

Abstract
Few biological systems permit rigorous testing of how changes in DNA sequence give rise to adaptive phenotypes. In this study, we sought a simplified experimental system with a detailed understanding of the genotype-to-phenotype relationship that could be altered by environmental perturbations. We focused on plasmid fitness, i.e., the ability of plasmids to be stably maintained in a bacterial population, which is dictated by the plasmid's replication and segregation machinery. Although plasmid replication depends on host proteins, the type II plasmid partitioning (Par) machinery is entirely plasmid encoded and relies solely on three components: parC, a centromere-like DNA sequence, ParR, a DNA-binding protein that interacts with parC, and ParM, which forms actin-like filaments that push two plasmids away from each other at cell division. Interactions between the Par operons of two related plasmids can cause incompatibility and the reduced transmission of one or both plasmids. We have identified segregation-dependent plasmid incompatibility between the highly divergent Par operons of plasmids pB171 and pCP301. Genetic and biochemical studies revealed that the incompatibility is due to the functional promiscuity of the DNA-binding protein ParRpB171, which interacts with both parC DNA sequences to direct plasmid segregation, indicating that the lack of DNA binding specificity is detrimental to plasmid fitness in this environment. This study therefore successfully utilized plasmid segregation to dissect the molecular interactions between genotype, phenotype, and fitness.
INTRODUCTION
In recent years, there has been appreciable progress in linking adaptive phenotypes to specific genetic alterations (reviewed in reference 1), thus shedding light on the molecular details of evolution. Two challenges, however, make it difficult to rigorously link mutations, through changes in biochemistry and cell biology, to gains in fitness: the complexity of living systems makes it difficult to define all the phenotypic outcomes of a given mutation, and accurate measurement of fitness in environments in which organisms have evolved is not always achievable. We therefore sought a simplified experimental system with a detailed understanding of the genotype-to-phenotype relationship that could be altered by environmental perturbations.
We focused specifically on the interactions between two proteins and one protein-binding DNA sequence that direct the segregation of a class of bacterial plasmids. Most plasmids are maintained at low copy numbers, and their successful transmission depends on regulated DNA replication and segregation mechanisms that ensure that each daughter cell receives at least one copy of the plasmid. Filament-driven plasmid segregation ensures accurate plasmid transmission at cell division, and unlike plasmid replication systems, this stabilization mechanism is completely plasmid encoded and, as far as we know, does not rely on host proteins. We utilized the type II partitioning (Par) system, first described for the R1 plasmid from Salmonella enterica (2), which relies on three components, all carried or encoded on a single operon (3): a centromere-like DNA sequence, parC, and two proteins, ParR (NP_957569.1), a DNA-binding protein that interacts with parC, and ParM (NP_957570.1), an ATPase that forms an actin-like filament (4–7). The binding of ParR to parC creates a complex that protects ParM filaments from depolymerization, and the growth of a filament between two plasmids can segregate them to either end of a rod-shaped cell, thus ensuring that each daughter receives at least one plasmid copy (8–10). The detailed analysis of the R1 Par operon is a framework to interpret the partitioning systems of other Par operons (11).
Interactions between genes and environments control the fitness of genotypes. We used incompatibility to manipulate the environment in which Par operons functioned. Two plasmids are compatible if the presence of one has no effect on the inheritance of the other and are incompatible if each interferes with the replication or segregation of the other. In our experimental system, we used compatible replication origins to focus attention on the role of the Par operon in incompatibility. The mixed-filament model for partitioning incompatibility hypothesizes that the elongation of one ParM filament with two different plasmids at each of its ends impairs the inheritance of both plasmids, eventually generating subpopulations of bacterial cells that contain only one of the two plasmids (reviewed in reference 12).
To study the evolution of biological specificity, we investigated the mechanism of segregation-dependent plasmid incompatibility. It has previously been shown that such incompatibility between the elements of a type 1 ParA/ParB segregation system is dictated by “discriminator contacts” between the centromere-like parS DNA sequence and the DNA-binding protein ParB (13, 14). In this study, we tested the compatibility of seven type II Par operons and detected strong incompatibility between two, namely, those from plasmids pB171 (NC_002142.1) (15) and pCP301 (NC_004851.1) (16), despite the substantial sequence divergence of their Par operons. Our experimental data have shown that ParR from pB171 (ParRpB171) binds to the parC sequence from both plasmids and that both interactions can direct plasmid segregation. Furthermore, our mathematical model supports the hypothesis that this molecular cross-reaction is the underlying mechanism that leads to plasmid incompatibility, consistent with the results found for the type I segregation system. This report therefore illuminates how genotype, phenotype, and environment interact to contribute to the success of a simple genetic element.
MATERIALS AND METHODS
Bacterial strains and plasmid construction.
Escherichia coli strain DH5α [fhuA2 lac(del)U169 phoA glnV44 Φ80′ lacZ(del)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17] was utilized in all experiments and manipulated using standard microbiology techniques. Plasmid construction details are in the supplemental material. Table S2 in the supplemental material summarizes the names and important features of all plasmids used in this study.
Plasmid stability and incompatibility assays.
DH5α cells were transformed with the pEMH606-derived resident plasmid and selected on chloramphenicol (Cm) plates. For qualitative plasmid stability assays, single transformants were grown to saturation in selective media, and 109 cells were plated directly onto LB plates containing 20 μg/ml X-Gal (5-bromo-4-chloro-3-indoyl-β-d-galactopyranoside) and incubated at 30°C for 3 to 4 days. Colonies were photographed without magnification. For quantitative plasmid stability assays, transformants were grown under selective conditions (LB plus Cm) to saturation and rediluted 10,000-fold into fresh media every 24 h, for 5 days, producing approximately 70 generations. Typically, on days 3, 4, and 5, cultures were sampled and plated onto (i) LB plates (nonselective) and (ii) LB-plus-Cm plates (selecting for plasmid). The counts on both plates were modeled as Poisson random variables with means as follows:
where ycgi represents the observed cell count on nonselective plate i from culture c at generation g, xcgi represents the observed cell count on selective plate i from culture c at generation g, Vcgi represents the volume of the plated sample on plate i from culture c at generation g, ρcg represents the cell density in culture c at generation g, and l represents the plasmid loss rate per generation.
Loss rate l was the parameter of interest; the generalized linear model framework, also known as a Poisson regression, gives joint maximum-likelihood estimates of l and ρcg, pooling information from samples taken across multiple time points (generations). As a nuisance parameter, ρcg was not reported. Estimates were made with the glm function in R (17).
For incompatibility tests, cells were initially transformed with the pEMH606-derived, low-copy-number “resident plasmid.” These cells were transformed using the high-copy-number (oriMB1) “challenge plasmid” carrying the kanamycin (Kan) resistance gene and were selected on LB-plus-Cm-plus-Kan plates. Individual transformants were grown for 90 min in doubly selective media (LB plus Cm plus Kan) and then transferred to LB plus Kan, which selects only for the challenge plasmid. Cultures were propagated and sampled for 70 generations, as described above. The loss rate of the resident plasmid was estimated by the maximum-likelihood method as detailed above using colony counts from LB-plus-Kan and LB-plus-Cm-plus-Kan plates.
For the half-sectored plasmid stability assay, cells were grown to saturation under selective conditions. Cultures were diluted and plated for single colonies on media containing X-Gal under nonselective conditions. Plates were incubated at 30°C for 2 days, and the half-sectored phenotype was scored for at least 500 colonies per plasmid tested.
Note on the plasmid incompatibility assay.
Fluctuations in resident plasmid copy number influence the loss rates detected for incompatibility assays. During the course of this work, it was brought to our attention that plasmid copy numbers are influenced by the growth phase of a bacterial culture (18, 19) and, therefore, that the length of time each culture is maintained in saturation impacts incompatibility levels. The data presented in each individual figure in this article were collected on the same day; therefore, the loss rates of all samples represented within the same figure can be directly compared. However, because experiments were not controlled for the length of time cultures were maintained at saturation between dilutions, quantitative differences between figures in loss rates reported are less reliable.
Red fluorescent protein (RFP) reporter assays.
DH5α cells were transformed with 2 independent clones of each plasmid containing the parC mKate fluorescence reporter construct. Three colonies were selected from each transformation plate and grown under selective conditions in the presence of ampicillin to the exponential phase. Fluorescence was monitored for each cell population using an LSRII flow cytometer (BD Biosciences) and mean fluorescence recorded.
Electrophoretic mobility shift assays.
parC DNA sequences were amplified and purified on mini-spin columns (Qiagen, Limburg, Holland); parCpCP301 was amplified from pEMH608 with oEMH414 and oEMH294, parCpB171 was amplified from pEMH617 with oEMH314 and oEMH294, and parCR1 was amplified from pEMH607 with oEMH548 plus oEMH294 and pUC18-derived DNA using primers oEMH549 and oEMH329. Purified ParR proteins were diluted in binding buffer (ParRpB171 binding buffer, 10 mM Tris-Cl [pH 7.5], 50 mM NaCl, 50 mM KCl, 1 mM EDTA [pH 8.0], 1 mM dithiothreitol [DTT], 2 mM MgCl2; ParRpCP301 binding buffer, 10 mM Tris-Cl [pH 7.5], 50 mM KCl, 1 mM EDTA [pH 8.0], 1 mM DTT, 2 mM MgCl2, 10% glycerol). parC DNA (2 nM) was incubated with increasing concentrations of ParR proteins in a final reaction volume of 15 μl, and binding was allowed to proceed for 30 min at room temperature. Gel loading buffer was added to each reaction, and the complete volume of each sample was loaded onto prerun 7.5% Tris-Cl gels (Bio-Rad). Gels were run at 80 V for 3 h at 4°C, and DNA was visualized by staining gels with SYBR green stain (Invitrogen) for 20 min and scanning stained gels on a Typhoon scanner (excitation wavelength of 488 nm and visualization parameters compatible with Alexa Fluor 488 dyes).
Simulating plasmid segregation.
The detailed model for our simulations is described in the supplemental material. We used first-order chemical kinetics to model association and dissociation of parC and preformed ParR complexes, making the assumptions that ParR and parC binding was slow relative to the capture of these complexes by ParM filaments and elongation of ParM filaments and that each ParR complex that was bound to DNA associated only with its homologous ParM filaments. The simulation was performed using Gillespie's stochastic simulation algorithm and a stimulation period that was long enough to ensure that the last state of a dividing cell was sampled from the steady-state distribution of plasmids and filaments. When a cell divided, all the plasmids that were associated with one type of ParR were equally partitioned between the two daughter cells; if the number of plasmids was odd, the last plasmid was randomly assigned to one of the daughters. All the plasmids that had not associated with ParR were randomly assigned to daughter cells, mimicking binomial partition. All ParR and parC complexes were assumed to form at the same rate, and their dissociation rates were estimated from analyzing the ability of ParR to inhibit the expression of fluorescent proteins from parC.
His-tagged ParR protein purification.
BL21(DE3) cells were transformed with C-terminally His-tagged ParRpB171 (pEMH535) or ParRpCP301(pEMH536) plasmids. A single transformant from each plate was grown to saturation in selective media and was used to inoculate 1.5 liters of fresh LB media. This culture was grown at 37°C to final optical densities at 600 nm (OD600) of ∼0.65 (ParRpCP301) and ∼0.4 (ParRpB171) and then transferred to 18°C. After 15 min, protein expression was induced by the addition of 0.4 mM IPTG (isopropyl-β-d-thiogalactopyranoside) and the culture was incubated for a further 20 h. Cells were pelleted at 3,000 × g for 20 min and stored overnight at −80°C. Cells were thawed on ice and resuspended in 30 ml lysis buffer (50 mM Tris-Cl [pH 9.0], 400 mM KCl, 1 mM phenylmethylsulfonyl fluoride [PMSF], 0.05% β-mercaptoethanol; for ParRpCP301, pH 9.0; for ParRpB171, pH 8.0). Benzonase (Sigma, MO) was added to reach a final concentration of 100 U/ml, and a French press was used to lyse the cells. The lysate was collected by centrifugation at 30,000 × g for 1 h and was incubated with a 2-ml bed volume of nickel-nitrilotriacetic acid (Ni-NTA) resin (Qiagen, Limburg, Holland) for 30 min at 4°C. The resin was then washed four times with 20 ml wash buffer (50 mM Tris-Cl [pH 9.0], 400 mM KCl, 0.05% β-mercaptoethanol; for ParRpCP301, pH 9.0; for ParRpB171, pH 8.0) containing increasing concentrations (10 mM, 25 mM, 50 mM, and 100 mM) of imidazole. His-tagged ParR proteins were eluted in 5 ml of wash buffer containing 300 mM imidazole. Subsequently, the ParR-purified proteins were desalted using Econo-Pac 10DG columns (Bio-Rad) and eluted in 1 ml buffer S (50 mM Tris-Cl [pH 7.5], 250 mM NaCl, 1 mM EDTA, 1 mM DTT, 10% glycerol). For both ParR proteins, the approximate yield was 1 mg. ParRpCP301 was determined to be >90% pure by SDS-PAGE, whereas ParRpB171 was less pure due to the presence of a lower-molecular-weight contaminating band at approximately 30% of the intensity of ParRpB171.
RESULTS
Plasmid partition incompatibility as a model system.
We began by creating a system that would allow us to look for segregation-based plasmid incompatibility. Figure 1A depicts the R1 segregation system. ParM filaments are stabilized when each end interacts with one copy of the complex formed by the binding of multiple ParR dimers to parC DNA. To quantify the effect of Par operons on plasmid segregation, we engineered a plasmid (pEMH606) that used the replication origin of plasmid F1, lacked its own partitioning system, and carried β-galactosidase (LacZ) and chloramphenicol (Cm) resistance genes, allowing us to monitor its segregation both qualitatively (on media containing X-Gal, a chromogenic LacZ substrate) and quantitatively (by following the appearance of chloramphenicol-sensitive cells) (Fig. 1B). Consistent with previous studies (3), an R1-partitioning (Par) operon stabilized pEMH606 approximately 103-fold (Fig. 1C).
FIG 1.

Establishing a plasmid system, based on plasmid partitioning, to study molecular adaptation. (A) Schematic of the structure of a functional example of R1 partition machinery: the molecular machine encoded by the Par operon that separates two R1 plasmids. (B) Map of the low-copy-number mini-F1 plasmid, pEMH606, which served as the backbone of the resident plasmids, showing the presence of chloramphenicol resistance (CmRes) and β-galactosidase (LacZ) genes, the origin of replication, oriF1, and the location where the type II partitioning operons were inserted. (C) Qualitative and quantitative plasmid stability and incompatibility assays to monitor (i) the ability of the R1 Par operon (blue) to stabilize pEMH606 (black) and (ii) the destabilization of a resident plasmid, made by cloning the R1 Par operon into pEMH606, by a high-copy-number challenge plasmid that also carries the R1 Par operon (red). Error bars represent the 95% confidence intervals derived from two biological replicates, assayed in triplicate, and loss rates are provided with standard deviations (SD). (D) The relationship between genotype, phenotype, and fitness of the plasmid segregation system.
To determine the influence of its biological environment on pEMH606 stability, we introduced a second plasmid into the same cell and examined its effect on the segregation of pEMH606. The additional plasmid uses a higher-copy-number replication origin (oriMB1 from pMB1), which is fully compatible with oriF1 and confers kanamycin resistance. We call this the challenge plasmid and the lower-copy-number plasmid, which confers chloramphenicol resistance and carries oriF1, the resident plasmid. Because the two replication origins are fully compatible, we can determine the effect of the challenge plasmid on the segregation of the resident plasmid by monitoring the loss rate of the resident plasmid in cells that are selected to maintain the challenge plasmid (see Fig. S1 in the supplemental material). Figure 1C shows both the qualitative and quantitative results of the incompatibility produced when both the resident plasmid and challenge plasmid carried the R1 Par operon: the challenge plasmid was destabilized at a level greater than 6-fold by the challenge plasmid carrying an intact segregation system but was unaffected when the challenge plasmid lacked a Par operon. This result demonstrates that we can measure the segregation incompatibility between plasmids and thus determine the relationship between genotype, phenotype, and fitness in a biologically and biochemically defined model system (Fig. 1D).
Plasmid partition incompatibility exists between two highly divergent partitioning operons.
To investigate the linkage between genotype, phenotype, and fitness, we looked for cases of segregation incompatibility between homologous type II Par operons. Using the PFAM database entry for ParR (PF10784), we selected eleven “seed sequences,” identified their associated ParM and parC sequences, and cloned the entire operon into the resident plasmid; for ParM, their amino acid identity with R1 ParM ranged from 21% (pSK41) to 47% (pCP-933T), and their pairwise identity with each other ranged from 17% to 99%. The host species for each of these plasmids, as well as that for the R1 plasmid, is listed in Table S1 in the supplemental material. Three operons (from plasmids pCP301, pAPEC, and pCT-MX3) behaved like R1 and conferred 10-fold stabilization. Three others (from pKPN4, pB171, and CP-933T_UT) stabilized the challenge plasmid to a lesser extent, and five (from pSK41, pADAP, pCoo, CP-933T, and R478) were nonfunctional (Fig. 2A). The phylogenetic relationship between the amino acid sequences of ParM, the most conserved component of these operons, is shown in Fig. 2A.
FIG 2.

Limited plasmid partition incompatibility detected between type II partitioning homologs. (A) A maximum-likelihood-based phylogenetic tree of the ParM amino acid sequence of the type II partitioning operons utilized in this study, indicating the stability of resident plasmids containing them. Bootstrap values are shown. (B) A matrix displaying the results of quantitative compatibility assays for the indicated combinations of partitioning operons. Increasingly red squares indicate the levels of partition incompatibility observed. (C) Qualitative and quantitative incompatibility assay results showing the mutual incompatibility between ParpB171 and ParpCP301 plasmids. CMR, chloramphenicol resistance plasmid. (D) Direct detection of resident plasmid loss in the first cell division of a colony by the formation of half-sectored colonies for β-galactosidase expression. The plasmid pairs tested are indicated.
Next we sought to understand why particular heterologous Par operons failed to improve plasmid segregation or stabilized plasmids less well than the R1 par operon. We hypothesized that these defective Par operons (i) had acquired debilitating or loss-of-function mutations during the course of their evolution, (ii) require a host-specific factor(s) for partitioning function not found in E. coli, (iii) are inadequately expressed in E. coli, or (iv) have an indirect, unexpected effect on oriF1 replication, thereby destabilizing the resident plasmid. Our data showed that the Par operons from pB171, pKPN4, pSK41, and pCoo did not negatively impact that stability of the mini-F1 plasmid lacking any partitioning operon, indicating that oriF1 replication was unaffected (not shown). We also found that expressing both ParR and ParM proteins at high levels from an additional plasmid did not assuage the impaired stabilization of pEMH606 by either pB171 or pKPN4 Par operons (not shown). This limited analysis might suggest that the suboptimal stabilization of pEMH606 by particular heterologous Par operons indicates that the products of these operons cannot function or be expressed in E. coli or, alternatively, that, in the absence of continued strong selection, they have accumulated mutations that reduce their ability to promote accurate partition. The observation that pB171 contains two partitioning operons is consistent with the second possibility (20).
We tested segregation compatibility by making resident and challenge plasmids with each of the seven functional operons, except pCT-MX3, which we were unable to clone into the high-copy-number plasmid. Every operon was strongly self-incompatible (Fig. 2B), but most pairs of operons were fully compatible (Fig. 2B). There were two types of exceptions. Resident plasmids carrying the pKPN4 or CP-933T_UT Par operons were destabilized by the majority of other Par operons. These plasmids were also destabilized by a challenge plasmid that lacks any partitioning sequence, so we conclude that their worse segregation was not due to interactions between the different Par operons. The molecular basis for this effect was not investigated further. The other interaction was specific incompatibility between the pCP301, pB171, and pAPEC Par operons. Because the Par operons of pAPEC and pB171 are almost identical and pB171 has been previously studied (20–22), we focused on the incompatibility between pCP301 and pB171 for the remainder of this study. The Par operons of pCP301 and pB171 mutually destabilize each other, raising the loss rate of the resident plasmid from ≤1% to about 5% (Fig. 2C).
We tested one potential caveat to our results, which depend on propagating a bacterial culture for 70 generations. If losing the resident plasmid allowed cells to divide faster, we would overestimate the rate of plasmid loss. To test this possibility, we used a method that directly detects the cell divisions in which plasmids were lost. When a colony arises from a single cell, a plasmid loss event at the first division produces a half-sectored colony: one half of the colony contains the plasmid, and one half does not. Because the resident plasmid carries the LacZ-encoding gene, the half of the colony that contains the plasmid would turn blue on X-Gal plates and the other half would remain white. Missegregation events that occur at a later division event produce colonies that are less than 50% white. With ParpB171 on the resident plasmid and ParpCP301 on the challenge plasmid, ∼10% of the colonies were half-sectored, and the reciprocal experiment gave ∼6% half-sectored colonies (Fig. 2D). These results show that the incompatibility between pB171 and pCP301 is not due to faster growth of cells that have lost plasmids and suggest that the serial transfer method slightly underestimates plasmid loss rates.
If plasmid incompatibility results from different plasmids being segregated from each other by the same ParM filaments, both plasmids from an incompatible pair must possess parC. To test this assertion, we made challenge plasmids that expressed ParR and ParM but lacked parC. These parC-less challenge plasmids did not destabilize ParpB171 or ParpCP301 resident plasmids (see Fig. S2 in the supplemental material), showing that incompatibility depends on the presence of parC, supporting the hypothesis that it arises through mixed pairing.
pB171 ParR protein binds productively to the pCP301 parC DNA sequence.
To investigate the mechanism of partition incompatibility between ParpB171 and ParpCP301 plasmids, we engineered a series of six chimeric Par operons containing all combinations of ParM (pB171 gene identifier [ID] = 1238680, pCP301 = 1237997), ParR (gene ID pB171 = 1238681, pCP301 = 1237996) and parC from both plasmids (Fig. 3A). We reasoned that any chimera that stabilized a resident plasmid must have a functional parC and ParR interface and a functional ParR and ParM interface. Figure 3A shows that a chimera with ParR and ParM from pB171 and ParC from pCP301 (M171R171C301) fully stabilized a resident plasmid. None of the other chimeras reduced the plasmid loss rate below 7%, and one, composed of pB171 ParR and parC and pCP301 ParM (M301R171C171), made the resident plasmid segregate more poorly than it would have if all the plasmid molecules moved independently of each other (binomial partition).
FIG 3.

Cross talk between the pB171 and pCP301 Par operons occurs at the ParR and parC interface. (A) A chimera with parCpCP301, ParRpB171, and ParMpB171 is stable. Data represent the results of qualitative and quantitative plasmid loss assays for resident plasmids carrying each of the 6 indicated chimeric operons as well as pB171 and pCP301 Par operons. (B) ParRpB171 and ParMpB171 stabilize resident plasmids containing only parCpCP301. Data represent the results of quantitative plasmid loss assays for a resident plasmid carrying only parCpCP301 in the presence of a rescue plasmid expressing either green fluorescent protein (GFP) (Par−) or ParRMpB171 from a strong constitutive promoter. Error bars represent the 95% confidence intervals derived from two biological replicates, assayed in triplicate. (C) ParRpB171 and ParRpCP301 each cross repress the other's parC. Genetic structure of eight RFP reporters, with RFP expressed from either the pCP301 parC promoter (Pr_pCP301; red bars) or the pB171 parC promoter (Pr_pB171; blue bars). Data represent the mean fluorescence of each strain normalized to that of the strain containing the appropriate, nonrepressed RFP reporter. Error bars represent the 95% confidence intervals derived from six independent transformants of each reporter plasmid. (D) pCP301 and pB171 each bind the other's parC in vitro. Data represent the results of a gel shift assay to assess the ability of purified His-tagged ParRpB171 and His-tagged ParRpCP301 protein to bind to both parCpB171 and parCpCP301 DNA. Lanes marked with a star contain equal amounts of purified ParRpB171 (8 μM) and a 2 nM concentration of the indicated DNA molecule. pUC18-derived DNA and parCR1 act as nonspecific control DNA sequences.
The behavior of the M171R171C301 chimera suggests that ParRpB171 can bind to parCpCP01, generating a protein and DNA complex that interacts with ParMpB171 filaments to direct segregation. Therefore, we predicted that expressing only ParRpB171 and ParMpB171 from a second plasmid could stabilize a resident plasmid that carries only parCpCP301. Figure 3B verifies this prediction, as the loss rate of the parCpCP301 plasmid decreased from 7.4%/generation to 2.2%/generation when pB171 ParR and ParM proteins were coexpressed from a medium-strength promoter (PRNAI) in the same cell.
To assess ParRpB171 and parCpCP01 binding in vivo, we exploited the findings that parC contains the Par promoter and that ParR binding to the parC represses transcription (23). We placed the gene for mKate (24), a monomeric RFP, downstream of parC and monitored mKate expression by fluorescence-activated cell sorting. In the absence of any ParR protein, parCpCP301 drove strong mKate expression (Fig. 3C, upper panel), but cloning ParRpCP301 or ParRpB171 downstream of mKate reduced fluorescence 30- or 3-fold, respectively. A mutant of ParRpB171 that eliminates DNA binding (parR-K6E) (22) alleviated repression (Fig. 3C, upper panel).
The interaction between heterologous ParR and parC is not reciprocal: the M171R171C301 chimera fully stabilized the resident plasmid, whereas the M301R301C171 chimera, carrying parCpB171 DNA and encoding ParRpCP301 and ParMpCP301 proteins, did not. This result suggests that ParRpCP301 cannot bind to parCpB171 and support segregation. To probe this hypothesis, we designed similar mKate reporter constructs to probe transcriptional repression but instead drove expression from parCpB171. Figure 3C (lower panel) shows that both ParRpB171 and ParRpCP301 produced roughly 2-fold repression of parCpB171, suggesting that the requirements for transcriptional repression and plasmid segregation are not identical.
We next sought to verify the specificity of ParR and parC binding in vitro using a gel-shift assay. We purified His-tagged ParRpB171 (NP_053129.1) and ParRpCP301 (NP_858327.1) and monitored their ability to slow the electrophoretic gel mobility of parC DNA. Consistent with our analysis of transcriptional repression, both versions of ParR bound to both versions of parC. Neither ParRpB171 nor ParRpCP301 displayed detectable affinity for parC from a third plasmid (R1) or a control sequence derived from a standard cloning vector (pUC18) (Fig. 3D and data not shown).
A mixed-filament model can explain pCP301 and pB171 partition incompatibility.
In the mixed-filament model of partitioning incompatibility, a plasmid with parCpCP301 and a plasmid with parCpB171, both bound by ParRpB171, would be segregated from each other by a ParMpB171 filament (Fig. 4A). The presence of multiple plasmids and multiple ParM filaments in small cells makes a rigorous test for these mixed filaments impossible. We therefore turned to simulation to ask whether formation of mixed filaments could explain our experimental data.
FIG 4.

A mixed-filament model can explain plasmid partition incompatibility. (A) Schematic of the proposed mixed filaments formed in cells containing both ParpCP301 and ParpB171 plasmids, illustrating the “incorrect” binding of ParRpB171 to parCpCP301. (B) A two-step kinetic model for pB171 and pCP301 incompatibility based on the relative in vivo binding affinities of each ParR protein complex to both parC sequences. See the supplemental material for details. (C) Output from the simulations of plasmid partition incompatibility for the indicated combinations of plasmids, indicating the dependence of the resident plasmid (copy number = 3) loss rate on the copy number of the challenge plasmid. The region corresponding to the experimentally determined mean copy number of the oriMB1 challenge plasmid is shaded. Error bars represent 95% confidence intervals. (D) The dependence of the kinetic model for pCP301 and pB171 incompatibility on k−2, the dissociation rate of ParRpB171 binding to parCpCP301. The resident plasmid and challenge plasmid copy numbers were set at 3 and 15, respectively, and the experimentally approximated value for k−2 is highlighted. Error bars represent the 95% confidence interval.
The model, shown in Fig. 4B, makes three key assumptions. The first is that preformed ParR complexes bind with first-order kinetics to parC and that the binding rate is the same for all possible interactions. As a result, the different affinities of ParR complexes for parC are determined only by differences in the off rate with which ParR dissociates from parC. The second is that ParM filaments can interact only with their cognate ParR complexes, and the third is that filament formation and extension along the long axis of the cell are instantaneous. Under these assumptions, the pattern of segregation is determined by which parC-containing plasmid molecules associate with a given ParR.
We used transcriptional repression to estimate the relative affinities of different forms of ParR for parC. Employing the reporter plasmid with RFP driven by parC, described earlier, we expressed ParRpB171 or ParRpCP301 constitutively from a second plasmid and monitored fluorescence. Assuming that repression correlates with the level of ParR bound to parC and that ParR is not a limiting factor in the cell, these data yield the best approximation of the relative binding affinities for each pair of ParR and parC complexes (see Fig. S3 in the supplemental material), allowing us to assign relative off rates for the unbinding of different ParR proteins and parC sequences. Given these rates, we simulated partition incompatibility for 10,000 cells in parallel, using the Gillespie algorithm (25). At the end of the simulation, we paired plasmids bound by the same ParR protein and partitioned them to the two daughter cells, with each cell receiving one member of the pair. Unbound plasmids were assigned randomly to both cells with equal probabilities.
Figure 4C indicates the simulated loss rates for different combinations of resident and challenge plasmids. The resident plasmid was present at 3 copies per cell, and the copy numbers of the challenge plasmid differed. As the number of challenge plasmids increases, it gets harder for the resident plasmid to form an unmixed filament, and the loss rate of the resident plasmid rises. When two plasmids have the same Par operon (maximally incompatible), the resident plasmid's loss rate approaches the binomial loss rate at high copy numbers of the challenge plasmid.
We compared the simulation results to our experimental data. Quantitative PCR revealed that our resident plasmid was at about 3 copies/chromosome terminus (see Fig. S4 in the supplemental material), and the oriMB1 challenge plasmid is known to be maintained at ∼15 copies/chromosome terminus (26). These values and our model predict a resident plasmid loss rate for ParpCP301 and ParpB171 plasmids of about 8%/generation, which is higher than the in vivo rate of about 5%/generation. This difference could reflect multiple factors, including greater discrimination in the formation of the complex that directs segregation, compared to our transcriptional repression measurements, and underestimation of the plasmid copy number.
Next, we investigated how robust the kinetic model was with respect to changes in the dissociation rate of the complex between ParRpB171 and parCpCP301 (k−2), the critical interaction that gives rise to our mixed filament. Over a 10-fold range, variation in k−2 switches the segregation of the resident plasmid from efficient (k−2 = 10/s) to barely better than binomial (k−2 = 1/s), indicating that this model of incompatibility is sensitive to k−2. Our estimate of k−2, 1.9/s, lies within this range. Furthermore, the modeled loss rate is insensitive to changes in the rate constant k−3, the dissociation constant of ParRpCP301 and parCpB171 complexes near these parameter values (see Fig. S5 in the supplemental material). This provides us with confidence that, similarly to the in vivo behavior, k−2 is the decisive parameter that governs incompatibility between ParpCP301 and ParpB171 plasmids.
Partition incompatibility reflects the interaction between pB171 ParR and pCP301 parC.
The mixed-filament model for partition incompatibility makes testable predictions. The first is that a challenge plasmid that carries only parC can destabilize a resident plasmid with an intact Par operon, as was first shown for R1 (27). We predict that ParpB171 will be sensitive to a parCpCP301 challenge plasmid, because ParRpB171 can bind to parCpCP301, producing mixed filaments linking resident and challenge plasmids. In contrast, ParpCP301 will be resistant to a parCpB171 challenge plasmid because the binding of ParRpCP301 to parCpB171 is not productive for segregation. Figure 5A confirms these predictions: on its own, each ParpB171 plasmid and ParpCP301 plasmid is destabilized by its own parC on challenge plasmids, and ParpB171 is destabilized by a parCpCP301 challenge plasmid, but ParpCP301 resists a parCpB171 challenge plasmid.
FIG 5.
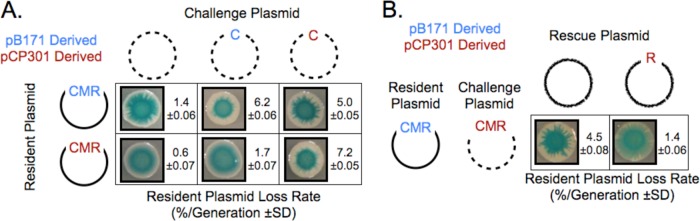
Partition incompatibility depends on competition between ParRpB171 and ParRpCP301 for binding to parCpCP301. (A) Binding of ParRpB171 to parCpCP301 causes segregation incompatibility. Data represent the results of a quantitative and qualitative plasmid compatibility assay comparing a challenge plasmid carrying parCpB171 to a challenge plasmid carrying parCpCP301 with respect to the stability of ParpB171 or ParpCP301 resident plasmids. (B) Overexpression of ParRpCP301 protects a ParpB171 resident plasmid from incompatibility. Data represent the results of quantitative and qualitative plasmid compatibility assays showing the incompatibility between a resident ParpB171 plasmid and a challenge ParpCP301 plasmid under conditions of constitutive expression of either GFP (Par− plasmid) or ParRpCP301 from a third plasmid.
The second prediction from our competition model is that incompatibility will be affected by changing the expression of the proteins involved in cross talk. In particular, we predict that increasing levels of ParRpCP301 will help a resident plasmid with ParpCP301 resist a challenge from a ParpB171 plasmid: more ParRpCP301 makes it harder for ParRpB171 to bind parCpCP301 and thus avoids the formation of mixed filaments. Figure 5C confirms the prediction: adding a third plasmid that expressed ParRpCP301 stabilized the ParpCP301 resident plasmid 3-fold.
DISCUSSION
We used bacterial plasmid segregation to define the interactions between genotype, phenotype, and fitness. The partitioning machinery is simple, is entirely encoded by the plasmid, has minimal interactions with host proteins, and directly controls the propagation and thus the fitness of the plasmid. Related plasmids are analogous to related species and are from a common origin, with sequence similarities and differences in their segregation machinery affecting the degree of cross-reactivity. Compatible plasmids can coexist within a single cell and can be thought of as occupying different niches, whereas incompatible plasmids occupy the equivalent of overlapping niches and compete with each other. This competition means that the addition of an incompatible challenge plasmid alters the environment, phenotype, and fitness of a resident plasmid. Specifically, we uncovered the molecular basis of the mutual incompatibility between the Par operons derived from plasmids pB171 and CP301: the DNA-binding protein, ParRpB171, can bind to the centromere-like parC of both plasmids, showing that accurate segregation in a cell with multiple plasmids depends on the specificity of the interaction between ParR and parC.
ParR plays two roles: it interacts with DNA to control the expression of the Par operon and forms a protein-DNA complex that binds to and modulates the dynamics of ParM filaments. In vivo analysis of transcriptional repression and in vitro analysis of DNA binding both show cross-reactivity between ParR and parC of the two plasmids. However, at the phenotypically critical level of plasmid segregation, the cross-reaction is unidirectional: binding of ParRpB171 to parCpCP301 can direct plasmid segregation, whereas the reciprocal reaction, the binding of ParRpCP301 to parCpB171, does not.
The molecular biology of ParR suggests reasons why cross-reactivity could be different for different aspects of plasmid behavior. Like ParRR1, ParRpB171 is a dimer in solution and binds cooperatively to its own parC DNA (20, 28). In both R1 and pB171, multiple ParR dimers bind to parC, making a ring-like protein-DNA complex (22, 29). In R1, this complex interacts with and stabilizes the ends of ParM filaments, and current models for filament elongation invoke multiple interactions between the ParM filament and the ParR and ParC complex (9, 30). We speculate that the inability of the parCpB171 and ParRpCP301 complex to support partitioning is caused by too few ParRCP301 dimers binding to parCpB171.
Examining the sequence of parC does not offer an easy explanation for the ability of ParRpB171 to direct the segregation of a plasmid bearing parCpCP301. The sequence homology between pB171 and pCP301 parC is weak (see Fig. S6 in the supplemental material), and the consensus sequences that are believed to mediate the binding of ParRpB171 to parCpB171 are missing from parCpCP301. Furthermore, the DNA-binding N-terminal regions of both ParRpB171 and ParRpCP301 show limited sequence identity (see Fig. S6) and are no more similar than the sequences of ParR proteins from compatible partitioning operons (22). Nevertheless, the ability of a mutation that prevents DNA binding to block the repressive activity of ParRpB171 with respect to both parCPB171 and parCpCP301 demonstrates that DNA binding is required for the biological activity of ParR. Interestingly, the discriminator contacts that define the segregation specificity of plasmids encoding the type I Par operon, such as PI, have been mapped to an interaction between two hexameric sequences in the parS DNA and a 16-amino-acid stretch in the C terminus of the ParB protein (13, 14, 31–33). For the P1 plasmid, both these regions are dispensable for parS and ParB binding but are essential for mediating P1 segregation specificity (13). The limited sequence homology between parC/ParRpB171 and parC/ParRpCP301 (22) (see Fig. S6) suggests that the type II Par systems do not encode a similar discriminator that can distinguish different parC sequences, but further analyses would be required to rigorously test this possibility. Alternatively, other features of parC may be required to form the structure that directs plasmid segregation, such as the ability of parC DNA to easily bend into a ring-like parC and ParR complex or the spacing between the repetitive binding sites that might influence cooperativity between bound ParR dimers.
Our reductionist system succeeds in defining the basis for plasmid fitness in a given environment. The combination of simulation and genetic data show that cross talk at a single molecular interface, between ParR and parC, can cause inaccurate plasmid segregation. The binding of ParRpB171 to parCpCP301 is a failure to distinguish “self” from “nonself” on the part of both operons and means that both Par operons have reduced fitness when they end up in the same cell. If we think of incompatible plasmid pairs such as pB171 and pCP301 as belonging to the same group or “species” of plasmid, we can use the evolution of partition compatibility as a toy model for species determinations by identifying and characterizing mutations in the Par operon that make a pair of incompatible partitioning operons compatible, potentially by promoting specificity and self-recognition.
Is there selection for the evolution of plasmid compatibility? Our analysis focused on seven Par operons. Of these, three showed mutual incompatibility (with one pair very closely related in sequence), two showed Par-independent incompatibility, and the remaining two, R1 and pCT-MX3, were compatible with each other and with all the remaining operons. The sample was too small to permit general conclusions. Without better ecological knowledge on how often in nature plasmids encounter other plasmids within the same cell, we cannot tell where the truth lies between two extremes: whether plasmid compatibility results from selection for the divergence of replication and segregation machinery in plasmids that regularly cohabit and compete with each other or whether compatibility is the result of genetic drift in a simple molecular machine. The strong incompatibility, despite the high sequence divergence between the Par operons of pCP301 and pB171, argues for the latter possibility, but a combination of plasmid ecology and molecular dissection will be needed to produce a definitive answer. Nonetheless, we have established a model system that can be used to select for changes in molecular interfaces and to ask how these changes alter the fitness of genetic elements.
Supplementary Material
ACKNOWLEDGMENTS
We thank P. Malkus, J.-I. Bouet, N. Firth, K. Gerdes, D. Taylor, and G. Jagura-Burdzy for plasmids.
The simulations in this article were run on the Odyssey cluster supported by the FAS Science Division Research Computing Group at Harvard University.
This work was supported by a Charles A. King Trust postdoctoral fellowship (E.M.H.) and NIH grant RO1-GM43987 (A.W.M).
We thank M. Müller, R. Losick, and M. Laub for comments on the manuscript.
Footnotes
Published ahead of print 9 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01811-14.
REFERENCES
- 1.Watt WB. 2013. Specific-gene studies of evolutionary mechanisms in an age of genome-wide surveying. Ann. N. Y. Acad. Sci. 1289:1–17. 10.1111/nyas.12139 [DOI] [PubMed] [Google Scholar]
- 2.Nordström K, Molin S, Aagaard-Hansen H. 1980. Partitioning of plasmid R1 in Escherichia coli. I. Kinetics of loss of plasmid derivatives deleted of the par region. Plasmid 4:215–227 [DOI] [PubMed] [Google Scholar]
- 3.Gerdes K, Molin S. 1986. Partitioning of plasmid R1. Structural and functional analysis of the parA locus. J. Mol. Biol. 190:269–279 [DOI] [PubMed] [Google Scholar]
- 4.Salje J, Gayathri P, Lowe J. 2010. The ParMRC system: molecular mechanisms of plasmid segregation by actin-like filaments. Nat. Rev. Microbiol. 8:683–692. 10.1038/nrmicro2425 [DOI] [PubMed] [Google Scholar]
- 5.van den Ent F, Moller-Jensen J, Amos LA, Gerdes K, Lowe J. 2002. F-actin-like filaments formed by plasmid segregation protein ParM. EMBO J. 21:6935–6943. 10.1093/emboj/cdf672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi CL, Claridge SA, Garner EC, Alivisatos AP, Mullins RD. 2008. Protein-nanocrystal conjugates support a single filament polymerization model in R1 plasmid segregation. J. Biol. Chem. 283:28081–28086. 10.1074/jbc.M803833200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salje J, Lowe J. 2008. Bacterial actin: architecture of the ParMRC plasmid DNA partitioning complex. EMBO J. 27:2230–2238. 10.1038/emboj.2008.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garner EC, Campbell CS, Mullins RD. 2004. Dynamic instability in a DNA-segregating prokaryotic actin homolog. Science 306:1021–1025. 10.1126/science.1101313 [DOI] [PubMed] [Google Scholar]
- 9.Gayathri P, Fujii T, Moller-Jensen J, van den Ent F, Namba K, Lowe J. 2012. A bipolar spindle of antiparallel ParM filaments drives bacterial plasmid segregation. Science 338:1334–1337. 10.1126/science.1229091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garner EC, Campbell CS, Weibel DB, Mullins RD. 2007. Reconstitution of DNA segregation driven by assembly of a prokaryotic actin homolog. Science 315:1270–1274. 10.1126/science.1138527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerdes K, Moller-Jensen J, Bugge Jensen R. 2000. Plasmid and chromosome partitioning: surprises from phylogeny. Mol. Microbiol. 37:455–466. 10.1046/j.1365-2958.2000.01975.x [DOI] [PubMed] [Google Scholar]
- 12.Bouet JY, Nordstrom K, Lane D. 2007. Plasmid partition and incompatibility–the focus shifts. Mol. Microbiol. 65:1405–1414. 10.1111/j.1365-2958.2007.05882.x [DOI] [PubMed] [Google Scholar]
- 13.Radnedge L, Davis MA, Austin SJ. 1996. P1 and P7 plasmid partition: ParB protein bound to its partition site makes a separate discriminator contact with the DNA that determines species specificity. EMBO J. 15:1155–1162 [PMC free article] [PubMed] [Google Scholar]
- 14.Sergueev K, Dabrazhynetskaya A, Austin S. 2005. Plasmid partition system of the P1par family from the pWR100 virulence plasmid of Shigella flexneri. J. Bacteriol. 187:3369–3373. 10.1128/JB.187.10.3369-3373.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tobe T, Hayashi T, Han CG, Schoolnik GK, Ohtsubo E, Sasakawa C. 1999. Complete DNA sequence and structural analysis of the enteropathogenic Escherichia coli adherence factor plasmid. Infect. Immun. 67:5455–5462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Liu H, Zhang X, Yang J, Yang F, Yang G, Shen Y, Hou Y, Jin Q. 2003. Complete DNA sequence and gene analysis of the virulence plasmid pCP301 of Shigella flexneri 2a. Sci. China C. Life. Sci. 46:513–521. 10.1360/02yc0060 [DOI] [PubMed] [Google Scholar]
- 17.Development Core Team R. 2013. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 18.Carapuça E, Azzoni AR, Prazeres DM, Monteiro GA, Mergulhão FJ. 2007. Time-course determination of plasmid content in eukaryotic and prokaryotic cells using real-time PCR. Mol. Biotechnol. 37:120–126. 10.1007/s12033-007-0007-3 [DOI] [PubMed] [Google Scholar]
- 19.Skulj M, Okrslar V, Jalen S, Jevsevar S, Slanc P, Strukelj B, Menart V. 2008. Improved determination of plasmid copy number using quantitative real-time PCR for monitoring fermentation processes. Microb. Cell Fact. 7:6. 10.1186/1475-2859-7-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ringgaard S, Ebersbach G, Borch J, Gerdes K. 2007. Regulatory cross-talk in the double par locus of plasmid pB171. J. Biol. Chem. 282:3134–3145. 10.1074/jbc.M609092200 [DOI] [PubMed] [Google Scholar]
- 21.Ebersbach G, Gerdes K. 2001. The double par locus of virulence factor pB171: DNA segregation is correlated with oscillation of ParA. Proc. Natl. Acad. Sci. U. S. A. 98:15078–15083. 10.1073/pnas.261569598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Møller-Jensen J, Ringgaard S, Mercogliano CP, Gerdes K, Löwe J. 2007. Structural analysis of the ParR/parC plasmid partition complex. EMBO J. 26:4413–4422. 10.1038/sj.emboj.7601864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jensen RB, Dam M, Gerdes K. 1994. Partitioning of plasmid R1. The parA operon is autoregulated by ParR and its transcription is highly stimulated by a downstream activating element. J. Mol. Biol. 236:1299–1309 [DOI] [PubMed] [Google Scholar]
- 24.Shcherbo D, Merzlyak EM, Chepurnykh TV, Fradkov AF, Ermakova GV, Solovieva EA, Lukyanov KA, Bogdanova EA, Zaraisky AG, Lukyanov S, Chudakov DM. 2007. Bright far-red fluorescent protein for whole-body imaging. Nat. Methods 4:741–746. 10.1038/nmeth1083 [DOI] [PubMed] [Google Scholar]
- 25.Gillespie DT. 2007. Stochastic simulation of chemical kinetics. Annu. Rev. Phys. Chem. 58:35–55. 10.1146/annurev.physchem.58.032806.104637 [DOI] [PubMed] [Google Scholar]
- 26.Covarrubias L, Cervantes L, Covarrubias A, Soberon X, Vichido I, Blanco A, Kupersztoch-Portnoy YM, Bolivar F. 1981. Construction and characterization of new cloning vehicles. V. Mobilization and coding properties of pBR322 and several deletion derivatives including pBR327 and pBR328. Gene 13:25–35 [DOI] [PubMed] [Google Scholar]
- 27.Dam M, Gerdes K. 1994. Partitioning of plasmid R1. Ten direct repeats flanking the parA promoter constitute a centromere-like partition site parC, that expresses incompatibility. J. Mol. Biol. 236:1289–1298 [DOI] [PubMed] [Google Scholar]
- 28.Møller-Jensen J, Borch J, Dam M, Jensen RB, Roepstorff P, Gerdes K. 2003. Bacterial mitosis: ParM of plasmid R1 moves plasmid DNA by an actin-like insertional polymerization mechanism. Mol. Cell 12:1477–1487. 10.1016/S1097-2765(03)00451-9 [DOI] [PubMed] [Google Scholar]
- 29.Hoischen C, Bussiek M, Langowski J, Diekmann S. 2008. Escherichia coli low-copy-number plasmid R1 centromere parC forms a U-shaped complex with its binding protein ParR. Nucleic Acids Res. 36:607–615. 10.1093/nar/gkm672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schumacher MA, Glover TC, Brzoska AJ, Jensen SO, Dunham TD, Skurray RA, Firth N. 2007. Segrosome structure revealed by a complex of ParR with centromere DNA. Nature 450:1268–1271. 10.1038/nature06392 [DOI] [PubMed] [Google Scholar]
- 31.Youngren B, Radnedge L, Hu P, Garcia E, Austin S. 2000. A plasmid partition system of the P1-P7par family from the pMT1 virulence plasmid of Yersinia pestis. J. Bacteriol. 182:3924–3928. 10.1128/JB.182.14.3924-3928.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dabrazhynetskaya A, Brendler T, Ji X, Austin S. 2009. Switching protein-DNA recognition specificity by single-amino-acid substitutions in the P1 par family of plasmid partition elements. J. Bacteriol. 191:1126–1131. 10.1128/JB.01358-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dabrazhynetskaya A, Sergueev K, Austin S. 2005. Species and incompatibility determination within the P1par family of plasmid partition elements. J. Bacteriol. 187:5977–5983. 10.1128/JB.187.17.5977-5983.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.