

SUMMARY
Accumulating evidence indicates that human natural killer (NK) cells develop in secondary lymphoid tissue (SLT) through a so-called “stage 3” developmental intermediate minimally characterized by a CD34-CD117+CD94- immunophenotype that lacks mature NK cell function. This stage 3 population is heterogeneous, potentially composed of functionally distinct innate lymphoid cell (ILC) types that includes interleukin-1 receptor (IL-1R1) positive, IL-22-producing ILC3s. Whether human ILC3s are developmentally related to NK cells is a subject of ongoing investigation. Here we show that antagonism of the aryl hydrocarbon receptor (AHR) or silencing of AHR gene expression promotes differentiation of tonsillar IL-22-producing IL-1R1hi human ILC3s to CD56brightCD94+ IFN-gamma-producing cytolytic mature NK cells expressing eomesodermin (EOMES) and T-Box Protein 21 (TBX21 or TBET). Hence, AHR is a transcription factor that prevents human IL-1R1hi ILC3s from differentiating into NK cells.
INTRODUCTION
Natural killer (NK) cells are large granular lymphocytes whose roles in immunity include the production and release of immunomodulatory chemokines and cytokines as well as the direct cytolytic killing of malignant or pathogen-infected cells. NK cells are distinct from T and B lymphocytes in that NK cells do not rearrange T cell receptor or immunoglobulin receptor genes, and for many years NK cells were considered to represent the only non-T/B lymphocyte population (Spits et al., 2013; Walker et al., 2013). However, a wealth of recent data now indicate that NK cells represent only one subset of a much larger population of non-T/B lymphocytes now collectively described as innate lymphoid cells (ILCs) (Spits et al., 2013; Walker et al., 2013). ILC subsets vary in terms of their surface immunophenotypes, transcription factor expression, and functional attributes, and NK cells are currently classified as Group 1 ILCs. Non-NK Group 1 ILCs (designated ILC1 cells) have also been described (Bernink et al., 2013; Spits et al., 2013; Walker et al., 2013), and while non-NK ILC1s can produce IFN-γ, they are not cytolytic (Bernink et al., 2013) and do not express the transcription factor, eomesodermin (EOMES), which is selectively expressed in NK cells (Gordon et al., 2012; Klose et al., 2013; Spits et al., 2013). Given their diverse roles in immunity and human disease, gaining an understanding of how these various ILC populations develop is of high clinical relevance.
Within human secondary lymphoid tissue (SLT), NK cells appear to proceed through four discrete stages of maturity as they progress from oligopotent CD34+CD45RA+ progenitor cells to functionally competent CD56brightCD94+ NK cells (Freud et al., 2005; Freud et al., 2006). These four “lineage negative” (lacking CD3, CD14, and CD19 expression) lymphoid populations may be distinguished by their surface expression patterns of CD34, CD117, and CD94 such that stage 1 cells are CD34+CD117-CD94-, stage 2 cells are CD34+CD117+CD94-, stage 3 cells are CD34-CD117+CD94-, and stage 4 cells, which bear immunophenotypic and functional features that most closely resemble peripheral blood CD56bright NK cells, are CD34-CD117+/-CD94+ (Freud and Caligiuri, 2006). Stage 3 cells were originally classified as “immature NK cells” because unlike stage 1 and stage 2 cells they do not retain T cell or dendritic cell developmental potential ex vivo, yet in response to in vitro interleukin (IL)-15 stimulation or co-culture with autologous T cells or OP9 stroma, at least a subset of stage 3 cells differentiates into stage 4 NK cells (Freud and Caligiuri, 2006). In addition, stage 3 cells lack expression of certain receptors expressed by mature (stage 4) NK cells, and they also lack two hallmark functions of mature NK cells: the capacities to produce IFN-γ and to perform perforin-mediated cytotoxicity (Freud et al., 2006). Although the role of IL-15 in driving human NK cell development (Mrozek et al., 1996), survival (Cooper et al., 2002), and effector function (Carson et al., 1994) has been well documented, ex vivo culture assays show that stage 3 to stage 4 cell maturation in response to IL-15 is inefficient in vitro (Freud et al., 2006; Hughes et al., 2010). This suggests that the stage 3 population may be functionally heterogeneous and/or IL-15 on its own may be inadequate to drive optimal progression from stage 3 to stage 4 in vitro (Ahn et al., 2013; Freud et al., 2006; Hughes et al., 2010).
Several recent studies provide additional evidence to suggest that the stage 3 population, minimally defined as CD34-CD117+CD94-, may be comprised of a heterogeneous group of ILC subsets, potentially including bona fide stage 3 NK cell developmental intermediates that would fit into the aforementioned linear model of human NK cell development as well as other non-NK lineage ILC subsets that share the basic CD34-CD117+CD94- immunophenotype. In particular, the latter include Group 3 ILCs (ILC3s), which can express T-Box Protein 21 (TBX21 or TBET) and are defined by expression of the transcription factors, RAR-related orphan receptor C (RORC) and aryl hydrocarbon receptor (AHR) (Spits et al., 2013). According to the most recent classification of ILC subsets, ILC3s comprise at least two populations thought to be mutually exclusive in humans: 1) a population expressing natural cytotoxicity receptors (NCRs), including NKp44 and NKp46, as well as IL-1 receptor (IL-1R1), IL-23R, and IL-22 (Cella et al., 2009; Cella et al., 2010; Crellin et al., 2010; Hughes et al., 2010) – a population now referred to as NCR+ ILC3 (Spits et al., 2013; Walker et al., 2013); and 2) a lymphoid tissue-inducer (LTi) population expressing molecules required for the development of lymphoid tissues as well as CD117, CD127, CD161, IL-1R1, IL-23R, IL-22 and IL-17 but not CD56 or NCRs ((Spits et al., 2013; Walker et al., 2013) and the references within). Recent fate-mapping studies in mice suggest that the analogous NCR+ ILC3s and NK cells – which require expression of the transcription factors TBX21 and EOMES for development (Gordon et al., 2012; Intlekofer et al., 2005; Spits et al., 2013) – represent distinct lineages (Satoh-Takayama et al., 2010); however, adoptive transfer experiments show that some NCR+ ILC3s can adopt an NK-like phenotype in vivo (Lee et al., 2012; Qiu et al., 2012; Vonarbourg et al., 2010), potentially representing non-conventional NK cells or other ILC1 cells. Similar to these mouse data, clonal in vitro assays with human cells suggest some plasticity between the Group 3 and Group 1 ILC phenotypes (Cella et al., 2010; Crellin et al., 2010; Hughes et al., 2010). Therefore, whether an ILC3 phenotype is truly a transient attribute of at least some human NK cell precursors or whether ILC3s comprise a stable, mutually exclusive lineage apart from NK cells in humans is still unclear. The molecular mechanism(s) regulating these processes are also unknown.
AHR is a ligand-activated transcription factor (Hankinson, 1995; Swanson and Bradfield, 1993) that regulates differentiation of dendritic cells, regulatory T cells, TH17 cells, intraepithelial intestinal γδ T cells and, notably, mouse NCR+ ILC3s (Kadow et al., 2011; Kremer et al., 1994; Lee et al., 2012; Platzer et al., 2009; Quintana et al., 2008; Veldhoen et al., 2008). AHR is required for IL-22 production by TH17 cells (Veldhoen et al., 2008). AHR binds to halogenated aromatic hydrocarbons, and currently described ligands include naturally occurring dietary substances as well as synthetic substances and environmental pollutants (reviewed by (Denison and Nagy, 2003)). For example, upon exposure to light, the aromatic amino acid tryptophan can be metabolized in vitro to products including the AHR agonist, 6-formylindolo[3,2-b]carbazole (FICZ) (Rannug et al., 1995). Tryptophan is also metabolized to FICZ in vivo by skin keratinocytes upon UV light exposure (Wei et al., 1998). In contrast to many naturally occurring AHR antagonists which have been reported to act as either partial agonists or incomplete antagonists (reviewed by (Stejskalova et al., 2011)), the chemical CH-223191 lacks AHR agonist activity and efficiently antagonizes activation of AHR (Smith et al., 2011; Zhao et al., 2010).
In this study, we evaluated the impact of AHR modulation on the differentiation and maturation of a subset of human SLT-derived CD34-CD117+CD94- cells with constitutive IL-22 production and high expression of AHR and IL-1R1 ex vivo (previously referred to as stage 3 cells and herein referred to as IL-1R1hi ILC3s) (Freud et al., 2006; Hughes et al., 2010). Whereas in vitro stimulation of purified IL-1R1hi ILC3s with the AHR agonist, FICZ, acted to suppress differentiation into stage 4 mature NK cells, AHR blockade with CH-223191 or silencing of AHR gene expression via shRNA induced their differentiation into phenotypically and functionally mature NK cells. These data suggest AHR likely plays an important regulatory role in the differentiation of human ILC populations and provide new evidence that its constitutive expression in IL-1R1hi ILC3s prevents human NK cell differentiation. Furthermore, these data indicate that inhibition of AHR activity represents a potentially novel means by which the generation of cytotoxic, IFN-γ-producing NK cells may be achieved pharmacologically for clinical benefit.
RESULTS
IL-1β utilizes AHR to maintain the IL-22+ phenotype of human IL-1R1hi ILC3s
We purified fresh human stage 3 or IL-1R1hi ILC3s and CD56brightCD94+ stage 4 NK cells from SLT to assess transcription factor expression in each population ex vivo via Real-time RT-PCR. While RORC and AHR were both absent, TBX21 (TBET) and EOMES were highly expressed by CD56brightCD94+ stage 4 NK cells, providing additional support for their designation as NK cells (Spits et al., 2013). In contrast, IL-1R1hi ILC3s lacked expression of TBX21 and EOMES, but constitutively expressed RORC and AHR, the latter of which was present at levels at least 15.50 ± 1.22-fold greater than that detected in CD56brightCD94+ stage 4 NK cells (Fig. 1A).
Fig. 1. Human IL-1R1hi ILC3s express AHR and respond to AHR ligands in vitro.
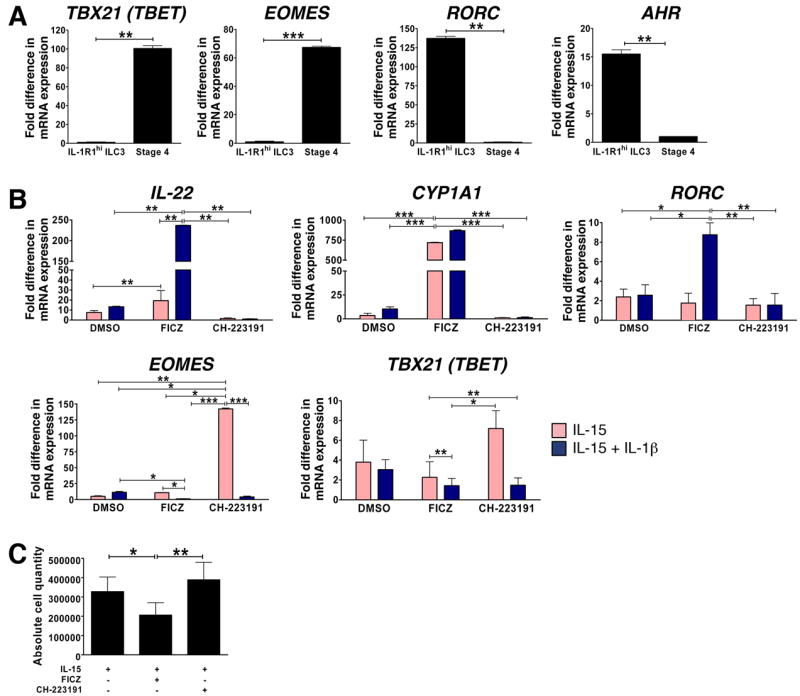
(A, B) Real-time RT-PCR was used to assess: (A) TBX21 (TBET), EOMES, RORC and AHR mRNA expression ex vivo in human IL-1R1hi ILC3s (Lin-CD34-CD117+CD94-IL-1R1hi) and stage 4 (Lin-CD34-CD117+/-CD94+) mature CD56bright NK cells, FACS-purified from SLT; or (B) IL-22, CYP1A1, RORC, EOMES, and TBX21 (TBET) mRNA expression after culture of IL-1R1hi ILC3s for 14 d in the presence of IL-15 (1 nM;
 ) or IL-15 + IL-1β (10 ng/ml; ■), plus either carrier alone (DMSO, 1 μl/ml), or carrier containing AHR agonist (FICZ, 300 nM) or AHR antagonist (CH-223191, 3 μM). For (A, B), gene expression levels were normalized to 18S mRNA, and relative quantification was performed using the ΔΔCt method. Y axis depicts fold difference in mRNA expression, quantified relative to the condition in which expression was lowest (arbitrarily normalized to 1). (C) Absolute number of viable cells generated from culture of IL-1R1hi ILC3s for 14 d with IL-15 plus DMSO, IL-15 plus AHR agonist FICZ, or IL-15 plus AHR antagonist CH-223191. Data in A, B and C presented as mean ± SEM (for A and B, n ≥ 4; for C, n = 7; * for P < 0.05, ** for P < 0.01, *** for P < 0.001). See also Fig. S1.
) or IL-15 + IL-1β (10 ng/ml; ■), plus either carrier alone (DMSO, 1 μl/ml), or carrier containing AHR agonist (FICZ, 300 nM) or AHR antagonist (CH-223191, 3 μM). For (A, B), gene expression levels were normalized to 18S mRNA, and relative quantification was performed using the ΔΔCt method. Y axis depicts fold difference in mRNA expression, quantified relative to the condition in which expression was lowest (arbitrarily normalized to 1). (C) Absolute number of viable cells generated from culture of IL-1R1hi ILC3s for 14 d with IL-15 plus DMSO, IL-15 plus AHR agonist FICZ, or IL-15 plus AHR antagonist CH-223191. Data in A, B and C presented as mean ± SEM (for A and B, n ≥ 4; for C, n = 7; * for P < 0.05, ** for P < 0.01, *** for P < 0.001). See also Fig. S1.
We therefore reasoned that IL-1R1hi ILC3s may respond to treatment with AHR ligands. We previously showed that these cells constitutively and selectively express abundant AHR and IL-22 mRNA relative to earlier and later stages of human NK cell development, and that exogenous IL-1β is required maintain this phenotype in the presence of the cell’s survival factor, IL-15 (Hughes et al., 2010). However, to prove that IL-1β mediates its effect on IL-22 gene expression via AHR we cultured IL-1R1hi ILC3s for 14 d in medium containing either IL-15 or IL-15 + IL-1β, plus either carrier alone or an equal volume of carrier containing an AHR agonist (FICZ) or antagonist (CH-223191) at concentrations previously shown to modulate IL-22 production in TH17 cells (Veldhoen et al., 2009). Indeed IL-1β-mediated expression of IL-22 was significantly enhanced in the presence of the AHR agonist FICZ, whereas IL-22 was significantly and completely abrogated in the presence of the AHR antagonist CH-223191, thus strongly suggesting IL-1β mediates this effect via AHR (Fig. 1B). As expected, culture in the presence of FICZ also induced expression of CYP1A1, indicating that AHR ligand binding and activation of downstream signaling occurred (Wei et al., 1998). Interestingly, the addition of IL-1β further increased the magnitude of CYP1A1 induction that occurred in the presence of FICZ. Likewise, the combination of FICZ and IL-1β also promoted greater retention of RORC (Fig. 1B). Thus there appears to be a synergistic interaction between AHR and IL-1β in regulating expression of these genes. In contrast, FICZ inhibited while IL-1β had little effect on expression of TBX21. Notably, blockade of AHR signaling with CH-223191 led to an increase in TBX21 expression while inhibiting expression of RORC and IL-22. Moreover, only in the presence of CH-223191 did we detect substantial induction of EOMES, a transcription factor that seems to be uniquely associated with the NK lineage among ILC subsets (Klose et al., 2013; Spits et al., 2013) (Fig. 1B). Thus, the presence of CH-223191 during culture of IL-1R1hi ILC3s promotes the acquisition of a TBX21+EOMES+RORClow phenotype, similar to that observed in freshly isolated stage 4 NK cells ex vivo.
To determine the effect of the AHR ligands on IL-1R1hi ILC3 survival, we cultured IL-1R1hi ILC3s for 14 d in the presence of IL-15 plus either carrier (DMSO) alone, or carrier containing the AHR agonist, FICZ, or the AHR antagonist, CH-223191 at their appropriate concentrations (Veldhoen et al., 2009). The addition of FICZ significantly reduced the total number of viable cells, while the presence of the CH-223191 slightly increased the total number of viable cells generated in vitro (Fig. 1C). Comparable results were obtained using an alternative AHR agonist (curcumin) and antagonist (StemRegenin1) (T.H., unpublished data). Thus, IL-15-mediated survival of this IL-1R1hi ILC3 population may itself be influenced by the expression of AHR. Further, these data show that the ability of CH-223191 to abrogate IL-1β-mediated expression of IL-22 via antagonism of AHR in IL-1R1hi ILC3s is not the result of cell death.
AHR prevents differentiation of human IL-1R1hi ILC3s into CD56brightCD94+ stage 4 NK cells
We next assessed expression of NK cell maturation markers following culture of IL-1R1hi ILC3s for 14 d with IL-15 and either carrier alone, or carrier containing FICZ or CH-223191. Culture of IL-1R1hi ILC3s in the presence of the AHR antagonist CH-223191 led to a significant increase in stage 4 NK cells as determined by the relative increase in surface density of CD56 expression, as well as a significantly higher percentage of cells expressing CD94 and having down-regulated CD117 when compared to cultures containing IL-15 and either DMSO or IL-15 and the AHR agonist, FICZ (Fig. 2A-E). In fact, the fraction of IL-1R1hi ILC3s expressing CD56, the mean fluorescence intensity of CD56, and the percentage of cells expressing CD94 were significantly lower in the presence of IL-15 plus the AHR agonist, FICZ, when compared to IL-1R1hi ILC3s cultured in IL-15 plus DMSO (Fig. 2A-E). Further, while IL-1R1hi ILC3s cultured in IL-15 plus DMSO increased or acquired expression of NKp44, NKp46, CD161, CD69, NKG2D, and granzyme B; IL-1R1hi ILC3s cultured in IL-15 plus the AHR agonist FICZ failed to increase expression or acquire these molecules (Fig. S1A). Thus, the AHR agonist not only appears to counter IL-15-mediated survival of IL-1R1hi ILC3s in culture, but also suppresses IL-15-mediated differentiation of IL-1R1hi ILC3s toward the stage 4 CD56bright NK cell phenotype.
Fig. 2. AHR prevents differentiation of human IL-1R1hi ILC3s to NK cells in vitro.
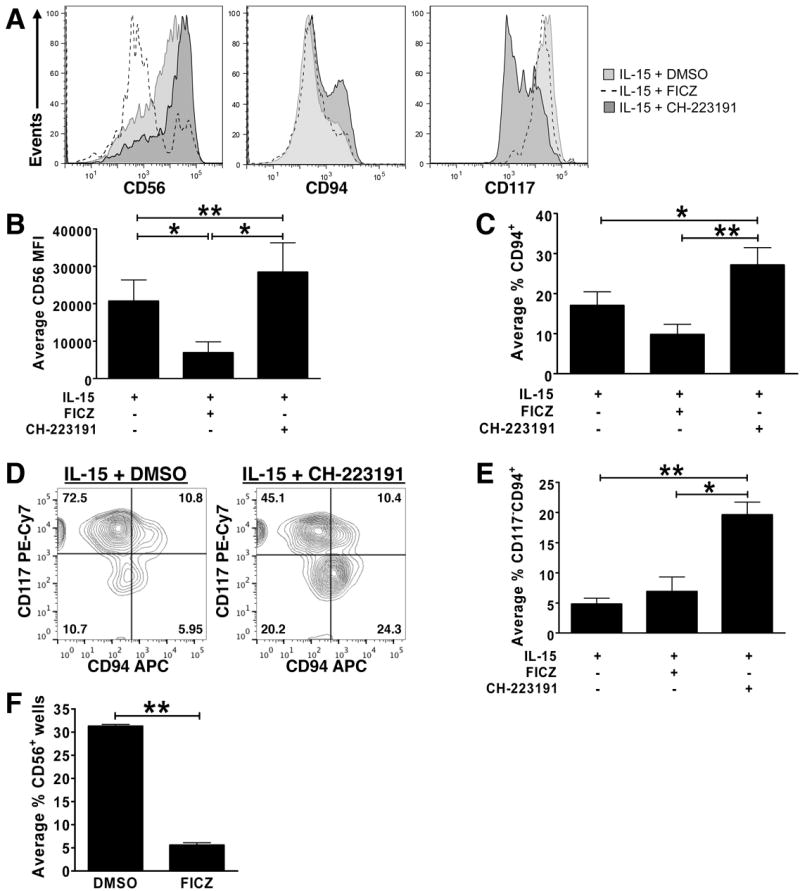
(A-E) IL-1R1hi ILC3s were FACS-purified from SLT, cultured for 14 d under indicated conditions, then assessed for expression of CD56, CD94, and CD117. Data in (A, D) depict staining in a representative donor (CH-223191,
 ; DMSO,
; DMSO,
 ; FICZ, dashed line; n ≥ 8). Data in (B, C, E) depict average: (B) mean fluorescence intensity (MFI) of CD56 surface expression (n = 12), (C) percentage of cells that acquired CD94 surface expression (n = 8), and (E) percentage of cells that lost CD117 and acquired CD94 (n = 4). Data presented as mean ± SEM; * for P < 0.05, ** for P < 0.005. (F) Single IL-1R1hi ILC3s were cultured on OP9-GFP+ stroma with IL-15 plus either DMSO carrier or AHR agonist FICZ. After 14 d, each of 60 replicate wells was individually assessed for CD56 surface expression via flow cytometry as previously described (Hughes et al., 2010). One hundred percent of the wells scored positive for at least some CD56 expression. Data represent mean ± SEM (**, P < 0.005; n = 2 donors).
; FICZ, dashed line; n ≥ 8). Data in (B, C, E) depict average: (B) mean fluorescence intensity (MFI) of CD56 surface expression (n = 12), (C) percentage of cells that acquired CD94 surface expression (n = 8), and (E) percentage of cells that lost CD117 and acquired CD94 (n = 4). Data presented as mean ± SEM; * for P < 0.05, ** for P < 0.005. (F) Single IL-1R1hi ILC3s were cultured on OP9-GFP+ stroma with IL-15 plus either DMSO carrier or AHR agonist FICZ. After 14 d, each of 60 replicate wells was individually assessed for CD56 surface expression via flow cytometry as previously described (Hughes et al., 2010). One hundred percent of the wells scored positive for at least some CD56 expression. Data represent mean ± SEM (**, P < 0.005; n = 2 donors).
To assess this at the single cell level, we FACS sorted IL-1R1hi ILC3s into wells seeded with OP9-GFP stroma, which is required for survival of IL-1R1hi ILC3s at the single cell level (Freud et al., 2006; Hughes et al., 2010). The OP9 stromal cell line promotes human NK cell differentiation (Cupedo et al., 2009), and others have shown that it produces mediators that antagonize the AHR pathway (Magnusson et al., 2013). Thus, it is possible that OP9 stroma promote survival and differentiation of IL-1R1hi ILC3s in part via production of an endogenous AHR antagonist. Because of this, we could not assess the effects of exogenous AHR antagonist on IL-1R1hi ILC3s at the single cell level, however we did perform a clonal assay comparing single IL-1R1hi ILC3s continuously cultured in the presence of OP9 cells, IL-15 and either carrier alone or the AHR agonist, FICZ. The number of wells in which clones were generated was similar for both culture conditions. As expected from our polyclonal cultures, wells with single viable IL-1R1hi ILC3s cultured with OP9 cells in IL-15 plus FICZ were found to have CD56 expression in only 5.60 ± 0.52% of wells, versus wells with single viable IL-1R1hi ILC3s cultured with OP9 cells in IL-15 and DMSO that had 31.29 ± 0.38% of wells with CD56 expression (Fig. 2F). Similar results were found for CD94 expression (Fig. S1B) and, as we previously noted (Hughes et al., 2010), compared to polyclonal cultures, a higher percentage of CD94+ cells were generated during our clonal assays which required co-culture on the OP9-GFP stromal cell line. These clonal data support the finding that an endogenous AHR antagonist is being produced by OP9-GFP stromal cells (Magnusson et al., 2013), while also further demonstrating that AHR and its agonist FICZ prevent differentiation of IL-1R1hi ILC3s to stage 4 mature NK cells in vitro. Collectively, our polyclonal and single cell data suggest that activation of constitutively expressed AHR with an AHR agonist suppresses IL-15-mediated differentiation of IL-1R1hi ILC3s toward the stage 4 CD56bright NK cell phenotype.
It should be noted that there was no significant surface expression of the Fc receptor, CD16, or killer cell immunoglobulin (Ig)-like receptors (KIR), among stage 4 mature CD56bright NK cells derived in vitro from IL-1R1hi ILC3s in the presence of the AHR antagonist, suggesting that inhibition of AHR cannot drive stage 4 CD56bright NK cells to stage 5 CD56dimCD16+KIR+/- NK cells in vitro (Fig. S1A) (Freud and Caligiuri, 2006; Freud et al., 2006). Indeed, stage 4 mature CD56bright NK cells isolated directly from SLT lacked detectable expression of AHR ((Hughes et al., 2010) and Fig. 1A).
AHR prevents the differentiation of human IL-1R1hi ILC3s into functional NK cells that produce IFN-γ
Freshly isolated IL-1R1hi ILC3s cannot produce IFN-γ ex vivo (Freud et al., 2006; Hughes et al., 2010). Given the upregulation of TBX21 and EOMES mRNA (Fig. 1B), as well as the acquisition of the stage 4 CD56bright NK cell immunophenotype by IL-1R1hi ILC3s that occurred in response to AHR antagonism (Fig. 2), we next investigated whether antagonism of AHR might promote differentiation to a functional NK cell as well. To this end, IL-1R1hi ILC3s freshly isolated from SLT were placed into culture for 14 d with IL-15 plus either carrier alone or carrier containing the AHR agonist or antagonist. On d 14, cells were counted, and an equal number of progeny generated in each culture condition were used for effector function assays detailed below.
To assess the capacity for monokine-induced IFN-γ production, an equal number of cells were incubated for 12 h in either media alone (“unstimulated”) or media containing IL-12, IL-15, and IL-18. Surface expression of NK maturation markers was assessed in conjunction with intracellular (IC) flow cytometric staining for IFN-γ protein (Fig. 3A-C). Cell-free supernatants were also assessed for IFN-γ secretion by ELISA (Fig. 3D).
Fig. 3. AHR prevents IL-1R1hi ILC3s from acquiring the capacity for IFN-γ production in vitro.
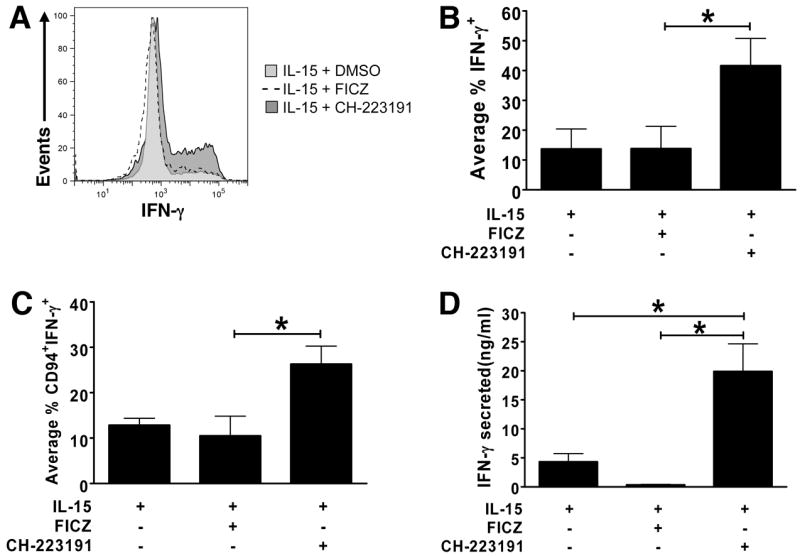
(A-D) IL-1R1hi ILC3s were FACS-sorted from SLT and cultured in conditions shown. After 14 d, an equal number of cells from each condition were stimulated for 12 h with IL-12, IL-15, and IL-18 then assessed for (A-C) intracellular (n = 3) or (D) secreted (n = 4) IFN-γ production. (A) Represents an individual donor (CH-223191,
; DMSO,
; FICZ, dashed line), while (B-D) depicts the average result from multiple donors, presented as mean ± SEM, * for P < 0.05.
Monokine stimulation induced only modest levels of IFN-γ expression by IL-1R1hi ILC3s cultured in the presence of IL-15 with carrier or IL-15 plus FICZ. In contrast, among the progeny of IL-1R1hi ILC3s generated during culture in IL-15 with the AHR antagonist CH-223191, monokine stimulation induced IFN-γ expression in a significantly greater percentage of cells (Fig. 3A-B), and also significantly increased IFN-γ production among the CD94+ fraction derived in vitro (Fig. 3C). ELISA assays indicated that supernatants from IL-1R1hi ILC3s cultured in vitro with IL-15 plus carrier or IL-15 plus AHR agonist contained scant amounts of IFN-γ (4.32 ± 1.39 ng/ml or ≤ 0.3 ± 0.08 ng/ml, respectively) following monokine stimulation; however, IL-1R1hi ILC3s cultured with IL-15 and AHR antagonist secreted significantly greater IFN-γ, averaging 19.9 ± 4.75 ng/ml when activated with IL-12, IL-15, and IL-18 (Fig. 3D). Though IL-1R1hi ILC3s cultured in vitro with IL-15 and carrier alone also produced some IFN-γ, the addition of AHR antagonist resulted in cells producing IFN-γ at levels similar to those seen in freshly isolated stage 4 CD56bright NK cells (Freud et al., 2006). Thus, activation of constitutively expressed AHR in human IL-1R1hi ILC3s prevents their differentiation into functional Group 1 ILCs or NK cells that produce IFN-γ.
AHR prevents the differentiation of human IL-1R1hi ILC3s to cytolytic NK cells
Human IL-1R1hi ILC3s also lack perforin and natural cytotoxicity against NK-sensitive K562 target cells ex vivo (Freud et al., 2006; Hughes et al., 2010). We investigated whether AHR signaling influences acquisition of cytolytic effector functions following culture of IL-1R1hi ILC3s in vitro. Culture in the presence of the AHR antagonist CH-223191 promoted in vitro acquisition of TNF-related apoptosis-inducing ligand (TRAIL), a surface receptor used by NK cells to induce apoptosis of target cells expressing appropriate cognate death receptors (Kashii et al., 1999). TRAIL expression was especially pronounced within the subset of IL-1R1hi ILC3s that had also acquired CD94 during culture in IL-15 plus CH-223191 (Fig. 4A). While there were twice as many CD117-CD94+ cells resulting from IL-1R1hi ILC3s cultured with IL-15 plus the AHR antagonist CH-223191 (Fig. 2E), perforin expression was only moderately increased in this population when compared to the CD94+ cells resulting from culture of IL-1R1hi ILC3s with IL-15 plus carrier alone (Fig. 4B). A standard 51Cr release cytotoxicity assay was performed using IL-1R1hi ILC3s cultured for 14 d with IL-15 plus carrier, IL-15 plus FICZ, or IL-15 plus CH-223191. Cells generated in the latter condition showed significantly and substantially greater lysis of K562 target cells compared to the two former conditions (Fig. 4C). Though IL-1R1hi ILC3s cultured in vitro with IL-15 and carrier were capable of low levels of target cell lysis, the addition of AHR antagonist boosted cytolytic activity significantly, resulting in killing at levels comparable to that of freshly isolated stage 4 NK cells (Freud et al., 2006). As expected, this cytolytic effect was even more pronounced in the CD94+ fraction (Fig. 4D). Thus, AHR inhibits IL-1R1hi ILC3s from acquiring cytolytic effector cell function typically found in stage 4 CD56bright NK cells.
Fig. 4. AHR prevents IL-1R1hi ILC3s from acquiring cytotoxic effector function in vitro.
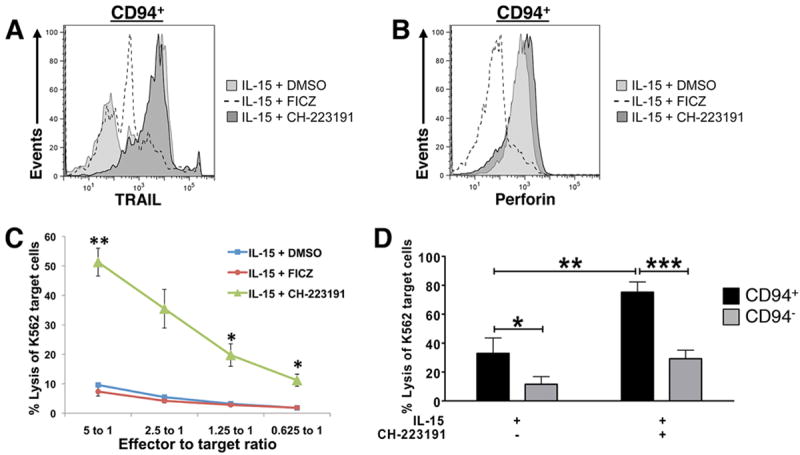
(A-D) IL-1R1hi ILC3s were FACS-sorted from SLT and cultured under the indicated conditions. (A, B) On d 14, flow cytometry was used to assess: (A) TRAIL and CD94 surface expression, or (B) intracellular perforin and CD94 surface expression. Histograms depict staining in a representative donor after gating on CD94+ lymphocytes (CH-223191,
; DMSO,
; FICZ, dashed line). For A and B, n ≥ 3. (C, D) Cytotoxic activity was assessed against the K562 target cell line using an equal number of (C) total progeny, or (D) FACS-sorted CD94+ and CD94- cells generated from IL-1R1hi ILC3s after 14 d in conditions indicated. Cells were plated in triplicate, and target cell lysis was assessed at (C) each effector:target (E:T) ratios indicated (n = 3), or (D) an E:T ratio of 5:1 (CD94+,■; CD94-,
; n = 4). Data in C, D presented as mean ± SEM, *** for P < 0.005, ** for P < 0.01, * for P < 0.05.
AHR mRNA knockdown promotes in vitro differentiation of IL-1R1hi ILC3s
To confirm the results derived from pharmacologic antagonism of AHR by genetic targeting, we used a lentiviral vector (PSIH1) expressing green fluorescent protein (GFP) either without (PSIH1-H1-copGFP, “empty vector”) or with (PSIH1-H1-copGFP-AHR, “AHR shRNA”) a short hairpin RNA (shRNA) directed against AHR using a previously described sequence (Boitano et al., 2010).
We first validated the vector’s ability to knock down AHR mRNA in the NKL human NK cell line, as the low number of primary IL-1R1hi ILC3s we were able to obtain and infect with lentiviral vector precluded validation in this population. As depicted in Fig. 5A, NKL cells expressed comparable levels of AHR prior to and following infection with empty lentiviral vector (AHR expression after infection with empty vector = 114.48 ± 12.42% of uninfected); however, when NKL cells were infected with lentiviral vector containing AHR shRNA, AHR transcript was diminished by 59.5% compared to expression seen following infection with empty vector.
Fig. 5. AHR knockdown via shRNA confirms that AHR prevents the differentiation of IL-1R1hi ILC3s to stage 4 mature NK cells.
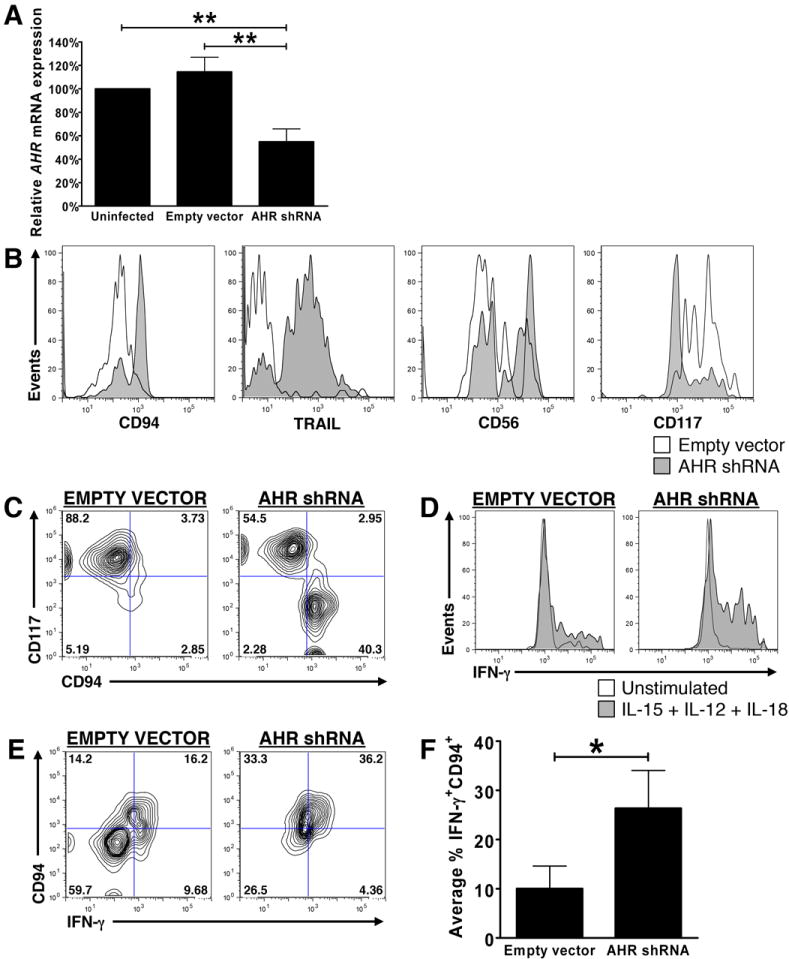
(A) AHR mRNA was quantified via Real-Time RT-PCR in the human NKL cell line prior to infection (left), and following infection with an empty lentiviral vector (PSIH1-H1-copGFP, “empty vector”; middle) or a lentiviral vector containing AHR shRNA (PSIH1-H1-copGFP-shAHR, “AHR shRNA”; right). Values shown are relative to AHR mRNA expression in uninfected NKL cells, arbitrarily set at 100%. Shown as mean ± SEM (n = 4; ** for P < 0.01). (B) Histograms depict IL-1R1hi ILC3s from representative donor infected with empty vector (□) or vector containing AHR shRNA (
), cultured for 14 d, then stained for expression of CD94, TRAIL, CD56 and CD117 on the surface of GFP+ cells. (C) Contour plots depict co-expression of CD94 and CD117 on the surface of GFP+ IL-1R1hi ILC3s from a representative donor infected with empty vector or vector containing AHR shRNA after 14 d of culture. (D-F) IL-1R1hi ILC3s were infected with empty vector or vector containing AHR shRNA, cultured for 14 d, then stimulated for 12 h with IL-12, IL-15, and IL-18, and stained for intracellular IFN-γ protein expression. (D) Histograms depict intracellular IFN-γ protein expression in unstimulated (□) or monokine-stimulated (
) GFP+ infected cells from a representative donor. (E, F) Monokine-induced IFN-γ within GFP+ infected cells co-expressing CD94 shown in (E) a representative donor, and (F) as mean result of multiple donors. Values in F calculated as described for Fig. 3B, C, and depicted as mean ± SEM. For B-F, n ≥ 3; * for P < 0.05.
We next used each lentiviral vector to infect FACS-sorted IL-1R1hi ILC3s from the same donor’s SLT, and cultured these two populations in identical conditions with IL-15 plus carrier (DMSO) alone for 14 d in vitro. Compared to IL-1R1hi ILC3s infected with empty vector, infection with lentivirus containing AHR shRNA increased the percentage of cells expressing CD94, TRAIL, and CD56, and decreased the percentage of cells expressing CD117 (Fig. 5B, C). These data are consistent with results obtained using the AHR antagonist CH-223191. Likewise, relative to IL-1R1hi ILC3s infected with empty vector, infection of cells with lentivirus containing AHR shRNA increased the percentage of cells that were positive for intracellular IFN-γ following monokine stimulation (Fig. 5D). This effect was especially pronounced among GFP+ cells that had also acquired CD94 during culture (Fig. 5E-F). Too few infected cells and high background staining precluded us from validating the acquisition of cytolytic function via 51Cr-release assay or staining for CD107a, respectively. Nonetheless, these data confirm our observations regarding the functional acquisition of IFN-γ production by IL-1R1hi ILC3s cultured in vitro in the presence of the AHR antagonist CH-223191, and support the notion that the AHR transcription factor prevents the phenotypic and functional differentiation of human IL-1R1hi ILC3s to stage 4 mature CD56bright NK cells.
DISCUSSION
The recent discovery and characterization of an ILC3 or Group 3 ILC population with immunophenotypic features overlapping those of CD34-CD117+CD94- stage 3 human NK cell developmental intermediate (Cupedo et al., 2009; Freud et al., 2006; Hughes et al., 2010; Hughes et al., 2009; Spits et al., 2013; Walker et al., 2013) have challenged us to further examine our original four-stage model of human NK cell development (Freud et al., 2006). Indeed because stage 3 immature NK cells or IL-1R1hi ILC3s neither produce IFN-γ nor display cytolytic activity ex vivo yet in bulk or single cell culture can give rise to functionally and phenotypically mature NK cells, Freud et al. originally characterized this population as committed immature NK cells (Freud et al., 2006). However, it is now known that a major fraction of the CD34-CD117+CD94- (stage 3) population constitutively expresses RORC, AHR, and IL-22, the latter of which is also secreted in response to cytokine stimulation (Cella et al., 2009; Cella et al., 2010; Hughes et al., 2010). In light of this, as well as fate-mapping studies in mice and some in vitro studies in humans, this IL-22-producing population is often considered to comprise a distinct lymphocyte lineage, separate from NK cells (Crellin et al., 2010; Satoh-Takayama et al., 2010). Nonetheless, some studies indicate plasticity between Group 1 and Group 3 ILC phenotypes (Ahn et al., 2013; Cella et al., 2010; Lee et al., 2012; Qiu et al., 2012; Vonarbourg et al., 2010), so whether IL-22-producing IL-1R1hi ILC3s cells comprise a stable ILC lineage distinct from NK cells or whether at least a fraction can serve as physiologic stage 3 NK cell precursors is a subject of ongoing debate and investigation. Further, the transcriptional machinery responsible for such plasticity or lack thereof has not been explored in humans.
In the current report we provide novel and compelling evidence that a single transcription factor, AHR, prevents IL-22-producing IL-1R1hi ILC3s from differentiating into CD56bright NK cells in the presence of IL-15. First we show that the IL-1R1hi ILC3 phenotype and expression of IL-22 in this population is dependent upon expression of AHR, which in turn is maintained by IL-1β. We next demonstrate that in the presence of IL-15, human IL-1R1hi ILC3s respond to antagonism of AHR – whether due to the pharmacologic AHR antagonism or via direct genetic targeting of the transcription factor – with quantitatively and qualitatively enhanced differentiation into stage 4 CD56bright NK cells in vitro. Specifically, inhibition of AHR resulted in diminished CD117 surface expression, increased CD94, CD56, and TRAIL expression along with a moderate increase in perforin expression, as well as enhanced functional maturation as evidenced by the acquisition of both IFN-γ production and cytotoxicity against tumor cell targets. AHR antagonism also inhibited RORC expression and promoted expression of the transcription factors, TBX21 (TBET) and EOMES. Notably, the expression of EOMES – recently shown to maintain maturation of murine NK cells (Gordon et al., 2012) and uniquely associated with NK cells rather than non-NK ILC1s (Klose et al., 2013; Spits et al., 2013) – was induced at high levels only in the presence of the AHR antagonist. Collectively, these data support the conclusion that AHR inhibits the differentiation of human IL-1R1hi ILC3s into functionally mature human NK cells in the presence of IL-15 in vitro.
Importantly, the data presented here are in agreement with recent findings in mice and humans that attest to the presence of lineage plasticity among ILC subsets (Bernink et al., 2013; Cella et al., 2010; Vonarbourg et al., 2010). For example, Vonarbourg et al. demonstrated that in the presence of IL-12, murine NCR+ ILC3s could downregulate expression of RORγt and adopt an NK cell-like phenotype (Vonarbourg et al., 2010). Cella et al. showed that culture in IL-2 could promote differentiation of human NCR+ ILC3s toward an IFN-γ-producing phenotype at the expense of an IL-22-producing phenotype in vitro (Cella et al., 2010). Likewise, Bernink et al. recently reported that human NCR+ ILC3s could serve as precursors to IFN-γ-producing non-NK ILC1s (Bernink et al., 2013). The data we present here provide additional evidence for potential lineage plasticity among human ILC subsets and support the notion that AHR prevents human IL-1R1hi ILC3s from giving rise to EOMES+, CD94+, IFN-γ-producing, cytolytic stage 4 CD56brightNK cells.
Interestingly, in light of fate-mapping studies in mouse models (Kiss et al., 2011; Satoh-Takayama et al., 2010; Vonarbourg et al., 2010), these human data call into new question what defines an NK cell. Should NK cells be defined by their molecular, phenotypic and functional attributes regardless of their derivation, or must they never have traversed a RORγt+ intermediate phenotype? The data presented here raise the possibility that at least human NK cells may be derived via two distinct pathways: one that bypasses and one that could potentially progress through an IL-1R1hi ILC3 intermediate stage. Whether this pathway (i.e. IL-1R1hi ILC3 → CD56bright NK via AHR blockade) occurs naturally in vivo in humans is still unclear and is a subject of ongoing investigation.
In light of our human data presented herein, it is notable that Shin et al. recently reported that NK cells derived from AHR-/- mice actually have diminished IFN-γ production and cytotoxicity, so these mice are more prone to developing tumors (Shin et al., 2013). In addition to the inherent species differences and the distinct differences in human and mouse NK cell developmental microenvironments that are now well established (Yu et al., 2013), it is known that AHR controls CD117 expression as demonstrated in γδ T cells (Kadow et al., 2011), in mouse RORγt+ cells (Kiss et al., 2011), and in human ILC3s (this report). It is also well know that mice lacking CD117 (c-Kit) or its ligand (KL) have poor NK cell development (Colucci and Di Santo, 2000; Shibuya et al., 1995). Thus it is conceivable if not likely that the genetic disruption of AHR in mice leads to low or absent CD117 which in turn leads to poor NK cell development, possibly through the elimination of a critical ILC3-like NK cell precursor.
When taken in the context of other recently published reports regarding ILC development and plasticity (Bernink et al., 2013; Cella et al., 2009; Crellin et al., 2010; Spits et al., 2013; Vonarbourg et al., 2010), our study further suggests that ILCs may be modified by cues from the microenvironment, the clinical relevance of which is not yet clear. Indeed ILC populations have been shown to regulate and are implicated in a host of pathological processes (Powell et al., 2012; Sonnenberg et al., 2012; Spits and Di Santo, 2011). Moreover, it is known that AHR-mediated gene regulation is highly context-dependent and cell type specific (Frericks et al., 2007). Notably, AHR has been shown to regulate gene transcription in many cell types – including those of the immune system (Kadow et al.; Kiss et al.; Platzer et al., 2009; Veldhoen et al., 2008) – which possess AHR-responsive elements in their promoters (Sun et al., 2004). In particular, AHR has been shown to regulate the development of the IL-22-producing TH17 lineage of T cells (Veldhoen et al., 2008), and at least some of AHR’s functions have been reported to be ligand binding-specific (Burbach et al., 1992). Importantly, the addition of AHR agonists – which trigger activity of AHR as a transcription factor (Wei et al., 1998) – to culture medium has been shown to enhance TH17 differentiation and IL-22 production (Quintana et al., 2008). Our data, which indicate that adding an AHR agonist to IL-1R1hi ILC3 cultures in the presence of IL-1β results in a substantial increase in IL-22 production, would support this earlier report in T cells.
AHR is also involved in metabolizing xenobiotics and drugs (Denison and Nagy, 2003) and has reported involvement in carcinogenesis, metabolically activating environmental pollutants to create carcinogens, such as benzo[a]pyrene (Shimizu et al., 2000). Epidemiological studies have shown that certain genetic polymorphisms – specifically, polymorphisms that result in a higher level of AHR activity – are associated with increased risk for developing certain types of cancers, and AHR is constitutively active in rapidly growing tumors and immortalized tumor cell lines (DiNatale et al., 2012). Recently, an endogenous tumor-promoting AHR agonist was discovered in the setting of glioma (Opitz et al., 2011), raising the possibility that endogenous AHR agonists could play a similar role in other types of cancer – promoting tumor cell survival, while impeding NK development thereby limiting tumor cell lysis. Indeed, we have previously shown that higher expression of IL-1β, which we show maintains AHR expression and the IL-22+ phenotype in IL-1R1hi ILC3s ((Hughes et al., 2010) and this report), is associated with an unfavorable prognosis in a subset of cytogenetically normal AML patients (Marcucci et al., 2008).
It has been long known that cues from the local microenvironment regulate NK cell development and acquisition of effector functions (Yu et al., 2013). It has also been previously reported that treatment with AHR antagonists in vitro may lower pro-growth and anti-apoptotic signals in several head and neck cancer cell lines (DiNatale et al., 2012). If this finding also holds true in vivo, dampening of AHR signaling may represent a useful strategy to combat certain cancers. Importantly, we observed that AHR antagonism seemed to be required for IL-1R1hi ILC3 induction of EOMES expression, and resulted in enhanced killing of tumor target cells – a finding which is in agreement with those of Gill et al., who used a mouse model to show that high levels of NK cell EOMES expression were associated with decreased tumor burden and increased rate of survival (Gill et al., 2012). Our data from this report would support the notion that pharmacologic inhibition of AHR signaling could promote differentiation of IL-1R1hi ILC3s to cytotoxic, IFN-γ-producing stage 4 mature NK cells, thus bolstering NK cell effector function, while simultaneously differentiating the tumor cell and increasing its susceptibility to apoptosis and/or inhibiting its survival, thus potentially reducing tumor burden via two different – but complementary – mechanisms. Because at least some of AHR’s functions are reported to be ligand binding-specific (Veldhoen et al., 2009), some have proposed the use of AHR as a unique “druggable” target for therapeutic immunomodulation (Murray et al., 2010).
We and others have previously demonstrated that exposure of human IL-1R1hi ILC3s to IL-15 and IL-1β expands this subset of IL-22-producing cells while maintaining expression of AHR (Cella et al., 2010; Hughes et al., 2010). Here we show that an AHR agonist prevents the upregulation of NCRs, granzyme B, and NK cell function in the presence of IL-15 and profoundly increases the cell’s expression of IL-22 via activation of AHR, and the addition of IL-1β accentuates this effect. It is thus possible that IL-1β and AHR work together to tightly repress IL-1R1hi ILC3 differentiation to stage 4 mature NK cells – that is to say, IL-1β induces AHR and thus helps expand IL-22-producing IL-1R1hi ILC3s in response to appropriate environmental stimuli when these cells are necessary for mucosal immunity. If IL-22-producing IL-1R1hi ILC3s are not desirable or necessary in certain inflammatory conditions, antagonism/blockade/downregulation of IL-1R1 and/or AHR could potentially prevent autoimmunity and consequent tissue damage by efficiently converting many IL-1R1hi ILC3s with TH17-like properties to stage 4 mature NK cells endowed with TH1-like properties and effector functions. Thus, IL-1β-mediated maintenance of AHR expression might not only serve to promote IL-22 production, but may also inadvertently allow easy access to a “kill switch” allowing these cells to differentiate to stage 4 NK cells upon exposure to an AHR antagonist and a second set of stimuli to quickly differentiate this population of IL-1R1hi ILC3s. Together, these data establish AHR as a transcription factor whose constitutive expression in IL-1R1hi ILC3s prevents their differentiation into NK cells.
MATERIALS AND METHODS
Isolation of human NK precursors from SLT
All protocols were approved by The Ohio State University (OSU) Institutional Review Board. Fresh, normal human tonsils were obtained, and NK developmental intermediates, including IL-1R1hi ILC3s, were isolated as described (Freud et al., 2006; Hughes et al., 2010). All populations were sorted to ≥95% purity with a FACS Aria II cell sorter (BD Biosciences).
Flow cytometry
All antibodies used for flow cytometry were purchased from BD Biosciences, except CD56 (Beckman Coulter) and CD94 (R&D Systems). Intracellular (IC) staining for perforin and IFN-γ was performed after surface staining. Cells were analyzed immediately as previously described (Freud et al., 2005) using an LSR II cytometer (BD Biosciences) and FlowJo software (Tree Star, Inc.). Non-specific staining was detected with an appropriately labeled isotype antibody.
Real-Time RT-PCR
mRNA was obtained by lysing a portion of each FACS-sorted population in approximately equal quantity (≥ 1×104 cells). RNA was isolated using RNeasy Micro Kit (QIAGEN), and cDNA was synthesized according to manufacturer’s instructions using MMLV reverse transcriptase kit (Invitrogen). Real-Time RT-PCR was performed on an ABI Prism 7900HT (Applied Biosystems). Taqman primer/probe sets for AHR, CYP1A1, and IL-22 purchased from Applied Biosystems. Detection of TBX21 and EOMES was performed using primers with sequences as previously described (Yu et al., 2006). Gene expression was normalized to 18S internal control, then analyzed by the ΔΔCt method (Fehniger et al., 1999).
Cell culture
FACS-purified IL-1R1hi ILC3s were cultured in a round-bottom 96-well plate (Costar) at a starting density of 25,000 cells/ml in α-MEM medium containing 10% FBS, penicillin G (100 μg/ml), and streptomycin (100 μg/ml) (Invitrogen), supplemented with recombinant human IL-15 (1 nM; Amgen) with or without IL-1β (10 ng/ml; Peprotech), and either carrier alone (DMSO, 1 μl/ml; Invitrogen) or carrier containing AHR agonist (FICZ, 300 nM; Enzo Life Sciences) or AHR antagonist (CH-223191, 3 μM; Calbiochem) at concentrations previously reported to modulate TH17 differentiation (Veldhoen et al., 2009). Half the medium was removed every 2-3 d and replaced with medium containing 2x cytokines. After 14 d of culture, cells were counted for viability with trypan blue prior to assays.
Single cell culture
A FACS Aria II was used to deposit single IL-1R1hi ILC3s directly into each well of a 96-well flat bottom plate containing GFP+ murine OP9 stromal cells in α-MEM medium containing 10% FBS and IL-15 plus either DMSO carrier or AHR agonist, FICZ. Half the medium was removed every 2-3 d and replaced with fresh medium containing 2x cytokines. After 14 d, each of 60 replicate wells was individually assessed via flow cytometry for human CD56 surface expression as previously described (Hughes et al., 2010). Wells were scored as positive when the proportion of GFP- lymphocytes expressing CD56 exceeded 2% (corresponding to level of background staining with isotype control mAb).
Effector function assays
FACS-sorted IL-1R1hi ILC3s were cultured as indicated for 14 d. Progeny of these cells were assessed either in bulk, or after FACS-sorting CD94+ and CD94- subsets generated during culture. Cells were counted, and an equal number of cells generated in each culture condition were used for each assay. Cytotoxicity assay against K562 targets was performed as described (Trotta et al., 1998).
IFN-γ production was measured as previously described, either via ELISA (Trotta et al., 2005), or via intracellular (IC) flow cytometric staining (Cooper et al., 2001). Results following 12 h stimulation with IL-12 + IL-15 + IL-18 were compared to parallel controls incubated in the absence of monokines. “% IFN-γ+” was calculated by subtracting percentage of cells producing IFN-γ in the absence of stimulation (value was < 1%) from those producing IFN-γ in response to monokine stimulation.
Lentiviral infection of primary human IL-1R1hi ILC3s
The lentiviral expression vector PSIH1-H1-copGFP was obtained from Systems Biosciences. To generate PSIH1-H1-copGFP-shAHR, hairpin oligos directed against AHR sequences described previously (Boitano et al., 2010) were phosphorylated, annealed, and ligated into predigested PSIH1-H1-copGFP vector. 293T cells were grown to 80% confluence, then transfected as previously described (Trotta et al., 2012). Infectious supernatant from PSIH1-H1-copGFP or PSIH1-H1-copGFP-shAHR retroviral transfection of the 293T cell line was collected after 48 h. IL-1R1hi ILC3s were FACS-sorted from SLT, and immediately subjected to 1 cycle of infection with PSIH1-H1-copGFP (“empty vector”) or PSIH1-H1-copGFP-shAHR (“AHR shRNA”) using previously published methods (Trotta et al., 2012), then cultured for 14 d in the presence of IL-15 plus DMSO carrier. After culture, phenotypic and functional analyses of infected cells were performed via flow cytometry by gating on GFP+ lymphocytes.
To confirm shRNA-mediated down-modulation of AHR expression, the NKL cell line (generous gift from Michael Robertson, Indiana University, Indianapolis, IN), which expressed AHR mRNA prior to infection, was infected in an identical manner with either PSIH1-H1-copGFP or PSIH1-H1-copGFP-shAHR. On d 5 of culture, infected NKL cells expressing GFP were FACS-sorted, and AHR mRNA was measured via Real-Time RT-PCR as described above.
Statistical analysis
For comparison of two conditions, data were analyzed using paired t-tests. Linear mixed models were used for analysis when using more than two treatment conditions. Two-way ANOVA was applied to test the interaction between CD94 and CH-223191. All tests were two sided. P < 0.05 was considered significant for single comparisons, and after adjustment for multiple comparisons.
Supplementary Material
HIGHLIGHTS.
IL-1β maintains a human population of IL-22+IL-1R1hi ILC3s in SLT by regulating AHR.
Inhibition of AHR in human IL-1R1hi ILC3s promotes NK cell differentiation in vitro.
Expression of AHR prevents human IL-1R1hi ILC3 differentiation to NK cells.
Acknowledgments
We acknowledge the Tissue Procurement Shared resource of the OSU Comprehensive Cancer Center for provision of fresh human tonsil specimens. We also thank Amgen for provision of IL-15. This work was supported by NCI grants NCI (CA095426, CA163205, CA16058 and CA068458 to M.A.C. and CA155521 to J.Y.). This work was supported by the Pelotonia Fellowship Program (T.H.).
Footnotes
AUTHOR CONTRIBUTION
T.H., A.G.F., J.Y., D.M.B., and M.C. designed the experiments and wrote the manuscript. J.Z. performed statistical analysis. T.H., E.L.B., R.T., S.M., S.D.S., K.K., Y.D., J.C., N.H., and C.M. carried out the experiments.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn YO, Blazar BR, Miller JS, Verneris MR. Lineage relationships of human interleukin-22-producing CD56+ RORgammat+ innate lymphoid cells and conventional natural killer cells. Blood. 2013;121:2234–2243. doi: 10.1182/blood-2012-07-440099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, Hreggvidsdottir HS, Heinsbroek SE, Legrand N, Buskens CJ, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol. 2013;14:221–229. doi: 10.1038/ni.2534. [DOI] [PubMed] [Google Scholar]
- Boitano AE, Wang J, Romeo R, Bouchez LC, Parker AE, Sutton SE, Walker JR, Flaveny CA, Perdew GH, Denison MS, et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 2010;329:1345–1348. doi: 10.1126/science.1191536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbach KM, Poland A, Bradfield CA. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc Natl Acad Sci U S A. 1992;89:8185–8189. doi: 10.1073/pnas.89.17.8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson WE, Giri JG, Lindemann MJ, Linett ML, Ahdieh M, Paxton R, Anderson D, Eisenmann J, Grabstein K, Caligiuri MA. Interleukin (IL) 15 is a novel cytokine that activates human natural killer cells via components of the IL-2 receptor. J Exp Med. 1994;180:1395–1403. doi: 10.1084/jem.180.4.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, Doherty JM, Mills JC, Colonna M. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 2009;457:722–725. doi: 10.1038/nature07537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M, Otero K, Colonna M. Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc Natl Acad Sci U S A. 2010;107:10961–10966. doi: 10.1073/pnas.1005641107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colucci F, Di Santo JP. The receptor tyrosine kinase c-kit provides a critical signal for survival, expansion, and maturation of mouse natural killer cells. Blood. 2000;95:984–991. [PubMed] [Google Scholar]
- Cooper MA, Bush JE, Fehniger TA, VanDeusen JB, Waite RE, Liu Y, Aguila HL, Caligiuri MA. In vivo evidence for a dependence on interleukin 15 for survival of natural killer cells. Blood. 2002;100:3633–3638. doi: 10.1182/blood-2001-12-0293. [DOI] [PubMed] [Google Scholar]
- Cooper MA, Fehniger TA, Ponnappan A, Mehta V, Wewers MD, Caligiuri MA. Interleukin-1beta costimulates interferon-gamma production by human natural killer cells. Eur J Immunol. 2001;31:792–801. doi: 10.1002/1521-4141(200103)31:3<792::aid-immu792>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Crellin NK, Trifari S, Kaplan CD, Cupedo T, Spits H. Human NKp44+IL-22+ cells and LTi-like cells constitute a stable RORC+ lineage distinct from conventional natural killer cells. J Exp Med. 2010;207:281–290. doi: 10.1084/jem.20091509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, Fibbe WE, Cornelissen JJ, Spits H. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat Immunol. 2009;10:66–74. doi: 10.1038/ni.1668. [DOI] [PubMed] [Google Scholar]
- Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- DiNatale BC, Schroeder JC, Perdew GH. Ah receptor antagonism inhibits constitutive and cytokine inducible IL6 production in head and neck tumor cell lines. Mol Carcinog. 2012;50:173–183. doi: 10.1002/mc.20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehniger TA, Shah MH, Turner MJ, VanDeusen JB, Whitman SP, Cooper MA, Suzuki K, Wechser M, Goodsaid F, Caligiuri MA. Differential cytokine and chemokine gene expression by human NK cells following activation with IL-18 or IL-15 in combination with IL-12: implications for the innate immune response. J Immunol. 1999;162:4511–4520. [PubMed] [Google Scholar]
- Frericks M, Meissner M, Esser C. Microarray analysis of the AHR system: tissue-specific flexibility in signal and target genes. Toxicol Appl Pharmacol. 2007;220:320–332. doi: 10.1016/j.taap.2007.01.014. [DOI] [PubMed] [Google Scholar]
- Freud AG, Becknell B, Roychowdhury S, Mao HC, Ferketich AK, Nuovo GJ, Hughes TL, Marburger TB, Sung J, Baiocchi RA, et al. A human CD34(+) subset resides in lymph nodes and differentiates into CD56bright natural killer cells. Immunity. 2005;22:295–304. doi: 10.1016/j.immuni.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Freud AG, Caligiuri MA. Human natural killer cell development. Immunol Rev. 2006;214:56–72. doi: 10.1111/j.1600-065X.2006.00451.x. [DOI] [PubMed] [Google Scholar]
- Freud AG, Yokohama A, Becknell B, Lee MT, Mao HC, Ferketich AK, Caligiuri MA. Evidence for discrete stages of human natural killer cell differentiation in vivo. J Exp Med. 2006;203:1033–1043. doi: 10.1084/jem.20052507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill S, Vasey AE, De Souza A, Baker J, Smith AT, Kohrt HE, Florek M, Gibbs KD, Jr, Tate K, Ritchie DS, Negrin RS. Rapid development of exhaustion and down-regulation of eomesodermin limit the antitumor activity of adoptively transferred murine natural killer cells. Blood. 2012;119:5758–5768. doi: 10.1182/blood-2012-03-415364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, Lindsten T, Reiner SL. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity. 2012;36:55–67. doi: 10.1016/j.immuni.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankinson O. The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- Hughes T, Becknell B, Freud AG, McClory S, Briercheck E, Yu J, Mao C, Giovenzana C, Nuovo G, Wei L, et al. Interleukin-1beta selectively expands and sustains interleukin-22+ immature human natural killer cells in secondary lymphoid tissue. Immunity. 2010;32:803–814. doi: 10.1016/j.immuni.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes T, Becknell B, McClory S, Briercheck E, Freud AG, Zhang X, Mao H, Nuovo G, Yu J, Caligiuri MA. Stage 3 immature human natural killer cells found in secondary lymphoid tissue constitutively and selectively express the TH 17 cytokine interleukin-22. Blood. 2009;113:4008–4010. doi: 10.1182/blood-2008-12-192443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, Mullen AC, Gasink CR, Kaech SM, Miller JD, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol. 2005;6:1236–1244. doi: 10.1038/ni1268. [DOI] [PubMed] [Google Scholar]
- Kadow S, Jux B, Zahner SP, Wingerath B, Chmill S, Clausen BE, Hengstler J, Esser C. Aryl hydrocarbon receptor is critical for homeostasis of invariant gammadelta T cells in the murine epidermis. J Immunol. 2011;187:3104–3110. doi: 10.4049/jimmunol.1100912. [DOI] [PubMed] [Google Scholar]
- Kashii Y, Giorda R, Herberman RB, Whiteside TL, Vujanovic NL. Constitutive expression and role of the TNF family ligands in apoptotic killing of tumor cells by human NK cells. J Immunol. 1999;163:5358–5366. [PubMed] [Google Scholar]
- Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, Diefenbach A. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science. 2011;334:1561–1565. doi: 10.1126/science.1214914. [DOI] [PubMed] [Google Scholar]
- Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y, Goppert N, Croxford AL, Waisman A, Tanriver Y, Diefenbach A. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature. 2013;494:261–265. doi: 10.1038/nature11813. [DOI] [PubMed] [Google Scholar]
- Kremer J, Gleichmann E, Esser C. Thymic stroma exposed to arylhydrocarbon receptor-binding xenobiotics fails to support proliferation of early thymocytes but induces differentiation. J Immunol. 1994;153:2778–2786. [PubMed] [Google Scholar]
- Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, Mantovani A, Kopan R, Bradfield CA, Newberry RD, Colonna M. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol. 2012;13:144–151. doi: 10.1038/ni.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson M, Sierra MI, Sasidharan R, Prashad SL, Romero M, Saarikoski P, Van Handel B, Huang A, Li X, Mikkola HK. Expansion on stromal cells preserves the undifferentiated state of human hematopoietic stem cells despite compromised reconstitution ability. PLoS One. 2013;8:e53912. doi: 10.1371/journal.pone.0053912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcucci G, Radmacher MD, Maharry K, Mrozek K, Ruppert AS, Paschka P, Vukosavljevic T, Whitman SP, Baldus CD, Langer C, et al. MicroRNA expression in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1919–1928. doi: 10.1056/NEJMoa074256. [DOI] [PubMed] [Google Scholar]
- Mrozek E, Anderson P, Caligiuri MA. Role of interleukin-15 in the development of human CD56+ natural killer cells from CD34+ hematopoietic progenitor cells. Blood. 1996;87:2632–2640. [PubMed] [Google Scholar]
- Murray IA, Morales JL, Flaveny CA, Dinatale BC, Chiaro C, Gowdahalli K, Amin S, Perdew GH. Evidence for ligand-mediated selective modulation of aryl hydrocarbon receptor activity. Mol Pharmacol. 2010;77:247–254. doi: 10.1124/mol.109.061788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- Platzer B, Richter S, Kneidinger D, Waltenberger D, Woisetschlager M, Strobl H. Aryl hydrocarbon receptor activation inhibits in vitro differentiation of human monocytes and Langerhans dendritic cells. J Immunol. 2009;183:66–74. doi: 10.4049/jimmunol.0802997. [DOI] [PubMed] [Google Scholar]
- Powell N, Walker AW, Stolarczyk E, Canavan JB, Gokmen MR, Marks E, Jackson I, Hashim A, Curtis MA, Jenner RG, et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity. 2012;37:674–684. doi: 10.1016/j.immuni.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Heller JJ, Guo X, Chen ZM, Fish K, Fu YX, Zhou L. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity. 2012;36:92–104. doi: 10.1016/j.immuni.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- Rannug U, Rannug A, Sjoberg U, Li H, Westerholm R, Bergman J. Structure elucidation of two tryptophan-derived, high affinity Ah receptor ligands. Chem Biol. 1995;2:841–845. doi: 10.1016/1074-5521(95)90090-x. [DOI] [PubMed] [Google Scholar]
- Satoh-Takayama N, Lesjean-Pottier S, Vieira P, Sawa S, Eberl G, Vosshenrich CA, Di Santo JP. IL-7 and IL-15 independently program the differentiation of intestinal CD3-NKp46+ cell subsets from Id2-dependent precursors. J Exp Med. 2010;207:273–280. doi: 10.1084/jem.20092029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya A, Nagayoshi K, Nakamura K, Nakauchi H. Lymphokine requirement for the generation of natural killer cells from CD34+ hematopoietic progenitor cells. Blood. 1995;85:3538–3546. [PubMed] [Google Scholar]
- Shimizu Y, Nakatsuru Y, Ichinose M, Takahashi Y, Kume H, Mimura J, Fujii-Kuriyama Y, Ishikawa T. Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A. 2000;97:779–782. doi: 10.1073/pnas.97.2.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Zhang L, Murillo-Sauca O, Kim J, Kohrt HE, Bui JD, Sunwoo JB. Modulation of natural killer cell antitumor activity by the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A. 2013;110:12391–12396. doi: 10.1073/pnas.1302856110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KJ, Murray IA, Tanos R, Tellew J, Boitano AE, Bisson WH, Kolluri SK, Cooke MP, Perdew GH. Identification of a high-affinity ligand that exhibits complete aryl hydrocarbon receptor antagonism. J Pharmacol Exp Ther. 2011;338:318–327. doi: 10.1124/jpet.110.178392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, Shibata N, Grunberg S, Sinha R, Zahm AM, et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science. 2012;336:1321–1325. doi: 10.1126/science.1222551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, et al. Innate lymphoid cells--a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13:145–149. doi: 10.1038/nri3365. [DOI] [PubMed] [Google Scholar]
- Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol. 2011;12:21–27. doi: 10.1038/ni.1962. [DOI] [PubMed] [Google Scholar]
- Stejskalova L, Dvorak Z, Pavek P. Endogenous and exogenous ligands of aryl hydrocarbon receptor: current state of art. Curr Drug Metab. 2011;12:198–212. doi: 10.2174/138920011795016818. [DOI] [PubMed] [Google Scholar]
- Sun YV, Boverhof DR, Burgoon LD, Fielden MR, Zacharewski TR. Comparative analysis of dioxin response elements in human, mouse and rat genomic sequences. Nucleic Acids Res. 2004;32:4512–4523. doi: 10.1093/nar/gkh782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson HI, Bradfield CA. The AH-receptor: genetics, structure and function. Pharmacogenetics. 1993;3:213–230. doi: 10.1097/00008571-199310000-00001. [DOI] [PubMed] [Google Scholar]
- Trotta R, Chen L, Ciarlariello D, Josyula S, Mao C, Costinean S, Yu L, Butchar JP, Tridandapani S, Croce CM, Caligiuri MA. miR-155 regulates IFN-gamma production in natural killer cells. Blood. 2012;119:3478–3485. doi: 10.1182/blood-2011-12-398099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotta R, Parihar R, Yu J, Becknell B, Allard J, 2nd, Wen J, Ding W, Mao H, Tridandapani S, Carson WE, Caligiuri MA. Differential expression of SHIP1 in CD56bright and CD56dim NK cells provides a molecular basis for distinct functional responses to monokine costimulation. Blood. 2005;105:3011–3018. doi: 10.1182/blood-2004-10-4072. [DOI] [PubMed] [Google Scholar]
- Trotta R, Puorro KA, Paroli M, Azzoni L, Abebe B, Eisenlohr LC, Perussia B. Dependence of both spontaneous and antibody-dependent, granule exocytosis-mediated NK cell cytotoxicity on extracellular signal-regulated kinases. J Immunol. 1998;161:6648–6656. [PubMed] [Google Scholar]
- Veldhoen M, Hirota K, Christensen J, O’Garra A, Stockinger B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J Exp Med. 2009;206:43–49. doi: 10.1084/jem.20081438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–109. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, Flach M, Bengsch B, Thimme R, Holscher C, et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity. 2010;33:736–751. doi: 10.1016/j.immuni.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells--how did we miss them? Nat Rev Immunol. 2013;13:75–87. doi: 10.1038/nri3349. [DOI] [PubMed] [Google Scholar]
- Wei YD, Helleberg H, Rannug U, Rannug A. Rapid and transient induction of CYP1A1 gene expression in human cells by the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole. Chem Biol Interact. 1998;110:39–55. doi: 10.1016/s0009-2797(97)00111-7. [DOI] [PubMed] [Google Scholar]
- Yu J, Freud AG, Caligiuri MA. Location and cellular stages of natural killer cell development. Trends Immunol. 2013;34:573–582. doi: 10.1016/j.it.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Wei M, Becknell B, Trotta R, Liu S, Boyd Z, Jaung MS, Blaser BW, Sun J, Benson DM, Jr, et al. Pro- and antiinflammatory cytokine signaling: reciprocal antagonism regulates interferon-gamma production by human natural killer cells. Immunity. 2006;24:575–590. doi: 10.1016/j.immuni.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Zhao B, Degroot DE, Hayashi A, He G, Denison MS. CH223191 is a ligand-selective antagonist of the Ah (Dioxin) receptor. Toxicol Sci. 2010;117:393–403. doi: 10.1093/toxsci/kfq217. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.