

Abstract
The observation that antagonists of the N-methyl-D-aspartate glutamate receptor (NMDAR), such as phencyclidine (PCP) and ketamine, transiently induce symptoms of acute schizophrenia had led to a paradigm shift from dopaminergic to glutamatergic dysfunction in pharmacological models of schizophrenia. The glutamate hypothesis can explain negative and cognitive symptoms of schizophrenia better than the dopamine hypothesis, and has the potential to explain dopamine dysfunction itself. The pharmacological and psychomimetic effects of ketamine, which is safer for human subjects than phencyclidine, are herein reviewed. Ketamine binds to a variety of receptors, but principally acts at the NMDAR, and convergent genetic and molecular evidence point to NMDAR hypofunction in schizophrenia. Furthermore, NMDAR hypofunction can explain connectional and oscillatory abnormalities in schizophrenia in terms of both weakened excitation of inhibitory -aminobutyric acidergic (GABAergic) interneurons that synchronize cortical networks and disinhibition of principal cells. Individuals with prenatal aberrations of NMDAR might experience the onset of schizophrenia towards the completion of synaptic pruning in adolescence, when network connectivity drops below a critical value. We conclude that ketamine challenge is useful for studying the positive, negative, and cognitive symptoms, dopaminergic and GABAergic dysfunction, age of onset, functional dysconnectivity, and abnormal cortical oscillations observed in acute schizophrenia.
Keywords: Ketamine, Glutamate, N-methyl-D-aspartate receptor, Schizophrenia
INTRODUCTION
Schizophrenia is a mental disorder characterized by both positive and negative symptoms, as well as social and occupational dysfunction. The positive symptoms of schizophrenia, such as delusions, hallucinations, disorganized speech, and catatonic behavior, are additions to or disturbances of normal, healthy function. The negative symptoms, such as avolition, alogia, and affective flattening, are deficits in normal, healthy function (American Psychiatric Association, 2000). Schizophrenia has a lifetime prevalence of 0.30–0.66% (McGrath et al., 2008), and patients live 12–15 years shorter than the average lifespan, a gap that is still rising (Saha et al., 2007).
The two neurotransmitter systems most frequently implicated in the pathogenesis of schizophrenia are those of dopamine and glutamate. The relationship between these neurotransmitter systems is complicated, as are efforts to disentangle the responsibility of each in generating the principal categories of schizophrenic symptoms. The observation that dopamine D2 receptor antagonists such as chlorpromazine alleviate psychosis lead to the dopamine hypothesis of schizophrenia, developed during the 1960s and 70s (Table 1) (Howes and Kapur, 2009). The dopamine hypothesis postulates that hyperdopaminergia is responsible for positive symptoms of schizophrenia (Lau et al., 2013). Later refinements to this theory have added a new caveat: striatal regions suffer from hyperdopaminergia, while prefrontal cortical regions suffer from hypodopaminergia (Lau et al., 2013; Howes and Kapur, 2009). The need for a mechanism explaining this differential subcortical and cortical dopamine dysfunction has been cited as a shortcoming of the dopamine hypothesis (Javitt, 2007); nevertheless, exponents of the dopamine hypothesis have formulated plausible theories to account for this discrepancy (Kapur, 2003; Grace, 1991). A notable example is the tonic/phasic model of dopamine release, in which the prefrontal cortex (PFC) regulates the tonic release of ambient dopamine, which serves to modulate the more familiar phasic, subcortical dopamine system responsive to behaviorally relevant stimuli. Schizophrenic hypofrontality is hypothesized to lessen tonic dopamine release, and phasic dopamine release is in turn increased as a homeostatic compensation mechanism (Grace, 1991).
TABLE 1.
Receptors involved in schizophrenia
| Receptor | Type | Notable ligands | Mediated effects | Ketamine affinity | Relevance to schizophrenia? |
|---|---|---|---|---|---|
| N-methyl-D-aspartate glutamate receptor (NMDAR) | Ionotropic |
|
|
Ki = 0.5 ± 0.15 μM | Established |
| Dopamine D2 receptor | Metabotropic |
|
|
Ki = 0.5 ± 0.2 μM (disputed) | Established |
| γ-aminobutyric acid A receptor (GABAAR) | Ionotropic |
|
|
|
Fairly well established |
| Sigma-1 receptor | Receptor chaperone |
|
|
Ki = 66.0 ± 10.0 μM | Postulated |
| Kappa receptor | Opioid |
|
|
Ki = 85.2 ± 26.0 μM | Postulated |
| Cannabinoid receptor type 1 (CB1) | Metabotropic |
|
|
|
Postulated |
| Muscarinic acetylcholine receptors | Metabotropic |
|
|
Ki = 45 μM (M1 receptor) Ki = 294 μM (M2 receptor) Ki = 246 μM (M3 receptor) |
Unknown |
A more commonly cited weakness of the dopamine hypothesis is the poor efficacy of D2 receptor antagonists in alleviating the negative and cognitive symptoms of schizophrenia (Javitt et al., 2012; Javitt, 2007). However, other evidence still allows for the relevance of dopamine to these symptoms by showing dopamine D1 and D2 receptors to be involved in working memory and psychosis, respectively (Goldman-Rakic et al., 2004: 1). Exponents of this theory draw on such lines of research as radiotracer positron emission tomography (PET) studies correlating changes in D1 receptor binding potential with cognitive symptoms (Kashima, 1991; Karlsson et al., 2002; Abi-Dargham et al., 2002) and reduced working memory performance in animal models following injection of D1 receptor antagonists into PFC (Sawaguchi and Goldman-Rakic, 1991).
The glutamate hypothesis of schizophrenia is a newer hypothesis, formulated roughly two decades ago, which implicates glutamergic dysfunction as a mechanism underlying both positive and negative symptoms, as well as cognitive dysfunction, in schizophrenia (Olney and Farber, 1995). The glutamate hypothesis is based on the observation that N-methyl-D-aspartate-type glutamate receptor (NMDAR) antagonists, such as phencyclidine (PCP) and ketamine, transiently induce symptoms and cognitive deficits characteristic of schizophrenia in humans (Thornberg and Saklad, 1996; Stone et al., 2008; Newcomer et al., 1999; Olney et al., 1999; Morris et al., 2005). For instance, single photon emission computed tomography (SPECT) imaging of healthy subjects shows a strong (r = 0.96) correlation between negative symptoms and ketamine-induced changes in the binding of an [123I]CNS-1261 radiotracer to NMDAR (Stone et al., 2008). Furthermore, interactions between dopamine D1 receptors and NMDAR might place dopaminergic dysfunction as a later step in a longer pathway rooted in NMDAR hypofunction (Roberts et al., 2010). The exact level of this interaction is uncertain; however, given the dysfunctional role of cortico-limbocortico-thalamic circuitry in schizophrenia (Tsai and Coyle, 2002) and the importance of dopamine and glutamate to these circuits, a systems level interaction is plausible. Before reviewing evidence supporting NMDAR antagonist models of schizophrenia, with an emphasis on ketamine as the safest human model, principles of glutamatergic neurotransmission, physiology of the NMDAR, and ketamine’s pharmacological mechanism of action will be reviewed.
Molecular Physiology and Pharmacology of NMDAR
Glutamate, an amino acid, is the principal excitatory neurotransmitter of the central nervous system (Fain, 1999). In addition to NMDAR, two other classes of ligand-gated ionotropic glutamate receptors have been described: α-amino-3-hyrdoxy-5-methyl-4-isoxazoleproprionic acid receptors (AMPAR) and kainate receptors. The NMDAR is an ionotropic receptor named after N-methyl-D-aspartate, a selective agonist of the receptor (Figure 1). The amino acid glycine is a co-agonist required for activation of NMDAR. At most synapses, glutamate is the agonist released from the presynaptic terminal, while ambient, micromolar concentrations of glycine are thought to activate the glycine binding site (Fain, 1999). Other agonists of the glycine binding site include D-serine and D-cycloserine. Activation of the glycine binding site is necessary but not sufficient for channel opening; it is an allosteric modulatory site regulating the time constant and desensitization rate of the channel (Javitt, 2007). Glycine type 1 (GLYT1) transporters and small neutral amino acid transporters (SNATs) are thought to modulate synaptic glycine and D-serine levels (Supplisson and Bergman, 1997; Mackenzie and Erickson, 2004; Nakanishi et al., 2001).
Figure 1. Pharmacology of NMDAR.
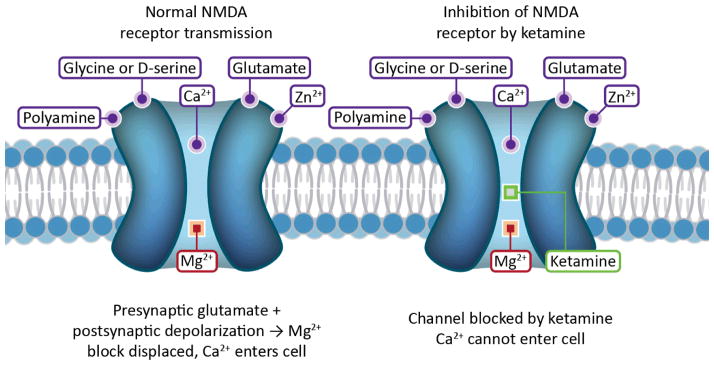
The NMDAR is an ionotropic glutamate receptor and nonselective cation channel with many ligands. Ketamine is one of several uncompetitive antagonists that block the channel at a PCP binding site within the pore. Glycine/D-serine and glutamate are coagonists that bind to the extracellular domain of the receptor. Both are necessary for channel opening; however, the NMDAR is rarely considered a glycine receptor because it is glutamate that is released from the presynaptic terminal, with micromolar background levels of glycine sufficing for channel activation. Polyamines, which modulate the effects of other ligands, and heavy metal cations, which can block or potentiate the channel, also have distinct binding sites. Mg2+ is a voltage dependent channel blocker that mediates the Hebbian mechanism of LTP.
NMDAR antagonists that block the glutamate or glycine binding sites are competitive antagonists, such as 2-amino-7-phosphonoheptanoic acid (AP7) (Fain, 1999). This review, however, concerns itself with the uncompetitive antagonists, as it is these pharmacological agents that reproduce symptoms of schizophrenia. Uncompetitive NMDAR antagonists bind to a PCP binding site within the pore of the ion channel. Such uncompetitive antagonists include, in order of descending affinity, dizocilpine (MK-801), PCP, and ketamine (Fain, 1999; Daniell, 1990).
NMDAR are also sensitive to heavy metal cations, such as Zn2+ and Cd2+, and as well as polyamines, protons, and redox agents. Critical to its role in the Hebbian mechanism of long-term potentiation (LTP), the NMDAR is clogged by an Mg2+ ion at resting membrane potentials. Sufficient depolarization of the postsynaptic cell removes the Mg2+ block, and Ca2+ influx through the NMDAR induces LTP. Thus, the NMDAR is necessary for the induction of LTP, a comprehensive delineation of which is beyond the scope of this review (Fain, 1999).
The biophysical properties of NMDAR are dependent on their subunit composition. NMDAR are heteromeric, composed of an NR1 subunit and modulatory subunits of the NR2 (NR2A-D) family (Fain, 1999). The NR3 (NR3A-B) family of modulatory subunits confers interesting properties on NMDAR: low Ca2+ permeability and insensitivity to channel blockers such as Mg2+, MK-801, and competitive antagonists. Furthermore, D-serine, normally an agonist of NMDAR, inhibits channels containing a subunit of the NR3 family (Chatterton et al., 2002).
Uses and Properties of Ketamine
Ketamine (2-chlorphenyl-2-methylamino-cyclohexanone), the uncompetitive NMDAR antagonist on which this review focuses, is a PCP derivative, first synthesized by Calvin Stevens of the Parke-Davis pharmaceutical company in 1962 (Rowland, 2005) and formally described in 1965 (Domino et al., 1965), eight years after the initial synthesis of PCP (Thornberg and Saklad, 1996). Because of PCP’s neurotoxicity and emergence reactions, such as hallucinations, confusion, and delirium, ketamine was intended as a safer alternative to PCP (Rowland, 2005). For similar ethical reasons, ketamine is preferred over PCP in pharmacological challenge of human subjects in neuroimaging or neuropsychological studies. Appropriately, we have chosen to focus our review on ketamine to inform researchers working with human subjects. Ketamine is recognized as a dissociative anesthetic (Corssen and Domino, 1966), with additional analgesic, amnesic, and possible fast acting antidepressant properties still under investigation (Aroni et al., 2009; Niesters et al., 2012; Rowland, 2005; Berman et al., 2000). Today, ketamine is used internationally as a general anesthetic in human and veterinary medicine (Rowland, 2005), but typically not used as a primary anesthetic. It is recognized by the World Health Organization as an essential medicine for any basic health care system (Anon, 2011). It is also a Schedule III controlled substance in the United States, due to its psychedelic properties and potential for abuse (Rowland, 2005). The legality of ketamine varies within the European Union, where it is controlled at the country level. It is a class C controlled substance in the United Kingdom, a controlled substance in Italy under Tables I – II, A of the DPR 309/90, and controlled as a stupéfiant, or narcotic, in France (Anon, 2008).
While the emergence reactions of ketamine are less severe than those of PCP, adverse reactions have been reported, including hallucinations, blurred vision, delirium, floating sensations, and vivid dreams. Hallucinations are reported at a frequency ranging from 5–30%. Notably, hallucinations are most common in patients over 16 years of age and rarest in children (Reich and Silvay, 1989). Indeed, the most frequent use of ketamine in the United States is for children (Rowland, 2005). These data support the ketamine model of schizophrenia, as the rarity of hallucinations in children younger than 16 years of age is consistent with the age of onset for schizophrenia, which is late-adolescence to early adulthood (Coyle et al., 2012).
Ketamine’s two enantiomers, (R)-ketamine and (S)-ketamine, have different pharmacological properties, as well as differential effects on psychiatric symptoms and cerebral metabolism in healthy volunteers (Oye et al., 1992; Vollenweider et al., 1997). (S)-ketamine has a higher affinity for the NMDAR PCP binding site than (R)-ketamine by a factor of 4–5 (Oye et al., 1992). Whereas (R)-ketamine does not significantly alter scores on psychiatric assessments, (S)-ketamine produces symptoms of depersonalization, derealisation, visual disturbances, thought disorders, and apathy (Vollenweider et al., 1997). PET imaging using [18F] fluorodeoxyglucose (FDG) shows (S)-ketamine to increase cerebral metabolic rates of glucose in many brain regions and (R)-ketamine to do generally the opposite. However, neither enantiomer significantly increases or decreases whole brain metabolism (Vollenweider et al., 1997).
Ketamine Pharmacology
Though ketamine is principally an NMDAR ligand, it also has varying affinities for many other receptors (Figure 3; Table 1). Perhaps the most notable of its many confusing and promiscuous affinities is its partial agonism, shared by PCP, for the high-affinity state of the D2 dopamine receptor, a confound that threatens the foundations of the glutamate hypothesis. According to the work of Kapur and Seeman (2002), ketamine’s affinity for D2 receptors (Ki = 0.5 ± 0.2 μM) is equal to its affinity for NMDAR. However, lack of replication elsewhere has rendered this finding contentious. Also of potential interest is ketamine’s weaker affinity for serotonin 5-HT2A receptors (Ki = 15 ± 5 μM) (Kapur and Seeman, 2002), which are implicated in visual hallucinations induced by serotoninergic agents (Kometer et al., 2013). Taken together, these facts distinguish a ketamine model of schizophrenia from the glutamate hypothesis. In fact, a ketamine model has potential to unify the competing glutamate and dopamine hypotheses into a single, multi-transmitter hypothesis that may also include other transmitter systems, herein reviewed.
Figure 3. Diverse effects of ketamine at many receptors.

Ketamine is a ligand of many diverse receptors. Ascertaining which receptors mediate which of ketamine’s effects is a challenge for a ketamine model of schizophrenia. Above, we have mapped out speculative, causal relationships between receptors and the effects of ketamine, underscoring the multi-transmitter nature of the ketamine model of schizophrenia. Note that multiple pathways may converge to cause positive symptoms. Effects of D1 dopamine receptors and NMDAR may also converge to cause cognitive symptoms; however, only receptors that interact directly with ketamine are pictured here.
Moving beyond the three aforementioned receptors, ketamine is a ligand for many other receptors and shows weak affinity for the sigma receptor (Ki = 66.0 ± 10.0 μM) (Oye et al., 1992; Smith et al., 1987). This fact holds possible relevance for the pathophysiology of schizophrenia (Debonnel, 1993; Hashimoto, 2010; Ishikawa and Hashimoto, 2010). The sigma receptor is a nonopioid receptor found in the endoplasmic reticulum (Hayashi and Su, 2004). The sigma-1 receptor (one of two subtypes), described as a “receptor chaperone,” is involved in mitochondrial Ca2+ signaling, neuroprotection, neuroplasticity, and neurite outgrowth, thus leading to the hypothesis that it is also necessary for optimal cognition and cerebral metabolism (Hayashi and Su, 2007; Ishikawa and Hashimoto, 2010). It has been demonstrated that sigma receptor ligands can protect dopaminergic cells from the excitotoxic effects of NMDA (but not ionomycin) in vitro, suggesting that sigma receptors regulate NMDAR (Shimazu et al., 2000). One postmortem study of schizophrenic brains revealed a reduction of sigma receptors in temporal cortex (Weissman et al., 1991), but a later study, which failed to replicate these findings, instead found an increase in sigma receptors in superior parietal cortex (Shibuya et al., 1992). Supporting the idea that schizophrenia patients suffer from a deficit in sigma receptors, mice that are repeatedly administered PCP show fronto-cortical (Ishima et al., 2009) and hippocampal (Kunitachi et al., 2009) reductions of the sigma-1 receptor.
SKF10047 is a sigma-1 receptor agonist that, like ketamine, induces schizophrenia-like symptoms (Masatomo et al., 2010). Furthermore, the antipsychotic drug haloperidol is known to bind to sigma receptors (Tam and Cook, 1984). These facts invite the question of whether sigma-1 receptor dysfunction, rather than or in addition to NMDAR dysfunction, can explain the effects of ketamine and the symptoms of schizophrenia. This question might be partially resolved with the consideration of two facts. Firstly, SKF10047 is also an NMDAR antagonist with affinity for the PCP binding site (Whittemore et al., 1997). Second, induction of schizophrenic symptoms and sigma-1 receptor activation are accomplished by different ketamine enantiomers (Aroni et al., 2009; Vollenweider et al., 1997; Oye et al., 1992). (S)-ketamine has negligible affinity for the sigma receptor, compared to the mild affinity of (R)-ketamine for sigma receptors (Oye et al., 1992). Clearly, sigma receptors and the PCP binding site of NMDAR share common ligands, and more research is required to ascertain what role, if any, sigma receptors play in the pathophysiology of schizophrenia.
Studies conducted in guinea-pig ileum preparation also show ketamine to be an agonist of the kappa opioid receptor (Hustveit et al., 1995), its affinity for which (Ki = 85.2 ± 26.0 μM) is comparable to that for sigma receptors (Smith et al., 1987). As with the sigma receptor, there is reason to implicate the kappa receptor in the pathophysiology of schizophrenia. Kappa receptor agonists have not only analgesic but also psychomimetic properties (Pfeiffer et al., 1986). Salvinorin A, in particular, is a kappa receptor agonist and the most potent naturally occurring hallucinogen. In contrast to most hallucinogens, salvinorin A does not activate or bind to the 5-HT2A serotonin receptor (Sheffler and Roth, 2003). Although findings have been mixed, many studies report a positive correlation between levels of dynorphins, endogenous kappa receptor ligands, in the cerebrospinal fluid of schizophrenia patients and psychotic symptoms (Tejeda et al., 2012). An abnormal laminar distribution of kappa receptors in the hippocampus has also been reported for schizophrenia patients (Royston et al., 1991). Finally, kappa receptor agonists administered to rats have varied effects on prepulse inhibition, which is reduced in schizophrenia, depending on which agonist is used (Tejeda et al., 2012). Further research is needed to demonstrate both causal relationships between kappa receptor dysfunction and schizophrenia and to measure the psychomimetic similarities between ketamine and kappa receptor agonists such as salvinorin A.
Besides NMDAR, sigma receptors, and kappa opioid receptors, the relevance to schizophrenia of ketamine’s interactions with other receptors is largely unknown. In the frog peripheral nervous system, ketamine blocks Na+ currents, but its washout also triggers spontaneous action potentials, implying that it modifies some Na+ channels it an excitable way (Benoit et al., 1986). At doses above what is used for general anesthesia, ketamine blocks human central nervous system Na+ channels (Frenkel and Urban, 1992). Ketamine has an inhibitory effect on Ca2+ currents in smooth (Yamakage et al., 1995) and cardiac (Baum and Tecson, 1991) muscle and also blocks both open and closed nicotinic acetylcholine receptors in vitro (Scheller et al., 1996). Furthermore, it shows affinity for the muscarinic acetylcholine receptor in guinea-pig ileum preparation (Hustveit et al., 1995; Hirota, 1996; Hirota et al., 2002). Besides the kappa opioid receptor, already discussed, ketamine is known also to have affinity for the delta and mu opioid receptors (Gupta et al., 2011; Hirota et al., 1999), synergistically enhancing the effects of opioids at the mu receptor (Gupta et al., 2011). Recently, the antidepressant effects of ketamine have been blocked in mice using NBQX, an AMPAR antagonist, suggesting that ketamine interacts with the AMPAR, though this interaction may or may not be direct. For instance, these data could be explained by the possibility that ketamine merely changes the relative throughputs of AMPAR and NMDAR. (Maeng et al., 2008).
Evidence for NMDAR Dysfunction
Although determining what proportion of ketamine’s analgesic and psychomimetic effects can be attributed to which receptors is arguably still a challenge for the ketamine model of schizophrenia, much evidence points to NMDAR dysfunction in schizophrenia. Having explored ketamine’s varied effects at other receptors, it is important to note that ketamine’s effects at NMDAR are complex and somewhat counterintuitive. While an NMDAR antagonist, evidence from magnetic resonance spectroscopy (MRS) and microdialysis has demonstrated that ketamine and other uncompetitive NMDAR antagonists have a net positive effect on excitatory transmission by inducing excessive release of glutamate (Rowland et al., 2005; Stone et al., 2012; Kim et al., 2011) and acetylcholine (Hasegawa et al., 1993; Giovannini et al., 1994). In ketamine challenge of humans subjects, pretreatment with the anticonvulsant lamotrigine, a Na+ channel blocker that reduces glutamate release, attenuates both many subjective effects and blood oxygen-level-dependent (BOLD) signal responses induced by ketamine (Deakin et al., 2008). Olney and colleagues (1999) have proposed that chronic over-release of excitatory neurotransmitters can explain both cognitive and behavioral symptoms of schizophrenia, as well as morphological changes and neurodegeneration in patients’ brains. Indeed, ketamine-induced release of glutamate in anterior cingulated cortex (ACC) is correlated with positive psychotic symptom scores in healthy human subjects (r = 0.72) (Stone et al., 2012). Furthermore, NMDAR antagonists play known roles in neurodegenration and excitotoxicity, including apoptosis of mature corticolimbic pyramidal cells (Zhou et al., 2007; Farber and Olney, 2003; Horváth et al., 1997; Wozniak et al., 1998), thus offering NMDAR hypofunction as a possible basis for brain atrophy observed in schizophrenia (Rais et al., 2012; Andreone, et al., 2007; Rais et al., 2008; Goldman et al., 2007; Ferrari et al., 2006).
Much molecular evidence points to NDMAR dysfunction in schizophrenia. Schizophrenic brains have reduced levels of a protein linked to NMDAR function and working memory (Karlsgodt et al., 2011), dystrobrevin-binding-protein-1 (dysbindin) (Talbot et al., 2004), and dysbindin mRNA (Weickert et al., 2004), in hippocampal formation and dorsolateral prefrontal cortex (DLPFC), respectively. Polymorphisms of DTNBP1 (Voisey et al., 2010), the gene encoding dysbindin, and GRIN2B (Li and He, 2007a), the gene encoding the NR2B subunit of NMDAR, have been associated with schizophrenia. The growth factor Neuregulin-1 (NRG1), its receptor, erbB4, and the scaffold protein known as postsynaptic density protein of 95 kDa (PSD-95) further suggest a link between NMDAR and schizophrenia. NRG1 inhibits prefrontal cortical NMDAR in humans, and schizophrenics have increased interactions between erbB4 and PSD-95 (Hahn et al., 2006).
Further molecular evidence for NMDAR dysfunction in schizophrenia comes from genes regulating the NMDAR co-agonist D-serine. D-amino acid oxidase (DAAO) is an enzyme that degrades D-serine by oxidation. Several single nucleotide polymorphisms (SNPs) in DAAO have been linked to schizophrenia (Chumakov et al., 2002). Additionally, G72 (also known as D-amino acid oxidase activator: DAOA) and G30, overlapping genes that interact with DAAO, have SNPs associated with schizophrenia (Li and He, 2007b: 30; Shinkai et al., 2007). Consistent with these associations, an early clinical trial of D-serine supplementation of antipsychotic regimens for schizophrenia treatment reported the following mean reductions of symptoms six weeks after baseline: Positive and Negative Syndrome Scale (PANSS) positive, 17.4%, p = 0.004; Scale for the Assessment of Negative Symptoms (SANS), 20.6%, p = 0.0004; PANSS cognitive, 11.6%, p = 0.004; Clinical Global Impression Scale (CGI), 45.8%, p = 0.004 (Tsai et al., 1998). Supporting these findings, meta-analyses (Singh and Singh, 2011; Tsai and Lin, 2010) have found that supplementing antipsychotic medication with D-serine alleviates the negative (p = 0.02; pooled effect size (ES) = 0.48), cognitive (p = 0.007; ES = 0.42), and total (p = 0.02; ES = 0.4) symptoms of schizophrenia (Tsai and Lin, 2010).
If the above genetic evidence is to be seen as inconclusive, more direct lines of evidence from in situ hybridization histochemistry (ISHH) and SPECT have directly established the presence of NMDAR hypofunction in schizophrenia patients (Law and Deakin, 2001; Pilowsky et al., 2006). A SPECT imaging study using [123I]CNS-1261 as an intrachannel NMDAR radiotracer found reduced binding in the left hippocampus of medication-free schizophrenia patients, the first in vivo evidence of NMDAR hypofunction in schizophrenia (Pilowsky et al., 2006). An earlier ISHH study of postmortem brain tissue from psychiatric patients found significant reductions in all psychiatric groups (schizophrenic, depressive, and bipolar) for GRIN1 mRNA encoding the NR1 subunit of NMDAR in the dentate gyrus. Unique to the schizophrenia cohort was the left-lateralization of this finding (Law and Deakin, 2001). While the similar findings in affective disorder cohorts initially seem to undermine the importance of NMADR hypofunction to schizophrenia, a recent, large genome-wide meta-analysis has found associations between SNPs at the same four loci and five different psychiatric disorders, including the three disorders examined in the aforementioned postmortem study (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013). This suggests a larger trend in which seemingly disparate psychiatric disorders share similar molecular dysfunctions.
Taken together, the above evidence not only supports NMDAR as the principal site of ketamine’s psychomimetic effects, but also favors the ketamine model of schizophrenia over competing models. Both adrenochrome (an autoxidative metabolite of epinephrine) (Smythies, 2002) and Δ-9-tetrahydrocannabinol (Δ-9-THC) (D’Souza et al., 2004), the main active ingredient in cannabis, can also induce schizophrenia-like symptoms, but neither of these models have accumulated as much evidence as the ketamine model. It remains possible, however, that these and other hypotheses of schizophrenia are compatible with and/or auxiliary to the ketamine model and the glutamate hypothesis. For instance, dreaming has been proposed as a novel model of schizophrenia due to similarities in psychological state, cortical connectivity, sensory gating, hypofrontality, and neuromodulation (Gottesmann, 2006), but it and the glutamate hypothesis are not necessarily mutually exclusive, as there is decreased release of glutamate in the nucleus accumbens during REM sleep (Lena et al., 2006).
Neurotransmitter Effects and Impact on Cognition
Of course, the main competitor to the glutamate hypothesis is the dopamine hypothesis, already introduced earlier in this review. As previously noted, ketamine models of schizophrenia are distinct from the glutamate hypothesis due to ketamine’s affinity for many non-glutamate receptors and its effect of increasing glutamate release. Nonetheless, it will be useful to herein compare the dopamine and glutamate hypotheses and delineate possible advantages of glutamatergic theories. Some formulations of the glutamate hypothesis postulate that the glutamatergic system exerts influence over the dopaminergic system, while other formulations postulate that D2 receptors regulate glutamate release at NMDAR synapses (Figure 3) (Kehrer et al., 2008; Olney et al., 1999). Pharmacological evidence suggests that NMDAR dysfunction underlies dopaminergic dysfunction in schizophrenia. PET has shown the severity of ketamine induced positive symptoms to be correlated with dopamine release in lateral prefrontal and anterior cingulate cortex (Aalto et al., 2005). Rats treated both chronically and subchronically with PCP at blood levels comparable to PCP psychosis in humans (mean concentrations of 69.3 ng/ml and 85.4 ng/ml for chronic and subchronic, respectively) show potentiation of amphetamine-induced prefrontal cortical and striatal dopamine release (Javitt et al., 2004). This enhanced sensitivity to the dopaminergic effects of amphetamine challenge is also characteristic of schizophrenia patients, as revealed by PET and SPECT imaging (Laruelle et al., 1996; Breier et al., 1997). These PCP-induced effects in rats are treatable with both glycine and NFPS, a glycine transport antagonist (Javitt et al., 2004). Similarly, MK-801 administered to rats increases dopaminergic (as well as serotonergic) activity in frontal cortex, nucleus accumbens, and striatum (Löscher et al., 1991).
Furthermore, NMDAR dysfunction easily accounts for many cognitive symptoms of schizophrenia, in part because NMDAR are ubiquitous in the cortex, whereas implicated dopamine receptors are more localized. Because NMDAR are necessary for LTP, their dysfunction offers a more straightforward explanation for learning and memory deficits in schizophrenia patients than dopaminergic models. The established role of NMDAR in mismatch negativity (MMN) generation can also predict and explain schizophrenia patients’ reduced MMN auditory event-related potentials (ERP) in auditory oddball paradigms, where an odd auditory stimulus is included in a series of repetitive tones (Javitt et al., 2012). In fact, MMN amplitude predicts the susceptibility of healthy volunteers to the effects of ketamine (but not the hallucinogen psilocybin) (Umbricht et al., 2002). Ketamine has been shown to reduce MMN generation in a visual oddball paradigm, and concurrent functional magnetic resonance imaging (fMRI) data show this reduction to be associated with reduced activation in sensory cortex (Musso et al., 2011), further suggesting that attentional deficits in schizophrenia are actually sensory in nature. This is hardly a trivial finding: by shifting the focus of pathology posteriorly from frontal circuits to sensory cortex, one exits the neurochemical “jurisdiction” of dopamine (Nieoullon, 2002), placing greater responsibility on glutamatergic transmission.
Returning anteriorly to frontal circuits, chronic PCP exposure induces hypofrontality in rats (Cochran et al., 2003). Pharmacological fMRI studies have also demonstrated that ketamine reduces ventromedial frontal activation, elucidating the neurochemical mechanism of this deactivation in the process (Deakin et al., 2008). Hypofrontality is a symptom of schizophrenia whose underlying cause is often assumed to be dopamine dysfunction (Liemburg et al., 2012; Coppa-Hopman et al., 2009; Nagai et al., 2010; Nieoullon, 2002); however, administration of lamotrigine prior to ketamine challenge in humans has shown that increased glutamate release, induced by katamine, explains observed frontal deactivation (Deakin et al., 2008). That changes in glutamate release so strongly affect data from functional imaging is perhaps unsurprising considering that cerebral metabolism and the BOLD signal are believed to strongly reflect glutamate cycling through astrocytes at excitatory synapses (Raichle and Mintun, 2006).
Finally, prefrontal hypodopaminergia models predict that during task-switching paradigms, schizophrenia patients should have increased switch cost (poorer performance immediately following switching) (Javitt et al., 2012). Yet instead, schizophrenia patients have switch costs equal to controls. Their performance is similar to that of monkeys administered ketamine: worse performance for incongruent stimuli than congruent stimuli (Wylie et al., 2010). Thus, some cognitive tasks that are independent of NMDAR might be unaffected in schizophrenia (Javitt et al., 2012).
Disruptions to Connectivity
The glutamate hypothesis is also an attractive model because it offers an explanation for schizophrenia’s age of onset in early adulthood (Kehrer et al., 2008). Explanations for this age of onset dating as far back as 30 years ago point to synaptic pruning, the programmed elimination of a large fraction of synaptic connections, that takes place in late adolescence, suggesting that schizophrenia may, in fact, be an over-pruning syndrome (Feinberg, 1982; Granger, 1996). On the basis of recent knowledge about NMDAR dysfunction in schizophrenia, this synaptic pruning need not necessarily be excessive or aberrant to trigger pathology. Rather, NMDAR dysfunction is perhaps a weakness that manifests itself during prenatal development, and the completion of synaptic pruning late in adolescence is the proverbial straw that, so to speak, breaks the camel’s back (Granger, 1996; Kehrer et al., 2008; Olney and Farber, 1997). This idea is supported by the fact that ketamine induced hallucinations are most common in persons older than 16 years of age (Reich and Silvay, 1989).
Recently, the roles of brain rhythms and neural synchrony in neurodevelopment and schizophrenia have also been examined. The dynamics of neural systems such as cortical networks are nonlinear (Breakspear, 2006). While a comprehensive summary of nonlinear systems theory is beyond the scope of this review, per se, some fundamental terms will be defined herein. Nonlinear systems are often multistable, meaning that several stable, steady states are possible (e.g., regular oscillations, quiescence, chaotic behavior, etc.). Control parameters are those which induce transitions between stable, steady states in dynamical systems when tuned past critical values. Such transitions are referred to as phase transitions or bifurcations (Stam, 2005). Viewing cortical networks in a nonlinear dynamics context, it has been suggested that the maturation of such networks during adolescent development occurs by a phase transition, and that a failure of networks to synchronize and integrate information signifies that a pathological phase transition has occurred (Uhlhaas and Singer, 2011). Could it be that glutamatergic connectivity is a control parameter for such a bifurcation? In support of this hypothesis, pruning connections in neural network models increases the dimensional complexity of simulated electroencephalogram (EEG) and simulates hallucinations in speech perception networks (Friston, 1996; Hoffman and McGlashan, 1997).
The idea of disturbed network connectivity in schizophrenia meshes well with characterizations of schizophrenia as a disconnection syndrome (Sporns, 2011), a very old hypothesis dating back to Wernicke (1906), who attributed psychosis to disruptions of association fibers, and Bleuler (1908), who chose the word schizophrenia to describe the splitting of the mind in the disorder. Almost 80 years later, Weinberger (1987) hypothesized that schizophrenic symptoms occur due to a lesion or developmental defect disconnecting a mesocortical-corticofugal feedback loop regulating dopamine activity. The same year, a PET study of schizophrenia patients reported fewer functional interactions between brain regions than controls, including reduced correlations between activation of anterior and posterior regions and between thalamic and cortical regions (Volkow et al., 1988). Global reductions in functional connectivity (i.e., temporal cross correlations) between brain regions have since been found using other modalities, such as fMRI and EEG (Calhoun et al., 2009). Graph theoretical measures of both functional and anatomical brain networks in schizophrenia patients have revealed inefficient connectivity in these networks. Anatomical networks in patients are more sparsely connected when analyzed both in terms of axonal connectivity and cortical thickness (an indirect measure of the former) (Zalesky et al., 2011; Bassett et al., 2008; Wang et al., 2011). Resting-state functional networks in patients show reduced connection density (“clustering”), fewer “hub” regions with very high interconnectivity, and reduced efficiency (“small-worldness”) of connections (Lynall et al., 2010).
Such weakened global connectivity in schizophrenia is consistent with the ubiquity of presumably dysfunctional NMDAR in cortex and the coincidence of synaptic pruning with the disease’s onset. However, the story is not so simple: the default mode network (DMN), a set of brain regions deactivated by cognitive tasks and forming a functional module in resting state functional networks observed with fMRI (Sporns, 2012), has increased connectivity in schizophrenia patients. What’s more, the extent of strengthened connectivity within the DMN is correlated with the severity of psychiatric symptoms (Whitfield-Gabrieli et al., 2009). Similarly, data from simultaneous EEG and transcranial magnetic stimulation (TMS) have shown that TMS-evoked waves of excitation propagate longer and farther in schizophrenia patients than controls (Frantseva et al., 2012).
While these findings initially seem to contradict both the disconnection hypothesis and current models of the role NMDAR play in schizophrenic pathology, this contradiction is largely resolved upon further examination of the function of NMDAR in cortical microcircuits. NMDAR drive the excitation of inhibitory γ-aminobutyric acidergic (GABAergic) interneurons, particularly parvalbumin-positive fast-spiking interneurons, which in turn both inhibit pyramidal cells and synchronize their oscillations (Homayoun and Moghaddam, 2007; Korotkova et al., 2010; Behrens et al., 2007). Indeed, both mice and cultured interneurons challenged with ketamine exhibit a loss of GABA markers such as the Ca2+ binding protein parvalbumin and the enzyme glutamic acid decarboxylase of 67 kDa (GAD67), which produces GABA from glutamate by decarboxylation (Behrens et al., 2007; Kinney et al., 2006). Similar finding have been reported in rat PFC in vivo after PCP challenge (Cochran et al., 2003; Amitai et al., 2012). Other such in vivo microdialysis studies of rats have also shown that both PCP and MK-801 increase extracellular GABA levels in medial PFC. Conversely, the GABAA receptor agonist muscimol alleviates PCP and MK-801 induced increases in extracellular dopamine in the medial PFC (Yonezawa et al., 1998). Another GABAA receptor agonist, TPA023, blocks ketamine induced working memory deficits in rhesus monkeys (Table 1) (Castner et al., 2010). Postmortem studies of PFC tissue from schizophrenic patients reinforce the above findings by revealing reduced numbers of GABAergic interneurons expressing GAD67 (Volk et al., 2000; Hashimoto et al., 2003), parvalbumin (Hashimoto et al., 2003), and GABA transporter-1 (GAT-1) mRNA (Volk et al., 2001). PFC chandelier cells, GABAergic neurons that regulate pyramidal cell output, have lower axon cartridge (terminal array) density in schizophrenia patients (Pierri et al., 1999).
In light of these facts, NMDAR hypofunction appears to disinhibit some cortical networks by inhibiting (Homayoun and Moghaddam, 2007) and inducing long term adaptations (Cochran et al., 2003; Amitai et al., 2012) in GABAergic interneurons (Figure 3). Appropriately, the more neutral term “dysconnectivity” is now applied to both pathological increases and decreases in network connectivity in schizophrenia (Pettersson-Yeo et al., 2011). As expected, ketamine challenge in both animals and humans alters functional connectivity in ways that mirror dysconnectivity in schizophrenia. Extracellular recordings in low Mg2+ mouse neocortical slices challenged with ketamine show decoupling beginning at a spatial scale of 4 mm, a significant finding in 42% of slices (Voss et al., 2012). Autoradiographic imaging, also conducted in mice, using a subanaesthetic dose of ketamine combined with a 14C-2-deoxyglucose tracer, reveals both increases and decreases in the functional connectivity of a neural circuit composed of the dorsal reticular thalamic nucleus, anteroventral/mediodorsal thalamic nuclei, and PFC (Dawson et al., 2013). Using the same radiotracer, similar findings, including dysconnectivity between PFC and hippocampus, have been found in rats treated subchronically with PCP (Dawson et al., 2012). These are especially noteworthy findings given that several lines of evidence converge to implicate the thalamic reticular nucleus in the pathology of schizophrenia (Ferrarelli and Tononi, 2011). In human resting-state fMRI, ketamine challenge increases global-based connectivity (GBC), i.e., functional correlations of each voxel with every other voxel. In particular, the increased GBC of six clusters are associated with positive symptoms, whereas constant or decreased GBC in the dorsal and medial striatum and thalamus is associated with negative symptoms (Driesen et al., 2013). Ketamine also weakens the anticorrelated relationship between a task-positive network and the DMN during a working memory task (Anticevic et al., 2012), mirroring the reduced DMN suppression during working memory tasks observed in schizophrenia patients and their first-degree relatives (Whitfield-Gabrieli et al., 2009). Other ketamine induced resting state network changes include reduced connectivity between both the DMN and the affective network with the dorsal medial PFC (Scheidegger et al., 2012), reduced connectivity between the auditory and somatosensory network and pain processing areas, and increased connectivity between both cerebellum and visual cortex with the medial visual network (Niesters et al., 2012).
Cortical Oscillations
Although fMRI is limited by slow temporal resolution, the faster temporal resolution of EEG has revealed abnormal oscillatory activity in schizophrenia, particularly in the gamma band (30–80 Hz) (Uhlhaas, 2013; Uhlhaas and Singer, 2011; Kehrer et al., 2008; Roopun et al., 2008). The gamma rhythm is principally generated by fast-spiking GABAergic interneurons (Table 1) (Hájos et al., 2004; Gloveli et al., 2005), whose output induces GABAA receptor mediated inhibitory postsynaptic potentials (IPSPs) in principal cells (Roopun et al., 2008). Interneurons are excited in unison, either by afferent fibers or reciprocal connections from principal cells (Metherate and Cruikshank, 1999; Traub et al., 1996; Roopun et al., 2008); the interneurons, in turn, have reciprocal inhibitory connections with one another, limiting their firing to a synchronous pattern (Roopun et al., 2008). Gamma rhythms are associated with short-term memory (Tallon-Baudry et al., 1998), working memory (Haenschel et al., 2009), selective attention (Tiitinen et al., 1993), primary sensory representation (Singer and Gray, 1995), cognitive control (Cho et al., 2006), social cognition (Williams et al., 2009), and integrative processing (Herrmann and Demiralp, 2005). Many reductions in the gamma rhythm have been reported in schizophrenia patients. As with the abnormalities of functional networks observed with fMRI, the nature of gamma band abnormalities is complicated; while gamma oscillations in schizophrenia are often reduced, increases have also been observed (Bucci et al., 2007). Like resting-state GBC, decreases are often associated with negative symptoms and increases are often associated with positive symptoms (Figure 3) (Kehrer et al., 2008; Bucci et al., 2007; Baldeweg et al., 1998; Spencer et al., 2008: 2). Visual-evoked gamma band oscillations have lower frequency in schizophrenia patients (Spencer et al., 2004), as well as reduced phase locking (Spencer et al., 2008), decreased interhemispheric phase coherence (in contradistinction to the increased anterior-posterior phase coherence seen in healthy controls), and increased latency to coherence changes (Spencer et al., 2003). All of these abnormalities suggest a mechanism by which visual information is not integrated both within visual cortex and across cortical networks. Schizophrenia patients have difficulty with visual perception tasks such as contour integration (Keane et al., 2012), suggesting faulty binding of visual information within visual cortex or even later in the dorsal visual stream, which may depend on gamma coherence. Furthermore, schizophrenia patients’ deficits in top-down visual processing (Dima et al., 2011: 300) could be explained by poor gamma coherence (and thus functional integration) between posterior, visual and anterior, cognitive networks.
Schizophrenia patients’ auditory steady-state responses (ASSR) are reduced in both phase locking and evoked power, and phase locking of the 40-Hz harmonic of the 20-Hz ASSR is positively correlated with positive symptoms (Spencer et al., 2008). Based on these data, it is reasonable to suspect aberrant, gamma-mediated neural computations in faulty auditory processing in schizophrenia, including auditory hallucinations. Similarly, cognitive control induced modulation of prefrontal gamma band activity, correlated with cognitive control demand and task performance in healthy controls, is not observed in schizophrenia patients (Cho et al., 2006). N-back tests of working memory induce excessive frontal gamma band activity in schizophrenia patients, whose performance is worse than that of healthy controls (Barr et al., 2010; Barr et al., 2011). One might draw a crude analogy between the schizophrenic PFC and an overworked engine: frontal networks in schizophrenia appear inflexible and inefficient, thus serving as weaker substrates for working memory. Facial emotion processing in schizophrenia patients reduces gamma synchrony relative to pre-stimulus baseline, yet patients have higher absolute gamma synchrony relative to controls (Williams et al., 2009). Here again, schizophrenia patients’ cortical circuits generate gamma synchrony, perhaps even excessively, but cannot appropriately modulate it. In a sense, the “inappropriateness” of gamma modulation is reminiscent of the inappropriate affect central to schizophrenia’s negative symptoms: both seem to reflect misplaced salience of stimuli, a disconnection with the external world, and an inefficient use of resources. If cognitive symptoms are driven by inappropriate gamma modulation, could it be that the negative and cognitive symptoms of schizophrenia are unified on a more abstract level? Abnormally low dopaminergic input to the PFC might deprive both emotional and task-related stimuli of their appropriate salience, unifying these symptoms on a causal level, as well. While this isomorphism between symptoms offers welcome simplicity, the assumption that gamma rhythmogenesis per se is unimpaired in schizophrenia is challenged by evidence from TMS studies that seek to elucidate effective cortical relationships. Schizophrenia patients have reduced gamma oscillations elicited by TMS over right premotor cortex in the resting state (Ferrarelli et al., 2008) and by 20-Hz repetitive TMS (rTMS) over DLPFC during an N-back working memory task (Barr et al., 2011). Moreover, rTMS of DLPFC suppresses frontal gamma oscillations in schizophrenia patients in as little as one session (Barr et al., 2011; Farzan et al., 2012). This ultimately suggests that inappropriate gamma modulation is a problem rooted not in prefrontal hypodopaminergia, but rather in faulty gamma rhythmogenesis, which is in turn rooted in abnormal glutamatergic and GABAergic transmission at NMDAR and GABAA receptors, respectively. Cortical networks in schizophrenia appear rigid and inflexible, as judged from their gamma band activity. By contrast, complex physiological systems often operate on the “edge of chaos,” meaning their control parameters are posed near critical values, allowing the system to quickly and flexibly transitions between many possible states (Fraiman et al., 2009; Tagliazucchi et al., 2012; Haimovici, et al., 2013; Sporns, 2011). In fact, the very complexity that lends the system necessary flexibility is due to a healthy degree of nonlinear chaos. Note that chaos in this context is defined as extreme sensitivity to initial conditions, a state in which effects are disproportional to their causes (Rickles 2007; Coffey, 1998; Breakspear, 2006). This is distinct from popular notions of chaos as randomness or genuine disorder. Loss of complexity (i.e., a transition to lower dimensional dynamics) in physiology is often pathological (Otero-Siliceo and Arriada-Mendicoa, 2003; Rickles, 2007; Manor and Lipsitz, 2013). For instance, a heartbeat that is too regular is linked to cardiac pathologies such as congestive heart failure (Poon and Merrill, 1997; Norris et al., 2008; Ho et al., 2011). By the same token, an epileptic seizure manifests itself as an interictal-ictal phase transition in which EEG dynamics shift from complex to highly regular (Stam, 2005; Rickles et al., 2007). Along these same lines, schizophrenic pathology might be due in part to reduced complexity and flexibility of gamma oscillations. Future studies should analyze schizophrenia patients’ EEG oscillations in the context of nonlinear dynamics and complexity science.
The gamma band is not the only frequency band with abnormal oscillatory activity in schizophrenia. Various abnormalities have been detected in the high beta (20–29 Hz), alpha, theta, and delta bands in schizophrenia patients (Uhlhaas and Singer, 2011; Roopun et al., 2008: 2; Uhlhaas, 2013; Uhlhaas and Singer, 2012). As we have chosen to focus this section on gamma oscillations, a detailed delineation of findings in lower frequency bands is not appropriate here. However, it is important to note that, as with the gamma band, schizophrenia patients’ oscillations in lower frequencies bands include both abnormal increases and decreases in power during both induced and resting oscillations (Uhlhaas and Singer, 2011). All of these brain rhythms undergo maturation in healthy humans during ages at which the onset of schizophrenia often occurs, implying that schizophrenia is a neurodevelopmental disorder involving inappropriate neural synchrony (Uhlhaas and Singer, 2011). Drawing on correlations between conscious awareness and increased coherence between distributed brain regions, Tononi and Edelman (2000) have suggested that schizophrenia is a disease of faulty reentrant interactions, defined as reciprocal signals between functionally segregated brain areas that allow for functional integration (Edelman and Tononi, 2000).
Like the above hypotheses, any theory drawn from these findings must attempt to make sense of varied and sometimes even contradictory data. Naturally, one might hope for insight from a ketamine model. At it turns out, the effects of ketamine on cortical rhythms are just as varied and context dependent as cortical rhythms observed in schizophrenia. Subanesthetic doses of ketamine and MK-801 both increase gamma power in rat neocortex (Pinault, 2008) and hippocampus (Ma and Leung, 2007) in vivo, yet contradictory, null results are reported for ketamine’s effect on hippocampal gamma power in vitro (Cunningham et al., 2006; Dickinson et al., 2003), suggesting gamma rhythms there are generated from a distant origin. By contrast, ketamine attenuates gamma rhythmogenesis in rat medial entorhinal cortex in vitro (Cunningham et al., 2006; Roopun et al., 2008). If hippocampal gamma rhythms are, in fact, generated locally, than an alternate explanation for the varied effects of ketamine on gamma rhythmogenesis is that NMDAR mediated excitation modulates gamma power differentially across cortical layers and regions. Might these differential effects also explain context dependent increases and reductions of gamma activity in schizophrenia? Curiously, ketamine administered to patients for general anesthesia prior to surgery increases the amplitude of the 40-Hz ASSR (Plourde et al., 1997), whereas in schizophrenia patients, the amplitude of 40-Hz ASSR is instead reduced (Hamm et al., 2011; Uhlhaas and Singer, 2011). Should this finding call into question the validity of the ketamine model or merely the appropriateness of using anesthetic doses of ketamine to model schizophrenic symptoms? A more recent study administering subanaesthetic doses of ketamine to healthy subjects shows that ketamine increases gamma oscillations and reduces delta oscillations in response to auditory clicks (Hong et al., 2010). Similarly, cortical slices bathed in kainate and challenged with ketamine show increases in gamma power in primary auditory cortex (Roopun et al., 2008). In rat visual cortex, both ketamine and PCP induce phase coupling of gamma oscillations across cortical layers and decelerate high-frequency gamma oscillations, providing a possible physiological basis for psychotic visual hallucinations (Anver et al., 2011). NMDAR antagonists that are nonspecific or specific to the NR2A subunit increase gamma power in rat cortex, whereas antagonists specific to other NMDAR subunits have weak effects (Kocsis, 2012). This finding implies that the NR2A subunit may have special significance to the etiology of NMDAR hypofunction and schizophrenia.
Because changes in gamma power are largely context dependent in schizophrenia, it is difficult to assess the fidelity with which ketamine replicates these changes in healthy subjects. Although the auditory response of gamma oscillations under ketamine challenge is opposite that usually observed in schizophrenia patients (Plourde et al., 1997; Hong et al., 2010), the general theta-to-gamma shift observed in schizophrenia is replicated using ketamine (Ehrlichman et al., 2009). Ketamine also increases the power of 20–29 Hz oscillations in the beta band in association cortex and PFC, a finding likely compatible with increased frontal and parietal resting beta oscillations in schizophrenia patients with auditory hallucinations (Roopun et al., 2008; Mulert et al., 2011; Lee et al., 2006).
Alterations of Brain Signaling Dynamics
Having considered many lines of research, it is appropriate to take a step back and view the problem from a larger perspective. Abnormalities of cortical oscillations in schizophrenia and other psychiatric illnesses are generally attributable to alterations of a parameter that transcends the ketamine model, the balance of excitation and inhibition (E/I-balance) (Uhlhaas, 2013; Uhlhaas and Singer, 2012; Kehrer et al., 2008; Anticevic et al., 2012). This parameter reflects interactions between two transmitter systems involved in NMDAR hypofunction: the glutamatergic and GABAergic systems. Neural synchrony subtly depends on E/I balance (Uhlhaas and Singer, 2012), and maturations of E/I balance during adolescence offers insight into schizophrenia’s age of onset and support for the neurodevelopmental hypothesis of schizophrenia (Uhlhaas and Singer, 2011; Uhlhaas, 2013). Moreover, E/I-balance is an important parameter for understanding emergent properties of cortical networks in light of Turing instability, the principle by which self-organized spatial patterns can emerge from a system with homogenously distributed sources of activation and inhibition (Turing, 1952). By reducing the conductance of excitatory cells onto inhibitory cells (as would happen after ketamine challenge) in a biophysical model of working memory, a simulated BOLD signal results that matches that of empirical data (Anticevic et al., 2012). It has been postulated that schizophrenia itself is a dynamical diseases resulting from the tuning of a control parameter past a critical value (an der Heiden, 2006; Breakspear, 2006; Friston, 1996), and indeed, schizophrenia patients show nonlinearity in their EEG time series (Carlino et al., 2012; Lee et al., 2001). Future dynamical models of schizophrenia might attempt explain the onset of symptoms using E/I-balance as a control parameter.
Conclusions, Limitations, and Future Directions
The ketamine model of acute schizophrenia is useful for explaining the positive and negative symptoms, cognitive deficits, dopaminergic dysfunction, GABAergic dysfunction, age of onset, functional dysconnectivity, and abnormal cortical oscillations observed in schizophrenia in terms of NMDAR hypofunction and interactions with D2, opioid, and other receptors (Figure 3). Although the ketamine model has strong explanatory and epistemic value, it is not without its weaknesses. Perhaps due in part to ketamine’s promiscuity, some discrepancy exists between ketamine intoxication and schizophrenic symptoms. Because visual disturbances—induced by ketamine—are rare in established (chronic) schizophrenia and hallucinatory voices—common in established schizophrenia—are rarely induced by ketamine, the hallucinatory pattern of ketamine better matches that of acute schizophrenia, in which hallucinatory symptoms are still developing, than chronic schizophrenia (Javitt, 2007). As mentioned earlier, ASSR under ketamine challenge do not match those of schizophrenia patients, and in vitro ketamine challenge induces spatially differential modulations of gamma power that are difficult to interpret. Moreover, methodological problems exist with interpretations of phase synchronization in sensor space; future studies should aim to analyze phase synchronization in source space using source reconstruction (Uhlhaas, 2013). Beyond these smaller issues, there is still a need to integrate the ketamine model with other compatible pharmacological models of schizophrenia. For instance, Δ-9-THC induces positive and negative symptoms of schizophrenia (D’Souza et al., 2004), transiently disturbs glutamate and GABA release in adolescence, and cannabis use increases the risk of developing schizophrenia (Table 1) (Bossong and Niesink, 2010). How can these findings be unified with the ketamine model? Future studies should look at different pharmacological manipulations within the same group of subjects. Relatively few studies have challenged the same subjects with both ketamine and, for instance, amphetamine to simultaneously test the glutamate and dopamine hypotheses (Javitt, 2007). More comparisons between ketamine and salvinorin A, another dissociative with similar properties, are also greatly needed. Finally, although some authors have already framed schizophrenia in the context of nonlinear dynamics by suggesting that phase transitions might be important for understanding its pathogenesis (an der Heiden, 2006; Uhlhaas and Singer, 2011; Breakspear, 2006; Friston, 1996), few studies have actually examined schizophrenia in a nonlinear systems context. Future investigations should look for nonlinearity and evidence of bifurcations after ketamine infusion in the EEG of healthy volunteers.
Figure 2. Chemical Structures of (S)-ketamine and Phencyclidine.
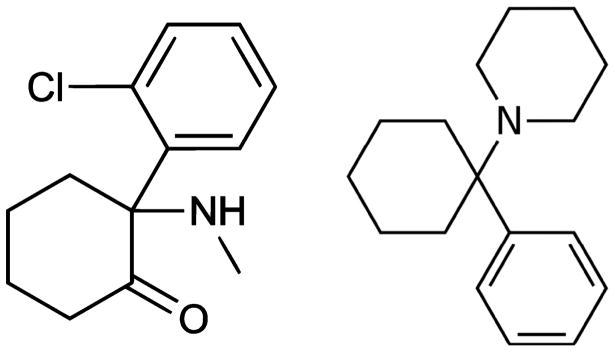
(S)-ketamine, left, is the more potent of ketamine’s two enantiomers. It is known as 2-chlorphenyl-2-methylamino-cyclohexanone by International Union of Pure and Applied Chemists (IUPAC) nomenclature. Phencyclidine (PCP, right) and ketamine share a binding site within the pore of the NMDAR and induce similar effects. Both chemicals are dissociative anesthetics and share structural similarities such as aromaticity.
Acknowledgments
Funding Acknowledgement: This work was supported by a National Alliance for Medical Image Computing (www.namic.org) sub-award to J.D.V.H. (2U54EB005149) and a R41NS081792 sub-award to J.D.V.H.
The authors wish to express their thanks to Vaughan Greer for his assistance with figure artwork. We also thank the faculty and staff of the Laboratory of Neuro Imaging and the Institute of Neuroimaging and Informatics at USC.
Footnotes
Conflict of interest
J.D.V.H. receives partial support as a consultant through an NIH STTR sub-award from Kitware, Inc., a medical imaging processing and visualization company based in Clifton Park, NY and Carrboro, North Carolina, USA. This article does not advocate for or against the products of this company and is not intended to represent the views or mission of Kitware, Inc. The authors declare no other potential conflicts of interest.
Contributor Information
Joel Frohlich, Neuroscience Research Program, 1506D Gonda Center, University of California, Los Angeles Box 951761, Los Angeles, CA 90095-1761.
John Darrell Van Horn, Email: jvanhorn@usc.edu, The Institute for Neuroimaging and Informatics, Keck School of Medicine, University of Southern California, 2001 North Soto Street – SSB1-102, Los Angeles, CA 90032, Phone: (323) 442-7246.
References
- Aalto S, Ihalainen J, Hirvonen J, et al. Cortical glutamate-dopamine interaction and ketamine-induced psychotic symptoms in man. Psychopharmacology. 2005;182(3):375–383. doi: 10.1007/s00213-005-0092-6. [DOI] [PubMed] [Google Scholar]
- Abi-Dargham A, Mawlawi O, Lombardo I, et al. Prefrontal dopamine D1 receptors and working memory in schizophrenia. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22(9):3708–3719. doi: 10.1523/JNEUROSCI.22-09-03708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4. Washinton, D.C: American Psychiatric Association; 2000. text revision. [Google Scholar]
- Amitai N, Kuczenski R, Behrens MM, et al. Repeated phencyclidine administration alters glutamate release and decreases GABA markers in the prefrontal cortex of rats. Neuropharmacology. 2012;62(3):1422–1431. doi: 10.1016/j.neuropharm.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreone N, Tansella M, Cerini R, et al. Cerebral atrophy and white matter disruption in chronic schizophrenia. European archives of psychiatry and clinical neuroscience. 2007;257(1):3–11. doi: 10.1007/s00406-006-0675-1. [DOI] [PubMed] [Google Scholar]
- Anonymous. Substances and classifications table. European Monitoring Centre for Drugs and Drug Addiction; 2008. Available from: http://www.emcdda.europa.eu/html.cfm/index5175EN.html. [Google Scholar]
- Anonymous. WHO Model List of Essential Medicines, 17th list. World Health Organization; 2011. [accessed 23 June 2013]. Available from: http://www.who.int/medicines/publications/essentialmedicines/en/index.html. [Google Scholar]
- Anticevic A, Gancsos M, Murray JD, et al. NMDA receptor function in large-scale anticorrelated neural systems with implications for cognition and schizophrenia. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(41):16720–16725. doi: 10.1073/pnas.1208494109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anver H, Ward PD, Magony A, et al. NMDA receptor hypofunction phase couples independent γ-oscillations in the rat visual cortex. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2011;36(2):519–528. doi: 10.1038/npp.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroni F, Iacovidou N, Dontas I, et al. Pharmacological aspects and potential new clinical applications of ketamine: reevaluation of an old drug. Journal of clinical pharmacology. 2009;49(8):957–964. doi: 10.1177/0091270009337941. [DOI] [PubMed] [Google Scholar]
- Baldeweg T, Spence S, Hirsch SR, et al. Gamma-band electroencephalographic oscillations in a patient with somatic hallucinations. Lancet. 1998;352(9128):620–621. doi: 10.1016/S0140-6736(05)79575-1. [DOI] [PubMed] [Google Scholar]
- Barr MS, Farzan F, Tran LC, et al. Evidence for excessive frontal evoked gamma oscillatory activity in schizophrenia during working memory. Schizophrenia research. 2010;121(1–3):146–152. doi: 10.1016/j.schres.2010.05.023. [DOI] [PubMed] [Google Scholar]
- Barr MS, Farzan F, Arenovich T, et al. The effect of repetitive transcranial magnetic stimulation on gamma oscillatory activity in schizophrenia. PloS one. 2011;6(7):e22627. doi: 10.1371/journal.pone.0022627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett DS, Bullmore E, Verchinski BA, et al. Hierarchical organization of human cortical networks in health and schizophrenia. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28(37):9239–9248. doi: 10.1523/JNEUROSCI.1929-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum VC, Tecson ME. Ketamine inhibits transsarcolemmal calcium entry in guinea pig myocardium: direct evidence by single cell voltage clamp. Anesthesia and analgesia. 1991;73(6):804–807. doi: 10.1213/00000539-199112000-00022. [DOI] [PubMed] [Google Scholar]
- Behrens MM, Ali SS, Dao DN, et al. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science (New York, NY) 2007;318(5856):1645–1647. doi: 10.1126/science.1148045. [DOI] [PubMed] [Google Scholar]
- Benoit E, Carratu MR, Dubois JM, et al. Mechanism of action of ketamine in the current and voltage clamped myelinated nerve fibre of the frog. British Journal of Pharmacology. 1986;87(2):291–297. doi: 10.1111/j.1476-5381.1986.tb10817.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, et al. Antidepressant effects of ketamine in depressed patients. Biological psychiatry. 2000;47(4):351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- Bleuler E. Die Prognose der Dementia praecox. Allg Z Psychiatr. 1908;65:436–464. [Google Scholar]
- Bossong MG, Niesink RJM. Adolescent brain maturation, the endogenous cannabinoid system and the neurobiology of cannabis-induced schizophrenia. Progress in neurobiology. 2010;92(3):370–385. doi: 10.1016/j.pneurobio.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Breakspear M. The nonlinear theory of schizophrenia. The Australian and New Zealand journal of psychiatry. 2006;40(1):20–35. doi: 10.1080/j.1440-1614.2006.01737.x. [DOI] [PubMed] [Google Scholar]
- Breier A, Su TP, Saunders R, et al. Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(6):2569–2574. doi: 10.1073/pnas.94.6.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci P, Mucci A, Merlotti E, et al. Induced gamma activity and event-related coherence in schizophrenia. Clinical EEG and neuroscience: official journal of the EEG and Clinical Neuroscience Society (ENCS) 2007;38(2):96–104. doi: 10.1177/155005940703800212. [DOI] [PubMed] [Google Scholar]
- Calhoun VD, Eichele T, Pearlson G. Functional brain networks in schizophrenia: a review. Frontiers in human neuroscience. 2009;3:17. doi: 10.3389/neuro.09.017.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlino E, Sigaudo M, Pollo A, et al. Nonlinear analysis of electroencephalogram at rest and during cognitive tasks in patients with schizophrenia. Journal of psychiatry & neuroscience: JPN. 2012;37(4):259–266. doi: 10.1503/jpn.110030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castner SA, Arriza JL, Roberts JC, et al. Reversal of ketamine-induced working memory impairments by the GABAAalpha2/3 agonist TPA023. Biological psychiatry. 2010;67(10):998–1001. doi: 10.1016/j.biopsych.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Chatterton JE, Awobuluyi M, Premkumar LS, et al. Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature. 2002;415(6873):793–798. doi: 10.1038/nature715. [DOI] [PubMed] [Google Scholar]
- Cho RY, Konecky RO, Carter CS. Impairments in frontal cortical gamma synchrony and cognitive control in schizophrenia. Proceedings of the National Academy of Sciences of the United States of America; 2006. pp. 19878–19883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chumakov I, Blumenfeld M, Guerassimenko O, et al. Genetic and physiological data implicating the new human gene G72 and the gene for D-amino acid oxidase in schizophrenia. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(21):13675–13680. doi: 10.1073/pnas.182412499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran SM, Kennedy M, McKerchar CE, et al. Induction of metabolic hypofunction and neurochemical deficits after chronic intermittent exposure to phencyclidine: differential modulation by antipsychotic drugs. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2003;28(2):265–275. doi: 10.1038/sj.npp.1300031. [DOI] [PubMed] [Google Scholar]
- Coffey DS. Self-organization, complexity and chaos: the new biology for medicine. Nature medicine. 1998;4(8):882–885. doi: 10.1038/nm0898-882. [DOI] [PubMed] [Google Scholar]
- Coppa-Hopman R, Galle J, Pimkine D. D1 receptor antagonist-induced long-term depression in the medial prefrontal cortex of rat, in vivo: an animal model of psychiatric hypofrontality. Journal of psychopharmacology (Oxford, England) 2009;23(6):672–685. doi: 10.1177/0269881108091256. [DOI] [PubMed] [Google Scholar]
- Corssen G, Domino EF. Dissociative anesthesia: further pharmacologic studies and first clinical experience with the phencyclidine derivative CI-581. Anesthesia and analgesia. 1966;45(1):29–40. [PubMed] [Google Scholar]
- Coyle JT, Basu A, Benneyworth M, et al. Glutamatergic synaptic dysregulation in schizophrenia: therapeutic implications. Handbook of experimental pharmacology. 2012;(213):267–295. doi: 10.1007/978-3-642-25758-2_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross-Disorder Group of the Psychiatric Genomics Consortium. Smoller JW, Craddock N, et al. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381(9875):1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham MO, Hunt J, Middleton S, et al. Region-specific reduction in entorhinal gamma oscillations and parvalbumin-immunoreactive neurons in animal models of psychiatric illness. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26(10):2767–2776. doi: 10.1523/JNEUROSCI.5054-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza DC, Perry E, MacDougall L, et al. The psychotomimetic effects of intravenous delta-9-tetrahydrocannabinol in healthy individuals: implications for psychosis. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2004;29(8):1558–1572. doi: 10.1038/sj.npp.1300496. [DOI] [PubMed] [Google Scholar]
- Daniell LC. The noncompetitive N-methyl-D-aspartate antagonists, MK-801, phencyclidine and ketamine, increase the potency of general anesthetics. Pharmacology, biochemistry, and behavior. 1990;36(1):111–115. doi: 10.1016/0091-3057(90)90134-4. [DOI] [PubMed] [Google Scholar]
- Dawson N, Morris BJ, Pratt JA. Subanaesthetic Ketamine Treatment Alters Prefrontal Cortex Connectivity With Thalamus and Ascending Subcortical Systems. Schizophrenia bulletin. 2013;39(2):366–377. doi: 10.1093/schbul/sbr144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson N, Xiao X, McDonald M, et al. Sustained NMDA Receptor Hypofunction Induces Compromised Neural Systems Integration and Schizophrenia-Like Alterations in Functional Brain Networks. Cerebral cortex (New York, NY: 1991) 2012 doi: 10.1093/cercor/bhs322. [DOI] [PubMed] [Google Scholar]
- Deakin JFW, Lees J, McKie S, et al. Glutamate and the neural basis of the subjective effects of ketamine: a pharmaco-magnetic resonance imaging study. Archives of general psychiatry. 2008;65(2):154–164. doi: 10.1001/archgenpsychiatry.2007.37. [DOI] [PubMed] [Google Scholar]
- Debonnel G. Current hypotheses on sigma receptors and their physiological role: possible implications in psychiatry. Journal of psychiatry & neuroscience: JPN. 1993;18(4):157–172. [PMC free article] [PubMed] [Google Scholar]
- an der Heiden U. Schizophrenia as a dynamical disease. Pharmacopsychiatry. 2006;39(Suppl 1):S36–42. doi: 10.1055/s-2006-931487. [DOI] [PubMed] [Google Scholar]
- Dickinson R, Awaiz S, Whittington MA, et al. The effects of general anaesthetics on carbachol-evoked gamma oscillations in the rat hippocampus in vitro. Neuropharmacology. 2003;44(7):864–872. doi: 10.1016/s0028-3908(03)00083-2. [DOI] [PubMed] [Google Scholar]
- Dima D, Dillo W, Bonnemann C, et al. Reduced P300 and P600 amplitude in the hollow-mask illusion in patients with schizophrenia. Psychiatry research. 2011;191(2):145–151. doi: 10.1016/j.pscychresns.2010.09.015. [DOI] [PubMed] [Google Scholar]
- Domino EF, Chodoff P, Corssen G. Pharmacologic effects of CI-581, a new dissociative anesthetic, in man. Clinical pharmacology and therapeutics. 1965;6:279–291. doi: 10.1002/cpt196563279. [DOI] [PubMed] [Google Scholar]
- Driesen NR, McCarthy G, Bhagwagar Z, et al. Relationship of resting brain hyperconnectivity and schizophrenia-like symptoms produced by the NMDA receptor antagonist ketamine in humans. Molecular psychiatry. 2013 doi: 10.1038/mp.2012.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelman GM, Tononi G. A Universe of Consciousness: How Matter Becomes Imagination. New York: Basic Books; 2000. [Google Scholar]
- Ehrlichman RS, Gandal MJ, Maxwell CR, et al. N-methyl-d-aspartic acid receptor antagonist-induced frequency oscillations in mice recreate pattern of electrophysiological deficits in schizophrenia. Neuroscience. 2009;158(2):705–712. doi: 10.1016/j.neuroscience.2008.10.031. [DOI] [PubMed] [Google Scholar]
- Fain G. Eastern Economy Edition. New Delhi: Prentice-Hall of India; 1999. Molecular and Cellular Physiology of Neurons. [Google Scholar]
- Farber NB, Olney JW. Drugs of abuse that cause developing neurons to commit suicide. Brain research. Developmental brain research. 2003;147(1–2):37–45. doi: 10.1016/j.devbrainres.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Farzan F, Barr MS, Sun Y, et al. Transcranial magnetic stimulation on the modulation of gamma oscillations in schizophrenia. Annals of the New York Academy of Sciences. 2012;1265:25–35. doi: 10.1111/j.1749-6632.2012.06543.x. [DOI] [PubMed] [Google Scholar]
- Feinberg I. Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? Journal of psychiatric research. 1982;17(4):319–334. doi: 10.1016/0022-3956(82)90038-3. [DOI] [PubMed] [Google Scholar]
- Ferrarelli F, Massimini M, Peterson MJ, et al. Reduced evoked gamma oscillations in the frontal cortex in schizophrenia patients: a TMS/EEG study. The American journal of psychiatry. 2008;165(8):996–1005. doi: 10.1176/appi.ajp.2008.07111733. [DOI] [PubMed] [Google Scholar]
- Ferrarelli F, Tononi G. The thalamic reticular nucleus and schizophrenia. Schizophrenia bulletin. 2011;37(2):306–315. doi: 10.1093/schbul/sbq142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari MCL, Kimura L, Nita LM, et al. Structural brain abnormalities in early-onset schizophrenia. Arquivos de neuro-psiquiatria. 2006;64(3B):741–746. doi: 10.1590/s0004-282x2006000500008. [DOI] [PubMed] [Google Scholar]
- Fraiman D, Balenzuela P, Foss J, et al. Ising-like dynamics in large-scale functional brain networks. Physical review. E, Statistical, nonlinear, and soft matter physics. 2009;79(6 Pt 1):061922. doi: 10.1103/PhysRevE.79.061922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantseva M, Cui J, Farzan F, et al. Disrupted Cortical Conductivity in Schizophrenia: TMS-EEG Study. Cerebral cortex (New York, NY: 1991) 2012 doi: 10.1093/cercor/bhs304. [DOI] [PubMed] [Google Scholar]
- Frenkel C, Urban BW. Molecular actions of racemic ketamine on human CNS sodium channels. British journal of anaesthesia. 1992;69(3):292–297. doi: 10.1093/bja/69.3.292. [DOI] [PubMed] [Google Scholar]
- Friston KJ. Theoretical neurobiology and schizophrenia. British medical bulletin. 1996;52(3):644–655. doi: 10.1093/oxfordjournals.bmb.a011573. [DOI] [PubMed] [Google Scholar]
- Giovannini MG, Camilli F, Mundula A, et al. Glutamatergic regulation of acetylcholine output in different brain regions: a microdialysis study in the rat. Neurochemistry international. 1994;25(1):23–26. doi: 10.1016/0197-0186(94)90048-5. [DOI] [PubMed] [Google Scholar]
- Gloveli T, Dugladze T, Saha S, et al. Differential involvement of oriens/pyramidale interneurones in hippocampal network oscillations in vitro. The Journal of physiology. 2005;562(Pt 1):131–147. doi: 10.1113/jphysiol.2004.073007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman MB, Torres IJ, Keedy S, et al. Reduced anterior hippocampal formation volume in hyponatremic schizophrenic patients. Hippocampus. 2007;17(7):554–562. doi: 10.1002/hipo.20292. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, Castner SA, Svensson TH, et al. Targeting the dopamine D1 receptor in schizophrenia: insights for cognitive dysfunction. Psychopharmacology. 2004;174(1):3–16. doi: 10.1007/s00213-004-1793-y. [DOI] [PubMed] [Google Scholar]
- Gottesmann C. The dreaming sleep stage: a new neurobiological model of schizophrenia? Neuroscience. 2006;140(4):1105–1115. doi: 10.1016/j.neuroscience.2006.02.082. [DOI] [PubMed] [Google Scholar]
- Grace AA. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience. 1991;41(1):1–24. doi: 10.1016/0306-4522(91)90196-u. [DOI] [PubMed] [Google Scholar]
- Granger B. Synaptogenesis and synaptic pruning: role in triggering schizophrenia. Presse médicale (Paris, France: 1983) 1996;25(33):1595–1598. [PubMed] [Google Scholar]
- Gupta A, Devi LA, Gomes I. Potentiation of μ-opioid receptor-mediated signaling by ketamine. Journal of neurochemistry. 2011;119(2):294–302. doi: 10.1111/j.1471-4159.2011.07361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haenschel C, Bittner RA, Waltz J, et al. Cortical oscillatory activity is critical for working memory as revealed by deficits in early-onset schizophrenia. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29(30):9481–9489. doi: 10.1523/JNEUROSCI.1428-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn C-G, Wang H-Y, Cho D-S, et al. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nature medicine. 2006;12(7):824–828. doi: 10.1038/nm1418. [DOI] [PubMed] [Google Scholar]
- Haimovici A, Tagliazucchi E, Balenzuela P, et al. Brain organization into resting state networks emerges at criticality on a model of the human connectome. Physical review letters. 2013;110(17):178101. doi: 10.1103/PhysRevLett.110.178101. [DOI] [PubMed] [Google Scholar]
- Hájos N, Pálhalmi J, Mann EO, et al. Spike timing of distinct types of GABAergic interneuron during hippocampal gamma oscillations in vitro. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2004;24(41):9127–9137. doi: 10.1523/JNEUROSCI.2113-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm JP, Gilmore CS, Picchetti NAM, et al. Abnormalities of neuronal oscillations and temporal integration to low- and high-frequency auditory stimulation in schizophrenia. Biological psychiatry. 2011;69(10):989–996. doi: 10.1016/j.biopsych.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Kinoshita H, Amano M, et al. MK-801 increases endogenous acetylcholine release in the rat parietal cortex: a study using brain microdialysis. Neuroscience letters. 1993;150(1):53–56. doi: 10.1016/0304-3940(93)90106-u. [DOI] [PubMed] [Google Scholar]
- Hashimoto K. Role of sigma-1 receptors in neural plasticity and in antipsychotic action. Nihon shinkei seishin yakurigaku zasshi = Japanese journal of psychopharmacology. 2010;30(3):123–127. [PubMed] [Google Scholar]
- Hashimoto T, Volk DW, Eggan SM, et al. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23(15):6315–6326. doi: 10.1523/JNEUROSCI.23-15-06315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Su T-P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007;131(3):596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Su T-P. Sigma-1 receptor ligands: potential in the treatment of neuropsychiatric disorders. CNS drugs. 2004;18(5):269–284. doi: 10.2165/00023210-200418050-00001. [DOI] [PubMed] [Google Scholar]
- Herrmann CS, Demiralp T. Human EEG gamma oscillations in neuropsychiatric disorders. Clinical neurophysiology: official journal of the International Federation of Clinical Neurophysiology. 2005;116(12):2719–2733. doi: 10.1016/j.clinph.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Hirota K, Hashimoto Y, Lambert DG. Interaction of intravenous anesthetics with recombinant human M1 - M3 muscarinic receptors expressed in Chinese hamster ovary cells. Anesthesia and Analgesia. 2002;95:1607–1610. doi: 10.1097/00000539-200212000-00025. [DOI] [PubMed] [Google Scholar]
- Hirota K, Okawa H, Appadu BL, et al. Stereoselective interaction of ketamine with recombinant mu, kappa, and delta opioid receptors expressed in Chinese hamster ovary cells. Anesthesiology. 1999;90(1):174–182. doi: 10.1097/00000542-199901000-00023. [DOI] [PubMed] [Google Scholar]
- Hirota K. Ketamine: Its mechanism(s) of action and unusual clinical uses. British Journal of Anesthesia. 1996;77(4):441–444. doi: 10.1093/bja/77.4.441. [DOI] [PubMed] [Google Scholar]
- Ho Y-L, Lin C, Lin Y-H, et al. The prognostic value of non-linear analysis of heart rate variability in patients with congestive heart failure--a pilot study of multiscale entropy. PloS one. 2011;6(4):e18699. doi: 10.1371/journal.pone.0018699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman RE, McGlashan TH. Synaptic elimination, neurodevelopment, and the mechanism of hallucinated ‘voices’ in schizophrenia. The American journal of psychiatry. 1997;154(12):1683–1689. doi: 10.1176/ajp.154.12.1683. [DOI] [PubMed] [Google Scholar]
- Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27(43):11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong LE, Summerfelt A, Buchanan RW, et al. Gamma and delta neural oscillations and association with clinical symptoms under subanesthetic ketamine. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2010;35(3):632–640. doi: 10.1038/npp.2009.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horváth ZC, Czopf J, Buzsáki G. MK-801-induced neuronal damage in rats. Brain research. 1997;753(2):181–195. doi: 10.1016/s0006-8993(96)01290-5. [DOI] [PubMed] [Google Scholar]
- Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III--the final common pathway. Schizophrenia bulletin. 2009;35(3):549–562. doi: 10.1093/schbul/sbp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hustveit O, Maurset A, Oye I. Interaction of the chiral forms of ketamine with opioid, phencyclidine, sigma and muscarinic receptors. Pharmacology & toxicology. 1995;77(6):355–359. doi: 10.1111/j.1600-0773.1995.tb01041.x. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Hashimoto K. The role of sigma-1 receptors in the pathophysiology of neuropsychiatric diseases. Journal of Receptor, Ligand and Channel Research. 2010;3:25–36. [Google Scholar]
- Ishima T, Fujita Y, Kohno M, et al. Improvement of phencyclidine-induced cognitive deficits in mice by subsequent subchronic administration of fluvoxamine, but not sertraline. The Open Clinical Chemistry Journal. 2009;2:7–11. [Google Scholar]
- Javitt DC, Balla A, Burch S, et al. Reversal of phencyclidine-induced dopaminergic dysregulation by N-methyl-D-aspartate receptor/glycine-site agonists. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2004;29(2):300–307. doi: 10.1038/sj.npp.1300313. [DOI] [PubMed] [Google Scholar]
- Javitt DC, Zukin SR, Heresco-Levy U, et al. Has an angel shown the way? Etiological and therapeutic implications of the PCP/NMDA model of schizophrenia. Schizophrenia bulletin. 2012;38(5):958–966. doi: 10.1093/schbul/sbs069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC. Integrating the Neurobiology of Schizophrenia, International Review of Neurobiology. San Diego: Academic Press; 2007. Glutamate and Schizophrenia: Phencyclidine, N-methyl-D-aspartate receptors, and dopamine–glutamate interactions; pp. 70–96. [DOI] [PubMed] [Google Scholar]
- Kapur S, Seeman P. NMDA receptor antagonists ketamine and PCP have direct effects on the dopamine D2 and serotonin 5-HT2 receptors-implications for models of schizophrenia. Molecular psychiatry. 2002;7(8):837–844. doi: 10.1038/sj.mp.4001093. [DOI] [PubMed] [Google Scholar]
- Kapur S. Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. The American journal of psychiatry. 2003;160(1):13–23. doi: 10.1176/appi.ajp.160.1.13. [DOI] [PubMed] [Google Scholar]
- Karlsgodt KH, Robleto K, Trantham-Davidson H, et al. Reduced dysbindin expression mediates N-methyl-D-aspartate receptor hypofunction and impaired working memory performance. Biological psychiatry. 2011;69(1):28–34. doi: 10.1016/j.biopsych.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson P, Farde L, Halldin C, et al. PET study of D1 dopamine receptor binding in neuroleptic-naive patients with schizophrenia. The American journal of psychiatry. 2002;159(5):761–767. doi: 10.1176/appi.ajp.159.5.761. [DOI] [PubMed] [Google Scholar]
- Kashima H. Frontal dysfunction of chronic schizophrenia--the pros and cons in neuropsychological assessment. Yakubutsu, seishin, kōdō = Japanese journal of psychopharmacology. 1991;11(1):83–88. [PubMed] [Google Scholar]
- Keane BP, Silverstein SM, Barch DM, et al. The spatial range of contour integration deficits in schizophrenia. Experimental brain research. Experimentelle Hirnforschung. Expérimentation cérébrale. 2012;220(3–4):251–259. doi: 10.1007/s00221-012-3134-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrer C, Maziashvili N, Dugladze T, et al. Altered excitatory-inhibitory balance in the NMDA-hypofunction model of schizophrenia. Frontiers in molecular neuroscience. 2008;1:6. doi: 10.3389/neuro.02.006.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S-Y, Lee H, Kim H-J, et al. In vivo and ex vivo evidence for ketamine-induced hyperglutamatergic activity in the cerebral cortex of the rat: Potential relevance to schizophrenia. NMR in biomedicine. 2011;24(10):1235–1242. doi: 10.1002/nbm.1681. [DOI] [PubMed] [Google Scholar]
- Kinney JW, Davis CN, Tabarean I, et al. A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26(5):1604–1615. doi: 10.1523/JNEUROSCI.4722-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocsis B. Differential role of NR2A and NR2B subunits in N-methyl-D-aspartate receptor antagonist-induced aberrant cortical gamma oscillations. Biological psychiatry. 2012;71(11):987–995. doi: 10.1016/j.biopsych.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kometer M, Schmidt A, Jäncke L, et al. Activation of serotonin 2A receptors underlies the psilocybin-induced effects on α oscillations, N170 Visual-Evoked Potentials, and Visual Hallucinations. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33(25):10544–10551. doi: 10.1523/JNEUROSCI.3007-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkova T, Fuchs EC, Ponomarenko A, et al. NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron. 2010;68(3):557–569. doi: 10.1016/j.neuron.2010.09.017. [DOI] [PubMed] [Google Scholar]
- Kunitachi S, Fujita Y, Ishima T, et al. Phencyclidine-induced cognitive deficits in mice are ameliorated by subsequent subchronic administration of donepezil: role of sigma-1 receptors. Brain research. 2009;1279:189–196. doi: 10.1016/j.brainres.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Abi-Dargham A, Van Dyck CH, et al. Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(17):9235–9240. doi: 10.1073/pnas.93.17.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau C-I, Wang H-C, Hsu J-L, et al. Does the dopamine hypothesis explain schizophrenia? Reviews in the neurosciences. 2013;24(4):389–400. doi: 10.1515/revneuro-2013-0011. [DOI] [PubMed] [Google Scholar]
- Law AJ, Deakin JF. Asymmetrical reductions of hippocampal NMDAR1 glutamate receptor mRNA in the psychoses. Neuroreport. 2001;12(13):2971–2974. doi: 10.1097/00001756-200109170-00043. [DOI] [PubMed] [Google Scholar]
- Lee S-H, Wynn JK, Green MF, et al. Quantitative EEG and low resolution electromagnetic tomography (LORETA) imaging of patients with persistent auditory hallucinations. Schizophrenia research. 2006;83(2–3):111–119. doi: 10.1016/j.schres.2005.11.025. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Zhu YS, Xu YH, et al. Detection of non-linearity in the EEG of schizophrenic patients. Clinical neurophysiology: official journal of the International Federation of Clinical Neurophysiology. 2001;112(7):1288–1294. doi: 10.1016/s1388-2457(01)00544-2. [DOI] [PubMed] [Google Scholar]
- Lena I, Muffat S, Deschaux O. Dreaming and schizophrenia have the same neurochemical background. Journal of Sleep Research. 2006;13:141. [Google Scholar]
- Li D, He L. Association study between the NMDA receptor 2B subunit gene (GRIN2B) and schizophrenia: a HuGE review and meta-analysis. Genetics in medicine: official journal of the American College of Medical Genetics. 2007a;9(1):4–8. doi: 10.1097/01.gim.0000250507.96760.4b. [DOI] [PubMed] [Google Scholar]
- Li D, He L. G72/G30 genes and schizophrenia: a systematic meta-analysis of association studies. Genetics. 2007b;175(2):917–922. doi: 10.1534/genetics.106.061796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liemburg EJ, Knegtering H, Klein HC, et al. Antipsychotic medication and prefrontal cortex activation: a review of neuroimaging findings. European neuropsychopharmacology: the journal of the European College of Neuropsychopharmacology. 2012;22(6):387–400. doi: 10.1016/j.euroneuro.2011.12.008. [DOI] [PubMed] [Google Scholar]
- Löscher W, Annies R, Hönack D. The N-methyl-D-aspartate receptor antagonist MK-801 induces increases in dopamine and serotonin metabolism in several brain regions of rats. Neuroscience letters. 1991;128(2):191–194. doi: 10.1016/0304-3940(91)90258-u. [DOI] [PubMed] [Google Scholar]
- Lynall M-E, Bassett DS, Kerwin R, et al. Functional connectivity and brain networks in schizophrenia. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30(28):9477–9487. doi: 10.1523/JNEUROSCI.0333-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Leung LS. The supramammillo-septal-hippocampal pathway mediates sensorimotor gating impairment and hyperlocomotion induced by MK-801 and ketamine in rats. Psychopharmacology. 2007;191(4):961–974. doi: 10.1007/s00213-006-0667-x. [DOI] [PubMed] [Google Scholar]
- Mackenzie B, Erickson JD. Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Pflügers Archiv: European journal of physiology. 2004;447(5):784–795. doi: 10.1007/s00424-003-1117-9. [DOI] [PubMed] [Google Scholar]
- Maeng S, Zarate CA, Jr, Du J, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biological psychiatry. 2008;63(4):349–352. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Manor B, Lipsitz LA. Physiologic complexity and aging: Implications for physical function and rehabilitation. Progress in neuro-psychopharmacology & biological psychiatry. 2013;45(1):287–293. doi: 10.1016/j.pnpbp.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Saha S, Chant D, et al. Schizophrenia: a concise overview of incidence, prevalence, and mortality. Epidemiologic reviews. 2008;30:67–76. doi: 10.1093/epirev/mxn001. [DOI] [PubMed] [Google Scholar]
- Metherate R, Cruikshank SJ. Thalamocortical inputs trigger a propagating envelope of gamma-band activity in auditory cortex in vitro. Experimental brain research. Experimentelle Hirnforschung. Expérimentation cérébrale. 1999;126(2):160–174. doi: 10.1007/s002210050726. [DOI] [PubMed] [Google Scholar]
- Morris BJ, Cochran SM, Pratt JA. PCP: from pharmacology to modelling schizophrenia. Current opinion in pharmacology. 2005;5(1):101–106. doi: 10.1016/j.coph.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Mulert C, Kirsch V, Pascual-Marqui R, et al. Long-range synchrony of γ oscillations and auditory hallucination symptoms in schizophrenia. International journal of psychophysiology: official journal of the International Organization of Psychophysiology. 2011;79(1):55–63. doi: 10.1016/j.ijpsycho.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso F, Brinkmeyer J, Ecker D, et al. Ketamine effects on brain function--simultaneous fMRI/EEG during a visual oddball task. NeuroImage. 2011;58(2):508–525. doi: 10.1016/j.neuroimage.2011.06.045. [DOI] [PubMed] [Google Scholar]
- Nagai T, Kitahara Y, Shiraki A, et al. Dysfunction of dopamine release in the prefrontal cortex of dysbindin deficient sandy mice: an in vivo microdialysis study. Neuroscience letters. 2010;470(2):134–138. doi: 10.1016/j.neulet.2009.12.071. [DOI] [PubMed] [Google Scholar]
- Nakanishi T, Kekuda R, Fei YJ, et al. Cloning and functional characterization of a new subtype of the amino acid transport system N. American journal of physiology. Cell physiology. 2001;281(6):C1757–1768. doi: 10.1152/ajpcell.2001.281.6.C1757. [DOI] [PubMed] [Google Scholar]
- Newcomer JW, Farber NB, Jevtovic-Todorovic V, et al. Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 1999;20(2):106–118. doi: 10.1016/S0893-133X(98)00067-0. [DOI] [PubMed] [Google Scholar]
- Nieoullon A. Dopamine and the regulation of cognition and attention. Progress in neurobiology. 2002;67(1):53–83. doi: 10.1016/s0301-0082(02)00011-4. [DOI] [PubMed] [Google Scholar]
- Niesters M, Khalili-Mahani N, Martini C, et al. Effect of subanesthetic ketamine on intrinsic functional brain connectivity: a placebo-controlled functional magnetic resonance imaging study in healthy male volunteers. Anesthesiology. 2012;117(4):868–877. doi: 10.1097/ALN.0b013e31826a0db3. [DOI] [PubMed] [Google Scholar]
- Norris PR, Stein PK, Morris JA., Jr Reduced heart rate multiscale entropy predicts death in critical illness: a study of physiologic complexity in 285 trauma patients. Journal of critical care. 2008;23(3):399–405. doi: 10.1016/j.jcrc.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Olney JW, Farber NB. Discussion of Bogerts’ temporolimbic system theory of paranoid schizophrenia. Schizophrenia bulletin. 1997;23(3):533–536. doi: 10.1093/schbul/23.3.533. [DOI] [PubMed] [Google Scholar]
- Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Archives of general psychiatry. 1995;52(12):998–1007. doi: 10.1001/archpsyc.1995.03950240016004. [DOI] [PubMed] [Google Scholar]
- Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. Journal of psychiatric research. 1999;33(6):523–533. doi: 10.1016/s0022-3956(99)00029-1. [DOI] [PubMed] [Google Scholar]
- Otero-Siliceo E, Arriada-Mendicoa N. Is it healthy to be chaotic? Medical hypotheses. 2003;60(2):233–236. doi: 10.1016/s0306-9877(02)00378-x. [DOI] [PubMed] [Google Scholar]
- Oye I, Paulsen O, Maurset A. Effects of ketamine on sensory perception: evidence for a role of N-methyl-D-aspartate receptors. The Journal of pharmacology and experimental therapeutics. 1992;260(3):1209–1213. [PubMed] [Google Scholar]
- Pettersson-Yeo W, Allen P, Benetti S, et al. Dysconnectivity in schizophrenia: where are we now? Neuroscience and biobehavioral reviews. 2011;35(5):1110–1124. doi: 10.1016/j.neubiorev.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Pfeiffer A, Brantl V, Herz A, et al. Psychotomimesis mediated by kappa opiate receptors. Science (New York, NY) 1986;233(4765):774–776. doi: 10.1126/science.3016896. [DOI] [PubMed] [Google Scholar]
- Pierri JN, Chaudry AS, Woo TU, et al. Alterations in chandelier neuron axon terminals in the prefrontal cortex of schizophrenic subjects. The American journal of psychiatry. 1999;156(11):1709–1719. doi: 10.1176/ajp.156.11.1709. [DOI] [PubMed] [Google Scholar]
- Pilowsky LS, Bressan RA, Stone JM, et al. First in vivo evidence of an NMDA receptor deficit in medication-free schizophrenic patients. Molecular psychiatry. 2006;11(2):118–119. doi: 10.1038/sj.mp.4001751. [DOI] [PubMed] [Google Scholar]
- Pinault D. N-methyl d-aspartate receptor antagonists ketamine and MK-801 induce wake-related aberrant gamma oscillations in the rat neocortex. Biological psychiatry. 2008;63(8):730–735. doi: 10.1016/j.biopsych.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Plourde G, Baribeau J, Bonhomme V. Ketamine increases the amplitude of the 40-Hz auditory steady-state response in humans. British journal of anaesthesia. 1997;78(5):524–529. doi: 10.1093/bja/78.5.524. [DOI] [PubMed] [Google Scholar]
- Poon CS, Merrill CK. Decrease of cardiac chaos in congestive heart failure. Nature. 1997;389(6650):492–495. doi: 10.1038/39043. [DOI] [PubMed] [Google Scholar]
- Raichle ME, Mintun MA. Brain work and brain imaging. Annual review of neuroscience. 2006;29:449–476. doi: 10.1146/annurev.neuro.29.051605.112819. [DOI] [PubMed] [Google Scholar]
- Rais M, Cahn W, Schnack HG, et al. Brain volume reductions in medication-naive patients with schizophrenia in relation to intelligence quotient. Psychological medicine. 2012;42(9):1847–1856. doi: 10.1017/S0033291712000098. [DOI] [PubMed] [Google Scholar]
- Rais M, Cahn W, Van Haren N, et al. Excessive brain volume loss over time in cannabis-using first-episode schizophrenia patients. The American journal of psychiatry. 2008;165(4):490–496. doi: 10.1176/appi.ajp.2007.07071110. [DOI] [PubMed] [Google Scholar]
- Reich DL, Silvay G. Ketamine: an update on the first twenty-five years of clinical experience. Canadian journal of anaesthesia = Journal canadien d’anesthésie. 1989;36(2):186–197. doi: 10.1007/BF03011442. [DOI] [PubMed] [Google Scholar]
- Rickles D, Hawe P, Shiell A. A simple guide to chaos and complexity. Journal of epidemiology and community health. 2007;61(11):933–937. doi: 10.1136/jech.2006.054254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts BM, Seymour PA, Schmidt CJ, et al. Amelioration of ketamine-induced working memory deficits by dopamine D1 receptor agonists. Psychopharmacology. 2010;210(3):407–418. doi: 10.1007/s00213-010-1840-9. [DOI] [PubMed] [Google Scholar]
- Roopun AK, Cunningham MO, Racca C, et al. Region-specific changes in gamma and beta2 rhythms in NMDA receptor dysfunction models of schizophrenia. Schizophrenia bulletin. 2008;34(5):962–973. doi: 10.1093/schbul/sbn059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland LM. Subanesthetic ketamine: how it alters physiology and behavior in humans. Aviation, space, and environmental medicine. 2005;76(7 Suppl):C52–58. [PubMed] [Google Scholar]
- Rowland LM, Bustillo JR, Mullins PG, et al. Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: a 4-T proton MRS study. The American journal of psychiatry. 2005;162(2):394–396. doi: 10.1176/appi.ajp.162.2.394. [DOI] [PubMed] [Google Scholar]
- Royston MC, Slater P, Simpson MD, et al. Analysis of laminar distribution of kappa opiate receptor in human cortex: comparison between schizophrenia and normal. Journal of neuroscience methods. 1991;36(2–3):145–153. doi: 10.1016/0165-0270(91)90040-7. [DOI] [PubMed] [Google Scholar]
- Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Archives of general psychiatry. 2007;64(10):1123–1131. doi: 10.1001/archpsyc.64.10.1123. [DOI] [PubMed] [Google Scholar]
- Sawaguchi T, Goldman-Rakic PS. D1 dopamine receptors in prefrontal cortex: involvement in working memory. Science (New York, NY) 1991;251(4996):947–950. doi: 10.1126/science.1825731. [DOI] [PubMed] [Google Scholar]
- Scheidegger M, Walter M, Lehmann M, et al. Ketamine decreases resting state functional network connectivity in healthy subjects: implications for antidepressant drug action. PloS one. 2012;7(9):e44799. doi: 10.1371/journal.pone.0044799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller M, Bufler J, Hertle I, et al. Ketamine blocks currents through mammalian nicotinic acetylcholine receptor channels by interaction with both the open and the closed state. Anesthesia and Analgesia. 1996;83(4):830–836. doi: 10.1097/00000539-199610000-00031. [DOI] [PubMed] [Google Scholar]
- Sheffler DJ, Roth BL. Salvinorin A: the ‘magic mint’ hallucinogen finds a molecular target in the kappa opioid receptor. Trends in pharmacological sciences. 2003;24(3):107–109. doi: 10.1016/S0165-6147(03)00027-0. [DOI] [PubMed] [Google Scholar]
- Shibuya H, Mori H, Toru M. Sigma receptors in schizophrenic cerebral cortices. Neurochemical research. 1992;17(10):983–990. doi: 10.1007/BF00966825. [DOI] [PubMed] [Google Scholar]
- Shimazu S, Katsuki H, Takenaka C, et al. Sigma receptor ligands attenuate N-methyl-D-aspartate cytotoxicity in dopaminergic neurons of mesencephalic slice cultures. European journal of pharmacology. 2000;388(2):139–146. doi: 10.1016/s0014-2999(99)00852-3. [DOI] [PubMed] [Google Scholar]
- Shinkai T, De Luca V, Hwang R, et al. Association analyses of the DAOA/G30 and D-amino-acid oxidase genes in schizophrenia: further evidence for a role in schizophrenia. Neuromolecular medicine. 2007;9(2):169–177. doi: 10.1007/BF02685890. [DOI] [PubMed] [Google Scholar]
- Singer W, Gray CM. Visual feature integration and the temporal correlation hypothesis. Annual review of neuroscience. 1995;18:555–586. doi: 10.1146/annurev.ne.18.030195.003011. [DOI] [PubMed] [Google Scholar]
- Singh SP, Singh V. Meta-analysis of the efficacy of adjunctive NMDA receptor modulators in chronic schizophrenia. CNS drugs. 2011;25(10):859–885. doi: 10.2165/11586650-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Smith DJ, Bouchal RL, deSanctis CA, et al. Properties of the interaction between ketamine and opiate binding sites in vivo and in vitro. Neuropharmacology. 1987;26(9):1253–1260. doi: 10.1016/0028-3908(87)90084-0. [DOI] [PubMed] [Google Scholar]
- Smythies J. The adrenochrome hypothesis of schizophrenia revisited. Neurotoxicity research. 2002;4(2):147–150. doi: 10.1080/10298420290015827. [DOI] [PubMed] [Google Scholar]
- Spencer KM, Nestor PG, Niznikiewicz MA, et al. Abnormal neural synchrony in schizophrenia. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23(19):7407–7411. doi: 10.1523/JNEUROSCI.23-19-07407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer KM, Nestor PG, Perlmutter R, et al. Neural synchrony indexes disordered perception and cognition in schizophrenia. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(49):17288–17293. doi: 10.1073/pnas.0406074101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer KM, Niznikiewicz MA, Shenton ME, et al. Sensory-evoked gamma oscillations in chronic schizophrenia. Biological psychiatry. 2008;63(8):744–747. doi: 10.1016/j.biopsych.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer KM, Salisbury DF, Shenton ME, et al. Gamma-band auditory steady-state responses are impaired in first episode psychosis. Biological psychiatry. 2008;64(5):369–375. doi: 10.1016/j.biopsych.2008.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporns O. Discovering the Human Connectome. Cambridge, MA: MIT Press; 2012. [Google Scholar]
- Sporns O. Networks of the Brain. Cambridge, MA: MIT Press; 2011. [Google Scholar]
- Stam CJ. Nonlinear dynamical analysis of EEG and MEG: review of an emerging field. Clinical neurophysiology: official journal of the International Federation of Clinical Neurophysiology. 2005;116(10):2266–2301. doi: 10.1016/j.clinph.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Stone JM, Dietrich C, Edden R, et al. Ketamine effects on brain GABA and glutamate levels with 1H-MRS: relationship to ketamine-induced psychopathology. Molecular psychiatry. 2012;17(7):664–665. doi: 10.1038/mp.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JM, Erlandsson K, Arstad E, et al. Relationship between ketamine-induced psychotic symptoms and NMDA receptor occupancy: a [(123)I]CNS-1261 SPET study. Psychopharmacology. 2008;197(3):401–408. doi: 10.1007/s00213-007-1047-x. [DOI] [PubMed] [Google Scholar]
- Supplisson S, Bergman C. Control of NMDA receptor activation by a glycine transporter co-expressed in Xenopus oocytes. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1997;17(12):4580–4590. doi: 10.1523/JNEUROSCI.17-12-04580.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliazucchi E, Balenzuela P, Fraiman D, et al. Criticality in large-scale brain fMRI dynamics unveiled by a novel point process analysis. Frontiers in physiology. 2012;3:15. doi: 10.3389/fphys.2012.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot K, Eidem WL, Tinsley CL, et al. Dysbindin-1 is reduced in intrinsic, glutamatergic terminals of the hippocampal formation in schizophrenia. The Journal of clinical investigation. 2004;113(9):1353–1363. doi: 10.1172/JCI20425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallon-Baudry C, Bertrand O, Peronnet F, et al. Induced gamma-band activity during the delay of a visual short-term memory task in humans. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1998;18(11):4244–4254. doi: 10.1523/JNEUROSCI.18-11-04244.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam SW, Cook L. Sigma opiates and certain antipsychotic drugs mutually inhibit (+)-[3H] SKF 10,047 and [3H]haloperidol binding in guinea pig brain membranes. Proceedings of the National Academy of Sciences of the United States of America. 1984;81(17):5618–5621. doi: 10.1073/pnas.81.17.5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejeda HA, Shippenberg TS, Henriksson R. The dynorphin/κ-opioid receptor system and its role in psychiatric disorders. Cellular and molecular life sciences: CMLS. 2012;69(6):857–896. doi: 10.1007/s00018-011-0844-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornberg SA, Saklad SR. A review of NMDA receptors and the phencyclidine model of schizophrenia. Pharmacotherapy. 1996;16(1):82–93. [PubMed] [Google Scholar]
- Tiitinen H, Sinkkonen J, Reinikainen K, et al. Selective attention enhances the auditory 40-Hz transient response in humans. Nature. 1993;364(6432):59–60. doi: 10.1038/364059a0. [DOI] [PubMed] [Google Scholar]
- Tononi G, Edelman GM. Schizophrenia and the mechanisms of conscious integration. Brain research. Brain research reviews. 2000;31(2–3):391–400. doi: 10.1016/s0165-0173(99)00056-9. [DOI] [PubMed] [Google Scholar]
- Traub RD, Whittington MA, Stanford IM, et al. A mechanism for generation of long-range synchronous fast oscillations in the cortex. Nature. 1996;383(6601):621–624. doi: 10.1038/383621a0. [DOI] [PubMed] [Google Scholar]
- Tsai G, Coyle JT. Glutamatergic mechanisms in schizophrenia. Annual Review of Pharmacology and Toxicology. 2002;42:165–179. doi: 10.1146/annurev.pharmtox.42.082701.160735. [DOI] [PubMed] [Google Scholar]
- Tsai G, Yang P, Chung LC, et al. D-serine added to antipsychotics for the treatment of schizophrenia. Biological psychiatry. 1998;44(11):1081–1089. doi: 10.1016/s0006-3223(98)00279-0. [DOI] [PubMed] [Google Scholar]
- Tsai GE, Lin P-Y. Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia, a critical review and meta-analysis. Current pharmaceutical design. 2010;16(5):522–537. doi: 10.2174/138161210790361452. [DOI] [PubMed] [Google Scholar]
- Turing AM. The chemical basis of morphogenesis. Philosophical Transactions of the Royal Society, Series B, Biological Sciences. 1952;237(641):37–72. doi: 10.1098/rstb.2014.0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlhaas PJ. Dysconnectivity, large-scale networks and neuronal dynamics in schizophrenia. Current opinion in neurobiology. 2013;23(2):283–290. doi: 10.1016/j.conb.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. Neuronal dynamics and neuropsychiatric disorders: toward a translational paradigm for dysfunctional large-scale networks. Neuron. 2012;75(6):963–980. doi: 10.1016/j.neuron.2012.09.004. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. The development of neural synchrony and large-scale cortical networks during adolescence: relevance for the pathophysiology of schizophrenia and neurodevelopmental hypothesis. Schizophrenia bulletin. 2011;37(3):514–523. doi: 10.1093/schbul/sbr034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbricht D, Koller R, Vollenweider FX, et al. Mismatch negativity predicts psychotic experiences induced by NMDA receptor antagonist in healthy volunteers. Biological psychiatry. 2002;51(5):400–406. doi: 10.1016/s0006-3223(01)01242-2. [DOI] [PubMed] [Google Scholar]
- Voisey J, Swagell CD, Hughes IP, et al. A polymorphism in the dysbindin gene (DTNBP1) associated with multiple psychiatric disorders including schizophrenia. Behavioral and brain functions: BBF. 2010;6:41. doi: 10.1186/1744-9081-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk D, Austin M, Pierri J, et al. GABA transporter-1 mRNA in the prefrontal cortex in schizophrenia: decreased expression in a subset of neurons. The American journal of psychiatry. 2001;158(2):256–265. doi: 10.1176/appi.ajp.158.2.256. [DOI] [PubMed] [Google Scholar]
- Volk DW, Austin MC, Pierri JN, et al. Decreased glutamic acid decarboxylase 67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Archives of general psychiatry. 2000;57(3):237–245. doi: 10.1001/archpsyc.57.3.237. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wolf AP, Brodie JD, et al. Brain interactions in chronic schizophrenics under resting and activation conditions. Schizophrenia Research. 1988;1:47–53. doi: 10.1016/0920-9964(88)90039-4. [DOI] [PubMed] [Google Scholar]
- Vollenweider FX, Leenders KL, Oye I, et al. Differential psychopathology and patterns of cerebral glucose utilisation produced by (S)- and (R)-ketamine in healthy volunteers using positron emission tomography (PET) European neuropsychopharmacology: the journal of the European College of Neuropsychopharmacology. 1997;7(1):25–38. doi: 10.1016/s0924-977x(96)00042-9. [DOI] [PubMed] [Google Scholar]
- Voss LJ, Baas CH, Hansson L, et al. Investigation into the effect of the general anaesthetics etomidate and ketamine on long-range coupling of population activity in the mouse neocortical slice. European journal of pharmacology. 2012;689(1–3):111–117. doi: 10.1016/j.ejphar.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Wang Q, Deng W, Huang C, et al. Abnormalities in connectivity of white-matter tracts in patients with familial and non-familial schizophrenia. Psychological medicine. 2011;41(8):1691–1700. doi: 10.1017/S0033291710002412. [DOI] [PubMed] [Google Scholar]
- Weickert CS, Straub RE, McClintock BW, et al. Human dysbindin (DTNBP1) gene expression in normal brain and in schizophrenic prefrontal cortex and midbrain. Archives of general psychiatry. 2004;61(6):544–555. doi: 10.1001/archpsyc.61.6.544. [DOI] [PubMed] [Google Scholar]
- Weinberger DR. Implications of Normal Brain Development for the Pathogenesis of Schizophrenia. Archives of general psychiatry. 1987;44(7):660–669. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- Weissman AD, Casanova MF, Kleinman JE, et al. Selective loss of cerebral cortical sigma, but not PCP binding sites in schizophrenia. Biological psychiatry. 1991;29(1):41–54. doi: 10.1016/0006-3223(91)90209-5. [DOI] [PubMed] [Google Scholar]
- Wernicke C. Grundrisse der Psychiatrie. Leipzig, Germany: Thieme; 1906. [Google Scholar]
- Whitfield-Gabrieli S, Thermenos HW, Milanovic S, et al. Hyperactivity and hyperconnectivity of the default network in schizophrenia and in first-degree relatives of persons with schizophrenia. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(4):1279–1284. doi: 10.1073/pnas.0809141106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittemore ER, Ilyin VI, Woodward RM. Antagonism of N-methyl-D-aspartate receptors by sigma site ligands: potency, subtype-selectivity and mechanisms of inhibition. The Journal of pharmacology and experimental therapeutics. 1997;282(1):326–338. [PubMed] [Google Scholar]
- Williams LM, Whitford TJ, Nagy M, et al. Emotion-elicited gamma synchrony in patients with first-episode schizophrenia: a neural correlate of social cognition outcomes. Journal of psychiatry & neuroscience: JPN. 2009;34(4):303–313. [PMC free article] [PubMed] [Google Scholar]
- Wozniak DF, Dikranian K, Ishimaru MJ, et al. Disseminated corticolimbic neuronal degeneration induced in rat brain by MK-801: potential relevance to Alzheimer’s disease. Neurobiology of disease. 1998;5(5):305–322. doi: 10.1006/nbdi.1998.0206. [DOI] [PubMed] [Google Scholar]
- Wylie GR, Clark EA, Butler PD, et al. Schizophrenia patients show task switching deficits consistent with N-methyl-d-aspartate system dysfunction but not global executive deficits: implications for pathophysiology of executive dysfunction in schizophrenia. Schizophrenia bulletin. 2010;36(3):585–594. doi: 10.1093/schbul/sbn119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakage M, Hirshman CA, Croxton TL. Inhibitory effects of thiopental, ketamine, and propofol on voltage-dependent Ca2+ channels in porcine tracheal smooth muscle cells. Anesthesiology. 1995;83(6):1274–1282. doi: 10.1097/00000542-199512000-00018. [DOI] [PubMed] [Google Scholar]
- Yonezawa Y, Kuroki T, Kawahara T, et al. Involvement of gamma-aminobutyric acid neurotransmission in phencyclidine-induced dopamine release in the medial prefrontal cortex. European journal of pharmacology. 1998;341(1):45–56. doi: 10.1016/s0014-2999(97)01435-0. [DOI] [PubMed] [Google Scholar]
- Zalesky A, Fornito A, Seal ML, et al. Disrupted axonal fiber connectivity in schizophrenia. Biological psychiatry. 2011;69(1):80–89. doi: 10.1016/j.biopsych.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Welsh AM, Chen D, et al. NMDA inhibitors cause apoptosis of pyramidal neurons in mature piriform cortex: evidence for a nitric oxide-mediated effect involving inhibitory interneurons. Neuropharmacology. 2007;52(7):1528–1537. doi: 10.1016/j.neuropharm.2007.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]