

Abstract
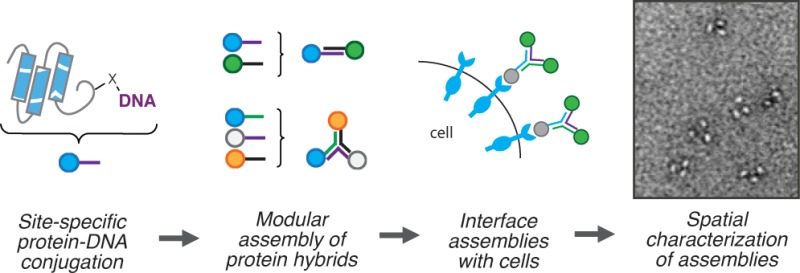
Expansion of antibody scaffold diversity has the potential to expand the neutralizing capacity of the immune system and to generate enhanced therapeutics and probes. Systematic exploration of scaffold diversity could be facilitated with a modular and chemical scaffold for assembling proteins, such as DNA. However, such efforts require simple, modular, and site-specific methods for coupling antibody fragments or bioactive proteins to nucleic acids. To address this need, we report a modular approach for conjugating synthetic oligonucleotides to proteins with aldehyde tags at either terminus or internal loops. The resulting conjugates are assembled onto DNA-based scaffolds with low nanometer spatial resolution and can bind to live cells. Thus, this modular and site-specific conjugation strategy provides a new tool for exploring the potential of expanded scaffold diversity in immunoglobulin-based probes and therapeutics.
DNA–protein conjugates can be assembled into nanoscale objects through the power of structural DNA nanotechnology. These motifs have the potential to revolutionize a number of biological and biomedical applications.1−3 One particular application of interest is antibody engineering using DNA scaffolds.4In vivo, antibodies of different classes have different biological activities and serve specialized roles during the immune response. An antibody’s class is determined by its constant region, or scaffold, which encodes the valency, effector functions, and higher-order architecture of the pendant variable domains. Expanding scaffold diversity in the antibody repertoire has the potential to expand the neutralizing capacity of the immune system, i.e. by delivering new effectors, increasing avidity, or modulating specificity. However, systematic exploration of antibody scaffold geometry, valency, and combinatorial binding capacity is difficult with protein-based scaffolds due to the challenges associated with protein design.5−7 DNA-based scaffolds, in contrast, are programmable and can combinatorially control the position and orientation of pendant proteins with nanometer resolution (Scheme 1A). Appropriately designed DNA scaffolds could assemble proteins that recognize specific combinations of receptors on cell surfaces,4 and even deliver protein-based therapeutics specifically to these cells.8,9
Scheme 1. Modular strategies for controlling antibody scaffold geometry using DNA–conjugates of aldehyde-tagged proteins. (A) (i) At least three unique functions can be incorporated on a Y-type (e.g. IgG) antibody scaffold. (ii) Protein–DNA conjugates can be used to selectively assemble Fab-bearing trimers. (iii) The DNA may be used as a handle for assembly of more complex scaffold architectures. (B) Attachment of oligonucleotides to aldehyde tags at protein termini or internal loops may proceed through at least four strategies: direct conjugation to DMT-protected aminooxy-modified DNA 1 or HIPS-modified DNA 2; or indirect conjugation through bifunctional polyethylene glycol 3 and subsequent copper-catalyzed triazole formation with hexynyl-modified DNA 4, or copper-free triazole formation to DBCO-modified DNA 5.
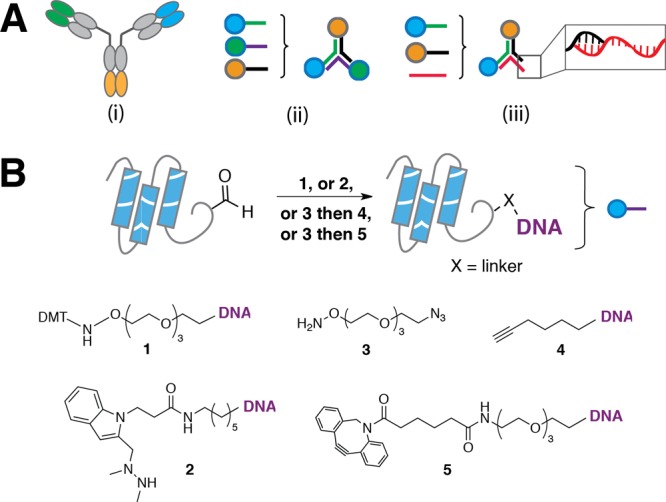
To fully realize the potential of using DNA scaffolds to expand the repertoire of antibody structure and function, more modular strategies for preparing DNA–protein conjugates are required. Ideal strategies would provide modularity in linkage chemistries, linkage site (e.g., termini or internal loops), and expression host. Aldehyde tagged proteins satisfy these requirements (Scheme 1B). Among peptide tags and self-ligating proteins,10−18 the aldehyde tag uniquely combines the advantages of a short consensus sequence (5 amino acids), a bioorthogonal handle which is amenable to conjugation through a number of chemical linkers, diverse prokaryotic or eukaryotic expression hosts, and compatibility with insertion at any position in a protein’s primary sequence.19−22
We explored the potential of aldehyde tagged proteins to form defined DNA–protein conjugates using a model substrate (maltose binding protein, MBP) bearing a C-terminal aldehyde tag expressed in E. coli. Coexpression of Formylglycine Generating Enzyme (FGE) leads to the post-translational conversion of the cysteine in the aldehyde tag consensus sequence (CxPxR) to formylglycine (Scheme S1). We also synthesized an oligonucleotide functionalized with a 5′-dimethoxytrityl (DMT)-protected aminooxy nucleophile, 1, from commercially available reagents. DMT-protection of the alkoxyamine stabilizes the product for storage but is rapidly deprotected in situ under the mildly acidic conjugation conditions. The resulting oxime product is observed as a higher molecular weight species by gel electrophoresis, and densitometry of the banding pattern indicated an 81% yield with respect to protein concentration (Figure 1A, lane 2). No conjugate was formed using a C → A mutation in the aldehyde tag consensus sequence (Figure S1). Thus, direct conjugation of aminooxy-modified DNA 1 to aldehyde-tagged proteins generates product efficiently using only commercially available reagents.
Figure 1.
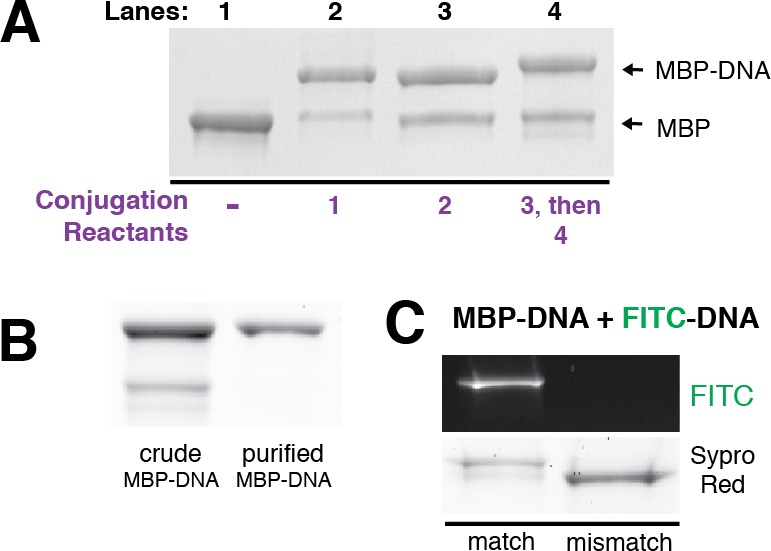
Modular and site-specific conjugation of oligonucleotides to aldehyde-tagged proteins. (A) SDS-PAGE analysis of crude reactions between aldehyde-tagged Maltose Binding Protein (MBP) and the indicated functionalized oligonucleotide (Scheme 1B). (B) MBP–DNA conjugates after purification by anion exchange chromatography. (C) MBP–DNA conjugates incubated with complementary and noncomplementary FITC–DNA and analyzed by SDS-PAGE.
Like other bioconjugation techniques such as thiol-maleimide coupling,23,24 the oxime linkage formed between 1 and an aldehyde-tagged protein is hydrolytically unstable upon long-term incubation in serum. This observation motivated the development of alternate conjugation strategies such as the Hydrazino-iso-Pictet–Spengler (HIPS) ligation.25 This recently reported reaction proceeds efficiently at near-physiological pH to form a stable covalent linkage with aldehyde tagged proteins. We therefore coupled the HIPS reagent to a 5′ amino-modified oligonucleotide and incubated the product 2 with aldehyde-tagged MBP at pH 5.5 to generate a DNA–protein conjugate in 62% yield (Figure 1A, lane 3). While the HIPS reagent must be synthesized prior to DNA conjugation, HIPS ligation proceeds at higher pH and forms a covalent and an irreversible C–C bond between DNA and protein.26
Additionally, we explored the potential to convert the formylglycine to a more reactive functionality for cases where more rapid coupling is required. Aldehyde bearing MBP was treated with an excess of a low molecular weight bifunctional linker 3 to introduce an azide group. Excess linker drives this reaction to completion and is easily removed by gel filtration due to its low molecular weight. Subsequent coupling with alkyne-modified DNA 4 occurred upon incubation with biocompatible copper stabilizing ligands such as BTTP,27 copper(II) sulfate, and sodium ascorbate with yields between 63% and 87% (Figure 1A, lane 4). Alkyne-modified DNA is inexpensive to synthesize in large quantities, allowing reaction scale-up and purification of the conjugate by anion exchange chromatography (Figures 1B, S2). The functionality and addressability of the DNA on the conjugate was verified by hybridizing it with a matching fluorescein isothiocyanate (FITC)-conjugated oligo (Figure 1C).
Conjugation of the azide-bearing protein with DNA can also proceed efficiently under copper-free conditions with dibenzocyclooctyne (DBCO)-modified DNA, 5. Incubation of azide-bearing MBP with 5 generated product in 79% yield with respect to protein (Figure S3). Together, this combination of four conjugation strategies provides flexible means of converting aldehyde-tagged proteins into DNA–protein conjugates with diverse physicochemical properties.
A key advantage of small peptides such as the aldehyde tag is that they can be used to prepare DNA–protein conjugates at either terminus or internal loops of immunoglobulins. For example, we inserted an aldehyde tag onto the C-terminus of a Fab raised against the Urokinase Plasminogen Activator Receptor (uPAR), an extracellular scaffold protein that regulates cell migration and invasion.28,29 After conversion of the formylglycine to an azide using the bifunctional linker 3, the product was conjugated to 4 using BTTP-stabilized click chemistry (Figure 2A). The resulting DNA–protein conjugate retained its ability to specifically bind uPAR on live cells. For example, an anti-uPAR Fab–DNA conjugate hybridized with a FITC-labeled oligonucleotide was able to efficiently label uPAR-expressing H1299 cells (Figure S4).
Figure 2.
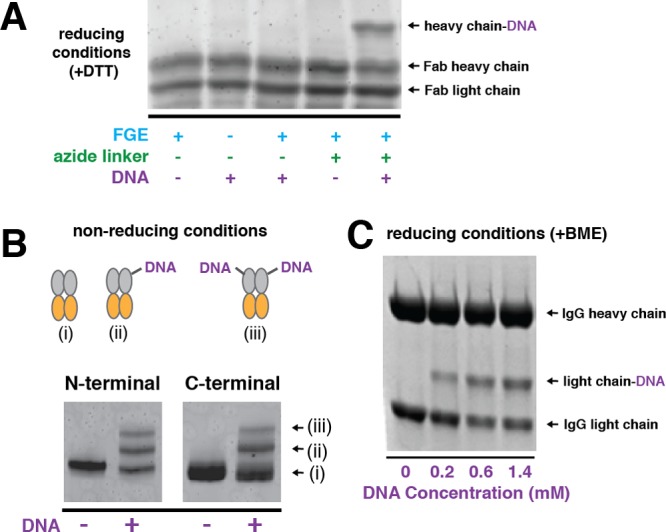
DNA conjugation to aldehyde-tagged immunogloblulins at either terminus or an internal loop. (A) SDS-PAGE analysis of DNA conjugated to the N-terminus of uPAR-binding Fab expressed in E. coli and then treated in vitro with FGE. Compounds 3 and then 4 were used to label the Fab with DNA. (B) DNA-conjugated to C-or N-terminal labeled Fc fragments expressed in FGE-expressing CHO cells using 3 and then 4. (C) DNA-conjugated to an internally labeled IgG expressed in FGE-expressing CHO cells using 1.
We also prepared glycosylated Fc fragments of a human IgG with the aldehyde tag inserted at the N- or C-terminus. Coexpression of these proteins with FGE in CHO cells yielded aldehyde-tagged protein with moderate levels of conversion. Transformation of the formylglycine to an alkyne using 3 and BTTP-stabilized click ligation to alkyne-modified oligonucleotides 4 generated a higher molecular weight species, consistent with formation of DNA–protein conjugates (Figure 2B). The lower yield of product observed for the C-terminally labeled site suggests that aldehyde reactivity depends on its placement within the primary sequence of the protein. Finally, we expressed a fully glycosylated IgG containing an aldehyde tag on an internal loop in FGE-expressing CHO cells. Incubation of different concentrations of aminooxy-modified DNA 1 with the IgG resulted in the appearance of a higher molecular weight band on a reducing SDS-PAGE gel that is consistent with a DNA-conjugated light chain (Figure 2C). Together, these experiments indicate the necessary modularity of tag placement and expression hosts when preparing DNA–protein conjugates for assembly into nanoscale geometries on DNA-scaffolds.
As our goal of synthesizing DNA–protein conjugates is to facilitate the assembly of proteins into antibody-like geometries, we explored the efficiency with which several simple DNA motifs mimicking the geometry of antibody scaffolds could be prepared from these reagents. We conjugated aldehyde-tagged MBP to 20 and 26 base oligonucleotides designed to self-assemble into dimers and trimers, respectively (Figure 3A). After hybridizing in PBS for 1 h at 25 °C, SDS-PAGE analysis indicated that both dimers and trimers assembled efficiently (Figure 3B).
Figure 3.
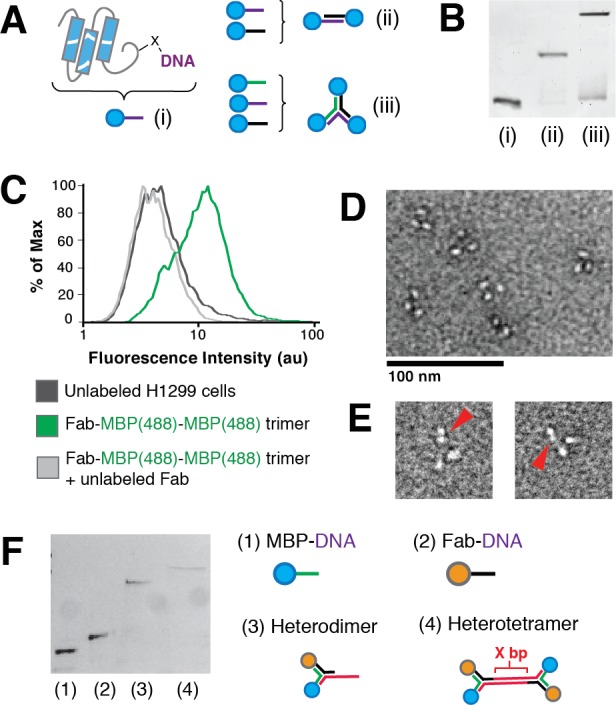
Modular assembly of protein-bearing DNA multimers and their interactions with live cells. (A) Assembly of protein–DNA conjugates into dimers and trimers based on oligonucleotide sequence. (B) SDS-PAGE of DNA-linked monomer (i), along with crude dimer (ii) and trimer (iii) assemblies. (C) Flow cytometry analysis of uPAR-expressing H1299 carcinoma cells incubated with DNA-scaffolded trimer bearing Fab and Alexa488-labeled MBP. (D) A field of negative-stained DNA-scaffolded MBP trimers imaged by transmission electron microscopy. (E) Fab–MBP–MBP trimers. The red arrow points to the Fab of the heterotrimeric construct. (F) SDS-PAGE of (1) MBP–DNA conjugate, (2) Fab–DNA conjugate, (3) a Fab and MBP bearing heterodimer, and (4) a Fab and MBP bearing heterotetramer using scaffolding strands with a variable length of X base pairs (here, x=16).
Additionally, DNA–protein conjugates assembled into small multiprotein motifs interacted efficiently with cell surfaces. For example, we used flow cytometry to analyze whether MBP/Fab heterotrimers retained their ability to interact with cells expressing uPAR. As a reporter for cell binding, we prepared an MBP–DNA conjugate modified with AlexaFluor-488 and assembled the resulting fluorescent MBP(488)–DNA conjugates with Fab–DNA to form heterotrimers. Incubation of the uPAR-expressing lung carcinoma cell line H1299 with the Fab-containing heterotrimer resulted in an increase in mean fluorescence in comparison to unlabeled cells (Figure 3C). To confirm that the heterotrimers were interacting specifically with uPAR, we preincubated the H1299 cells with nonfluorescent monomeric anti-uPAR Fab prior to addition of the Fab-MBP(488)-MBP(488) trimer and saw no increase in fluorescence. Similarly, fluorescence of Human Embryonic Kidney (HEK) cells, which do not express uPAR, was unchanged after incubation with the protein heterotrimer (Figure S5).
We examined the spatial organization of DNA-scaffolded proteins by negative stain transmission electron microscopy. Individual molecules of MBP in protein trimers were easily identifiable as light spots with a dark halo on a salt-and-pepper background (Figure 3D). In contrast to MBP, antibody fragments have more distinct features that can be identified as one of two distinct shapes (Figure S6). Thus, in heterotrimers, a single Fab protein was identifiable alongside an MBP dimer (Figure 3E). We calculated the distance between the center of individual proteins and the trimer centroid as 8.12 nm and measured an average spacing of 7.03 nm ±1.5 nm (s.d.) consistent with our estimate (Figure S7). The relatively large standard deviation in our measurements may indicate some conformational flexibility of the DNA scaffold. Additional spatial control and rigidity might be achieved using DNA motifs with longer persistence lengths, such as the double crossover motif.30,31 Moreover, the ability to modularly insert rigid or shorter chemical linkers would provide additional spatial control in these nanostructures.
Finally, because scaffold valency plays a central role in the immune system (IgG vs IgA vs IgM), we explored the hierarchical assembly of simple trimer motifs into higher-order structures. For example, we used an unmodified oligonucleotide as one arm of the trimer motif to assemble MBP–Fab dimers into tetramers, where the distance between each dimer could be varied based on the length of the unmodified scaffold DNA strands (Figure 3F). Elaboration of this simple strategy would allow for the assembly of scaffold protein assemblies of considerably higher valency and complexity.
In conclusion, we describe a simple and modular method for conjugating and then assembling multiple proteins onto DNA scaffolds. Our approach utilizes the aldehyde tag, which is genetically incorporated into the primary sequence of proteins expressed in both bacterial and mammalian expression systems. We tested four bioconjugation reactions that generate site-specific DNA–protein conjugates in moderate to excellent yield. The variety of strategies for conjugation of DNA to aldehyde-tagged proteins provides flexibility in linker chemistry and geometry, and is accessible to individuals with varying levels of synthetic expertise. Moreover, the ability to insert the aldehyde tag at both protein termini or in an internal loop will provide the potential for orientational control of proteins on DNA scaffolds. DNA–protein conjugates can be modularly assembled into dimeric and trimeric nanostructures resembling antibody scaffolds and interfaced with living cells. Transmission electron microscopy verified that the DNA scaffolds arranged proteins as predicted. These motifs can also be assembled hierarchically into structures of greater complexity. We anticipate expanding our DNA scaffold libraries to generate large collections of macromolecular assemblies varying in valency and architecture that may have novel activities as nanoscale probes or antibodies with unique specificities and biological activities.
Acknowledgments
We thank Prof. Charles Craik for the uPAR Fab plasmid and Dr. Natalia Sevillano Tripero for help with Fab purification. We thank Jason Hudak for the bifunctional linker and Prof. Peng Wu for the ligand BTTP. We thank Robyn Barfield and Greg deHart for providing research materials related to FGE and MBP production. We thank Prof. Yifan Cheng and Dr. Agustin Avila-Sakar for EM training. We thank Profs. Kevan Shokat, Pam England, Danica Fujimori, and Jack Taunton for sharing instruments and facilities. We thank the members of the Gartner lab for helpful comments on the manuscript and advice on experiments. This work is partially supported by the Achievement Rewards for College Scientists Foundation Fellowship and the Genentech Foundation Fellowship to S.I.L., a UCSF CTSI-SOS pilot grant, a NIGMS Systems Biology Center Grant (P50 GM081879), and the Kimmel Family Foundation.
Supporting Information Available
Figures and experimental methods. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): D. R. Rabuka and J. M. McFarland are CSO and an employee of Redwood Bioscience, Inc., respectively.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Pinheiro A. V.; Han D.; Shih W. M.; Yan H. Nat. Nanotechnol. 2011, 6, 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemeyer C. M. Angew. Chem., Int. Ed. 2010, 49, 1200–1216. [DOI] [PubMed] [Google Scholar]
- Simmel F. C. Curr. Opin. Biotechnol. 2012, 23, 516–521. [DOI] [PubMed] [Google Scholar]
- Kazane S. A.; Axup J. Y.; Kim C. H.; Ciobanu M.; Wold E. D.; Barluenga S.; Hutchins B. A.; Schultz P. G.; Winssinger N.; Smider V. V. J. Am. Chem. Soc. 2013, 135, 340–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter P. J. Exp. Cell Res. 2011, 317, 1261–1269. [DOI] [PubMed] [Google Scholar]
- Beck A.; Wurch T.; Bailly C.; Corvaia N. Nat. Rev. Immunol. 2010, 10, 345–352. [DOI] [PubMed] [Google Scholar]
- Kontermann R. MAbs 2012, 4, 182–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chari R. V. J. Acc. Chem. Res. 2008, 41, 98–107. [DOI] [PubMed] [Google Scholar]
- Alley S. C.; Okeley N. M.; Senter P. D. Curr. Opin. Chem. Biol. 2010, 14, 529–537. [DOI] [PubMed] [Google Scholar]
- Gautier A.; Juillerat A.; Heinis C.; Corrêa I. R.; Kindermann M.; Beaufils F.; Johnsson K. Chem. Biol. 2008, 15, 128–136. [DOI] [PubMed] [Google Scholar]
- Keppler A.; Gendreizig S.; Gronemeyer T.; Pick H.; Vogel H.; Johnsson K. Nat. Biotechnol. 2003, 21, 86–89. [DOI] [PubMed] [Google Scholar]
- Los G. V.; Encell L. P.; McDougall M. G.; Hartzell D. D.; Karassina N.; Zimprich C.; Wood M. G.; Learish R.; Ohana R. F.; Urh M.; Simpson D.; Mendez J.; Zimmerman K.; Otto P.; Vidugiris G.; Zhu J.; Darzins A.; Klaubert D. H.; Bulleit R. F.; Wood K. V. ACS Chem. Biol. 2008, 3, 373–382. [DOI] [PubMed] [Google Scholar]
- George N.; Pick H.; Vogel H.; Johnsson N.; Johnsson K. J. Am. Chem. Soc. 2004, 126, 8896–8897. [DOI] [PubMed] [Google Scholar]
- Chen I.; Howarth M.; Lin W.; Ting A. Y. Nat. Methods 2005, 2, 99–104. [DOI] [PubMed] [Google Scholar]
- Lin C.-W.; Ting A. Y. J. Am. Chem. Soc. 2006, 128, 4542–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antos J. M.; Miller G. M.; Grotenbreg G. M.; Ploegh H. L. J. Am. Chem. Soc. 2008, 130, 16338–16343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Suárez M.; Baruah H.; Martínez-Hernández L.; Xie K. T.; Baskin J. M.; Bertozzi C. R.; Ting A. Y. Nat. Biotechnol. 2007, 25, 1483–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovrinovic M.; Spengler M.; Deutsch C.; Niemeyer C. M. Mol. BioSyst. 2005, 1, 64–69. [DOI] [PubMed] [Google Scholar]
- Carrico I. S.; Carlson B. L.; Bertozzi C. R. Nat. Chem. Biol. 2007, 3, 321–322. [DOI] [PubMed] [Google Scholar]
- Hudak J. E.; Barfield R. M.; de Hart G. W.; Grob P.; Nogales E.; Bertozzi C. R.; Rabuka D. Angew. Chem., Int. Ed. 2012, 51, 4161–4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P.; Shui W.; Carlson B. L.; Hu N.; Rabuka D.; Lee J.; Bertozzi C. R. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 3000–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabuka D.; Rush J. S.; deHart G. W.; Wu P.; Bertozzi C. R. Nat. Protoc. 2012, 7, 1052–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alley S. C.; Benjamin D. R.; Jeffrey S. C.; Okeley N. M.; Meyer D. L.; Sanderson R. J.; Senter P. D. Bioconjugate Chem. 2008, 19, 759–765. [DOI] [PubMed] [Google Scholar]
- Shen B.-Q.; Xu K.; Liu L.; Raab H.; Bhakta S.; Kenrick M.; Parsons-Reponte K. L.; Tien J.; Yu S.-F.; Mai E.; Li D.; Tibbitts J.; Baudys J.; Saad O. M.; Scales S. J.; McDonald P. J.; Hass P. E.; Eigenbrot C.; Nguyen T.; Solis W. A.; Fuji R. N.; Flagella K. M.; Patel D.; Spencer S. D.; Khawli L. A.; Ebens A.; Wong W. L.; Vandlen R.; Kaur S.; Sliwkowski M. X.; Scheller R. H.; Polakis P.; Junutula J. R. Nat. Biotechnol. 2012, 30, 184–189. [DOI] [PubMed] [Google Scholar]
- Agarwal P.; van der Weijden J.; Sletten E. M.; Rabuka D.; Bertozzi C. R. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P.; Kudirka R.; Albers A. E.; Barfield R. M.; de Hart G. W.; Drake P. M.; Jones L. C.; Rabuka D. Bioconjugate Chem. 2013, 24, 846–851. [DOI] [PubMed] [Google Scholar]
- Wang W.; Hong S.; Tran A.; Jiang H.; Triano R.; Liu Y.; Chen X.; Wu P. Chem.—Asian J. 2011, 6, 2796–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duriseti S.; Goetz D. H.; Hostetter D. R.; LeBeau A. M.; Wei Y.; Craik C. S. J. Biol. Chem. 2010, 285, 26878–26888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund I. K.; Illemann M.; Thurison T.; Christensen I. J.; Høyer-Hansen G. Curr. Drug Targets 2011, 12, 1744–1760. [DOI] [PubMed] [Google Scholar]
- Sa-Ardyen P.; Vologodskii A. V.; Seeman N. C. Biophys. J. 2003, 84, 3829–3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J.; Lukeman P. S.; Sherman W. B.; Micheel C.; Alivisatos A. P.; Constantinou P. E.; Seeman N. C. Biophys. J. 2008, 95, 3340–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.