

Abstract
Recent highly expected structural characterizations of agonist-bound and antagonist-bound beta-2 adrenoreceptor (β2AR) by X-ray crystallography have been widely regarded as critical advances to enable more effective structure-based discovery of GPCRs ligands. It appears that this very important development may have undermined many previous efforts to develop 3D theoretical models of GPCRs. To address this question directly we have compared several historical β2AR models versus the inactive state and nanobody-stabilized active state of β2AR crystal structures in terms of their structural similarity and effectiveness of use in virtual screening for β2AR specific agonists and antagonists. Theoretical models, incluing both homology and de novo types, were collected from five different groups who have published extensively in the field of GPCRs modeling; all models were built before X-ray structures became available. In general, β2AR theoretical models differ significantly from the crystal structure in terms of TMH definition and the global packing. Nevertheless, surprisingly, several models afforded hit rates resulting from virtual screening of large chemical library enriched by known β2AR ligands that exceeded those using X-ray structures; the hit rates were particularly higher for agonists. Furthemore, the screening performance of models is associated with local structural quality such as the RMSDs for binding pocket residues and the ability to capture accurately most if not all critical protein/ligand interactions. These results suggest that carefully built models of GPCRs could capture critical chemical and structural features of the binding pocket thus may be even more useful for practical structure-based drug discovery than X-ray structures.
Keywords: GPCRs modeling, crystallography, beta-2 adrenoreceptor, agonist-bound, antagonist-bound, molecular docking, enrichment factor
Introduction
Structure based drug discovery (SBDD) has become a major strategy in identifying novel leads for important biological targets. SBDD enabled well documented discovery of several approved drugs, e.g. dorzolamide and imatinib. Typically, the 3D structures of biomolecules obtained by the means of X-ray crystallography and NMR spectroscopy are needed for the purpose of virtual screening (VS), whose performance is strongly affected by the quality of biomolecular structure, especially with respect to binding site description. When no experimentally determined structures are available, theoretical models based on either homology(comparative) or de novo modeling approaches are employed instead17, 21, 34. However, there were some debates about the accuracy and applicability of theoretical models10, 11, 20, 28, 30, 40 in SBDD. In principle, the success of theoretical modeling is typically measured by how close the models could reproduce experimental structures, which implies that the latter are regarded as inherently more appropriate choice for SBDD applications.
G-protein coupled receptors (GPCRs) is a protein family where theoretical models have been used widely because of GPCRs’ importance as targets for many drugs, on one hand, and the lack of experimental structures, until recently, on the other hand. The experimental effort to characterize the 3D structure of GPCRs had been seriously hindered by membrane related issues8. A large number of theoretical models have been employed in the past decades22, 24, 35, 47, 54, 59, 64 for VS often yielding reasonable results7, 12, 18, 19, 33. However, it was fully expected that the availability of the experimental structure of any GPCRs would substantially enhance the efficacy of SBDD efforts. Thus, the recent characterizations of the crystal structures of human beta-2 adrenergic GPCR (β2AR) in both agonist-bound49, 52 and antagonist-bound states13, 50, 51, cleared the way for the validation of previous theoretical models, as well as provided critical data for building homology based models of other GPCRs as the most obvious structural template.
It has been shown that human β2AR features a structurally conserved rhodopsin-like 7TM core, but there exist novel structural features that had not been identified previously. It remained unclear as to whether these structural divergences would affect the outcome of VS studies. Dissimilarity of theoretical models relative to the crystal structure should lead to relatively poorer performance of the former in docking and scoring of known ligands; however, this general expectation should not necessarily be regarded as a law. One should take into account that some of the theoretical models are actually manually refined with known medicinal chemistry data and therefore, there is at least a possibility that theoretical models may be even more suitable for drug discovery by VS than the crystal structure.
In this study, we have addressed this, both scientifically and pragmatically, important question directly. We have compared the X-ray structure of β2AR vs. several previously built theoretical models in terms of their respective ability to recover known β2AR ligands (both agonists and antagonists) from a large external compound library in VS experiments. None of these models were generated in our group to ensure objective and unbiased comparisons. Furthermore, although our group has developed both scoring functions63 and virtual screening protocols44, for the same reasons we restricted ourselves to using several popular commercial docking and scoring tools developed elsewhere. Thus, by design, this study lacked any user biases concerning preferred theoretical models or most familiar computational tools to emulate the situation that is most commonly faced by the majority of molecular modeling practitioners both in academia and industry.
There have been previous studies on comparing homology models among themselves or even with the crystal structure but to the best of our knowledge nobody approached the question poised herein as retrospectively and broadly as we did. For instance, Bissantz et al employed three human GPCRs models derived from rhodopsin for virtual screening using multiple docking programs and scoring functions10. Their work proved that homology models are suitable for VS but there was no comparison to the crystal structure. After the crystal structure of human β2AR was published, Costanzi reported two studies where carazolol was docked both into two rhodopsin-based homology models of human β2AR as well as into its X-ray structure of inactive state16, 57. The models afforded high accuracy of the docking poses and ideal enrichment for both antagonists and agonist, especially after incorporating the biochemical data to adjust the orientation of the binding pocket residues. However, these studies were limited to their in-house models and lacked of global structural comparison. More recently, Fan et al reported that for 27 out of the 38 protein targets, the consensus enrichment for multiple homology models was better than or comparable to that of both the holo- and the apo- X-ray structures20. However, that study was focused on soluble protein targets and applied a single homology model building tool that employed X-ray characterized structural templates. In contrast, all models included herein were built before the β2AR crystal structures became available as possible templates. Michino et al. and Kufareva et al. recently reported two large-scale assessments of current GPCR modeling efforts and molecular docking capacities37, 41. While their studies provided important insights on how to improve current structure prediction and docking techniques to reproduce the X-ray structures, our studies focus primarily on the pragmatic question as to whether computational models of GPCRs how they could be used successfully for structure based drug discovery.
We have carried out a systematic, retrospective study on a large collection of published human β2AR theoretical models and evaluated their structural accuracies and virtual screening performances in comparison with three crystal structures, i.e., 2RH1 (released by the RCSB Protein Data Bank (PDB) on Oct. 30, 200713), 3D4S (released by RCSB PDB on Jun. 17, 200826), and the latest agonist-bound structure 3P0G (released by RCSB PDB on Jan. 19, 201149). Two other structures, 2R4R and 2R4S50, were not employed because they came from the same source as 2RH1 but with lower resolutions. Another agonist-bound b2 adrenoceptor structure, 3PDS with an irreversible disulfide bond between ligand and complex, was not used because its conformation is more close to the inactive state52. As shown in Table I, we collected eight independently published theoretical models of human β2AR including both apo and holo structures. Both agonist and antagonist bound models were included to account for any structural features associated with functional activity. Furthermore, we incorporated both homology and de novo models to cover these two major types of GPCRs modeling.
Table I.
The synonym of eight human β2AR theoretical models employed in this study.
| Apo model | Source | Holo model | Source |
|---|---|---|---|
| AM1 | By G. Vriend, a homology model(7) based on the crystal structure of bovine rhodopsin | CM1 | By T. Lybrand, a de novo model(1, 5) bound by aminoflisopolol (β2AR antagonist) |
| AM2 | By A. Sali, a homology model(6) based on the crystal structure of bovine rhodopsin | CM2 | By T. Lybrand, a de novo model(1, 5) bound by TA2005 (β2AR agonist) |
| AM3 | By J. Skolnick, a hybrid model(2) combined threading and ab initio methods | CM3 | By W. Goddard, a de novo model(3, 4) bound by butoxamine (β2AR antagonist) |
| AM4 | By W. Goddard, a de novo model(3) based on first principles methods | CM4 | By W. Goddard, a de novo model(3, 4) bound by salbutamol (β2AR agonist) |
Surprisingly, we found that some of the theoretical models displayed better or comparable VS performances than the crystallographic structures. This study by no means undermines the extreme significance of the X-ray structures of β2AR as well as other GPCRs13, 29, 45, 46, 60, 62 in understanding the intricate details of GPCRs structure in relation to its function nor in the significance of X-ray structures for SBDD. Nevertheless, it most certainly testifies to the importance of intelligent computational modeling approaches especially those incorporating comprehensive medicinal chemistry knowledge of receptor/ligand complex for structure based virtual screening.
Materials and Methods
Structural similarity analysis
In addition to crystal structures of inactive state (PDB ID: 2RH1) and active state (PDB ID: 3P0G) of human β2AR, the structure of bovine rhodopsin (PDB ID: 1U19) in dark state was also included in the analysis because it had been used as the major template for all β2AR homology models. The structural similarity was assessed in three aspects, i.e. the accuracy of the boundary definitions for each transmembrane (TM) helixes, the backbone root-mean-square-deviation (RMSD) for TM regions, and the Cα RMSD of the binding pocket residues. The numbering of amino acids followed the conventions set by Weinstein et al58. The highly conserved residues embedded in each TM region were employed as anchors for the alignment. Each theoretical model was structurally aligned against 2RH1, 3P0G or 1U19 by individual TM helix as well as the whole TM bundle. The RMSDs were calculated using the entire lengths of the corresponding segments of 2RH1.
Screening libraries
Active seeds
To include most known binders, we collected 57 known antagonists of human β2AR reported in DrugBank61 and GLIDA43 databases as active seeds. All antagonists have sub-micromolar potency and can be found in additional external databases, such as PDSP Ki53, PubChem5 and KEGG31. A few of them are β2AR specific (e.g. butoxamine and aminoflisopolol) while others can act on both β1AR and β2AR. In the meantime, we compiled the agonist set of thirteen full agonists for human β2AR from the DrugBank database.
Decoys set I: PDSP binding decoys plus WDI drug-like decoys
As our initial effort to establish a reasonable decoy dataset for virtual screening, we employed the World Drug Index (WDI) database version 20041 since most of its compounds are drug-like. The original collection of 59,000 molecules was first cleaned by removing metals, salts and fragments, then filtered to eliminate unqualified compounds according to Lipinski’s rule of five and later extensions of this rule25. The remaining collection of ca. 38,000 compounds was further reduced to a diverse subset of 374 compounds using MOE2007.09. In addition, 12 binding decoys with similar chemical scaffolds but poor binding affinity (Ki > 10μM) were selected from the PDSP Ki database and merged into the WDI diverse subset. In the end, 57 human β2AR antagonists and 13 agonists were seeded amongst 386 decoys separately to constitute two different screening libraries.
Decoys set II: ChEMBL binding decoys plus Schrodinger drug-like decoys
In an effort to avoid the bias brought by one single screening library, we built another decoys set which includes 1000 drug-like decoys23 from Schrodinger web portal and 916 known nonbinders from ChEMBL database (https://www.ebi.ac.uk/chembl/). The size of this decoys set is fairly large and the ratio of actives to decoys is (r=33.6 for β2AR antagonists) close to the criteria set by the DUD database (r=36.0). The ChEMBL binding decoys were selected based on the standards similar to the ones in the decoys set I for PDSP database but of large number.
Decoys set III: Subset of II containing N+ only
Like other amine GPCRs families, β2AR’s ligands are special in that they normally contain a positively charged nitrogen at the side chain portion, which is a key ingredient of the binding event. To ensure that the power of current scoring function is based upon factors other than this simple structural feature, we created the 3rd decoys set by applying this filter (N+ present) to the decoys set II. In the end, there were 652 compounds left with positively charged nitrogen.
Molecular docking methods
We employed three popular docking programs, i.e. Glide4.01, AutoDock4.0 and eHiTS6.2, to evaluate systematically the screening performance of structural models.
Glide4.01
The calculations by Glide version 4.0123 was carried out using Schrodinger Suite 2007. The targeted protein and theoretical models were prepared through Protein Preparation module with the default setting and assigned with the OPLS 2001 force field atom types and partial charges. The screening databases were prepared within the LigPrep module and the ionization states of each molecule were calculated as to be compatible with the pH value of 7.0±2.0. All molecules were subjected to energy minimization with MMFFs force field before the docking computation. For X-ray structure and holo models, the center of the grid box was selected as the center of bound ligands. For apo models, their binding pockets were first aligned to that of 2RH1 and the center of co-crystallized carazolol was chosen. The proper size of the enclosing box was not set to be fixed but determined by the extent of the bound ligand. The Glide SP scoring function was used to rank the docking poses and the top-ranked poses for each database molecule were saved for post-docking analysis.
AutoDock4.0
We prepared the targeted protein and docking parameters for AutoDock version 4.027, 42 using the AutoDockTools graphic interface. Explicit hydrogen atoms were added to the receptor structures while atom types and partial charges were assigned to generate the pdbqt receptor files. The database molecules were prepared using the ‘prepare_ligand4.py’ script to merge non-polar hydrogen atoms and define flexible torsions. The center and dimension of the enclosing boxes were defined to include the whole binding pockets, similar to those in the Glide docking. The genetic algorithm were employed during the docking with a start population size of 150 individuals and 20 runs combined with a maximum number of 12,500,000 energy evaluation for each molecule. Other parameters for genetic algorithm were kept by the default value.
eHiTS6.2
The eHiTS version 6.265 was used through the CheVi user interface. Protein preparations, such as protonation state determination for residues, hydrogen atoms addition and partial charge assignment, were actually not needed since eHiTS’ docking and scoring are based on the prior training data of its knowledge base of 97 protein families. The grid box was assigned automatically using the bound ligand’s SDF file as the CLIP file. We employed the default settings for eHiTS docking and ranked the database molecule based on its lowest eHiTS score.
In addition, we added eight more scoring functions of different types by rescoring the top-ranked poses generated by Glide4.01, to ensure an unbiased and complete comparison. The multiple scoring functions in Sybyl8.04 CScore module and OpenEye2 FRED 2.2.4 were applied, including Chemscore, D_score, Gold_score, PMF, Chemgauss3, PLP, Screenscore and Shapegauss. The consensus scores were also used for the above scoring functions through the rank by rank strategy.
Assessment of virtual screening performance
To measure the efficiency of virtual screening we used the following conventional parameters: the enrichment factor and the receiver operating characteristic (ROC) curve that characterizes the ability if a method to recover known ligands among the top-scored screening molecules. The enrichment factor follows the most popular definition as to how many more seed compounds (i.e., known ligands) were found within a defined “early recognition” fraction of the ranked list relative to a random distribution:
| (1) |
where Hscr is the number of target-specific seeds recovered at a specific % level of the database; Htot is the total number of seeds for the target; Dscr is the number of compounds screened at a specific % level of the database; Dtot is the total number of compounds of the database. The ROC curve is generated by plotting the sensitivity (Se) vs. (1 – specificity (Sp)) for a binary classifier system as its discrimination threshold is varied. In the case of virtual screening for recovering the ith known active from the inactive decoys, the Sei and Spi are defined as follows:
| (2) |
| (3) |
The area under the ROC curve (AUC) is the metric that is widely accepted for assessing the likelihood that a screening method assigns a higher rank to known actives than to inactive compounds. The AUC values at a specific percentage of the ranked database are calculated from the following equation:
| (4) |
Here n is the total number of known actives in the screening database. One additional parameter, the yield, is also employed as the percentage of true hits retrieved by the virtual screening method:
| (5) |
Cluster analysis of binding profiles
To closely evaluate the key receptor/ligand interaction patterns, we employed the LigX module14 in MOE2007.09 to analyze the crystal structure of β2AR/carazolol complex and the docking poses generated by Glide4.01. For each antagonist, the top-ranked docking pose with the highest score was selected. Two major types of interactions that contribute to protein/ligand binding affinities were considered, i.e. hydrogen bonds (donor or acceptor) and non-bonded weak interactions. The score to assess the hydrogen bond is based on a scale of 0 to 100% that indicates the probability of being a geometrically perfect hydrogen bond while the score for non-bonded weak interaction is the pairwise distance between residue and ligand atoms. In our studies, we took the default parameters in which 4.5 angstrom is the cutoff for weak interactions and 2.5 angstrom is the closest distance between any residue/ligand atom pairs. The original score was normalized; thus, the values of the modified scores were between 0 and 1, which is proportional to the interaction intensities. To better visualize the binding patterns of docked poses for each theoretical models and crystal structures, the LigX scores were transformed into heat maps and clustered using the R statistical package48. We applied the hierarchical clustering with the Ward linkage algorithm; thus, the patterns of interaction between 57 human β2AR antagonists and residues in the active sites of three different structural models would be expected to be similar if the respective clusters are similar.
In addition, we have exploited the Protein Ligand Interaction Fingerprints (PLIF, also available in MOE2007.09) for the same purpose. PLIF can identify and score major protein/ligand interactions, including hydrogen acceptor from sidechain, hydrogen donor to sidechain, hydrogen acceptor from backbone, hydrogen donor to backbone, ionic attractions and surface contacts. For each docking pose, the PLIF fingerprints ranging from 30 to 50 bits were generated. The relative frequencies of each identified fingerprint can be then used to produce fingerprint significance chart, which is based on the hypothesis that ‘if the bit is set, then the compound is active’.
Results and Discussion
Structural comparison of theoretical models and X-ray structures of β2AR
Prior to VS experiments, we analyzed the similarity between theoretical models and two X-ray structures, i.e. 2RH1 and 1U19 deposited to the Protein Data Bank9. All theoretical models used in this study are listed in Table I; each model was aligned against the X-ray structures to evaluate relative definitions of transmembrane (TM) helices, their conformations, and relative orientation. The X-ray structure of bovine rhodopsin (1U19) was also included in addition to β2AR because the former had been used as a common template for GPCRs homology modeling. In doing so, we were interested to explore if the failure to predict the structural conservation and/or divergence from the template structure may cause poor VS results for the theoretical models.
Our initial efforts focused on evaluating the accuracy of TM helical boundaries as defined by models vs. X-ray structures. For this purpose we have employed MOE 2007.09 software3 to annotate the secondary structural elements in the 3D structures. We found that the MOE module assigned boundaries either at exact positions or only one amino acid apart to over 90% of the TM segments of 2RH1 and 1U19 in comparison with those in the PDB header. Thus, the MOE software was deemed reliable in identifying the helical boundaries; the results of applying MOE to the six theoretical models and two crystal structures (2RH1 and 1U19) are summarized in Figure 1a and Table II. From the alignment of the eight structures, it can be seen that the apo models (AM1-AM3) perform better than the holo models (CM1-CM3) in terms of accuracy of TM assignment. The location and length of the TM helixes for all three AM models are consistently close to those in the rhodopsin structure (1U19), with the only exception that TM6 and TM7 in the AM1 model are shorter than the corresponding helices in the crystal structures. This observation can be easily rationalized since AM1 and AM2 models are solely based on homology modeling whereas AM3 is a hybrid model developed with a combination of both threading and ab initio methods. In all cases, the crystal structures of bovine rhodopsin were used as a template for model building. Furthermore, we have concluded that individual TM helixes were very close in terms of helical length and relative orientation when compared to crystal structures of bovine rhodopsin and human β2AR13. Thus, not surprisingly, given the methods used for model building, the secondary structural elements for all three AM models were found to be assigned very accurately as compared to their homologous experimental structures.
Figure 1.
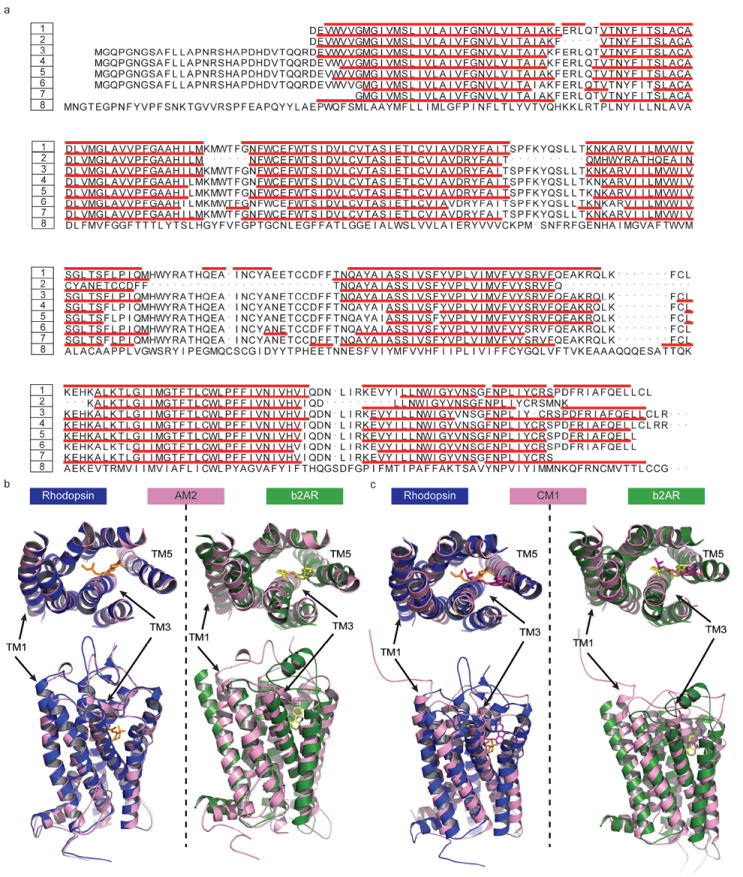
The structural similarity analysis of theoretical models in comparison with crystal structures. (a) The secondary structure assignment for TM segments of six theoretical models and two crystal structures (2RH1 and 1U19). The numbers and their corresponding structures are coded by 1:2RH1, 2:AM1, 3:AM2, 4:AM3, 5:CM1, 6:CM2, 7:CM3, 8:1U19. The remaining two models, i.e., AM4 and CM4, share similar backbone structures to CM3 with pairwise RMSD of TMs less than 0.4 Ǻ. Therefore only CM3 is included in the sequence alignment plot. The red bars indicate the helical structure elements identified by MOE. (b) The structural superposition of the theoretical models AM2 (rendered in pink) to 2RH1 (rendered in dark green) and 1U19 (rendered in blue). Note that the most structurally divergent TM regions are indicated. (c) The structural superposition of the theoretical models CM1 (rendered in pink) to 2RH1 (rendered in dark green) and 1U19 (rendered in blue). Note that the most structurally divergent TM regions are indicated as well.
Table II.
Comparisons for six β2AR theoretical models versus two crystal structures in terms of TM helical boundaries and RMSD. The bordering residues for each TM region are identified by MOE and annotated using a specialized numbering rule. The RMSDs for each model’s TM were calculated using the true length of the corresponding segments of 2RH1 and 1U19 (in italic).
| Segment | 2RH1 | AM1 | AM2 | AM3 | CM1 | CM2 | CM3 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| 1U19 | start/end | RMSD | start/end | RMSD | start/end | RMSD | start/end | RMSD | start/end | RMSD | start/end | RMSD | |
| TMI | 1.30/1.59 | 1.29/1.60 | 2.77Ǻ | 1.29/1.59 | 3.02Ǻ | 1.28/1.59 | 3.01Ǻ | 1.31/1.59 | 3.18Ǻ | 1.32/1.58 | 3.19Ǻ | 1.35/1.56 | 5.12Ǻ |
| 1.29/1.59 | 0.30Ǻ | 0.30Ǻ | 0.83Ǻ | 2.93Ǻ | 3.03Ǻ | 6.90Ǻ | |||||||
| TMII | 2.38/2.67 | 2.38/2.67 | 1.76Ǻ | 2.38/2.67 | 1.59Ǻ | 2.38/2.67 | 1.90Ǻ | 2.37/2.65 | 3.19Ǻ | 2.37/2.65 | 3.09Ǻ | 2.45/2.64 | 4.38Ǻ |
| 2.38/2.67 | 0.32Ǻ | 0.37Ǻ | 0.68Ǻ | 2.40Ǻ | 2.36Ǻ | 4.31Ǻ | |||||||
| TMIII | 3.21/3.55 | 3.22/3.55 | 4.32Ǻ | 3.22/3.54 | 2.09Ǻ | 3.23/3.54 | 1.67Ǻ | 3.22/3.55 | 2.57Ǻ | 3.23/3.55 | 2.56Ǻ | 3.27/3.47 | 6.99Ǻ |
| 3.21/3.54 | 0.34Ǻ | 0.37Ǻ | 1.08Ǻ | 2.21Ǻ | 2.25Ǻ | 5.00Ǻ | |||||||
| TMIV | 4.39/4.62 | N/A | 1.95Ǻ | 4.39/4.61 | 1.94Ǻ | 4.39/4.62 | 1.67Ǻ | 4.40/4.57 | 5.21Ǻ | 4.41/4.57 | 5.43Ǻ | 4.38/4.68 | 3.47Ǻ |
| 4.39/4.62 | 0.32Ǻ | 0.49Ǻ | 1.06Ǻ | 5.00Ǻ | 5.02Ǻ | 4.60Ǻ | |||||||
| TMV | 5.36/5.68 | 5.34/5.62 | 2.37Ǻ | 5.35/5.62 | 3.48Ǻ | 5.35/5.70 | 2.76Ǻ | 5.41/5.67 | 2.17Ǻ | 5.41/5.67 | 2.13Ǻ | 5.37/5.58 | 6.85Ǻ |
| 5.31/5.62 | 0.43Ǻ | 0.38Ǻ | 1.00Ǻ | 2.04Ǻ | 2.06Ǻ | 3.59Ǻ | |||||||
| TMVI | 6.29/6.60 | 6.33/6.60 | 1.67Ǻ | 6.33/6.59 | 3.80Ǻ | 6.27/6.60 | 1.96Ǻ | 6.28/6.59 | 4.24Ǻ | 6.28/6.59 | 4.21Ǻ | 6.38/6.59 | 8.12Ǻ |
| 6.25/6.59 | 0.32Ǻ | 9.11Ǻ | 0.80Ǻ | 3.63Ǻ | 3.69Ǻ | 12.54Ǻ | |||||||
| TMVII | 7.32/7.55 | 7.38/7.56 | 3.82Ǻ | 7.32/7.51 | 1.65Ǻ | 7.33/7.55 | 2.35Ǻ | 7.32/7.55 | 4.08Ǻ | 7.32/7.55 | 4.20Ǻ | 7.34/7.55 | 3.93Ǻ |
| 7.32/7.55 | 0.38Ǻ | 1.01Ǻ | 1.37Ǻ | 3.34Ǻ | 3.42Ǻ | 2.64Ǻ | |||||||
| TMI-VII | 2.47Ǻ | 3.19Ǻ | 2.25Ǻ | 3.59Ǻ | N/A | 4.41Ǻ | |||||||
| 1.15Ǻ | 1.88Ǻ | 1.38Ǻ | 3.20Ǻ | N/A | 3.83Ǻ | ||||||||
| ECLII | N/A | 13.43Ǻ | 13.19Ǻ | 7.00Ǻ | 7.18Ǻ | 19.00Ǻ | |||||||
| N/A | 1.32Ǻ | 1.81Ǻ | 13.72Ǻ | 13.96Ǻ | 19.70Ǻ | ||||||||
| Binding pocket | 3.71Ǻ | 3.39Ǻ | 3.58Ǻ | 2.33Ǻ | 2.40Ǻ | 5.64Ǻ | |||||||
The accuracies of TM helix boundaries assignments for CM models were less satisfactory. In general, seven TM helices in all three CM models were shorter than expected with the largest disagreements located at TM1, TM4 and TM5 (cf. Figure 1a and Table II). The CM3 model gave the largest deviation in terms of the percentage of correctly defined TM helical boundaries. Moreover, it had the shortest lengths for individual TM helices. For instance, it was eight residues shorter for TM1, fourteen for TM3 and eleven for both TM5 and TM6 in comparison with the β2AR crystal structure. One possible explanation is that all three CMs are de novo models, generated without any template structure. Both the Lybrand (CM1, 2) and the Goddard (CM3, 4) groups employed the standard alpha helix as a starting point and calculated the intrinsic tilt/kink and relative orientation of the TM helical bundle purely based on the physical considerations. If the rhodopsin structure is not employed as a reference, the secondary structure assignments could be affected by many factors, such as the type of phospholipid used in the MD simulations employed as part of model refinement in studies by the Goddard group22, 54.
It should be noted that the engineered modification of the wild type protein using a segment of T4 lysozyme to replace most residues of IL3 introduced an artifact in the crystal structure (2RH1) of human β2AR. This modification led to altering the boundaries of IL3, thus affecting the correct locations of both the TM5 terminus and the start of the TM6. The accurate definition of these two boundaries is less important in the comparison of structural similarities between theoretical models and the crystal structures. However, the accuracy of predicting the TM4 terminus and the beginning of TM5 is critical considering the functional roles of EL2 in both rhodopsin and human β2AR6, 55. As can be seen from Figure 1a and Table II among the three AM models, AM3 has the highest accuracy (one residue error) for the segment between TM4 and TM5 (EL2) followed by AM2 and AM1. CM3 model is comparable to AM2 while CM1 and CM2 had much larger errors with respect to TM helix assignments.
The seven TM helices of each theoretical model were superimposed onto respective helices of β2AR as well as the rhodopsin structure, and the backbone pairwise RMSD of individual respective TM helices was calculated (Figures 1b, 1c and Table II). As expected, the homology models (AMs) are generally more similar to the rhodopsin structure than to the β2AR structure. The RMSDs of most helices in AM1-3 range from 0.30 Ǻ to 1.00 Ǻ as compared to bovine rhodopsin where the RMSDs are as big as 1.60 Ǻ to 3.80 Ǻ when aligned against the human β2AR. For the whole TMs bundle, the RMSDs are 1.15 Ǻ to 1.88 Ǻ with respect to rhodopsin and 2.25 Ǻ to 3.19 Ǻ with respect to β2AR. In comparison, the de novo models (CMs) deviate more significantly from both crystal structures. The RMSDs of most helixes in CM1/CM2 are in the same range of 2.00 Ǻ - 5.50 Ǻ when aligned against both rhodopsin and β2AR. For CM3, the RMSD increases to 2.60 Ǻ and 7.00 Ǻ, respectively. Similarly, the RMSDs of TMs bundle for CM1 are 3.20 Ǻ when aligned against the rhodopsin and 3.59 Ǻ for β2AR. For CM3, the corresponding RMSDs are 3.83 Ǻ and 4.41 Ǻ (cf. Table II).
Consistently, the AMs models were observed to be aligned well to the rhodopsin structure (cf. AM2 in Figure 1b as an illustrative example). They deviated from the β2AR structure with the noticeable shifts for TM1, TM3 and TM5, although the secondary structure assignment for these regions was relatively accurate. The large RMSD differences for these three TMHs were obviously due to the differences between rhodopsin and β2AR crystal structures (7TMs backbone RMSD of 2RH1 vs. 1U19 is 1.85 Å). It was indeed reported in the original publication on β2AR crystal structure13 that there is a noticeable shift in TM1 of β2AR relative to bovine rhodopsin, primarily at the extracellular portion which tilts away from the TM bundle compared to bovine rhodopsin13, 51. The long N-terminal fragment could not be observed in both crystal structures, but it could cause large flexibility/variation in the assignment of TM1 boundaries, especially in the upper helical region. TM5 has a proline-induced kink at conserved positions along the transmembrane segments, which is believed to be responsible for the structural rearrangements required for the GPCRs activation32, 36, 39, 58. The subtle difference in the activation status of the current β2AR structure (2RH1, bound to an inverse agonist carazolol) may lead to the structural diversification at the kink region, in terms of the amplitude of motion and rotation degree. Notably TM3 and TM5 constitute half of the binding pockets for the co-crystallized carazolol13 (Figure 1b).
Unlike AM models, three CMs deviate from both bovine rhodopsin and β2AR in a similar way (cf. CM1 in Figure 1c as the representative case). A large discrepancy can be found at TM1, 4, 6 and 7 for CM1/CM2 and TM1, 3, 5 and 6 for CM3. Here the similar reasoning used in the analysis of AMs can be applied to TM1 because the N-terminal fragment was not considered as part of model building and optimization. Interestingly, the RMSD of CM1’s TM4 is as large as 5.00 Ǻ with respect to rhodopsin and 5.21 Ǻ when aligned against β2AR. It is surprising since TM4 seems the easiest one to model. Among all seven TMs, TM4 is the shortest and the most orthogonal to the plane of the phospholipid membrane. This observation indicates the limitation of computational protocols employed in developing the CM models in their ability to reproduce the conformation of this ‘anchor’ helix in the TM bundle. For TM5, 6 and 7, one of the common shared features is the proline-induced kink. The comparisons between β2AR models and the crystal structures highlight the difficulty associated with the accurate modeling of this unique structural feature of GPCRs.
Comparison of VS performance for theoretical models and X-ray structures of β2AR
Figure 2 shows the enrichment factor plot (a,c,e) and ROC curves (b,d,f) of 57 known β2AR antagonists against decoys by three docking methods. The yield plot is shown in since it essentially delivers the same information as the ROC curves. The detailed statistical parameters characterizing the VS performance, such as the maximum EF (EFmax), ROC AUC and the recently proposed Boltzmann-Enhanced Discrimination of ROC (BEDROC)15, 56, are summarized in Table III. All four holo models as well as the β2AR structure were used in the VS study. Because of the poor performance of the apo models in general during virtual screening, we only showed the data for the relatively better ones (AM1 and AM3) to represent the AMs group. In many cases, such as AM3 in Figures 2d and 2f, the ROC curve is close to the random expectation (the diagonal line). It is understandable because the side chain rotamers of binding pocket residues in the AM models had not been optimized in the way it was done for holo models.
Figure 2.
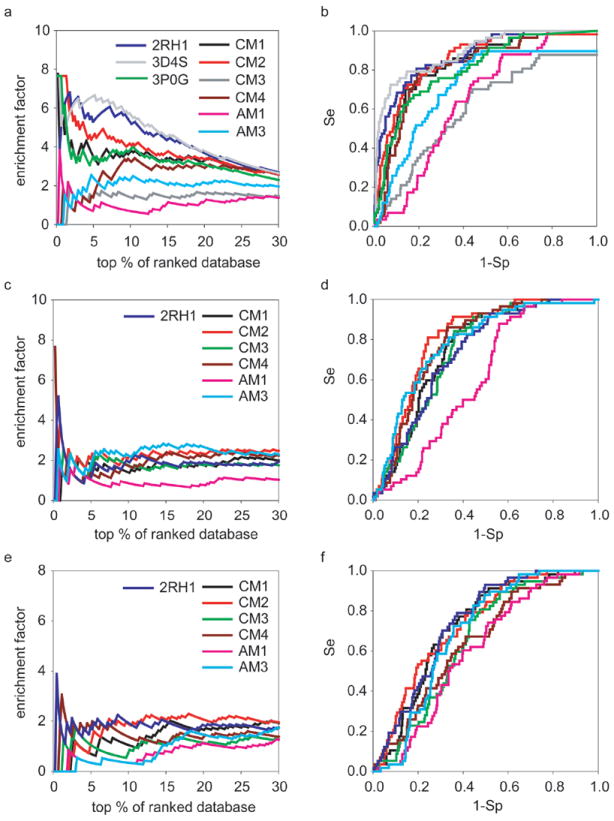
The docking performance of six theoretical models in comparison to three crystal structures 2RH1, 3D4S and 3P0G. The enrichment (a, c, e) and ROC curves (b, d, f) of fifty-seven known β2AR antagonists with the decoys set I by three docking methods. The annotations are (a, b) Glide4.01 (c, d) AutoDock4.0 and (e, f) eHiTS6.2.
Table III.
The enrichment factors (EF) and yields of fifty-seven known β2AR antagonists by theoretical models versus two crystal structures with decoys set I (PDSP binding decoys plus WDI drug-like decoys).
| Structure/Model | Docking method | EF at 1% db | EF at 5% db | EF at 25% db | % db to find 25% seeds | Yield 25% db | EFmax | % db EFmax occurred | ROC AUC | AUC at 25% db | α=20 | BEDROC α=53.6 | α=100 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2RH1 | Glide4.01 | 5.83 | 5.30 | 3.08 | 4.74 | 77.19 | 6.80 | 1.81 | 0.88 | 0.11 | 0.72 | 0.75 | 0.74 |
| AutoDock4.0 | 3.89 | 1.06 | 1.75 | 11.74 | 43.86 | 0.68 | 5.18 | 0.73 | 0.05 | ||||
| eHiTS6.2 | 1.94 | 1.77 | 1.82 | 13.54 | 45.61 | 3.89 | 0.45 | 0.74 | 0.05 | ||||
| 3D4S | Glide4.01 | 6.12 | 6.66 | 3.17 | 4.05 | 79.31 | 7.65 | 0.22 | 0.89 | 0.11 | 0.77 | 0.80 | 0.79 |
| 3P0G | Glide4.01 | 5.74 | 3.33 | 2.69 | 7.21 | 67.24 | 7.66 | 0.68 | 0.81 | 0.08 | 0.50 | 0.52 | 0.62 |
|
| |||||||||||||
| CM1 | Glide4.01 | 5.83 | 3.53 | 2.87 | 6.77 | 71.93 | 7.77 | 1.35 | 0.83 | 0.08 | 0.50 | 0.52 | 0.58 |
| AutoDock4.0 | 0.00 | 1.77 | 2.17 | 13.77 | 54.39 | 2.59 | 1.35 | 0.76 | 0.04 | ||||
| eHiTS6.2 | 0.00 | 0.71 | 1.75 | 14.67 | 43.86 | 2.03 | 15.58 | 0.72 | 0.04 | ||||
| CM2 | Glide4.01 | 7.66 | 4.52 | 2.90 | 5.63 | 72.41 | 7.66 | 1.35 | 0.86 | 0.09 | 0.61 | 0.72 | 0.83 |
| AutoDock4.0 | 3.83 | 2.09 | 2.48 | 11.49 | 62.07 | 3.83 | 0.90 | 0.81 | 0.06 | ||||
| eHiTS6.2 | 0.00 | 1.04 | 2.14 | 12.16 | 53.45 | 2.30 | 18.02 | 0.73 | 0.05 | ||||
| CM3 | Glide4.01 | 0.00 | 1.41 | 1.54 | 17.61 | 38.60 | 1.94 | 0.90 | 0.61 | 0.04 | 0.20 | 0.15 | 0.11 |
| AutoDock4.0 | 1.94 | 1.41 | 1.82 | 14.90 | 45.61 | 2.59 | 1.35 | 0.75 | 0.05 | ||||
| eHiTS6.2 | 0.00 | 1.06 | 1.26 | 23.25 | 31.58 | 1.79 | 2.93 | 0.65 | 0.03 | ||||
| CM4 | Glide4.01 | 1.91 | 1.39 | 2.90 | 2.70 | 72.41 | 3.42 | 10.59 | 0.82 | 0.07 | 0.34 | 0.19 | 0.13 |
| AutoDock4.0 | 1.91 | 1.39 | 2.34 | 12.61 | 58.62 | 7.66 | 0.23 | 0.78 | 0.05 | ||||
| eHiTS6.2 | 1.91 | 1.74 | 1.52 | 16.89 | 37.93 | 3.06 | 1.13 | 0.65 | 0.04 | ||||
| AM1 | Glide4.01 | 1.91 | 0.70 | 1.31 | 20.27 | 32.76 | 3.83 | 0.45 | 0.65 | 0.03 | 0.14 | 0.17 | 0.22 |
| AutoDock4.0 | 1.91 | 1.04 | 1.03 | 22.75 | 25.86 | 1.91 | 0.90 | 0.61 | 0.03 | ||||
| eHiTS6.2 | 0.00 | 0.35 | 0.97 | 25.90 | 24.14 | 1.42 | 37.61 | 0.60 | 0.03 | ||||
| AM3 | Glide4.01 | 0.00 | 2.44 | 2.07 | 10.36 | 51.72 | 2.55 | 4.73 | 0.72 | 0.06 | 0.28 | 0.20 | 0.14 |
| AutoDock4.0 | 1.91 | 1.74 | 2.34 | 10.81 | 58.62 | 2.83 | 14.64 | 0.78 | 0.07 | ||||
| eHiTS6.2 | 0.00 | 0.35 | 1.45 | 17.57 | 36.21 | 1.81 | 32.43 | 0.69 | 0.03 | ||||
Among all four CMs models, those from the Lybrand group (CM1, CM2) achieved better enrichment than the models from the Goddard group (CM3, CM4). In most cases, the CM2 model yielded comparable results to the β2AR crystal structure. In the screening by Glide4.01, CM2 model gave higher EF at the very early phase (0% - 2% of ranked database, cf. Figure 2a and Table III). After that, it remained up to one unit lower than the crystal structure until converging with the latter at the 22% of ranked database. In terms of ROC AUC, CM2 reached the value of 0.86, close to AUC of 0.88 for the crystal structure. Based on the BEDROC metric, CM2 was similar to 2RH1 when α was small (α=20) while showing better performance when α was large (α=53.6 or 100). We note that Glide4.01 gave better VS results in this study compared to AutoDock4.0 and eHiTS6.2. Thus, we placed more emphasis on the screening data/docking poses generated by Glide4.01. Nevertheless, the results obtained with both AutoDock4.0 and eHiTS6.2 also highlighted the impressive performance of CM2. As demonstrated by both types of plots in Figures 2c-f and most criteria in Table III, the CM2 afforded VS results superior to the crystal structure when using these two docking programs. Therefore, practically speaking, these results suggest that the use of crystal structure is not advantageous in terms of VS performance when the scoring function is not highly accurate. On the other hand, it reflects on the extreme sensitivity of the docking approaches to small structural variations. As mentioned above, CM3 and CM4 had poorer performance than CM1 and CM2 but were comparable to AM1 and AM3 models in this case (cf. Table III).
The crystal structure of β2AR represents an inactive state of the receptor because it is bound to the inverse agonist, carazolol13, 38. Thus, it may be considered unfair to compare the crystal structure of β2AR with theoretical models as applied to agonist screening, especially when the models were created to capture known data on agonists. However, for comparison purposes, we did explore the possible utility of 2RH1 for screening for agonists. The screening results are summarized in Figures 3 and Table IV. As expected, the CM2 model showed the best performance to enrich for thirteen β2AR agonists. With Glide4.01 method, the CM2 model could recover 100% of seed agonists at the 15% of ranked databases and its maximum EF could be as high as 36.09. Thus, it excelled over 2RH1 greatly in terms of these two parameters of VS performance. Taking into account the data for the antagonist virtual screening, we shall conclude that CM2 model demonstrated remarkable performance as a model of choice for virtual screening for both agonists and antagonists.
Figure 3.
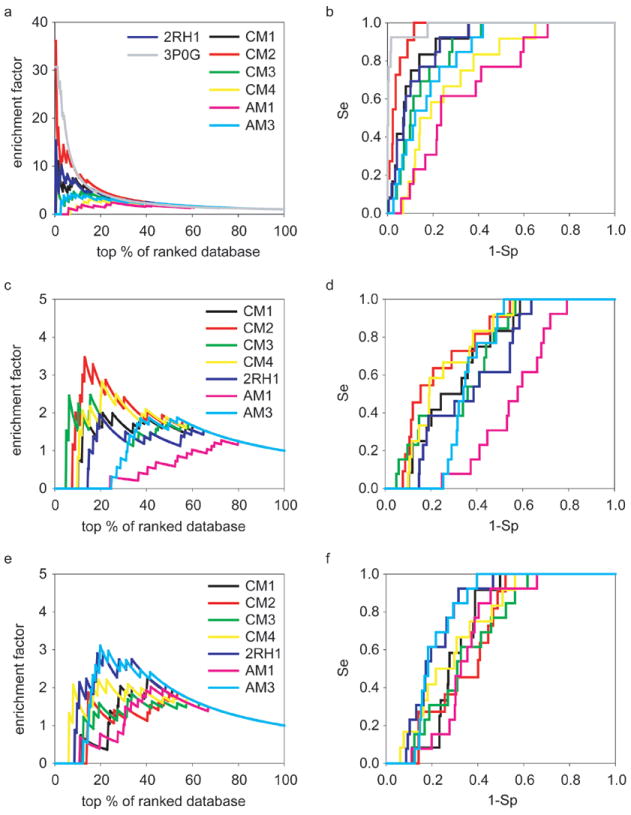
The enrichments and ROC curves of thirteen known β2AR agonists with the decoys set I during the screening against six theoretical models and two crystal structures (2RH1 and 3P0G). The annotations are (a, b) Glide4.01 (c, d) AutoDock4.0 and (e, f) eHiTS6.2.
Table IV.
The enrichment factors (EF) and yields of thirteen known β2AR agonists by six theoretical models versus the crystal structure 2RH1 with decoys set I (PDSP binding decoys plus WDI drug-like decoys).
| Structure/Model | Docking method | % db to find 25% agonists | EF at 25% db | Yield at 25% db | Max.EF | % db EFmax occurred | AUC | AUC at 25% db |
|---|---|---|---|---|---|---|---|---|
| 2RH1 | Glide4.01 | 3.01 | 3.38 | 84.62 | 15.35 | 0.50 | 0.90 | 0.15 |
| AutoDock4.0 | 18.55 | 1.53 | 38.46 | 1.97 | 19.55 | 0.63 | 0.03 | |
| eHiTS6.2 | 13.78 | 2.46 | 61.54 | 2.92 | 21.05 | 0.79 | 0.06 | |
|
| ||||||||
| CM1 | Glide4.01 | 3.77 | 3.65 | 91.67 | 11.06 | 0.75 | 0.91 | 0.15 |
| AutoDock4.0 | 12.31 | 1.99 | 50.00 | 2.03 | 12.31 | 0.70 | 0.04 | |
| eHiTS6.2 | 23.37 | 1.33 | 33.33 | 2.27 | 40.45 | 0.70 | 0.02 | |
| CM2 | Glide4.01 | 1.51 | 3.97 | 100.00 | 36.09 | 0.25 | 0.96 | 0.20 |
| AutoDock4.0 | 11.08 | 2.53 | 63.64 | 3.47 | 13.10 | 0.77 | 0.08 | |
| eHiTS6.2 | 14.61 | 1.08 | 27.27 | 1.87 | 53.40 | 0.66 | 0.03 | |
| CM3 | Glide4.01 | 7.77 | 3.07 | 76.92 | 15.35 | 0.50 | 0.86 | 0.11 |
| AutoDock4.0 | 15.04 | 1.53 | 38.46 | 2.48 | 15.54 | 0.69 | 0.06 | |
| eHiTS6.2 | 19.30 | 1.23 | 30.77 | 1.83 | 33.58 | 0.66 | 0.03 | |
| CM4 | Glide4.01 | 12.31 | 2.32 | 58.33 | 3.26 | 15.33 | 0.75 | 0.07 |
| AutoDock4.0 | 26.13 | 2.32 | 58.33 | 2.83 | 20.60 | 0.74 | 0.05 | |
| eHiTS6.2 | 15.33 | 1.99 | 50.00 | 2.21 | 18.84 | 0.72 | 0.05 | |
| AM1 | Glide4.01 | 17.04 | 2.46 | 61.54 | 2.48 | 24.81 | 0.69 | 0.05 |
| AutoDock4.0 | 44.11 | 0.31 | 7.69 | 1.27 | 72.68 | 0.44 | 0.00 | |
| eHiTS6.2 | 30.08 | 0.61 | 15.38 | 2.02 | 41.85 | 0.66 | 0.02 | |
| AM3 | Glide4.01 | 7.27 | 3.07 | 76.92 | 4.23 | 7.27 | 0.83 | 0.10 |
| AutoDock4.0 | 31.33 | 0.31 | 7.69 | 1.87 | 41.10 | 0.64 | 0.00 | |
| eHiTS6.2 | 15.79 | 2.76 | 69.23 | 3.11 | 19.80 | 0.78 | 0.05 | |
Figure 4, Table V and Table VI showed the results of virtual screening for antagonists and agonists with decoys sets II and III. We observed that the relative performance between crystal structures and theoretical models with new sets is similar to that observed in our previous experiments, although the absolute performance of each protein model varied by the choice of decoy sets. Notably the CM2 model was consistently ranked comparable to three experimental structures to enrich fifty-seven β2AR antagonists (cf. Figure 4 and Table V). And remarkably it outperformance structures 2RH1 and 3D4S for thirteen β2AR agonists on most aspects of screening, and is comparable to the structure of 3P0G at the late phase of screening (> 25% db, cf. Figure 4 and Table VI). Thus we believe that the conclusions we drew from the decoys set I are valid and expandable to other screening libraries, thus appear to be general.
Figure 4.
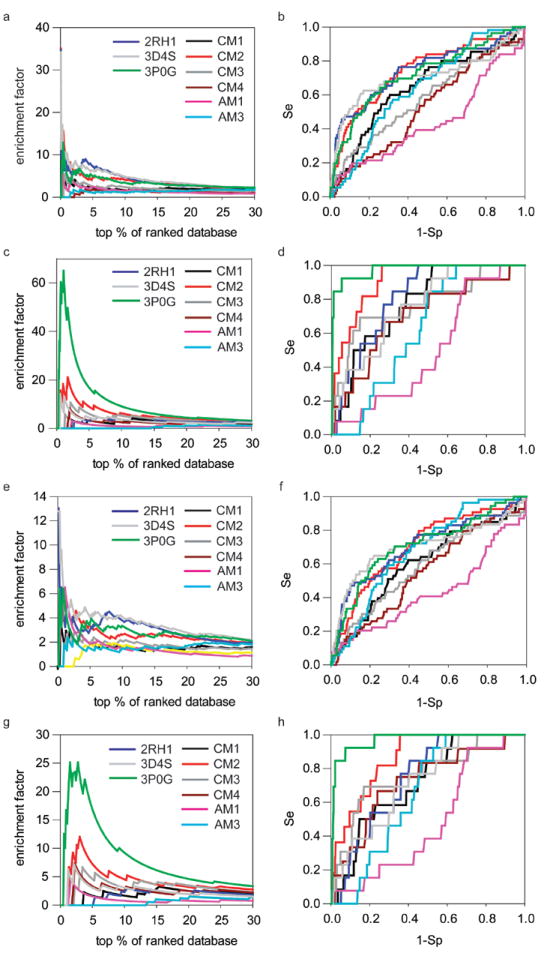
The docking performance of six theoretical models in comparison to three crystal structures 2RH1, 3D4S and 3P0G with the decoys sets II and III. The enrichment (a) and ROC curves (b) of fifty-seven known β2AR antagonists and the enrichment (c) and ROC curves (d) of thirteen known β2AR agonists with the decoys set II. The enrichment (e) and ROC curves (f) of fifty-seven known β2AR antagonists and the enrichment (g) and ROC curves (h) of thirteen known β2AR agonists with the decoys set III.
Table V.
The EFs and yields of 57 β2AR antagonists by theoretical models versus three crystal structures with decoys set II (ChEMBL binding decoys plus Schrodinger drug-like decoys). The data from decoys set III (subset of II containing N+ only) were marked in italic.
| Structure/Model | EF at 1% db | EF at 5% db | EF at 25% db | % db to find 25% seeds | Yield at 25% db | EFmax | % db EFmax occurred | ROC AUC | AUC at 25% db | α=20 | BEDROC α=53.6 | α=100 | Seeds Num. | Decoys Num. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2RH1 | 6.95 | 7.02 | 2.47 | 3.35 | 61.82 | 34.75 | 0.05 | 0.74 | 0.08 | 0.32 | 0.23 | 0.21 | 55 | 1856 |
| 3.72 | 3.25 | 2.09 | 6.96 | 52.83 | 13.02 | 0.14 | 0.71 | 0.07 | 0.31 | 0.28 | 0.33 | 53 | 637 | |
| 3D4S | 10.42 | 6.27 | 2.49 | 3.29 | 62.50 | 34.14 | 0.05 | 0.71 | 0.09 | 0.33 | 0.26 | 0.26 | 56 | 1856 |
| 5.48 | 4.02 | 2.54 | 5.79 | 64.81 | 12.80 | 0.14 | 0.70 | 0.08 | 0.37 | 0.37 | 0.44 | 54 | 637 | |
| 3P0G | 5.15 | 4.90 | 2.41 | 4.63 | 60.71 | 12.86 | 0.42 | 0.72 | 0.07 | 0.24 | 0.18 | 0.17 | 56 | 1865 |
| 5.51 | 3.93 | 2.35 | 7.78 | 59.26 | 6.43 | 0.29 | 0.71 | 0.06 | 0.26 | 0.25 | 0.28 | 54 | 640 | |
|
| ||||||||||||||
| CM1 | 3.51 | 2.17 | 1.95 | 12.47 | 49.09 | 7.03 | 0.26 | 0.64 | 0.04 | 0.14 | 0.10 | 0.10 | 55 | 1878 |
| 1.88 | 1.88 | 1.57 | 16.21 | 39.62 | 4.38 | 0.43 | 0.61 | 0.03 | 0.17 | 0.17 | 0.18 | 53 | 644 | |
| CM2 | 6.68 | 4.60 | 2.28 | 5.25 | 57.14 | 35.05 | 0.05 | 0.74 | 0.08 | 0.28 | 0.22 | 0.23 | 56 | 1907 |
| 4.89 | 3.26 | 2.12 | 9.80 | 53.70 | 13.04 | 0.14 | 0.71 | 0.06 | 0.28 | 0.30 | 0.35 | 54 | 650 | |
| CM3 | 3.57 | 2.50 | 1.45 | 11.42 | 36.36 | 5.49 | 0.66 | 0.58 | 0.04 | 0.15 | 0.11 | 0.10 | 55 | 1906 |
| 1.66 | 2.15 | 1.50 | 14.94 | 37.74 | 2.95 | 2.56 | 0.59 | 0.04 | 0.18 | 0.15 | 0.13 | 53 | 650 | |
| CM4 | 0.00 | 1.77 | 0.98 | 22.67 | 25.00 | 1.84 | 3.87 | 0.52 | 0.03 | 0.08 | 0.03 | 0.01 | 56 | 1907 |
| 0.00 | 1.81 | 1.10 | 21.31 | 27.78 | 1.92 | 8.66 | 0.55 | 0.03 | 0.12 | 0.06 | 0.02 | 54 | 650 | |
| AM1 | 1.75 | 2.48 | 0.84 | 27.46 | 21.43 | 5.84 | 0.31 | 0.44 | 0.03 | 0.12 | 0.09 | 0.08 | 56 | 1907 |
| 6.52 | 1.96 | 0.89 | 28.84 | 22.22 | 6.52 | 1.14 | 0.43 | 0.03 | 0.18 | 0.23 | 0.28 | 54 | 650 | |
| AM3 | 0.00 | 1.05 | 1.84 | 17.01 | 46.43 | 1.85 | 24.15 | 0.64 | 0.03 | 0.08 | 0.04 | 0.02 | 56 | 1907 |
| 1.63 | 1.41 | 1.92 | 16.05 | 48.15 | 2.37 | 1.56 | 0.68 | 0.03 | 0.15 | 0.12 | 0.10 | 54 | 650 | |
Table VI.
The EFs and yields of 13 β2AR agonists by six theoretical models versus three crystal structures with decoys set II (ChEMBL binding decoys plus Schrodinger drug-like decoys). The data from decoys set III (subset of II containing N+ only) were marked in italic.
| Structure/Model | EF at 1% db | EF at 2% db | % db to find 25% agonists | EF at 25% db | Yield at 25% db | EFmax | % db EFmax occurred | AUC | AUC at 25% db | α=20 | BEDROC α=53.6 | α=100 | Seeds Num. | Decoys Num. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2RH1 | 0.00 | 0.00 | 6.26 | 2.46 | 61.54 | 4.92 | 6.26 | 0.81 | 0.06 | 0.16 | 0.04 | 0.01 | 13 | 1856 |
| 0.00 | 0.00 | 10.46 | 2.15 | 53.85 | 2.94 | 10.46 | 0.74 | 0.04 | 0.09 | 0.01 | 0.00 | 13 | 637 | |
| 3D4S | 7.57 | 7.57 | 6.10 | 1.84 | 46.15 | 14.38 | 0.54 | 0.76 | 0.06 | 0.22 | 0.13 | 0.10 | 13 | 1856 |
| 0.00 | 3.85 | 8.92 | 1.84 | 46.15 | 7.14 | 2.15 | 0.72 | 0.05 | 0.19 | 0.11 | 0.08 | 13 | 637 | |
| 3P0G | 60.83 | 41.82 | 0.59 | 3.99 | 100.00 | 65.01 | 1.06 | 0.98 | 0.18 | 0.79 | 0.66 | 0.55 | 13 | 1865 |
| 14.35 | 23.44 | 1.38 | 3.98 | 100.00 | 25.12 | 1.53 | 0.97 | 0.17 | 0.72 | 0.54 | 0.41 | 13 | 640 | |
|
| ||||||||||||||
| CM1 | 0.00 | 0.00 | 8.94 | 2.33 | 58.33 | 4.28 | 11.69 | 0.77 | 0.06 | 0.14 | 0.04 | 0.01 | 12 | 1878 |
| 0.00 | 0.00 | 12.04 | 2.29 | 58.33 | 3.25 | 15.40 | 0.72 | 0.05 | 0.10 | 0.02 | 0.01 | 12 | 644 | |
| CM2 | 17.44 | 17.88 | 1.56 | 3.60 | 90.91 | 21.13 | 1.72 | 0.91 | 0.12 | 0.40 | 0.25 | 0.16 | 11 | 1907 |
| 0.00 | 8.58 | 2.57 | 3.26 | 81.82 | 12.02 | 3.03 | 0.86 | 0.09 | 0.32 | 0.19 | 0.11 | 11 | 650 | |
| CM3 | 0.00 | 7.38 | 3.49 | 2.76 | 69.23 | 10.80 | 2.14 | 0.77 | 0.09 | 0.25 | 0.12 | 0.06 | 13 | 1906 |
| 0.00 | 0.00 | 5.43 | 2.77 | 69.23 | 6.65 | 3.47 | 0.76 | 0.08 | 0.22 | 0.09 | 0.04 | 13 | 650 | |
| CM4 | 0.00 | 8.20 | 7.30 | 2.28 | 58.33 | 8.42 | 1.98 | 0.70 | 0.05 | 0.16 | 0.07 | 0.03 | 12 | 1907 |
| 0.00 | 3.94 | 6.50 | 2.66 | 66.67 | 7.36 | 2.27 | 0.72 | 0.05 | 0.18 | 0.09 | 0.04 | 12 | 650 | |
| AM1 | 0.00 | 0.00 | 41.77 | 0.92 | 23.08 | 3.28 | 2.34 | 0.50 | 0.02 | 0.06 | 0.03 | 0.01 | 13 | 1907 |
| 0.00 | 3.64 | 42.84 | 0.92 | 23.08 | 4.64 | 1.66 | 0.48 | 0.02 | 0.07 | 0.05 | 0.04 | 13 | 650 | |
| AM3 | 0.00 | 0.00 | 21.88 | 1.22 | 30.77 | 1.69 | 49.95 | 0.62 | 0.01 | 0.01 | 0.00 | 0.00 | 13 | 1907 |
| 0.00 | 0.00 | 20.21 | 1.21 | 30.77 | 1.78 | 47.51 | 0.65 | 0.01 | 0.02 | 0.00 | 0.00 | 13 | 650 | |
The possible explanation of the better performance of CM1/CM2 models is that Lybrand et al exploited many site-directed mutagenesis data during the model optimization24, 35. The important receptor/ligand interactions had been turned into distance restraints that were applied explicitly to specific atoms of both the receptor and its ligands during molecular dynamics simulations24. In comparison, CM3 and CM4 models from Goddard et al did not employ such information22, 54; their models were built by optimizing the target/ligand interaction using physical force field. Obviously, the differences in the type of data utilized for theoretical model building and optimization can largely affect the accuracy of binding pocket modeling, and consequently, the model performance in virtual screening experiments. In order to evaluate the similarity between binding pockets of individual models, we superimposed Cα atoms of key residues inside the pocket with their counterparts in 2RH1. The binding pocket was defined by residues found within 4 Å of the co-crystallized carazolol. Carazolol was merged into the binding sites of all models as defined by the alignment. As shown in Figure 5 (A-H), the CM2 binding pocket (Figure 5B) is most similar to that of 2RH1 with respect to both the ligand pose and the position of residues interacting with the ligand. The RMSD for its Cα atoms was 2.40 Ǻ while the one for CM1 was 2.33 Ǻ (cf. Table II). These two models also reproduced the contacts of carazolol with residues Ser2035.42, Asn3127.39 and Phe193 . For three AMs models, the RMSDs ranged from 3.39 Ǻ to 3.71 Ǻ. CM3 and CM4 models had the largest deviation (RMSD = 5.64 Ǻ), as can also be seen in Figures 5C and 5D.
Figure 5.
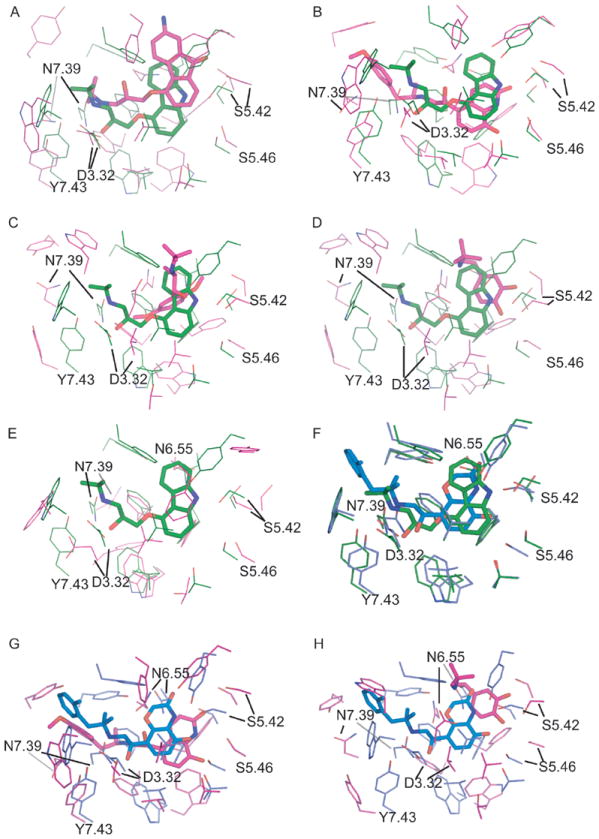
The comparison of the binding pockets of theoretical models versus the β2AR crystal structures of inactive state (2RH1; A:CM1, B:CM2, C:CM3, D:CM4, E:AM1) and active state (3P0G; F:2RH1, G:CM2, H:CM4). The active sites were superimposed by Ca atoms of key binding site residues of 2RH1 (W3.28, D3.32, V3.33, V3.36, T3.38, F5.32, Y5.38, S5.42, S5.43, S5.46, W6.48, F6.51, F6.52, N6.55, Y7.35, N7.39, Y7.43). The crystal structures 2RH1 and 3P0G are colored in green and blue respectively, while the model structures are colored in pink.
Furthermore, the close inspection of the top-ranked docking poses of all seed antagonists showed that the interactions between the antagonists and the binding site of the CM2 model were largely in agreement with the site-directed mutagenesis data. The protonated nitrogen in most β2AR antagonists formed salt bridges with ASP1133.32 and Asn3127.39; the amide hydroxyl group formed hydrogen bonds with Ser2035.42, Ser2045.43 or Ser2075.46. Another important interaction was formed between antagonists and Phe193 of EL2, i.e., the residue that was also found to interact with carazolol within the crystallographic structure of β2AR13, 24, 51. It should be pointed out that CM2/CM1 models include both extracellular and intracellular loops, whereas CM3/CM4 and AM1 models did not incorporate these regions22, 51.
To elucidate the molecular basis for dissimilar virtual screening performance of different CM2 and CM3 models in comparison with 2RH1 we have conducted the cluster analysis of the binding profiles of all 57 antagonists docked to the respective binding sites. Binding profiles reflected the strengths of interaction between antagonists and active site residues. Importantly, there were significant differences in the weak interaction patterns of CM2 (Figure 6b) and CM3 (Figure 6c) in comparison with 2RH1 (Figure 6a). The major clusters formed by CM2 in the region of conserved residues matched well to those found in 2RH1, suggesting a critical role of weak interactions between binding site and antagonists. In comparison, clusters formed by CM3 were scarce and many key interactions were missing, especially for residues Trp2866.48, Phe2896.51, Phe2906.52 and Asn2936.55 of the binding pocket. This analysis indicates that the binding pocket of CM2 was well-organized and similar to that of 2RH1 whereas the CM3 binding pocket was formed by somewhat different residues, with the key residues found in the binding site of the X-ray structure were inaccessible to the bound antagonists. The cluster profile of hydrogen bonding pattern was less informative as some prominent patterns at 2RH1 such as the ones with Asn3127.39 were absent at both CM2 and CM3. Notably, Ser2045.43 and Ser2075.46 of CM2 were found to be hydrogen bonded to ca. 15 antagonists, but the same pattern was not observed with either CM3 or 2RH1.
Figure 6.

The cluster analysis of the antagonists binding profile. (Upper panel, a-c) The comparison of weak interaction profiles of fifty-seven antagonists of human β2AR with 2RH1 (a), CM2 (b) and CM3 (c). (Lower panel, d-f) The comparison of hydrogen bonding (HBond) profiles of fifty-seven antagonists of human β2AR with 2RH1 (d), CM2 (e) and CM3 (f). The weak and HBond interactions were identified/scored by LigX module in MOE2007.09 and marked as X.1 for HBond donors and X.2 for HBond acceptors. In the case that one residue forms two HBonds to the ligand, the interactions were labeled as X(I).X or X(II).X, in which the X(I).X had the better score. The upper block in each map contains the binding pocket residues of 2RH1 while the lower block contains other interacting residues. Each point in the maps represents the identified interactions and was shaded by their respective scores, wherever darker indicates higher score and thus greater interaction strength. All the points in the map had been reorganized using hierarchical clustering by interaction scores.
During the course of our studies, Kobilka et al. experimentally characterized two new structures of β2AR, one in a nanobody-stabilized active state and another in complex with an irreversible agonist. In comparison with the structures of inactive state of β2AR, the agonist-binding pockets showed fairly subtle changes, with the differences at the hydrogen bonding contacts with Ser2035.42 and Ser2075.46 residues49, 52. To determine whether these minor changes will increase the receptor’s selectivity toward agonists, we carried out structure-based virtual screening studies using the nanobody-stabilized structure (PDB ID: 3P0G), and compared its performances with the inactive state structure as well as our collection of theoretical models. As expected, the structure of the active state showed better performances than its counterparts of the inactive state in enriching for thirteen agonists but less effective in enriching for fifty-seven antagonists (Figures 2a-b, 3a-b). With Glide4.01 docking protocol, the active state structure could recover 100% of seed agonists at the 25% of ranked databases with the maximum EF as high as 30.69. On the other hand, the active state model of CM2 showed comparable performances to the 3P0G structure in terms of EF and AUC metrics (cf. Table III and Table IV). As mentioned above, the de novo model of CM2 had captured the critical agonist-protein interaction at Ser2075.46 (cf. Figure 5G). This piece of evidence provides another illustration that the properly optimized theoretical models can provide a sensible and accurate description of the binding pocket of the active state structure, and therefore they can be employed reliably for structure-based virtual screening of β2AR agonists.
To summarize our observations, we established that theoretical models of GPCRs built by knowledge-based approaches can achieve similar if not better VS performance as experimental structures from X-ray crystallographic studies. This somewhat surprising observation is reassuring with respect to using carefully developed theoretical models of protein structures for SBDD.
Conclusions
In this study we have addressed the long-standing debate about the structural accuracy and applicability of theoretical models vs. X-ray structures of GPCRs for SBDD. We have carried out a systematic study on a large collection of historical human β2AR theoretical models and evaluated their structural accuracies and screening performances in comparison with recent agonist-bound and antagonist-bound crystal structures. We have shown that there exists a discrepancy between global structural accuracies of β2AR theoretical models and their screening performances. In general, β2AR theoretical models differ largely from the crystal structure in terms of TMHs definition and global packing while many can achieve the same performance in virtual screening and as demonstrated elsewhere16, pose predictions. Our analysis indicates that the binding pockets of models showing the best performance are well-organized and they also align well to active sites in the crystal structures. The key interactions of residues in the active site with the bound antagonists were found to be preserved in models that were built and refined taking into account the site-directed mutagenesis and other experimental data (i.e. the CM1 and CM2 models). Our results emphasize that knowledge-based approaches result in structural models that can achieve the same or even better performance in virtual screening as those built with X-ray crystallographic data. Also, we must stress that our studies address very specific (i.e. β2AR or GPCRs) and pragmatic question (i.e. virtual screening) concerning the use of protein models vs. experimental structures for virtual screening. Our observations by no means undermine the critical importance of experimental structures for understanding protein structure-function relationships as well as the role that crystal structures serve as a critical reference for evaluating the accuracy of predicted protein/ligand interactions.
Figure 7.
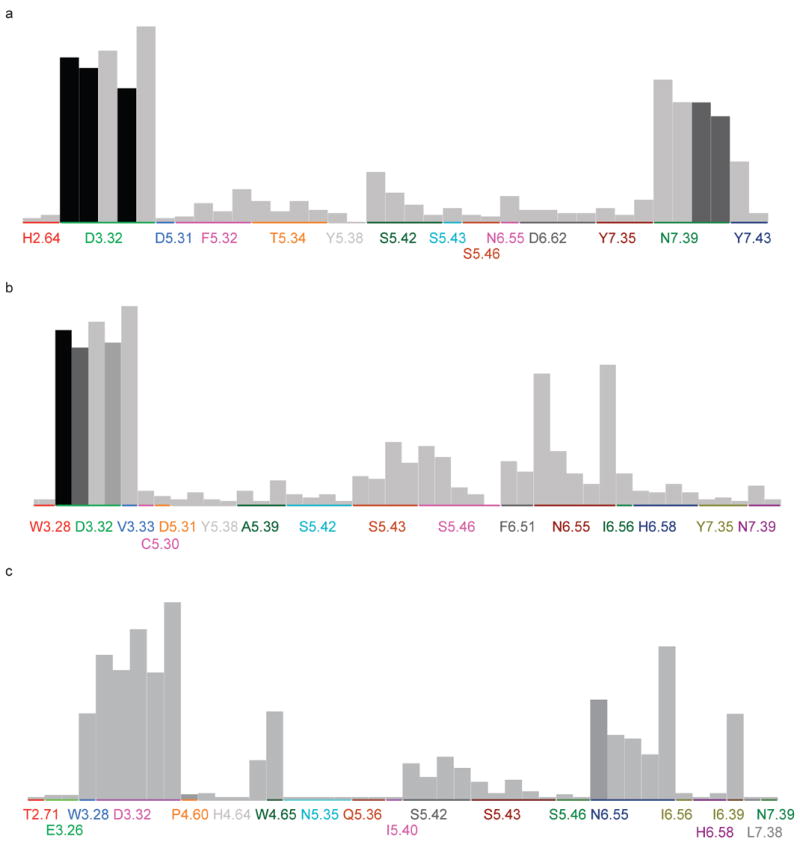
The significance chart for PLIF fingerprints generated from docking poses against (a) β2AR crystal structure (2RH1), (b) CM2 model and (c) CM3 model. The higher the bar (individual fingerprint bit) is, more frequently this type of interaction occurs in the dataset and of higher probability it contributes to the activities. The shade of the color indicates the significance of the particular bit to the actives, which is based on the hypothesis that ‘if the bit is set, then the compound is active’. The residues are randomly colored and several bars of the same residue indicate that they have different types of contacts.
Acknowledgments
We would like to thank Drs. Gert Vriend, Andrej Sali, Jeffrey Skolnick, Terry P. Lybrand, and Nagarajan Vaidehi for sharing the coordinates of their respective theoretical models of beta-2 adrenergic receptor. We are also grateful to Dr. Brian K. Kobilka for helpful discussions. Finally, we acknowledge the access to the computing facilities at the ITS Research Computing Division of the University of North Carolina at Chapel Hill.
Grant sponsor: National Institutes of Health, USA; Grant numbers: GM066940, P20-HG003898, G12 RR003048;
Grant sponsor: UNC-CH University Research Council; Grant number: A3-12988
References
- 1.World Drug Index (WDI) 2004 [Google Scholar]
- 2.OpenEye scientific software. 2006 [Google Scholar]
- 3.MOE. Chemical Computing Group; 2007. [Google Scholar]
- 4.sybyl8.0. 2007 [Google Scholar]
- 5.Austin CP, Brady LS, Insel TR, Collins FS. NIH Molecular Libraries Initiative. Science. 2004;306:1138–1139. doi: 10.1126/science.1105511. [DOI] [PubMed] [Google Scholar]
- 6.Avlani VA, Gregory KJ, Morton CJ, Parker MW, Sexton PM, Christopoulos A. Critical role for the second extracellular loop in the binding of both orthosteric and allosteric G protein-coupled receptor ligands. J Biol Chem. 2007;282:25677–25686. doi: 10.1074/jbc.M702311200. [DOI] [PubMed] [Google Scholar]
- 7.Becker OM, Marantz Y, Shacham S, Inbal B, Heifetz A, Kalid O, Bar-Haim S, Warshaviak D, Fichman M, Noiman S. G protein-coupled receptors: in silico drug discovery in 3D. Proc Natl Acad Sci U S A. 2004;101:11304–11309. doi: 10.1073/pnas.0401862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Becker OM, Shacham S, Marantz Y, Noiman S. Modeling the 3D structure of GPCRs: advances and application to drug discovery. Curr Opin Drug Discov Devel. 2003;6:353–361. [PubMed] [Google Scholar]
- 9.Berman HM, J W, Z F, G G, TN B, H W, IN S, PE B. The protein data bank. Nucleic acids research. 2000;28:235. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bissantz C, Bernard P, Hibert M, Rognan D. Protein-based virtual screening of chemical databases. II. Are homology models of G-Protein Coupled Receptors suitable targets? Proteins. 2003;50:5–25. doi: 10.1002/prot.10237. [DOI] [PubMed] [Google Scholar]
- 11.Bordogna A, Pandini A, Bonati L. Predicting the accuracy of protein-ligand docking on homology models. J Comput Chem. 2011;32:81–98. doi: 10.1002/jcc.21601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen JZ, Wang J, Xie XQ. GPCR structure-based virtual screening approach for CB2 antagonist search. J Chem Inf Model. 2007;47:1626–1637. doi: 10.1021/ci7000814. [DOI] [PubMed] [Google Scholar]
- 13.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clark AM, Labute P. 2D depiction of protein-ligand complexes. J Chem Inf Model. 2007;47:1933–1944. doi: 10.1021/ci7001473. [DOI] [PubMed] [Google Scholar]
- 15.Clark RD, Webster-Clark DJ. Managing bias in ROC curves. J Comput Aided Mol Des. 2008;22:141–146. doi: 10.1007/s10822-008-9181-z. [DOI] [PubMed] [Google Scholar]
- 16.Costanzi S. On the applicability of GPCR homology models to computer-aided drug discovery: a comparison between in silico and crystal structures of the beta2-adrenergic receptor. J Med Chem. 2008;51:2907–2914. doi: 10.1021/jm800044k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ellison KE, Gandhi G. Optimising the use of beta-adrenoceptor antagonists in coronary artery disease. Drugs. 2005;65:787–797. doi: 10.2165/00003495-200565060-00006. [DOI] [PubMed] [Google Scholar]
- 18.Evers A, Klabunde T. Structure-based drug discovery using GPCR homology modeling: successful virtual screening for antagonists of the alpha1A adrenergic receptor. J Med Chem. 2005;48:1088–1097. doi: 10.1021/jm0491804. [DOI] [PubMed] [Google Scholar]
- 19.Evers A, Klebe G. Successful virtual screening for a submicromolar antagonist of the neurokinin-1 receptor based on a ligand-supported homology model. J Med Chem. 2004;47:5381–5392. doi: 10.1021/jm0311487. [DOI] [PubMed] [Google Scholar]
- 20.Fan H, Irwin JJ, Webb BM, Klebe G, Shoichet BK, Sali A. Molecular Docking Screens Using Comparative Models of Proteins. J Chem Inf Model. 2009 doi: 10.1021/ci9003706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fanelli F, De Benedetti PG. Computational modeling approaches to structure-function analysis of G protein-coupled receptors. Chem Rev. 2005;105:3297–3351. doi: 10.1021/cr000095n. [DOI] [PubMed] [Google Scholar]
- 22.Freddolino PL, Kalani MY, Vaidehi N, Floriano WB, Hall SE, Trabanino RJ, Kam VW, Goddard WA., III Predicted 3D structure for the human beta 2 adrenergic receptor and its binding site for agonists and antagonists. Proc Natl Acad Sci U S A. 2004;101:2736–2741. doi: 10.1073/pnas.0308751101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 24.Furse KE, Lybrand TP. Three-dimensional models for beta-adrenergic receptor complexes with agonists and antagonists. J Med Chem. 2003;46:4450–4462. doi: 10.1021/jm0301437. [DOI] [PubMed] [Google Scholar]
- 25.Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem. 1999;1:55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- 26.Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, Velasquez J, Kuhn P, Stevens RC. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge-based desolvation. J Comput Chem. 2007;28:1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 28.Ivanov AA, Barak D, Jacobson KA. Evaluation of homology modeling of G-protein-coupled receptors in light of the A(2A) adenosine receptor crystallographic structure. J Med Chem. 2009;52:3284–3292. doi: 10.1021/jm801533x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kairys V, Fernandes MX, Gilson MK. Screening drug-like compounds by docking to homology models: a systematic study. J Chem Inf Model. 2006;46:365–379. doi: 10.1021/ci050238c. [DOI] [PubMed] [Google Scholar]
- 31.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katritch V, Reynolds KA, Cherezov V, Hanson MA, Roth CB, Yeager M, Abagyan R. Analysis of full and partial agonists binding to beta2-adrenergic receptor suggests a role of transmembrane helix V in agonist-specific conformational changes. J Mol Recognit. 2009;22:307–318. doi: 10.1002/jmr.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katritch V, Rueda M, Lam PC, Yeager M, Abagyan R. GPCR 3D homology models for ligand screening: lessons learned from blind predictions of adenosine A2a receptor complex. Proteins. 2010;78:197–211. doi: 10.1002/prot.22507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klabunde T, Hessler G. Drug design strategies for targeting G-protein-coupled receptors. Chembiochem. 2002;3:928–944. doi: 10.1002/1439-7633(20021004)3:10<928::AID-CBIC928>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 35.Kontoyianni M, DeWeese C, Penzotti JE, Lybrand TP. Three-dimensional models for agonist and antagonist complexes with beta 2 adrenergic receptor. J Med Chem. 1996;39:4406–4420. doi: 10.1021/jm960241a. [DOI] [PubMed] [Google Scholar]
- 36.Kota P, Reeves PJ, Rajbhandary UL, Khorana HG. Opsin is present as dimers in COS1 cells: identification of amino acids at the dimeric interface. Proc Natl Acad Sci U S A. 2006;103:3054–3059. doi: 10.1073/pnas.0510982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kufareva I, Rueda M, Katritch V, Stevens RC, Abagyan R. Status of GPCR modeling and docking as reflected by community-wide GPCR Dock 2010 assessment. Structure. 2011;19:1108–1126. doi: 10.1016/j.str.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lefkowitz RJ, Sun JP, Shukla AK. A crystal clear view of the beta2-adrenergic receptor. Nat Biotechnol. 2008;26:189–191. doi: 10.1038/nbt0208-189. [DOI] [PubMed] [Google Scholar]
- 39.Liang Y, Fotiadis D, Filipek S, Saperstein DA, Palczewski K, Engel A. Organization of the G protein-coupled receptors rhodopsin and opsin in native membranes. J Biol Chem. 2003;278:21655–21662. doi: 10.1074/jbc.M302536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McGovern SL, Shoichet BK. Information decay in molecular docking screens against holo, apo, and modeled conformations of enzymes. J Med Chem. 2003;46:2895–2907. doi: 10.1021/jm0300330. [DOI] [PubMed] [Google Scholar]
- 41.Michino M, Abola E, Brooks CL, III, Dixon JS, Moult J, Stevens RC. Community-wide assessment of GPCR structure modelling and ligand docking: GPCR Dock 2008. Nat Rev Drug Discov. 2009;8:455–463. doi: 10.1038/nrd2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry. 1998;19:1639–1662. [Google Scholar]
- 43.Okuno Y, Yang J, Taneishi K, Yabuuchi H, Tsujimoto G. GLIDA: GPCR-ligand database for chemical genomic drug discovery. Nucleic Acids Res. 2006;34:D673–D677. doi: 10.1093/nar/gkj028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oloff S, Zhang S, Sukumar N, Breneman C, Tropsha A. Chemometric analysis of ligand receptor complementarity: identifying Complementary Ligands Based on Receptor Information (CoLiBRI) J Chem Inf Model. 2006;46:844–851. doi: 10.1021/ci050065r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le T, I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 46.Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 47.Pieper U, Eswar N, Davis FP, Braberg H, Madhusudhan MS, Rossi A, Marti-Renom M, Karchin R, Webb BM, Eramian D, Shen MY, Kelly L, Melo F, Sali A. MODBASE: a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 2006;34:D291–D295. doi: 10.1093/nar/gkj059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.R Development Core Team. R: A language and environment for statistical computing. Vienna: 2008. [Google Scholar]
- 49.Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 51.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 52.Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SG, Choi HJ, Devree BT, Sunahara RK, Chae PS, Gellman SH, Dror RO, Shaw DE, Weis WI, Caffrey M, Gmeiner P, Kobilka BK. Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature. 2011;469:236–240. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roth BL, Lopez E, Patel ES, Kroeze WK. The Multiplicity of Serotonin Receptors: Uselessly diverse molecules or an embarrasment of riches? Neuroscientist. 2006;6:252–262. [Google Scholar]
- 54.Spijker P, Vaidehi N, Freddolino PL, Hilbers PA, Goddard WA., III Dynamic behavior of fully solvated beta2-adrenergic receptor, embedded in the membrane with bound agonist or antagonist. Proc Natl Acad Sci U S A. 2006;103:4882–4887. doi: 10.1073/pnas.0511329103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stern PS, Yu L, Choi MY, Jurenka RA, Becker L, Rafaeli A. Molecular modeling of the binding of pheromone biosynthesis activating neuropeptide to its receptor. J Insect Physiol. 2007;53:803–818. doi: 10.1016/j.jinsphys.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 56.Truchon JF, Bayly CI. Evaluating virtual screening methods: good and bad metrics for the “early recognition” problem. J Chem Inf Model. 2007;47:488–508. doi: 10.1021/ci600426e. [DOI] [PubMed] [Google Scholar]
- 57.Vilar S, Ferino G, Phatak SS, Berk B, Cavasotto CN, Costanzi S. Docking-based virtual screening for ligands of G protein-coupled receptors: not only crystal structures but also in silico models. J Mol Graph Model. 2011;29:614–623. doi: 10.1016/j.jmgm.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Visiers I, Ballesteros JA, Weinstein H. Three-dimensional representations of G protein-coupled receptor structures and mechanisms. Methods Enzymol. 2002;343:329–371. doi: 10.1016/s0076-6879(02)43145-x. [DOI] [PubMed] [Google Scholar]
- 59.Vriend G. WHAT IF: a molecular modeling and drug design program. J Mol Graph. 1990;8:52–6. 29. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 60.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–D672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang S, Golbraikh A, Tropsha A. Development of quantitative structure-binding affinity relationship models based on novel geometrical chemical descriptors of the protein-ligand interfaces. J Med Chem. 2006;49:2713–2724. doi: 10.1021/jm050260x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Y, Devries ME, Skolnick J. Structure modeling of all identified G protein-coupled receptors in the human genome. PLoS Comput Biol. 2006;2:e13. doi: 10.1371/journal.pcbi.0020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zsoldos Z, Reid D, Simon A, Sadjad SB, Johnson AP. eHiTS: a new fast, exhaustive flexible ligand docking system. J Mol Graph Model. 2007;26:198–212. doi: 10.1016/j.jmgm.2006.06.002. [DOI] [PubMed] [Google Scholar]