

Abstract
Helicases are ubiquitous enzymes that are required for virtually all processes in DNA and RNA metabolism including replication, repair, recombination, transcription, and translation. The mechanisms for helicase-catalyzed unwinding of double-stranded DNA or remodeling of RNA have been the subject of intense investigation for more than two decades. The central function of these enzymes is to transduce the energy available from ATP binding and hydrolysis to alter the conformation of nucleic acids. Specific interactions between helicases and nucleic acids have been investigated by chemical approaches in which the nucleic acid substrate has been modified in order to provide specific insight into the enzymatic mechanism.
Keywords: DNA unwinding, Peptide Nucleic Acid, Methylphosphonate, Abasic site, biotinylated DNA, FRET
1. Introduction
Helicases are enzymes that transduce the energy of ATP hydrolysis to unzip double-stranded nucleic acid or remodel secondary structure in single-stranded nucleic acid 1-3. Practically every process in DNA or RNA metabolism requires one or more helicases including DNA replication, repair, recombination, transcription, RNA processing and other activities. The enzymatic activity of helicases can be tightly coupled to the action of polymerases, nucleases, single-stranded binding proteins, and other proteins, but the activity is not dependent on such interactions. Helicases are considered molecular motor proteins, because they transduce chemical energy into mechanical energy. The medical relevance of helicases is primarily due to many forms of cancer which have been associated with mutations in genes encoding helicases 4. Helicases are categorized into six superfamilies based on sequence analysis 5. The members of some superfamilies form oligomeric structures, often hexamers, which function as large molecular machines 6. Other superfamilies exist as monomers or dimers, but retain the fundamental components of a molecular motor 3;7. The motor functions in general terms by coupling different protein conformations to movement along DNA as a function of ATP binding and hydrolysis. Differences have been noted in the mechanisms proposed for each superfamily, but during the past two decades, researchers have uncovered an underlying energy transduction efficiency of one ATP hydrolyzed per one base pair unwound. While there are exceptions to this observation, it appears that many helicases adhere to this standard. A noteworthy exception are those helicases referred to as DEAD box helicases that unwind short sequences of dsRNA in the absence of translocation 8;9.
Knowledge of the structural, chemical, and kinetic mechanism of helicase activity has steadily increased over the past two decades. Our lab has focused much of our effort on a helicase from bacteriophage T4 termed Dda for DNA-dependent-ATPase 10;11. The specific role of Dda in T4 multiplication is not certain, but evidence for its involvement in DNA origin recognition 12, replication 13, and recombination 14 has been reported. Dda is a member of superfamily 1B, along with other prokaryotic members such as RecD and TraI, and eukaryotic members such as human DNA helicase B and Pif1. Dda has served as an excellent model for this subclass of helicases because of its stability and robust activity in biochemical experiments.
This review primarily focuses on work from our lab that incorporates chemical modifications on DNA as probes for studying helicase activity. Many different chemical modifications are available commercially. Some of the early work in which DNA was modified in relation to helicase studies focused on DNA damage and the strand specificity observed in helicase assays. For example, the Hurley and Kodadek labs prepared oligonucleotide substrates using CC-1065, which is a groove binding molecule that reacts at a specific position with DNA 15. This team found that Dda exhibited strand specific inhibition by CC-1065 which provided a strong indication that helicases can track along one strand while sterically excluding the complementary strand.
2. DNA Helicases move along ssDNA with a directional bias. Application of biotin-labeled oligonucleotides
One of the major questions in helicase enzymology was whether these enzymes could move along ssDNA with a directional bias. DNA unwinding studies had established that helicases preferred either a 5′-overhang or a 3′-overhang adjacent to duplex DNA, and such data supported the idea that a directional bias existed 16. However, it was pointed out that some models for DNA unwinding could account for the need for a particular overhang in the absence of a directional bias on ssDNA 17. The basic argument was that a helicase needed only an asymmetric ss/dsDNA junction to move into the duplex, but if given only ssDNA, no net translocation would occur. In order to address this question, we developed a new assay to study helicase activity on single-stranded DNA so that a ss/dsDNA junction was not part of the substrate.
Modification of DNA with biotin became commercially available in the early 1990s. The Benkovic laboratory used biotin-labeled oligonucleotides to provide protein bumpers that allowed assembly of the T4 processivity factor and the T4 DNA polymerase on a short primer-template 18. The structure of the processivity factor is ring-shaped which wraps around the DNA. The streptavidin was placed onto the end of the duplex, which prevented the processivity factor from sliding off the end of the DNA. Inspired by the work with the DNA polymerase holoenzyme, we utilized biotin-labeled oligonucleotide substrates to show that a ring-shaped hexameric helicase also wraps around the DNA, albeit only on one of the strands 19. A DNA fork substrate was prepared in which a biotin-label was placed into the duplex region of one or the other strand of the fork. We found that the helicase was inhibited from unwinding the duplex only when the biotin-streptavidin complex blocked the 5′-to-3′ strand, but not the 3′-to-5′ strand. The dependence of inhibition on the placement of the biotin-streptavidin block strongly supported a mechanism whereby the hexameric helicase translocated on one strand of the duplex which passed through the central channel of the hexamer, whereas the second strand was excluded and passed along the outside of the hexamer. This strand-exclusion model has been advanced through work from multiple laboratories and appears to apply to many of the hexameric helicases 20-22.
We later applied the streptavidin-biotin bumper idea towards the question of directional biased translocation on single-stranded DNA. Dda helicase was used as the model system to test whether a helicase could increase the rate at which streptavidin dissociates from biotin (Figure 1). The rate of dissociation of streptavidin from various biotinylated oligonucleotides was determined in the presence of helicase using an electrophoretic mobility shift assay. Placement of the biotin label on the 5′-end of an oligonucleotide resulted in no displacement of streptavidin. However, placement of biotin on the 3′-end of the oligo followed by incubation with Dda, ATP, and Mg+2 resulted in rapid dissociation of streptavidin (Figure 1). Dda was found to significantly enhance the dissociation rate of streptavidin from biotin-labeled oligonucleotides in an ATP dependent reaction. Helicase-catalyzed dissociation of streptavidin from the 3′-end of a biotin-labeled 62-mer oligonucleotide occurred with a first order rate of 7.9 s-1. This is more than one million-fold faster than the spontaneous dissociation rate. There was no enhancement of streptavidin dissociation from the 5′-biotin-labeled oligonucleotide by either Dda or gp41 helicase. Dda's ability to dislodge streptavidin from the 3′-biotin suggests that helicases are capable of imparting a force during translocation. The difference in displacement between the 5′ and 3′-ends of the oligonucleotide is also consistent with a strong, 5′-to-3′ directional in translocation on ssDNA. Other helicases are also capable of streptavidin displacement, but only from one end or the other, dependent on whether the helicase translocates 5′-to-3′ or 3′-to-5′ 23. The concept that helicases remove proteins from DNA has been developed as a major role for this class of enzymes which applies to RNA helicases as well.
Figure 1. Oligonucleotides with biotin labels on the 3′-end or 5′-end have revealed directional biased activity of helicases on DNA.
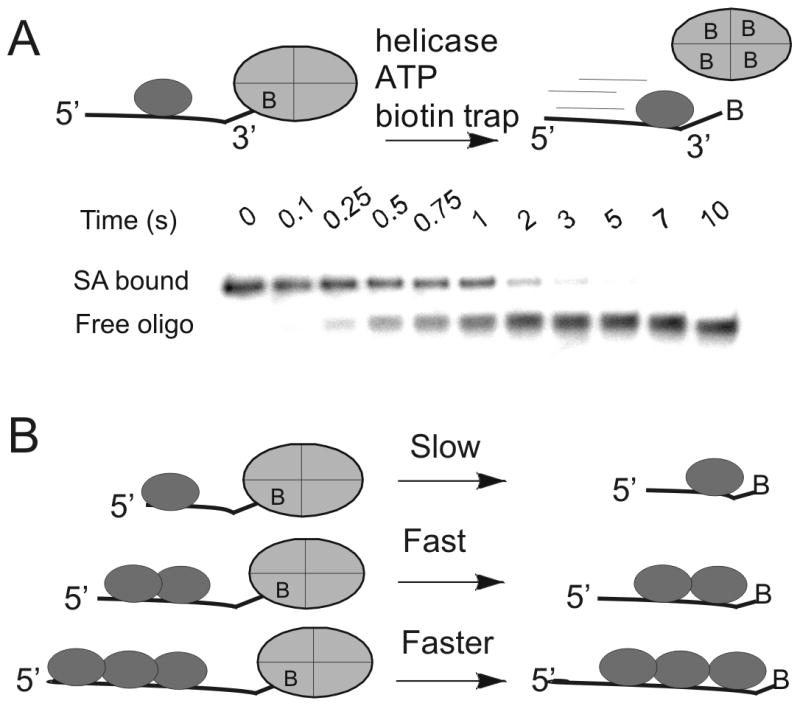
A. The action of a helicase on an oligonucleotide substrate can lead to rapid dissociation of streptavidin from biotin-labeled DNA. A native polyacrylamide gel can separate the streptavidin-bound DNA from free DNA. Helicases have been found capable of displacing streptavidin from either the 3′-end or the 5′-end, depending on the directional bias of the particular helicase. B) The rate of displacement of streptavidin from biotin-labeled DNA increases with increasing length of the ssDNA. In the case of Dda helicase, multiple molecules of the enzyme appear to function together by pushing in the same direction due to directionally biased translocation on ssDNA.
The mechanism of streptavidin displacement was investigated by varying the length of the oligonucleotide substrate. We found that a monomeric form of Dda can catalyze streptavidin displacement; however, activity increases when multiple helicase molecules are bound to the biotinylated oligonucleotide 24. The enhanced activity is the consequence of the directional bias in translocation of individual helicase monomers. When the lead molecule stalls, protein-protein interactions allow trailing molecules to push the lead molecule, resulting in greatly enhanced ability to dislodge streptavidin from biotinylated DNA (Figure 1B). This idea that multiple molecular motors can function together on the same DNA strand has yet to be evaluated in vivo. The general ability of helicases to disrupt protein-DNA interactions appears to be an important role in their biological function.
3. Strand-specific chemical modifications to DNA provide evidence for a tracking of helicases along one of the DNA strands
Most helicases require a single-stranded DNA overhang adjacent to duplex DNA in order to initiate unwinding. The strand containing the overhang is referred to as the tracking strand whereas the complementary strand is referred to as the displaced strand (Figure 2). After helicase-catalyzed DNA unwinding, the displaced strand can be trapped by a complementary strand that is typically added to the reaction mixture along with the ATP. The resulting single-stranded DNA can be separated from the parent duplex by native polyacrylamide gel electrophoresis to provide the relative amounts of product over time. This helicase assay has been utilized routinely by investigators over the past two decades.
Figure 2. Standard helicase assay for measuring DNA uwinding using oligonucleotide substrates.
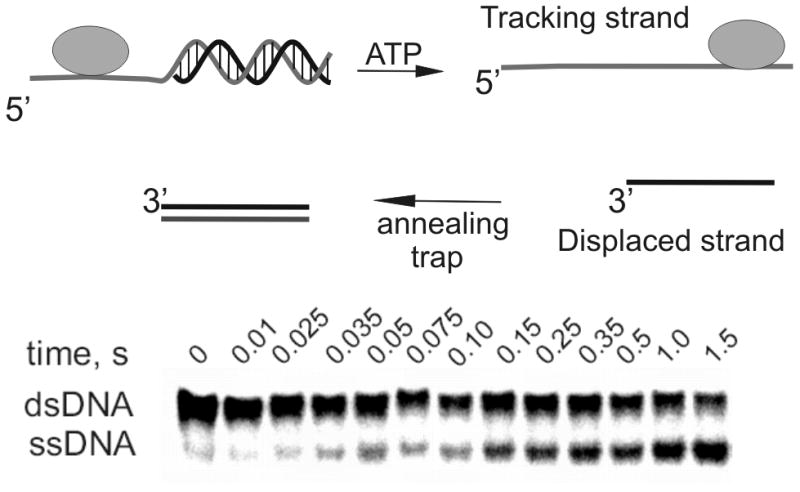
A partial duplex is needed because most helicases require ssDNA for binding to the substrate. In the presence of ATP hydrolysis, the helicase moves along the tracking strand, separating the duplex. The displaced strand can reanneal with the tracking strand, so third strand can be added to the reaction to serve as an annealing trap. The original substrate will migrate more slowly through native polyacrylamide than the ssDNA product.
As mentioned in the introduction, strand-specific modification of an oligonucleotide substrate by CC-1065 was used to explore the strand preference for helicase activity (Figure 3A). When CC-1065 was covalently linked to the tracking strand, helicase inhibition was observed. However, covalent attachment of CC-1065 to the displaced strand led to little or no inhibition 15. These experiments were amongst the earliest indicators that helicases can track along one strand, stripping off the complementary strand due in part to steric interactions. Translocation along one strand is only part of the overall mechanism for DNA unwinding by Dda. Specific interactions between the helicase and the DNA at the single-stranded/double-stranded DNA junction are also needed 25.
Figure 3. DNA adducts and DNA mimics as probes for helicase mechanisms.

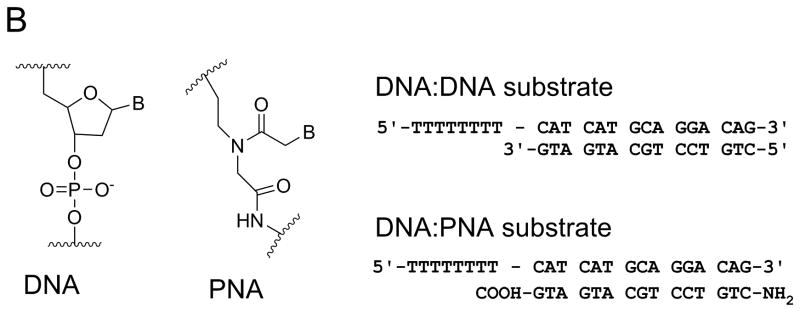
A. CC-1065, a potent antitumor drug, was used to prepare helicase substrates (1) and (2). Sequence selective covalent reaction between the N3 position of adenine and the cyclopropyl group of CC-1065 allowed preparation of adducted DNA on the tracking strand (substrate 1) or the displaced strand (substrate 2). When CC-1065 was attached to the tracking strand, as in substrate1, the ability of Dda helicase to unwind the duplex was strongly inhibited. However, when CC-1065 was attached to the displaced strand, as in substrate 2, little or no inhibition was observed with Dda helicase. B. Peptide nucleic acid (PNA) contains a peptide-like backbone to which the bases are attached. Substrates were prepared in which the entire displaced strand of DNA was replaced with a strand of PNA. The resulting DNA:PNA hybrid was readily unwound by Dda helicase at the same rate as the DNA:DNA substrate.
The nature of enzyme-substrate interactions between helicases and duplex DNA substrates was investigated by replacing the entire displaced strand of the duplex substrate with a strand of PNA; thereby creating a DNA-PNA hybrid (Figure 3B). PNA is a DNA mimic capable of forming duplex structures with DNA according to Watson-Crick base pairing rules 26. PNA contains a N-(2-aminoethyl) glycine backbone in place of the deoxyribose phosphates (Figure 3B). The PNA-DNA hybrids have higher melting temperatures than their DNA-DNA counterparts. Dda helicase was able to unwind the DNA-PNA substrate at similar rates as the DNA-DNA substrates 27. The results indicate that the rate-limiting step for unwinding is relatively insensitive to the chemical nature of the displaced strand and the thermal stability of oligonucleotide substrates. The results are consistent with the conclusion that translocation on ssDNA is sufficient to melt the duplex. Translocation activity on ssDNA appears to be the primary activity of this enzyme, with DNA unwinding being a consequence of the enzyme stripping away the displaced strand.
Another application of PNA in the study of helicases is in steady-state kinetic analyses. Measurement of steady-state rates of unwinding of double-stranded oligonucleotides is hampered due to rapid reannealing of the single-stranded DNA products. Including an oligonucleotide trapping strand in the reaction mixture that can hybridize with the displaced strand can prevent reannealing as illustrated in Figure 2. However, helicases bind to single-stranded DNA, therefore the additional oligonucleotide can sequester the enzyme, leading to slower observed rates during a multi-turnover kinetics experiment. To circumvent this problem, the oligonucleotide that serves as a trap can be replaced with a strand of PNA 28. Fluorescence polarization was used to determine that a 15mer PNA strand does not bind to Dda helicase which allowed steady-state kinetic parameters of unwinding catalyzed by Dda to be determined by using PNA as an annealing trap. In the presence of 250 nM substrate and 1 nM Dda, the rate of unwinding in the presence of the DNA trapping strand was 0.30 nM s-1 whereas the rate was 1.34 nM s-1 in the presence of the PNA trapping strand. PNA prevents reannealing of single-stranded DNA products, but does not sequester the helicase.
Helicases function in a variety of processes in which DNA secondary structure must be removed. A large body of work now proves that DNA can fold into quadruplex structures in vivo, and that helicases are needed to unfold these structures during DNA replication, transcription and other processes. Quadruplex DNA is formed in specific sequences that contain runs of guanine. Four guanine nucleotides interact through Hoogsteen base pairing to form a tetrad, and multiple tetrads stack together to form a stable quadruplex 29. In order to study helicase-catalyzed unfolding of these structures, the PNA trapping strategy has been applied to capture the quadruplex after unfolding. A PNA trapping strand was applied for determining the rate of unfolding quadruplex DNA substrate with several different helicases 30. In this case, the PNA served to trap the single-stranded oligonucleotide after action of the helicase but prior to refolding of the quadruplex. As with the DNA unwinding experiment, the presence of the PNA did not impede the action of the helicase as might be observed if a DNA strand was used as the trapping agent.
4. PNA and other chemical modifications placed into the tracking strand reveal important protein-nucleic acid interactions
The relative influence of specific chemical interactions between helicase and substrate over a series of multi-step catalytic events has been explored by placing chemical perturbations into the tracking strand. Such chemical modifications allow specific types of interactions to be examined. Initially, a 5′ DNA-PNA-DNA 3′ chimera was synthesized, thereby, conferring both a loss of charge and altering the conformational flexibility of the oligonucleotide (Figure 4A). DNA unwinding of this substrate by Dda was dependent on the number of Dda molecules bound to the substrate 31. Under conditions in which the DNA substrate concentration exceeded the helicase concentration, only one molecule of helicase could bind per substrate molecule. Under these conditions (excess substrate concentration) the DNA-PNA-DNA chimera was not unwound. The PNA residue prevented one helicase molecule from translocating through the duplex. However, if the concentration of helicase was in large excess of the DNA substrate, multiple molecules of helicase could bind to the ssDNA tracking strand and the chimera was readily unwound (Figure 4B). The results were reminiscent of the functional cooperativity mechanism that we observed for streptavidin displacement, ie, multiple helicase molecules work together when bound to the same substrate molecule 24. A single methylphosphonate residue placed into the tracking strand also lead to little or no product when one molecule of Dda was bound to the substrate (Figure 4A). This illustrates the importance of electrostatic interactions between the helicase and the DNA backbone. However, multiple Dda molecules bound to the substrate could bypass the methylphosphonate.
Figure 4. DNA modifications placed into the tracking strand of a helicase substrate.

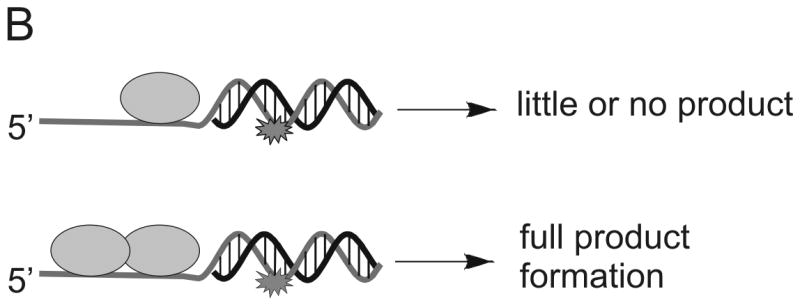
A) A single DNA modification containing a PNA residue, an abasic site mimic, or a methylphosphonate was placed into the tracking strand. Each modification served to disrupt possible interactions between the enzyme and the DNA. B) DNA unwinding by Dda helicase was inhibited by each modification, but multiple molecules of Dda bound to the same substrate could move past the DNA adduct (see Figure 1B).
One possible reason why multiple Dda molecules can move passed a DNA lesion is that the probability of bypass is enhanced, simply because there are multiple opportunities. If this were the case, then we would expect the rate of unwinding to be slower, reflecting the fact that the first ‘attempt’ at unwinding failed, whereas subsequent attempts by the additional Dda molecules succeeded. Importantly, when multiple Dda molecules are present, the rate is not changed when comparing the damaged DNA with the undamaged DNA. This implies that the bypass of the lesion is not due to an increase in the number of chances for bypass, but instead is due to a change in the activity of the lead molecule of helicase. The second molecule of Dda bound to the substrate may enable bypass of the lesion by pushing the lead molecule through or by binding to the lead molecule and holding it onto the DNA (Figure 4B).
A stable, abasic site structural mimic was introduced into the tracking strand which served to disrupt the hydrogen bonding lattice, the intra-molecular base stacking interactions, as well as possible inter-molecular base stacking interactions between aromatic amino acid side chains and the substrate (Figure 4A). DNA unwinding was greatly reduced for the substrate containing the abasic mimic which was initially surprising because electrostatic interactions between the helicase and the DNA appeared to be of primary importance 31. More recently, the x-ray crystal structure of Dda bound to ssDNA was solved (Figure 5A) 25. The expected helicase domains were present, similar to many other helicases in this superfamily 32-35. However, domain 1B (shown in red in Figure 5A) contained a ‘triple stacking’ interaction made up of a proline, phenylalanine, and an incoming nucleobase (Figure 5B). The DNA unwinding ability of Dda was completely lost upon mutation of the Phenylalanine residue to alanine, illustrating the importance of this stacking interaction 25. The structure provided an explanation for why the abasic site mimic in a DNA substrate would cause such strong inhibition of DNA unwinding. Dda helicase utilizes this stacking interaction to ‘sweep’ DNA through the active site due to conformational changes that occur with ATP binding and hydrolysis.
Figure 5. X-ray crystallographic structure of Dda helicase bound to ssDNA.
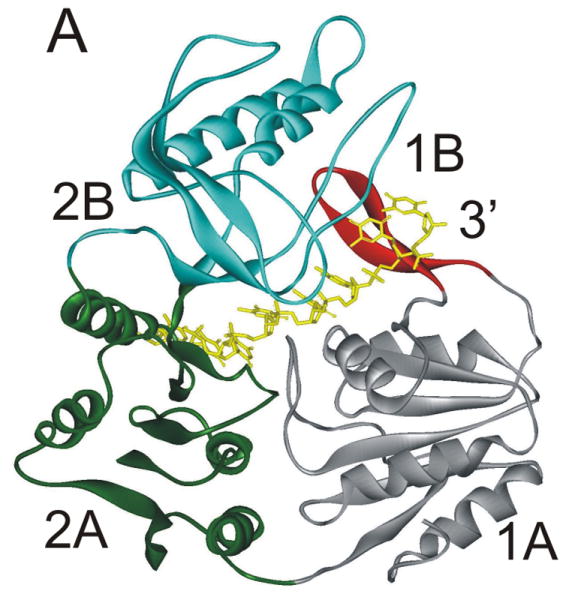
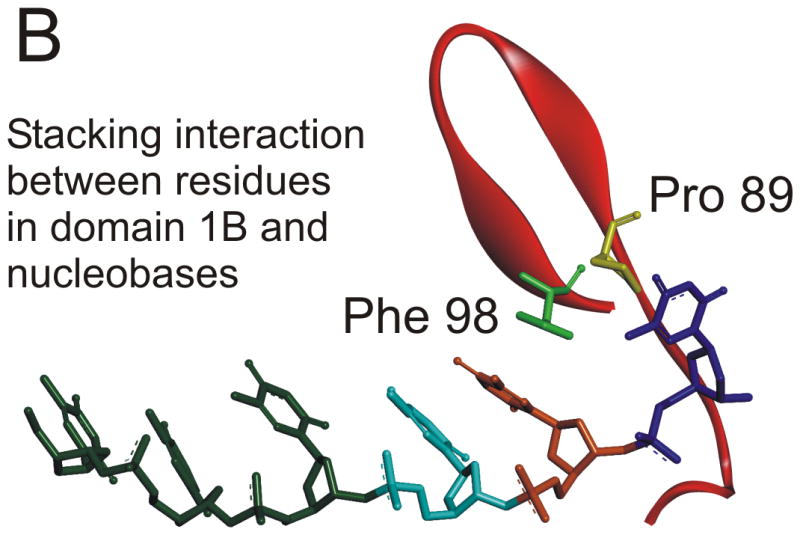
A) Dda helicase contains four minimal domains. Domains 1A and 2A are RecA-like domains associated with all helicases in superfamily 1. Domain 2B forms part of the cleft through which ssDNA is bound. Domain 1B is referred to as the ‘pin’ domain, and is responsible for physically separated the two strands of duplex DNA. The ssDNA shown is the tracking strand. B) Domain 1B shown expanded with specific amino acids forming a stacking interaction with one of the nucleobases. Mutation of Phe98 to Ala results in a loss of DNA unwinding activity.
Other chemical modifications have been introduced into the tracking strand for study in other systems. The Pyle lab used biosynthetic methods to create a library of RNA substrates in which a phosphorothioate replaced the normal phosphodiester backbone. Cleavage of the backbone at the modified sites produced RNA substrates of varying length that were used to determine the kinetic and physical step size of the NS3 helicase from the Hepatitis C virus. Results supported a relatively large step 18 bp. The surprisingly large step size was further explored by placement of polygycol spacers into the tracking strand. NS3 was able to function on such substrates which supported the conclusion that this enzyme exhibits an unusual large kinetic (and perhaps physical) step size 5;36.
Helicase-catalyzed DNA unwinding through stepwise movement of the enzyme eventually results in a duplex that contains too few base pairs to remain hybridized and therefore spontaneously melts to form ssDNA. The length of the duplex that melts is critical to determining the rate for DNA unwinding. However, the exact number of base pairs that can melt can vary from one helicase to the next due to subtle differences in the way that the DNA is bound and unwound. The sequence of DNA can also impact spontaneous melting. We devised a method to measure the number of base pairs that spontaneously melt by taking advantage of the methylphosphonate residue (Figure 4A). Based on the fact that a single methylphosphonate can disrupt Dda helicase unwinding activity, we designed a series of substrates to measure spontaneous melting 37. A methylphosphonate was placed into the tracking strand at varying positions which would allow Dda to traverse varying lengths through the duplex prior to encountering the DNA modification (Figure 6). Upon reaching the methylphosphonate, the enzyme will stop, but if the remaining duplex DNA is unstable, then it will spontaneously melt. By examining the quantity of helicase-catalyzed product with the different substrates containing methylphosphonate, we determined that 8 base pairs can spontaneously melt at 25 °C, which allowed us to determine the rate of DNA unwinding to be 250 bp/s.
Figure 6. DNA unwinding substrates containing a methylphosphonate (MeP) at varying positions.

Placement of MeP at different positions within the tracking strand of a helicase substrate was used to determine the number of base pairs that spontaneously melted during helicase-catalyzed unwinding. The loss of the negative charge on the DNA backbone due to the MeP was found to stop DNA unwinding activity of Dda helicase. Therefore, if melting was observed for a particular substrate in the presence of Dda helicase, the number of base pairs remaining in that duplex could serve as an estimate for spontaneous melting 37.
5. Fluorescently labeled DNA substrates for kinetic studies
A large number of fluorescence-based helicase assays have been developed through the years. The earliest reported made use of Förster Resonance Energy Transfer (FRET) by labeling the two strands of the duplex with appropriate donor and acceptor probes 38. The Kodadek lab used a fluorescein-labeled oligonucleotide hybridized to a coumarin-labeled oligo to illustrate a FRET based helicase assay (Figure 7A). Upon helicase-catalyzed separation of the strands, fluorescence quenching is removed leading to an easily measureable signal. This assay has advantages over the discontinuous gel-based assay because the entire kinetic curve can be obtained in one continuous experiment. FRET has also been applied to study the binding orientation of the hexameric helicase DnaB to a DNA fork where quantitative distance measurements provided evidence that ssDNA passed through the central channel of the hexamer 39. A different fluorescent approach using the fluorescent nucleotide, 2-aminopurine, was developed in the Benkovic lab 40. 2-aminopurine exhibits a strong fluorescence change upon hybridizing with a complementary oligonucleotide. Hence, it produces a signal during helicase-catalyzed DNA unwinding. Compared to large, bulky fluorescent probes, this assay has the advantage of causing essentially no disruption in the structure of the duplex because 2-aminopurine hydrogen bonds to thymidine to maintain the overall B-form structure of DNA.
Figure 7. Fluorescently-labeled oligonucleotides for kinetic analysis of DNA unwinding and translocation on ssDNA.
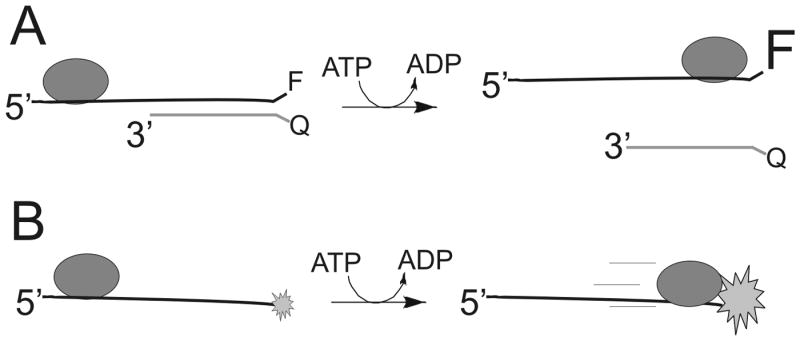
A. In the example shown, a fluorescent probe is placed onto the end of a DNA substrate and the complementary strand contains a fluorescence quencher. Upon helicase-catalyzed separation of the strands, an increase in fluorescence can be observed. B. Fluorescently-labeled oligonucleotides serve as a substrate for measuring translocation on ssDNA. When a helicase translocates to the end of the DNA, a change in fluorescence can be observed due to the interaction between the helicase and the fluorescent probe. The time required to reach the end of different length oligonucleotides can be used to determine the rate of translocation.
Measurement of the rate of helicase translocation on ssDNA using a fluorescence assay was reported by Dillingham, Webb, and Wigley 41. 2-aminopurine was attached to the end of an oligonucleotide and stopped-flow spectroscopy was used to measure the change in fluorescence that occurs upon binding of a helicase to the fluorescent nucleotide (Figure 7B). By measuring the fluorescence change from DNA substrates of varying length, the rate of movement was determined. The quantum yield of 2-aminopurine is relatively low, so other fluorescent probes have been applied to measure translocation of helicases on single stranded DNA 42.
The kinetic analysis of fluorescence data obtained for translocation and/or DNA unwinding has been carefully developed by the Lohman laboratory 43-45. Kinetic models to describe DNA unwinding and translocation based on the idea of a step-wise mechanism for movement of helicases have been proposed (Figure 8). The number of steps depends on the length of DNA so that longer substrates produce a longer lag phase for product formation. Kinetic analysis of the lag phase, the rate of product formation, and the amplitude of product formation can be analyzed globally to determine the kinetic constant for unwinding, ku, and dissociation, kd, for each step in the reaction. Over the past decade, the various fluorescence-based helicase assays have been heavily utilized in single-molecule experiments 46-49. Single-molecule approaches have revealed intermediate steps in the mechanisms of helicase activity that could not be readily examined using ensemble approaches.
Figure 8. Kinetic scheme for studying step-wise movement of helicases along DNA.
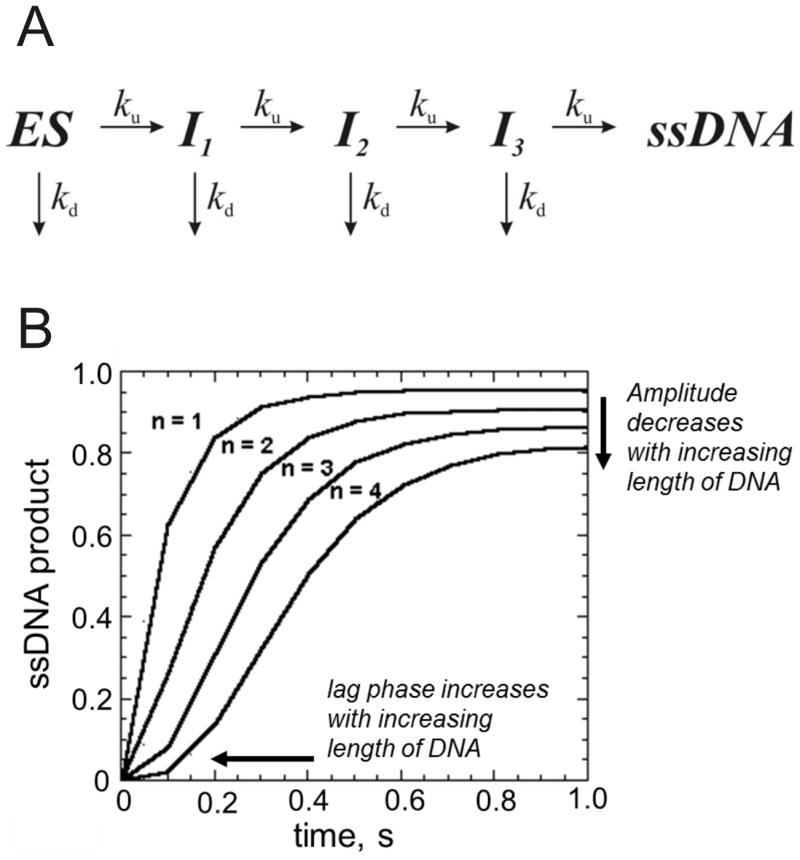
A. Helicase bound to DNA (ES) moves along the DNA substrate in a step-wise fashion, producing an intermediate state (I) after each step. The helicase can dissociate from the DNA at each step or continue to move along the DNA, eventually reaching the end. The number of steps needed to reach the end is dependent on the length of the DNA and the size of the step. B. A hypothetical progress curve for four different DNA substrates of varying length. Substrate 1 requires only one step in order to be unwound by a helicase. Substrate 2 requires two steps, substrate 3 requires three steps, and substrate 4 requires four steps. A lag phase increases as the number of steps increases. The progress curves can be analyzed according to the model in panel A to provide the number of steps, the step size, and the rate constants for unwinding and dissociation associated with each step.
6. A triply-labeled DNA substrate for identifying sites of protein-DNA interaction
In the absence of structural data, methods for identifying sites of protein-DNA interactions have been developed utilizing modified oligonucleotides. In one approach, three different chemical labels were added to the oligonucleotide (Figure 9) 50. A bromo-deoxyuridine label can be placed at any position in the DNA to serve as a photoactivatable crosslinker. This agent readily forms chemical bonds at amino acids near its binding site in the presence of UV light at the appropriate wavelength. The approach for using the tri-labeled oligonucleotide is shown in Figure 9B. In step 1, a protein-DNA complex is exposed to UV light (308 nm) to activate the bromo-deoxyuridine crosslinker. Crosslinked protein can be separated from uncrosslinked protein by SDS-PAGE. The diagram shows protein markers in the first lane and the crosslinked protein (red) or uncrosslinked protein (grey) in the second lane. Protein that is crosslinked to DNA will migrate more slowly than uncrosslinked protein due to the added mass of the DNA. The fluorescent probe can aid in identifying the crosslinked protein band by fluorescence imaging. In step 2, protease digestion of the protein labeled with DNA produces peptides, some of which are crosslinked to the DNA. In step 3, the DNA-peptide, crosslinked species can be isolated through the biotin label by capture with streptavidin beads. Enzymatic digestion (step 4) of the DNA by DNase I will produce a peptide (or peptides) that can be identified by mass spectrometric sequencing (step 5). Further purification of these peptides by HPLC can be performed if needed prior to analysis mass spectrometry. The combined approach can enable identification of the site of interaction of DNA on a protein. In the case of helicases, multiple DNA binding sites on the surface of the enzyme might be identified. The ability to place the chemical probes at any location along an oligonucleotide sequence allows specific regions of a DNA substrate to be explored for interaction with the helicase.
Figure 9. Identification of protein-nucleic acid interaction sites by crosslinking with tri-labeled oligonucleotide.
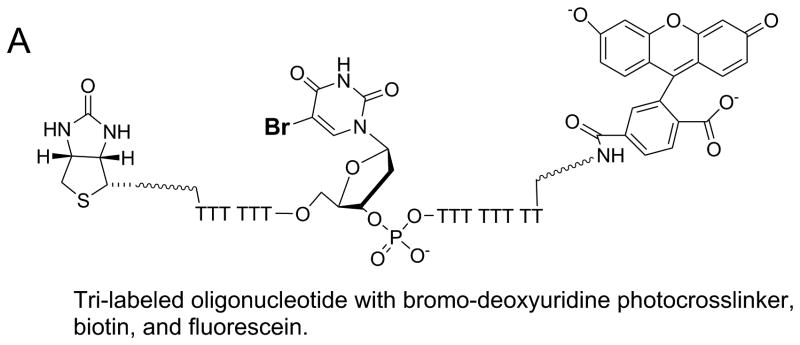
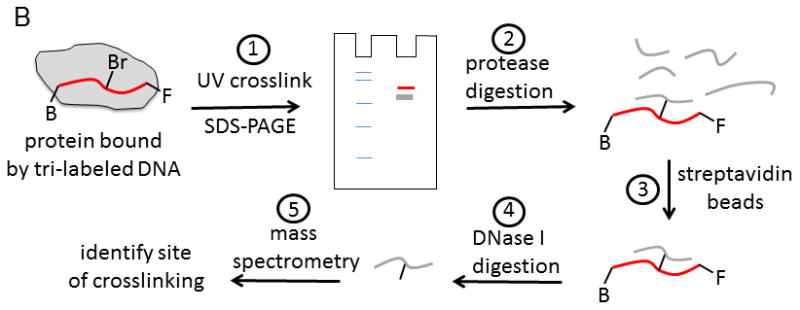
A. Oligonucleotide with three modifications for identification of protein-DNA interaction sites. A photoactivatable probe, bromo-deoxyuridine, can form covalent bonds with nearby amino acids upon exposure to the appropriate UV wavelength. The biotin label can be used to assist in isolation of the resulting DNA-protein crosslinks. The fluorescein label can aid in identification and isolation of peptide-DNA crosslinks for eventual characterization by mass spectrometry. B. Outline for steps involved in applying the tri-labeled oligonucleotide for the identification of DNA binding sites on proteins. Photocrosslinking of the DNA to the protein is followed by a series of steps to isolate the specific peptide that was involved in the crosslinking reaction. Mass spectrometry is used to identify the peptide. See text for more details of the protocol.
6. Conclusion
This review has focused on applications applied primarily to the study of Dda helicase, but many other labs have applied similar experimental methods. Dozens of modifications of DNA, many of which are commercially available, can provide an array of experimental approaches to probe the mechanism(s) of helicase activity. Chemical modifications in DNA substrates have revealed the strand specific nature of interaction between helicases and duplex DNA. By placing probes into the tracking strand, Dda helicase was found to move along ssDNA with a directional bias exerting force on proteins in its path, and skipping over lesions when multiple helicase molecules align on the DNA. Modifications placed in the displaced strand revealed that specific chemical interactions between Dda and the displaced strand are not required for DNA unwinding, although a DNA fork is a modestly better substrate than one that contains only a ssDNA overhang. Application of chemical modifications to DNA for studying helicase mechanisms will continue to expand with the increasing opportunities brought by new DNA chemistries. Future work may focus on how helicases engage DNA in the context of chromatin or through multi-protein complexes. Additionally, the role of epigenetic modifications on DNA unwinding by helicases has yet to be examined.
Acknowledgments
This work was supported by a grant from the NIH, NIGMS R01GM098922 to K.D.R. I thank Alicia Byrd for helpful comments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Byrd AK, Raney KD. Front Biosci(Landmark Ed) 2012;17:2070. doi: 10.2741/4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilhooly NS, Gwynn EJ, Dillingham MS. Front Biosci(Schol Ed) 2013;5:206. doi: 10.2741/s367. [DOI] [PubMed] [Google Scholar]
- 3.Lohman TM, Tomko EJ, Wu CG. Nat Rev Mol Cell Biol. 2008;9:391. doi: 10.1038/nrm2394. [DOI] [PubMed] [Google Scholar]
- 4.Brosh RM., Jr Nat Rev Cancer. 2013;13:542. doi: 10.1038/nrc3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gorbalenya AE, Koonin EV. Current Opinion in Structural Biology. 1993;3:419. [Google Scholar]
- 6.Donmez I, Patel SS. Nucleic Acids Res. 2006;34:4216. doi: 10.1093/nar/gkl508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fairman-Williams ME, Guenther UP, Jankowsky E. Curr Opin Struct Biol. 2010;20:313. doi: 10.1016/j.sbi.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jankowsky E, Putnam A. Methods Mol Biol. 2010;587:245. doi: 10.1007/978-1-60327-355-8_18. [DOI] [PubMed] [Google Scholar]
- 9.Linder P, Jankowsky E. Nat Rev Mol Cell Biol. 2011;12:505. doi: 10.1038/nrm3154. [DOI] [PubMed] [Google Scholar]
- 10.Eoff RL, Raney KD. Biochem Soc Trans. 2005;33:1474. doi: 10.1042/BST0331474. [DOI] [PubMed] [Google Scholar]
- 11.Mackintosh SG, Raney KD. Nucleic Acids Res. 2006;34:4154. doi: 10.1093/nar/gkl501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brister JR. J Mol Biol. 2008;377:1304. doi: 10.1016/j.jmb.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Formosa T, Burke RL, Alberts BM. Proc Natl Acad Sci U S A. 1983;80:2442. doi: 10.1073/pnas.80.9.2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kodadek T, Alberts BM. Nature. 1987;326:312. doi: 10.1038/326312a0. [DOI] [PubMed] [Google Scholar]
- 15.Maine IP, Sun D, Hurley LH, Kodadek T. Biochemistry. 1992;31:3968. doi: 10.1021/bi00131a012. [DOI] [PubMed] [Google Scholar]
- 16.Matson SW, George JW. J Biol Chem. 1987;262:2066. [PubMed] [Google Scholar]
- 17.Lohman TM. J Biol Chem. 1993;268:2269. [PubMed] [Google Scholar]
- 18.Kaboord BF, Benkovic SJ. Proc Natl Acad Sci U S A. 1993;90:10881. doi: 10.1073/pnas.90.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raney KD, Carver TE, Benkovic SJ. J Biol Chem. 1996;271:14074. doi: 10.1074/jbc.271.24.14074. [DOI] [PubMed] [Google Scholar]
- 20.Egelman EH, Yu X, Wild R, Hingorani MM, Patel SS. Proc Natl Acad Sci U S A. 1995;92:3869. doi: 10.1073/pnas.92.9.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Enemark EJ, Joshua-Tor L. Nature. 2006;442:270. doi: 10.1038/nature04943. [DOI] [PubMed] [Google Scholar]
- 22.Graham BW, Schauer GD, Leuba SH, Trakselis MA. Nucleic Acids Res. 2011;39:6585. doi: 10.1093/nar/gkr345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morris PD, Byrd AK, Tackett AJ, Cameron CE, Tanega P, Ott R, Fanning E, Raney KD. Biochemistry. 2002;41:2372. doi: 10.1021/bi012058b. [DOI] [PubMed] [Google Scholar]
- 24.Byrd AK, Raney KD. Nat Struct Mol Biol. 2004;11:531. doi: 10.1038/nsmb774. [DOI] [PubMed] [Google Scholar]
- 25.He X, Byrd AK, Yun MK, Pemble CW, Harrison D, Yeruva L, Dahl C, Kreuzer KN, Raney KD, White SW. Structure. 2012;20:1189. doi: 10.1016/j.str.2012.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nielsen PE, Egholm M, Buchardt O. Bioconjug Chem. 1994;5:3. doi: 10.1021/bc00025a001. [DOI] [PubMed] [Google Scholar]
- 27.Tackett AJ, Morris PD, Dennis R, Goodwin TE, Raney KD. Biochemistry. 2001;40:543. doi: 10.1021/bi002122+. [DOI] [PubMed] [Google Scholar]
- 28.Nanduri B, Eoff RL, Tackett AJ, Raney KD. Nucleic Acids Res. 2001;29:2829. doi: 10.1093/nar/29.13.2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murat P, Balasubramanian S. Curr Opin Genet Dev. 2013;25C:22. doi: 10.1016/j.gde.2013.10.012. [DOI] [PubMed] [Google Scholar]
- 30.Bharti SK, Sommers JA, George F, Kuper J, Hamon F, Shin-ya K, Teulade-Fichou MP, Kisker C, Brosh RM., Jr J Biol Chem. 2013;288:28217. doi: 10.1074/jbc.M113.496463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eoff RL, Spurling TL, Raney KD. Biochemistry. 2005;44:666. doi: 10.1021/bi0484926. [DOI] [PubMed] [Google Scholar]
- 32.Saikrishnan K, Griffiths SP, Cook N, Court R, Wigley DB. EMBO J. 2008;27:2222. doi: 10.1038/emboj.2008.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saikrishnan K, Powell B, Cook NJ, Webb MR, Wigley DB. Cell. 2009;137:849. doi: 10.1016/j.cell.2009.03.036. [DOI] [PubMed] [Google Scholar]
- 34.Singleton MR, Dillingham MS, Wigley DB. Annu Rev Biochem. 2007;76:23. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 35.Velankar SS, Soultanas P, Dillingham MS, Subramanya HS, Wigley DB. Cell. 1999;97:75. doi: 10.1016/s0092-8674(00)80716-3. [DOI] [PubMed] [Google Scholar]
- 36.Beran RK, Bruno MM, Bowers HA, Jankowsky E, Pyle AM. J Mol Biol. 2006;358:974. doi: 10.1016/j.jmb.2006.02.078. [DOI] [PubMed] [Google Scholar]
- 37.Eoff RL, Raney KD. Nat Struct Mol Biol. 2006;13:242. doi: 10.1038/nsmb1055. [DOI] [PubMed] [Google Scholar]
- 38.Houston P, Kodadek T. Proc Natl Acad Sci U S A. 1994;91:5471. doi: 10.1073/pnas.91.12.5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jezewska MJ, Rajendran S, Bujalowska D, Bujalowski W. J Biol Chem. 1998;273:10515. doi: 10.1074/jbc.273.17.10515. [DOI] [PubMed] [Google Scholar]
- 40.Raney KD, Sowers LC, Millar DP, Benkovic SJ. Proc Natl Acad Sci U S A. 1994;91:6644. doi: 10.1073/pnas.91.14.6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dillingham MS, Wigley DB, Webb MR. Biochemistry. 2002;41:643. doi: 10.1021/bi011137k. [DOI] [PubMed] [Google Scholar]
- 42.Fischer CJ, Maluf NK, Lohman TM. J Mol Biol. 2004;344:1287. doi: 10.1016/j.jmb.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 43.Fischer CJ, Lohman TM. J Mol Biol. 2004;344:1265. doi: 10.1016/j.jmb.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 44.Lucius AL, Maluf NK, Fischer CJ, Lohman TM. Biophys J. 2003;85:2224. doi: 10.1016/s0006-3495(03)74648-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tomko EJ, Fischer CJ, Lohman TM. Methods. 2010;51:269. doi: 10.1016/j.ymeth.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ha T, Kozlov AG, Lohman TM. Annu Rev Biophys. 2012;41:295. doi: 10.1146/annurev-biophys-042910-155351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee KS, Balci H, Jia H, Lohman TM, Ha T. Nat Commun. 2013;4:1878. doi: 10.1038/ncomms2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Myong S, Rasnik I, Joo C, Lohman TM, Ha T. Nature. 2005;437:1321. doi: 10.1038/nature04049. [DOI] [PubMed] [Google Scholar]
- 49.Bianco PR, Brewer LR, Corzett M, Balhorn R, Yeh Y, Kowalczykowski SC, Baskin RJ. Nature. 2001;409:374. doi: 10.1038/35053131. [DOI] [PubMed] [Google Scholar]
- 50.Tang Y, Chen Y, Lichti CF, Hall RA, Raney KD, Jennings SF. BMC Bioinformatics. 2005;6(Suppl 2):S9. doi: 10.1186/1471-2105-6-S2-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]