation was rooted with the native ensemble facing east.
3. Results and Discussion
Conformational landscapes of the 10 proteins can be conveniently depicted as networks of nodes and edges that illustrate folding properties of each peptide. These representations are shown in Figure 2A. The native ensembles as defined in Methods are colored green, though the maps’ layouts themselves were created without preknowledge of native or nonnative substate assignments. All nonnative substates are colored according to their computed f̅nat values.
Properties of Transition Networks
Many general phenomenological aspects of protein folding are visible in these abstractions in addition to features that distinguish the folding behavior of specific polypeptides. The prevalence of large substates within the highly interconnected native ensemble (see Table 2), for example, reflects the loss of entropy a folding peptide experiences upon attaining the energetically favorable folded conformation (cluster size and widths given in SI Figures S4 and S5). Second, folding facilitators (blue substates) are as expected mostly proximal to the native ensemble due to being conformationally very native-like.83 The topological isolation of the native ensemble, especially for TRP, WW, PG, A3D, and LAMDA, suggests modularity optimization effectively classifies the folded and unfolded ensembles without invoking any protein-specific parameters. A particularly evident separation between the two ensembles characterized the transition maps of WW, PG, and A3D, all of which had less than one percent of nonnative edges connecting the nonnative and native ensembles (range for all proteins: 0.5–20.9%, see lnn→n/lnn in Table 2). Higher values of this measure indicate less homogeneous folding pathways,84 most evident in HMDM and PB transition maps.
Large kinetic traps, transition bottlenecks, and facilitators, among other topological motifs, are unequally prominent among the networks. Several of the maps depict a nonnative ensemble of freely interconverting structures that form no apparent energetically coherent substates (min f̅nat values for WW, PG, and A3D are −0.6,–0.7, and −0.6, respectively), whereas substantial kinetic traps characterize the nonnative ensembles of TRP, BBL, PB, and HMDM (min f̅nat = −1.8,–1.6,–4.2, and −1.7, Table 2). The distribution of frustration scores for each peptide is shown in Figure 3. We also compared τnat and f̅nat values to corresponding quantities computed for phantom (i.e., synthesized) networks in SI Figure S7. The comparison reveals that the degree distribution inherent to the transition network of each peptide is sufficient input for approximating τnat and f̅nat in generated networks.
However, a more quantitative analysis is necessary to reveal the specific conformational features, or structural parameters, that are responsible for the frustration scores unique to each protein’s unfolded ensemble. We focus primarily on properties of kinetic traps because facilitators inherently border a native/nonnative delineation that is convenient but imposed; any conformational differences between facilitators and native substates are likely to be subtle with regard to the structural parameters used here.83 We first address clustering properties that could be thought to cause negative outlier f̅nat values and then discuss the structural features that indeed correlate with kinetic frustration.
Kinetic Frustration Is Not a Clustering Artifact
As shown in Figure 3, most substates within the nonnative ensemble are kinetically neutral; their individual presence in the FEL has little impact on the expected transit times of other substates. Importantly, these substates need not be small, or have few constituent conformers. While substates with substantial positive or negative frustration scores tend to be above average in size, the converse is not true (SI Figure S4). That kinetic traps, as identified through f̅nat, must contain a substantial number of conformers reflects our intuition that kinetic traps represent local minima with stabilizing intramolecular interactions in the nonnative FEL.85 Substate size (i.e., number of members) as a descriptive trait can be contrasted with substate width (i.e., the mean pairwise RMSD of any two of its members), which provides an approximation of local entropy. If kinetic traps presented increasing substate width as f̅nat values became more extreme, we could conclude that f̅nat values are actually artifacts of the clustering step. In this scenario larger and larger peripheral regions of the configurational state space are unfairly grouped together during clustering, resulting in artificially exaggerated f̅nat values. We observed, in contrast, that kinetic traps display decreasing width values, suggesting they represent genuine local energy minima (SI Figure S5).
Properties of Kinetic Traps
Frustration scores directly reflect the transition topology. Having discussed that clusters in our networks are well-formed, we next investigate conformational causes of this observed topology. Specifically, are there general structural features that cause kinetic traps?86 We selected five structural parameters that share the desirable properties of normalizability and interpretability, and we computed them for all substates (all nonnative and native trajectory frames) in the transition networks. Definitions for native contacts, Qn, nonnative contacts, Qnn, native secondary structure Hn, and nonnative secondary structure Hnn are given in Methods, and our fifth structural parameter was standard RMSD (against the native conformation). Figure 4 illustrates the relationship between f̅nat and fractional contacts (Qnn or Qn) for all nonnative substates within the conformational landscape of LAMDA. The relationship observed confirms our expectation that kinetic traps display more nonnative contacts than the nonnative ensemble in general. The presence of interresidue contacts, both native and nonnative, is normalized against the corresponding quantity observed in the native ensemble. Mean values for these features are shown with dashed horizontal lines, gray for the entire nonnative ensemble, green for the entire native ensemble. For LAMDA, we conclude that the enrichment of nonnative contacts among kinetically frustrated substates is one hypothesis for the appearance of outlying red substates in LAMDA’s transition network (Figure 2, white arrow).
Figure 4.

Structural features in the nonnative ensemble are related
to kinetic
frustration. Average intrasubstate Qnn (C) and Qn (D) values for LAMDA are
plotted against frustration scores, f̅nat, showing that nonnative contacts are associated with kinetic
frustration. Structural parameter values, Qn and Qnn included, are normalized against
the average corresponding values within the native ensemble. Marker
widths are scaled according to substate populations, and error bars
indicate one standard deviation. Dashed lines show normalized average
values for the nonnative (gray) and native (green) ensembles. Cumulative
sums, (A) sQnn =  (Qnn – Q̅nn) and (B) sQn =
(Qnn – Q̅nn) and (B) sQn =  (Qn – Q̅n), (see main text) show the propensity
of structural features to be more associated with negative or positive f̅nat values. When integrated these curves
yield the bias values, β, that allow quantitative comparison
between proteins (Figure 5 and SI Table S1). Color values along the curve correspond
to substate color values in Figure 2.
(Qn – Q̅n), (see main text) show the propensity
of structural features to be more associated with negative or positive f̅nat values. When integrated these curves
yield the bias values, β, that allow quantitative comparison
between proteins (Figure 5 and SI Table S1). Color values along the curve correspond
to substate color values in Figure 2.
In normalizing Qn and Qnn against their respective values in the native ensemble, we can evaluate their correlative relationship to frustration scores and then compare among protein systems. We thus quantify whether a feature is more enriched among kinetic traps or facilitators with a bias value
for any feature F with mean value F̅ (within the nonnative ensemble), where f indexes the ascendingly sorted f̅nat values. Bias values convey whether feature F is more enriched for negative or positive f̅nat values (Figure 4A and B), where the negative sign allows us to compare with standard linear correlation, which we performed with the addition of weighted substate size (SI Table S1). Bias values for all five structural parameters are presented in Figure 5. Values near zero indicate the structural parameter is not strongly associated with kinetic frustration. Large positive or negative values indicate a strong relationship. We performed permutation tests to check the statistical significance of these bias values (SI Table S1). As we would expect, native secondary structure and native contacts frequently have positive bias values (TRP βQn is the only statistically significant exception), indicating that facilitator substates contain many native-like structural features, whereas kinetic traps do not. Significant nonnative secondary structure bias values were observed for BBA, PG, A3D, and LAMDA (βHnn = −0.13,–0.19,–0.12, and −0.10 respectively), and significant nonnative contacts bias values were observed for TRP, BBA, BBL, PG, A3D, LAMDA (βQnn = −0.33,–0.13,–0.18,–0.15,–0.13, and −0.28, respectively). Because RMSD-to-native is so commonly invoked as a distance metric for how far a simulation has progressed, we also computed bias values for RMSD, which were negative and statistically significant for all proteins except TRP, VHP, and PB. Especially for BBA, WW, BBL, HMDM, and A3D, nonspecific structural deformity, the characteristic summarized by RMSD, appears more associated with kinetic frustration than the specific structural features tested. VHP and PB simulations did not present statistically significant bias values for any structural parameters, perhaps due to modularity optimization defining the native ensemble too inclusively for these networks (see SI Figure S2).
Figure 5.

Comparison of β values for five structural parameters. Frustration bias values relate structural features to kinetic frustration. Negative values indicate the structural feature is strongly associated with kinetic frustration, that is, slowing transition to the native state for that protein. Positive values indicate the feature is associated with states that facilitate attainment of the native state. The RMSD distance from the native conformation has the largest negative bias value for BBA, WW, BBL, HMDM, and A3D. Nonnative contacts have the largest negative biases for TRP, PB, and LAMDA. Nonnative secondary structure, Hnn, is the most biased structural parameter only for PG. Some bias values close to zero are not statistically significant (SI Table S1), indicating the structural parameter has little kinetic impact on folding for that protein.
Visualizing Kinetic Traps
Conformational ensembles consisting of snapshots from the most kinetically frustrated substates are shown in Figure 6, rendered with PyMOL.87 The 5% of frames that were members of substates with the most negative frustration scores were aligned to their collective mean structure and represented as ensembles (topological context shown in SI Figure S8). Then the native conformation (see Methods) was added, aligned, and shown in a thicker tube representation. These ensembles illustrate properties of the nonnative ensemble that were suggested by the transition networks in Figure 2. The diffuse nonnative ensembles of WW, PG, and A3D, for example, have few stabilizing nonnative interactions, so even their most kinetically frustrated states appear almost completely unstructured (Figure 6A). In contrast, the nonnative transition maps with more topological isolation among inhibitor substates, especially BBL and PG, show much more homogeneity in the respective structural ensembles. BBL’s bias for Qnn was −0.18, suggesting that the nonnative tertiary structure apparent in the ensemble is responsible for the cluster of kinetic traps evident in the network (Figure 2 black arrow). The kinetic trap ensemble for TRP shows that the peptide can get conformationally ‘stuck’ in a nonnative but stabilized geometry. Although the nonnative configuration in the ensemble and the superimposed native state have very different backbone geometries, the relative compactness of the former may explain why TRP presented a statistically significant negative βQn, a property not observed for any other peptide. Peptides with simple contact topologies have been shown in lattice models to allow more interplay between native and nonnative contacts,88 consistent with our findings on TRP. That stabilizing interactions generally may be responsible for kinetic traps is suggested by Figure 6B, which shows that kinetic traps commonly have smaller widths (lower average pairwise RMSD of constituent members) than the nonnative ensemble.
Summary
To compare the folding properties of 10 protein sequences, we have exploited both quantitative and interpretive aspects of network models of protein folding. Our definition of kinetic frustration is grounded in graph theoretic principles while being consistent with qualitative definitions of kinetic features, such as kinetic traps. The method thus allows direct comparison between temporal folding behaviors and conformational features, the latter summarized by five standard structural metrics that were normalized against their prevalence in the native ensemble. While nonnative intramolecular interactions or nonnative secondary structure formation have been recognized as contributing factors to misfolding89,90 or folding rate reductions,91,92 we quantified the influence of these structural malformations through a normative process that requires no prior domain knowledge of the protein of interest. Specifically, folding for TRP, VHP, PB, PG, and LAMDA was most kinetically frustrated by deformations other than that characterized by RMSD, suggestive of stabilizing forces that trap a folding protein in a semistructured but nonnative conformation. These details were resolvable because we chose to perturb individual substates rather than substate collections within the transition networks.29 We additionally observed that phantom networks constructed by mimicking gross topological attributes of the observed networks mostly reproduced emergent kinetic properties (τnat and f̅nat) (SI Figure S7). If transition networks directly reflect the kinetic barriers, traps, and pathways caused by conformational fluctuations, as argued, then further topological properties can hopefully be linked to more nuanced categories of structural deformation. Certainly, subjective concepts such as misfolded intermediates, unstructured intermediates, and kinetic traps, often invoked in the literature of misfolding pathologies,19,93−95 can especially benefit from this type of quantification since simulations are increasingly sampling the distant or rare FEL regions where these events occur.
Acknowledgments
The authors thank Dr. Arvind Ramanathan for helpful discussions and D.E. Shaw Research for graciously sharing MD simulation data. A.J.S. was a predoctoral trainee supported by National Institutes of Health (NIH) T32 training grant T32 EB009403 as part of the HHMI-NIBIB Interfaces Initiative. This work was supported by the National Institutes of Health (grants 1R01GM105978 and 5R01GM099738).
Supporting Information Available
One table and eight figures. This material is available free of charge via the Internet at http://pubs.acs.org/.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Levinthal C. J. Chim. Phys. 1968, 65, 44–45. [Google Scholar]
- Anfinsen C. B. Science 1973, 181, 223–230. [DOI] [PubMed] [Google Scholar]
- Fersht A. R. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 14122–14125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande V. S.; Baker I.; Chapman J.; Elmer S. P.; Khaliq S.; Larson S. M.; Rhee Y. M.; Shirts M. R.; Snow C. D.; Sorin E. J.; Zagrovic B. Biopolymers 2003, 68, 91–109. [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K.; Piana S.; Dror R. O.; Shaw D. E. Science 2011, 334, 517–520. [DOI] [PubMed] [Google Scholar]
- Freddolino P. L.; Schulten K. Biophys. J. 2009, 97, 2338–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R. B. Curr. Opin. Struct. Biol. 2012, 22, 52–61. [DOI] [PubMed] [Google Scholar]
- Frauenfelder H.; Sligar S. G.; Wolynes P. G. Science 1991, 254, 1598–1603. [DOI] [PubMed] [Google Scholar]
- Frauenfelder H.; Parak F.; Young R. D. Annu. Rev. Biophys. Biophys. Chem. 1988, 17, 451–479. [DOI] [PubMed] [Google Scholar]
- Chan H. S.; Dill K. A. Proteins 1998, 30, 2–33. [DOI] [PubMed] [Google Scholar]
- Karplus M. Nat. Chem. Biol. 2011, 7, 401–404. [DOI] [PubMed] [Google Scholar]
- Ueda Y.; Taketomi H.; Go̅ N. Biopolymers 1978, 17, 1531–1548. [Google Scholar]
- Plotkin S. S. Proteins 2001, 45, 337–345. [DOI] [PubMed] [Google Scholar]
- Oakley M. T.; Wales D. J.; Johnston R. L. J. At. Mol. Opt. Phys. 2012, 2012, 1–9. [Google Scholar]
- Lei H.; Wu C.; Liu H.; Duan Y. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 4925–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementi C.; Plotkin S. S. Protein Sci. 2004, 13, 1750–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward S.; Milner-White E. J. Proteins 2008, 71, 415–425. [DOI] [PubMed] [Google Scholar]
- Pappu R. V.; Srinivasan R.; Rose G. D. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 12565–12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neudecker P.; Robustelli P.; Cavalli A.; Walsh P.; Lundstrom P.; Zarrine-Afsar A.; Sharpe S.; Vendruscolo M.; Kay L. E. Science 2012, 336, 362–366. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Ganguly D.; Chen J. PLoS Comput. Biol. 2012, 8, e1002353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn J. E.; Millett I. S.; Jacob J.; Zagrovic B.; Dillon T. M.; Cingel N.; Dothager R. S.; Seifert S.; Thiyagarajan P.; Sosnick T. R.; Hasan M. Z.; Pande V. S.; Ruczinski I.; Doniach S.; Plaxco K. W. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 12491–12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millett I. S.; Doniach S.; Plaxco K. W. Adv. Protein Chem. 2002, 62, 241–262. [DOI] [PubMed] [Google Scholar]
- Knott M.; Best R. B. PLoS Comput. Biol. 2012, 8, e1002605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogne P.; Ozdowy P.; Richter C.; Saxena K.; Schwalbe H.; Kuhn L. T. PLoS One 2012, 7, e41301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei H.; Su Y.; Jin L.; Duan Y. Biophys. J. 2010, 99, 3374–3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galzitskaya O. V.; Glyakina A. V. Proteins 2012, 80, 2711–2727. [DOI] [PubMed] [Google Scholar]
- Rao F.; Caflisch A. J. Mol. Biol. 2004, 342, 299–306. [DOI] [PubMed] [Google Scholar]
- Bowman G. R.; Pande V. S. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 10890–10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson A.; Brooks C. L. III J. Am. Chem. Soc. 2013, 135, 4729–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go̅ N. Annu. Rev. Biophys. Bioeng. 1983, 12, 183–210. [DOI] [PubMed] [Google Scholar]
- Bryngelson J. D.; Onuchic J. N.; Socci N. D.; Wolynes P. G. Proteins 1995, 21, 167–195. [DOI] [PubMed] [Google Scholar]
- Shea J.-E.; Onuchic J. N.; Brooks C. L. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 12512–12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R. B.; Hummer G.; Eaton W. A. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 17874–17879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim P. S.; Baldwin R. L. Annu. Rev. Biochem. 1990, 59, 631–660. [DOI] [PubMed] [Google Scholar]
- Clementi C.; Nymeyer H.; Onuchic J. N. J. Mol. Biol. 2000, 298, 937–953. [DOI] [PubMed] [Google Scholar]
- Jain A.; Hegger R.; Stock G. J. Phys. Chem. Lett. 2010, 1, 2769–2773. [Google Scholar]
- Ganguly D.; Zhang W.; Chen J. Mol. Biosyst. 2012, 8, 198–209. [DOI] [PubMed] [Google Scholar]
- Chiang T.-H.; Hsu D.; Latombe J.-C. Bioinformatics 2010, 26, i269–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodera J. D.; Singhal N.; Pande V. S.; Dill K. A.; Swope W. C. J. Chem. Phys. 2007, 126, 155101. [DOI] [PubMed] [Google Scholar]
- Kemeny J. G.; Snell J. L.. Finite Markov Chains; Springer Verlag: New York, 1976. [Google Scholar]
- Zhang Z.; Shan T.; Chen G. Phys. Rev. E 2013, 87, 012112. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Julaiti A.; Hou B.; Zhang H.; Chen G. Eur. Phys. J. B 2011, 84, 691–697. [Google Scholar]
- Hinrichs N.Algorithms for Building Models of Molecular Motion from Simulations. Ph.D. thesis, Stanford University, Stanford, CA, 2007. [Google Scholar]
- Weber J. K.; Pande V. S. Biophys. J. 2012, 102, 859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangha A. K.; Keyes T. J. Phys. Chem. B 2009, 113, 15886–15894. [DOI] [PubMed] [Google Scholar]
- Oliveira R. J.; Whitford P. C.; Chahine J.; Wang J.; Onuchic J. N.; Leite V. B. P. Biophys. J. 2010, 99, 600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellmer T.; Henry E. R.; Hofrichter J.; Eaton W. A. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 18320–18325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson A.; Brooks C. L. J. Chem. Theory Comput. 2012, 8, 3044–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trefethen L.; Bau D.. Numerical Linear Algebra; SIAM: Philadelphia, 1997. [Google Scholar]
- Ferrara P.; Apostolakis J.; Caflisch A. Proteins 2000, 39, 252–260. [DOI] [PubMed] [Google Scholar]
- Jang H.; Hall C. K.; Zhou Y. Biophys. J. 2002, 83, 819–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caflisch A.; Hamm P. Curr. Phys. Chem. 2012, 2, 4–11. [Google Scholar]
- Silva D.-A.; Bowman G. R.; Sosa-Peinado A.; Huang X. PLoS Comp. Bio. 2011, 7, e1002054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng N.-j.; Zheng W.; Gallicchio E.; Levy R. M. J. Am. Chem. Soc. 2011, 133, 9387–9394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wensley B. G.; Batey S.; Bone F. A. C.; Chan Z. M.; Tumelty N. R.; Steward A.; Kwa L. G.; Borgia A.; Clarke J. Nature 2010, 463, 685–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shental-Bechor D.; Smith M. T. J.; Mackenzie D.; Broom A.; Marcovitz A.; Ghashut F.; Go C.; Bralha F.; Meiering E. M.; Levy Y. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 17839–17844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y.; Ming D. PLoS One 2014, 9, e87719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortajarena A. L.; Yi F.; Regan L. ACS Chem. Biol. 2008, 3, 161–166. [DOI] [PubMed] [Google Scholar]
- Said G.; Grippon S.; Kirkpatrick P. Nat. Rev. Drug. Discovery 2012, 11, 185–186. [DOI] [PubMed] [Google Scholar]
- Butterfoss G. L.; Kuhlman B. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 49–65. [DOI] [PubMed] [Google Scholar]
- Joachimiak L. A.; Kortemme T.; Stoddard B. L.; Baker D. J. Mol. Biol. 2006, 361, 195–208. [DOI] [PubMed] [Google Scholar]
- Piana S.; Lindorff-Larsen K.; Shaw D. E. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 17845–17850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry E. R.; Best R. B.; Eaton W. A. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 17880–17885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchamp K. A.; Bowman G. R.; Lane T. J.; Maibaum L.; Haque I. S.; Pande V. S. J. Chem. Theory Comput. 2011, 7, 3412–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel V. D.; Guillaume J.-L.; Lambiotte R.; Lefebvre E. J. Stat. Mech. 2008, 2008, P10008. [Google Scholar]
- Ward J. H. Jr J. Am. Statist. Assoc. 1963, 58, 236–244. [Google Scholar]
- Beauchamp K. A.; McGibbon R.; Lin Y.-S.; Pande V. S. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 17807–17813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande V. S.; Beauchamp K.; Bowman G. R. Methods 2010, 52, 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayachandran G.; Vishal V.; Pande V. S. J. Chem. Phys. 2006, 124, 164902. [DOI] [PubMed] [Google Scholar]
- Leskovec J.; Lang K. J.; Mahoney M.. 2010, 631–640.
- Nymeyer H.; García A. E.; Onuchic J. N. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 5921–5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy Y.; Wolynes P. G.; Onuchic J. N. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lätzer J.; Shen T.; Wolynes P. G. Biochemistry 2008, 47, 2110–2122. [DOI] [PubMed] [Google Scholar]
- Du R.; Pande V. S.; Grosberg A. Y.; Tanaka T.; Shakhnovich E. S. J. Chem. Phys. 1998, 108, 334. [Google Scholar]
- Toofanny R. D.; Jonsson A. L.; Daggett V. Biophys. J. 2010, 98, 2671–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labesse G.; Colloc’h N.; Pothier J.; Mornon J.-P. CABIOS 1997, 13, 291–295. [DOI] [PubMed] [Google Scholar]
- Torchala M.; Chelminiak P.; Kurzynski M.; Bates P. A. BMC Syst. Biol. 2013, 7, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman G.; Beauchamp K.; Boxer G.; Pande V. J. Chem. Phys. 2009, 131, 124101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATLAB, version 7.14.0.739 (R2012a); The MathWorks Inc.: Natick, MA.
- Peng J. W. PLoS Comp. Bio. 2010, 6, e1001015. [Google Scholar]
- Chodera J. D.; Pande V. S. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 12969–12970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastian M.; Heymann S.; Jacomy M. ICWSM 2009, 361–362. [Google Scholar]
- García-Fandiño R.; Bernadó P.; Ayuso-Tejedor S.; Sancho J.; Orozco M. PLoS Comput. Biol. 2012, 8, e1002647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galzitskaya O. V.; Finkelstein A. V. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 11299–11304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsytlonok M.; Itzhaki L. S. Arch. Biochem. Biophys. 2012, 531, 14–23. [DOI] [PubMed] [Google Scholar]
- Sulkowska J. I.; Noel J. K.; Onuchic J. N. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 17783–17788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PyMOL, The PyMOL Molecular Graphics System; Schrodinger LLC: New York, 2010.
- Faísca P. F. N.; Nunes A.; Travasso R. D. M.; Shakhnovich E. I. Protein Sci. 2010, 19, 2196–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilloni C.; Schaal D.; Schweimer K.; Schwarzinger S.; De Simone A. Biophys. J. 2012, 102, 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Ding J. Proteins 2010, 78, 2090–2100. [DOI] [PubMed] [Google Scholar]
- Ferreon A. C. M.; Moran C. R.; Ferreon J. C.; Deniz A. A. Angew. Chem., Int. Ed. 2010, 49, 3469–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromiha M. M.; Selvaraj S. J. Mol. Biol. 2001, 310, 27–32. [DOI] [PubMed] [Google Scholar]
- Mulligan V. K.; Chakrabartty A. Proteins 2013, 81, 1285–1303. [DOI] [PubMed] [Google Scholar]
- Stanley C. B.; Perevozchikova T.; Berthelier V. Biophys. J. 2011, 100, 2504–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson C. M.; Karplus M. Curr. Opin. Struct. Biol. 1999, 9, 92–101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.