

Abstract
The transcriptional regulator GntR1 downregulates the genes for gluconate catabolism and pentose phosphate pathway in Corynebacterium glutamicum. Gluconate lowers the DNA binding affinity of GntR1, which is probably the mechanism of gluconate-dependent induction of these genes. In addition, GntR1 positively regulates ptsG, a gene encoding a major glucose transporter, and pck, a gene encoding phosphoenolpyruvate carboxykinase. Here, we searched for the new target of GntR1 on a genome-wide scale by chromatin immunoprecipitation in conjunction with microarray (ChIP-chip) analysis. This analysis identified 56 in vivo GntR1 binding sites, of which 7 sites were previously reported. The newly identified GntR1 sites include the upstream regions of carbon metabolism genes such as pyk, maeB, gapB, and icd, encoding pyruvate kinase, malic enzyme, glyceraldehyde 3-phosphate dehydrogenase B, and isocitrate dehydrogenase, respectively. Binding of GntR1 to the promoter region of these genes was confirmed by electrophoretic mobility shift assay. The activity of the icd, gapB, and maeB promoters was reduced by the mutation at the GntR1 binding site, in contrast to the pyk promoter activity, which was increased, indicating that GntR1 is a transcriptional activator of icd, gapB, and maeB and is a repressor of pyk. Thus, it is likely that GntR1 stimulates glucose uptake by inducing the phosphoenolpyruvate (PEP):carbohydrate phosphotransferase system (PTS) gene while repressing pyk to increase PEP availability in the absence of gluconate. Repression of zwf and gnd may reduce the NADPH supply, which may be compensated by the induction of maeB and icd. Upregulation of icd, gapB, and maeB and downregulation of pyk by GntR1 probably support gluconeogenesis.
INTRODUCTION
Corynebacterium glutamicum is a high-GC Gram-positive soil bacterium that is traditionally used for the industrial production of amino acids (1, 2). This bacterium can also be used for efficient production of lactate and succinate from sugar (3–5). Central carbon metabolism plays a pivotal role in the generation of energy for biological processes and in the supply of precursor molecules for biosynthesis of cell compounds. Therefore, the molecular basis of its regulation is of great interest for the development of new bioprocesses.
Since the genome sequence of C. glutamicum was determined (1, 6, 7), several transcriptional regulators of various carbon metabolism genes have been discovered, and it is likely that these regulators, such as SugR, RamB, RamA, GlxR, LldR, and GntR1 and GntR2 (GntR1/2), form a global regulatory network (8, 9). This regulatory system is distinct from the well-studied system in Escherichia coli or Bacillus subtilis. For example, the global catabolite repression mechanism mediated by E. coli cyclic AMP receptor protein (CRP) or B. subtilis CcpA has not been established in C. glutamicum. In contrast, C. glutamicum can simultaneously utilize multiple carbon sources (10, 11). Genome-wide analysis such as microarray or chromatin immunoprecipitation (ChIP)-chip analysis was conducted for the understanding of the regulatory network of these regulators. For example, ChIP-chip analysis of GlxR detected more than 200 in vivo binding regions in both noncoding and coding regions of the C. glutamicum genome (12), establishing that GlxR is a global transcriptional regulator.
GntR1/2 is responsible for the induction of gluconate utilization genes in C. glutamicum ATCC 13032 (13). Expression of the gntP and gntK genes is upregulated by disruption of both of the functionally redundant gntR1 and gntR2 genes. This result indicates that GntR1/2 represses the genes encoding gluconate permease (GntP) and gluconate kinase (GntK) as in the cases of GntR in E. coli and B. subtilis. In addition, the pentose phosphate pathway genes tkt, tal, zwf, opcA, and devB, which are clustered in the genome, and gnd are under the control of GntR1/2 (13, 14). Gluconate and glucono-δ-lactone reduce the DNA binding activity of GntR1/2 (13). Thus, it is believed that GntR1/2 senses the presence of gluconate and glucono-δ-lactone in the cell and that decreased GntR1/2 activity results in the induction of gluconate utilization genes.
A unique characteristic of C. glutamicum GntR1/2 is that it functions as a transcriptional activator of ptsG and ptsS, the genes that encode the phosphoenolpyruvate (PEP):carbohydrate phosphotransferase system (PTS) for uptake of glucose and sucrose, respectively (13). The dual functions of GntR1/2, repression of gluconate utilization and activation of the PTS-dependent sugar uptake, probably contribute to the simultaneous utilization of gluconate and glucose in C. glutamicum. Recently, it was reported that the pck gene, encoding phosphoenolpyruvate carboxykinase, is also transcriptionally activated by GntR1/2 (15). Microarray analysis revealed that disruption of gntR1/2 resulted in the upregulation of 19 genes and downregulation of 26 genes (13). However, the direct binding of GntR1/2 was demonstrated in only the seven promoter regions of the above target genes (gntK gntP, tkt operon, ptsG, ptsS, pckA, and gnd). Thus, the gene list of direct targets of GntR1/2 remains unclear.
The unique characteristic of GntR1/2, i.e., that the regulon is not limited to the gluconate utilization genes, prompted us to search for the new target of GntR1 in C. glutamicum R, the strain in which GntR2 is not encoded by the genome and disruption of gntR1 is enough to induce gnd expression (14). In this study, we searched the in vivo binding site of GntR1 by ChIP-chip analysis. We identified 56 binding regions, including all the seven sites previously identified as described above. Binding of GntR1 to four of the newly identified regions upstream of carbon metabolism genes, i.e., maeB, gapB, icd, and pyk, was confirmed by electrophoretic mobility shift assay (EMSA). Disruption of the GntR1 binding site reduced the activity of maeB, gapB, and icd promoters but enhanced that of the pyk promoter. These results suggest a new role for GntR1 in the coordination of utilization of different carbon sources in C. glutamicum.
MATERIALS AND METHODS
Media and growth conditions.
C. glutamicum R was grown aerobically at 33°C in nutrient-rich A medium (16) supplemented with 2% (wt/vol) glucose or gluconate. Bacterial growth was monitored by determining the optical density at 610 nm (OD610).
Bacterial strains.
The strains used in this study are listed in Table 1. C. glutamicum R was used as a wild-type (WT) strain. The strains having the icd, gapB, pyk, or maeB promoter-lacZ fusion gene (Picd-lacZ, PgapB-lacZ, Ppyk-lacZ, and PmaeB-lacZ, respectively) with or without mutation at GntR1 binding region, were constructed as described previously (17).
TABLE 1.
Strains used in this study
| Strain | Description | Reference or source |
|---|---|---|
| R | JCM 18229 wild-type strain | 7 |
| KT8 | R ΔgntR1 | 32 |
| TI01 | R with FLAG-tagged gntR1 | This study |
| YT402 | R with Picd-lacZ | This study |
| YT403 | R with PicdΔGntR1site-lacZ | This study |
| YT362 | R with PgapB-lacZ | This study |
| YT363 | R with PgapBΔGntR1site-lacZ | This study |
| YT406 | R with Ppyk-lacZ | This study |
| YT407 | R with PpykΔGntR1site-lacZ | This study |
| YT384 | R with PmaeB-lacZ | This study |
| YT399 | R with PmaeBΔGntR1site1-lacZ | This study |
| YT395 | R with PmaeBΔGntR1site2-lacZ | This study |
| YT397 | R with PmaeBΔGntR1site1,2-lacZ | This study |
Construction of plasmids used in this study.
Translational promoter-lacZ fusion vectors were constructed as follows. The promoter region and coding sequence for the initial 5 amino acids of icd, gapB, pyk, and maeB were amplified by PCR using primers EcoRV-icd–400F and EcoRV-icd-15R for icd, EcoRV-gapB–400F and EcoRV-gapB-15R for gapB, EcoRV-pyk–390F and EcoRV-pyk-15R for pyk, EcoRV-maeB–410F and EcoRV-maeB-15R for wild-type maeB, and EcoRV-maeB–410F and EcoRV-maeB-mut GntR1-site2R for the maeB promoter with the mutation at GntR1 binding site 2 (Table 2). The amplified fragment was digested with EcoRV and cloned into the DraI in the site of the pCRA741 reporter plasmid (16). Mutagenesis of the GntR1 binding sites was conducted as follows. The plasmid containing icd-lacZ, gapB-lacZ, pyk-lacZ, or maeB-lacZ was used as a template for inverse PCR using corresponding primer sets (Table 2). The amplified fragment was digested with BglII and self-ligated. The resultant plasmid was used to transform C. glutamicum R, and a recombinant cell with a kanamycin resistance marker was selected. Insertion of the promoter-lacZ fusion gene between cgR_0734 and cgR_0735 was confirmed by PCR using primers LlacZLR-4354F and Ind7insert-checkR or LlacZLR-6425R and Ind7insert-checkF.
TABLE 2.
Primers used in this study
| Primer | Sequence (5′–3′) | Purpose |
|---|---|---|
| JW102 | GCGGTGACCCGGGAGATCTGAATTC | ChIP-chip |
| LlacZ1291F | CGGTAGAATTCGAGCTCGGTACCT | EMSA |
| Cy3-llacZ-Ra | ACGTTGTAAAACGACGGGATC | EMSA |
| gnd-F | CCACCGACGCAGTCATCA | qRT-PCR |
| gnd-R | TGATGTCGCCTTCGTCCAT | qRT-PCR |
| 16S rRNA-F | TCGATGCAACGCGAAGAAC | qRT-PCR |
| 16S rRNA-R | GAACCGACCACAAGGGAAAAC | qRT-PCR |
| EcoRV-icd–400F | CGCGATATCGCCTCCATCACTGTGAAGTG | Promoter-lacZ construction |
| EcoRV-icd-15R | CGCGATATCAATGATCTTGGCCATGAGTCTC | Promoter-lacZ construction |
| EcoRV-gapB–400F | CGCGATATCGGTGTCCTCTCGCGCA | Promoter-lacZ construction |
| EcoRV-gapB-15R | CGCGATATCGATCAACGGCACCATGTCCTC | Promoter-lacZ construction |
| EcoRV-pyk–390F | CGCGATATCGTCTCCGCAGTAGTGTTTG | Promoter-lacZ construction |
| EcoRV-pyk-15R | CGCGATATCTCTATCCACGCCCATAAGCCT | Promoter-lacZ construction |
| EcoRV-maeB–410F | CGCGATATCAAGTGGGTATCGCGTCTGG | Promoter-lacZ construction |
| EcoRV-maeB-15R | CGCGATATCCAGGTCGATGGTCATATCATTTAGC | Promoter-lacZ construction |
| BglII-icd–246R | CGCAGATCTTCTTTTCTCCCGTGGAAGAGTAC | Promoter-lacZ construction |
| BglII-icd–240F | CGCAGATCTCAAGTCGAGCTCGCGG | Promoter-lacZ construction |
| BglII-gapB–289R | CGCAGATCTCATTGGGTTGAGATATGGGTAC | Promoter-lacZ construction |
| BglII-gapB–275F | CGCAGATCTTTGCGTAAGGCGGCAG | Promoter-lacZ construction |
| BglII-pyk–40R | CGCAGATCTCATTAATTCTCGATGAATCTTGC | Promoter-lacZ construction |
| BglII-pyk–34F | CGCAGATCTCCTGTGGCTTGAGGGGGA | Promoter-lacZ construction |
| EcoRV-maeB-mut GntR1-site2R | CGCGATATCCAGGTCGATGGTCATTAAGCGTAGCCTTGTTAATCGGTG | Promoter-lacZ construction |
| BglII-maeB–205R | CGCAGATCTCTTTTGGGTGGTTTTGGGGT | Promoter-lacZ construction |
| BglII-maeB–194F | CGCAGATCTCAAACAATTAAATTCATCACAAAACAC | Promoter-lacZ construction |
| LlacZLR-4354F | ATAACCGGGCAGGGGTCTAG | Promoter-lacZ construction |
| Ind7insert-checkR | GCGTCACGAACAACAGACAGC | Promoter-lacZ construction |
| LlacZLR-6425R | CGACGGCCAGTGCCAAGC | Promoter-lacZ construction |
| Ind7insert-checkF | CGAGACTGGAATTGAGGCTC | Promoter-lacZ construction |
| NheI-2434–1500–F5 | GCGGCTAGCGTGCACGGATTCCATGAAC | gntR1-FLAG construction |
| SalI-2434–1500-R4 | GCGGTCGACAAAGCCGAGGAAACAAGAGCAGG | gntR1-FLAG construction |
| C_gntR_FLAG-tag_nheI | CGCGCTAGCTTACTTGTCATCGTCATCCTTGTAGTCTGCTGCTCCGTTCAGCGTGCCCAGC | gntR1-FLAG construction |
| C_nheI_gnt3′_ver4 | GCGGCTAGCCGATATCGCTAAGAGCTTCAACGA | gntR1-FLAG construction |
Cy3 labeled at the 5′ end.
The gntR1-FLAG strain (TI01) was constructed as described previously (18). First, the DNA fragment containing the gntR1 region was amplified from the C. glutamicum R genome by PCR using primers NheI-2434–1500-F5 and SalI-2434–1500-R4 (Table. 2). The amplified fragment was digested with NheI and SalI and cloned into pCRA725. The resultant plasmid was used as a template for inverse PCR to add FLAG-tag (DYKDDDK) at the C terminus of gntR1 using primer sets C_gntR_FLAG-tag_nheI and C_nheI_gnt3′_ver4. The resultant plasmids were introduced into C. glutamicum, and single-crossover cells were isolated by using kanamycin resistance. Isolated cells were cultivated on medium supplemented with 10% sucrose, and double-crossover cells were isolated. The gene modifications were confirmed by DNA sequencing of the PCR products around the modified region.
ChIP-chip analysis.
Exponentially growing C. glutamicum cultures (35 ml) in A medium with 2% glucose were treated with formaldehyde (at a final concentration of 1%) and incubated for 15 min at room temperature. The cross-linking was quenched by addition of glycine (at a final concentration of 125 mM), and the culture was incubated for 5 min at room temperature. Cells were then collected by centrifugation, washed twice with Tris-buffered saline (20 mM Tris-HCl [pH 7.5], 150 mM NaCl), and stored at −80°C. Pellets were resuspended in 0.5 ml FLAG buffer (20 mM Tris-HCl [pH 8.0], 200 mM KCl, 5 mM MgCl2, 10% glycerol, 0.1% SDS) containing protease inhibitor cocktail (Roche Antiprotease Mini). The cell suspension was transferred to a 15-ml tube containing ∼300 mg of 0.1-mm Zirconia/Silika beads (Bio Spec). Cell disruption and genomic DNA shearing were conducted using Bioruptor UCD-250 (Cosmo Bio) under the following conditions: 10 cycles of 5 s on, 5 s off at high power at 4°C. The average size of sheared DNA was 200 to 500 bp. The cell lysate was collected and centrifuged (20,000 × g for 30 min at 4°C) to prepare the cell extract. The amount of total protein was measured by the Bio-Rad protein assay (Bio-Rad) using bovine serum albumin (BSA) as a standard. The cell extract was diluted with FLAG buffer containing protease inhibitor cocktail to reach 0.5 mg/ml total protein. A 150-μl fraction of the cell extract was saved for later analysis (reference DNA). The remainder was subjected to immunoprecipitation with 80 μl of anti-FLAG M2 agarose (Sigma). The mixture was incubated 90 min on a rotating platform at 4°C. The beads were washed five times with FLAG buffer. Immunoprecipitated complexes were eluted from beads by treatment with 240 μl of IP buffer with 100 ng/μl of 3× FLAG-peptide (Sigma). Cross-links of immunoprecipitated samples and of total DNA samples were reversed by incubation overnight at 65°C. Samples were then treated with RNase A and protease K for 2 h at 55°C. DNA was extracted with phenol-chloroform and purified with a QIAquick PCR purification minElute kit (Qiagen). DNA samples were blunted with T4 DNA polymerase, ligated to linkers, and amplified by PCR. Amplified DNAs from reference DNA and immunoprecipitated DNA were differentially labeled with cyanine 3 (Cy3) and Cy5, respectively, by using a CGH labeling kit (Life Technologies) according to the manufacturer's instructions.
We used the Agilent eArray platform (Agilent Technologies, Palo Alto, CA, USA) to design a C. glutamicum oligonucleotide microarray. Two sets of probes were designed. One was used for global gene expression analysis, and the other was for ChIP-chip analysis. For global gene expression analysis, a set of 60-mer oligonucleotide probes specific for all the open reading frames (ORFs) on both the chromosome and a plasmid pCGR1 was designed and synthesized in duplicate (for a total of 6,742 probes). For ChIP-chip analysis, a set of 60-mer oligonucleotide probes covering the entire chromosome (on average, one probe for every 170 bases), was designed (yielding 18,462 and 18,449 probes on the plus strand and the minus strand, respectively). The regions for RNA genes, including rRNA and tRNA, were excluded to avoid biased signals because these regions repeatedly contain similar sequences. In addition, the region highly transcribed often causes false positives in the ChIP-chip analysis (33). Each array contains both probe sets.
Hybridization was performed using the Agilent Oligo aCGH hybridization kit according to the manufacturer's manual (Agilent). Equal amounts (2.5 μg) of each labeled DNA were mixed and hybridized to microarrays in an Agilent Technologies microarray chamber at 65°C for 24 h in a rotating Agilent hybridization oven at 20 rpm. After hybridization, microarrays were washed with Oligo aCGH/ChIP-on-chip wash buffer 1 (Agilent) at room temperature for 5 min, with Oligo aCGH/ChIP-on-chip wash buffer 2 (Agilent Technologies) at 31°C for 5 min, with acetonitrile for 10 s, and with a stabilization and drying solution (Agilent) for 30 s at room temperature. Slides were scanned immediately after washing by placement within an Agilent ozone barrier slide cover with the Agilent DNA microarray scanner (G2505C) at a resolution of 5 μm using the two-color scan setting for the 4x44K (Agilent) array slides. Scanning was done in both channels, and the photomultiplier tube (PMT) was set to 100% for both Cy3 and Cy5 channels. The scanned images were quantified with Feature Extraction Software 10.5.5.1 (Agilent) using default parameters (protocol GE2_105_Dec08). Log2 ratios for each probe were determined as rProcessedSignal/gProcessedSignal (Cy5/Cy3). The entire procedure was carried out two times. Creation of plots of the log2 ratios against probe location, extraction of ChIP-chip peaks, and determination of genomic locations for the peaks were performed using MochiView v 1.45 (34). Peak extraction was performed using the following setting: for smoothing, smoothing window flank span, 500 bp; minimum weighted location count for inclusion, 1.01; maximum gap for data interpolation, 100 bp; for peak-finding setting, 1st pass filter; peak location width, 250 bp; minimum value for peak inclusion, 0.5; second-pass filter scan distance, 700 bp; minimum peak height change, 0.6; minimum distance between peak midpoint, 200 bp; number of significance sampling, 0. The chromosomal sequences corresponding to the ChIP-chip peaks were extracted and analyzed using multiple Em for motif elicitation (MEME)-ChIP (35) to define the GntR1 consensus sequence. The ChIP-chip peak regions were manually searched for the conserved sequence motif obtained (AWWGGTMRYACCWWT) to find additional consensus sequences. The enrichment factor for a given gene was defined as the log2 ratio of each peak. These data analyses were performed using one of two ChIP-chip data sets, and reproducibility was checked in the other data set.
Electrophoretic mobility shift assay.
The histidine-tagged GntR1 proteins was expressed in E. coli and purified as described previously (14). Briefly, overexpression of the histidine-tagged protein was achieved by using a cold shock-expressing vector pCold I system (TaKaRa). E. coli cells harboring the resultant plasmid were grown at 37°C in 100 ml of LB medium with ampicillin (50 ng/ml) to an OD610 of 0.5. Cultures were incubated for 30 min at 15°C, and then IPTG (isopropyl-β-d-thiogalactopyranoside) was added at a final concentration of 0.1 mM. After 24 h of culture, cells were harvested by centrifugation and suspended in 900 μl of His binding buffer (0.5 M NaCl, 20 mM Tris-HCl, 5 mM imidazole, pH 7.9) (Novagen), 100 μl of Fast Break cell lysis reagent (Promega), and 0.2 mg of lysozyme. The mixture was incubated for 15 min at room temperature, centrifuged for 5 min at 12,000 × g, and the supernatant was pooled. The histidine-tagged protein was purified using the His Bind resin and buffer kit (Novagen) according to the procedure specified by the manufacturer. Purification of histidine-tagged GntR1 was checked by SDS-PAGE and Coomassie brilliant blue (CBB) staining (see Fig. S3 in the supplemental material). A single band corresponding to the size of GntR1 (28 kDa) was observed.
EMSA was carried out in a total volume of 20 μl of binding buffer, consisting of 20 mM Tris-HCl (pH 7.9), 100 mM NaCl, 3 mM MgCl2, 0.1 mM EDTA, 0.1 mM dithiothreitol (DTT), 5% glycerol, and 50 μg/ml poly(dI-dC), and 50 μg/ml bovine serum albumin. The Cy3-labeled promoter DNA fragment was amplified by PCR using primers listed in Table 2 and genomic DNA of the wild-type strain or a strain carrying the mutated GntR1 binding site as the template. The DNA fragment (2 nM) was incubated with the purified GntR1 protein for 10 min at 25°C. The mixture was fractionated by electrophoresis on a native 5% polyacrylamide gel containing 5% glycerol in 0.5× Tris-borate EDTA (TBE; 45 mM Tris-borate, pH 8.0, 1 mM EDTA) at room temperature.
DNA microarray analysis.
C. glutamicum R wild-type cells and cells with gntR1 deleted were grown aerobically in nutrient-rich A medium supplemented with 2% (wt/vol) glucose. Total RNA was isolated from exponentially growing cells (OD610, 2.0) using the Nucleo Spin RNA (Macherey-Nagel). For transcriptome analysis, the cDNAs were reverse transcribed from 10 μg of total RNAs and labeled with cyanine 3 (Cy3) using the SuperScript Indirect cDNA labeling system (Life Technologies). Hybridization was performed using the gene expression hybridization kit according to the manufacturer's manual (Agilent). The labeled cDNA was hybridized to microarrays in an Agilent Technologies microarray chamber at 65°C for 17 h in a rotating Agilent hybridization oven. After hybridization, microarrays were washed with GE wash buffer 1 (Agilent) at room temperature for 1 min and with GE wash buffer 2 (Agilent) at 37°C for 1 min. Slides were scanned immediately after washing on the Agilent DNA microarray scanner (G2505C) at a resolution of 5 μm, using the single-color scan setting for 4x44k array slides. PMT was set to 100% for Cy3 channels. The scanned images were quantified with Feature Extraction Software 10.5.5.1 (Agilent) using default parameters (protocol GE1_105_Dec08). Feature extracted data were analyzed using GeneSpring GX v 12.0 software from Agilent. Normalization of the data was done in GeneSpring GX using the recommended percentile shift normalization (50th percentile). The resultant data were expressed as averages of mRNA ratios (ΔgntR1 mRNA/WT mRNA) from three independent experiments. Genes that showed significantly altered mRNA levels (P value of <0.02 in a Student t test), by a factor of 1.5 or more, were identified and are listed in Table S2 in the supplemental material.
β-Galactosidase assay.
For the β-galactosidase assay, C. glutamicum R was grown to an OD610 of 2.0, and 1 ml of culture samples was harvested and dissolved in 1 ml of Z buffer (Na2H or NaH2PO4 [pH 7.0], 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol) with 2% toluene to permeabilize the cell. β-Galactosidase activity was determined with permeabilized cells as described previously (17).
Microarray data accession number.
The microarray and ChIP-chip data have been submitted to the NCBI Gene Expression Omnibus (GEO) functional genomics data repository under the accession number GSE58633.
RESULTS
Identification of in vivo binding sites of GntR1.
A genome-wide search for the binding region of GntR1 was conducted by ChIP-chip analysis using a strain in which the chromosomally encoded gntR1 was modified to add the FLAG tag at its C terminus (strain TI01). TI01 and the wild-type strain showed the same gnd mRNA expression level, in contrast to the upregulation of this gene by gntR1 deletion, indicating that the FLAG-tagged GntR1 is functional in vivo (see Fig. S1 in the supplemental material). To determine the target region of GntR1, TI01 was grown in the nutrient-rich A medium supplemented with 2% (wt/vol) glucose, and the DNA fragments binding to GntR1 were collected. GntR1 is expected to be in the active form under the experimental conditions, because the expression of gnd mRNA is in a repressed state and deletion of gntR1 relieved this repression under the growth conditions (14). ChIP and microarray analysis was performed as described in Materials and Methods, and regions that were considered possible GntR1 target regions are listed in Table S1 in the supplemental material. Under the stricter conditions whereby the regions exhibiting enrichment factors of >2.0 were selected, we identified 56 regions that are very likely to be the in vivo GntR1 binding sites, and these regions were selected for further analysis. Of these regions, 51 sites are located upstream of coding sequences, suggesting that GntR1 regulates transcription of these genes. The identified 56 sites include the upstream regions of genes gntP, gntK, gnd, tkt, ptsG, ptsS, and pckA, the regions that GntR1 was shown to bind to in vitro (13–15).
A MEME (multiple Em for motif elicitation) (19) alignment of the putative GntR1 binding regions produced the conserved sequence 5′-AWWGGTMRYACCWWT-3′ (Fig. 1), which is found in all the regions identified in the ChIP-chip analysis. The sequence is a 6-3-6-bp inverted repeat with high conservation of T at the 6th position. The sequence is found in the GntR1 binding region within the gntK and gnd promoters that has been experimentally tested by DNase I footprinting assay (13, 14), strongly indicating that the motif is the recognition sequence for GntR1 binding.
FIG 1.
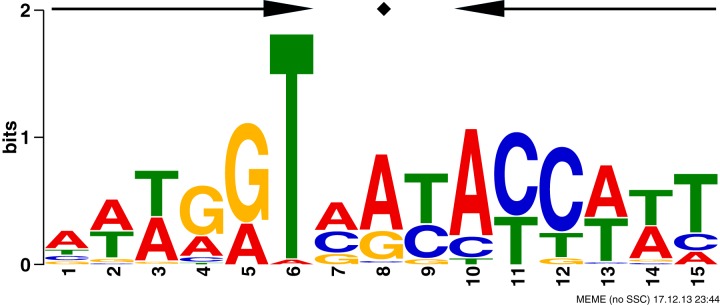
Prediction of GntR1 consensus binding motif. A conserved motif was identified using the MEME algorithm by analyzing the regions with enrichment factors of >2.0 in the ChIP-chip experiment. The size of each letter indicates the relative abundance at the respective position in the consensus matrix generated with MEME. The palindromic sequence is depicted by symmetrical arrows.
Transcriptome analysis of the effects of gntR1 gene deletion.
The results of ChIP-chip analysis indicate that GntR1 may be involved in the regulation of many more genes than previously assumed. Therefore, we conducted transcriptome analysis using a DNA microarray. C. glutamicum WT and a gntR1 deletion strain were grown in nutrient-rich A medium supplemented with 2% (wt/vol) glucose. Total RNA was extracted from the logarithmic grown cells and subjected to microarray analysis, and the mRNA expression levels in the gntR1 deletion strain were compared to those in the wild-type strain. Seventy genes were upregulated more than 1.5-fold, whereas 229 genes were downregulated more than 1.5-fold (see Table S2 in the supplemental material). Decreased expression of ptsG and pckA and increased expression of tkt, gnd, gntK, gntP, and gntV were observed as described previously (13–15). In addition to these genes, we found that the expression of many of the genes involved in carbon metabolism was changed; these genes are gapB, rbsK2, rbsK1, dctP2, ugpE, dhaS, maeB, dctA, dctM1, dctP1, bglF2, and bglF. In Table 3, carbon metabolism genes identified in ChIP-chip analysis are presented with transcriptome data. The genes that were highly enriched by ChIP tend to show a large change in the expression by deletion of gntR1, although expression of some of the ChIP-enriched genes was not significantly changed. Therefore, we further investigated the role of GntR1 on the expression of carbon metabolism genes that showed high enrichment by ChIP-chip with altered or unaltered mRNA levels in transcriptome data as follows.
TABLE 3.
Carbon metabolic genes identified in ChIP-chip analysisa
| Gene ID | Gene name | Enrichment factorb | DNA microarray ratioc | GntR1 binding motif (5′–3′) | Function |
|---|---|---|---|---|---|
| cgR_1513 | gnd | 5.66 | 3.65 | AAAGGTGTGACCATT | 6-Phosphogluconate dehydrogenase |
| cgR_2397 | gntV | 5.19 | 60.60 | ATTGGTACTATCATA | Gluconate kinase |
| cgR_1973 | pyk | 5.14 | 1.34 | ACAGGTACTACCATT | Pyruvate kinase |
| cgR_0261 | iolT1 | 5.06 | 0.74 | ATTGGTAATACTTAG | Metabolite transport protein |
| cgR_0402 | bglS | 5.01 | 10.72 | AAAGGGATTACCATT | Putative beta-glucosidase |
| cgR_2751 | pckA | 4.91 | 0.42 | AAAGGGATTACCATT | Phosphoenolpyruvate carboxykinase |
| cgR_2895 | maeB | 4.67 | 0.48 | AATGATATGACCATC | Malic enzyme |
| cgR_2262 | mdh | 4.64 | 0.85 | GATGGTGTTACCTTT | Malate dehydrogenase |
| cgR_1425 | ptsG | 4.54 | 0.29 | AAAAGTATTACCTTT | Phosphotransferase, glucose-specific enzyme II |
| cgR_1624 | tkt | 4.46 | 1.82 | AAAGGTGTGACCAAT | Transketolase |
| cgR_2806 | gntP | 4.32 | 2.79 | ATTAGTATGATCAAA | Gluconate permease |
| cgR_2497 | dctA | 4.25 | 4.17 | ATTAATATTACCTTT | Na+/H+-dicarboxylate symporter |
| cgR_1636 | gapA | 4.24 | 1.23 | ATTGGGATTACCATT | Glyceraldehyde-3-phosphate dehydrogenase |
| cgR_2454 | 4.20 | 9.01 | TTAGGTGTGATCTTT | Sugar phosphate isomerase/epimerase | |
| cgR_1038 | gapB | 4.14 | 0.31 | AAAGATGTGATCATT | Glyceraldehyde 3-phosphate dehydrogenase |
| cgR_2451 | 3.91 | 2.40 | ATTAGTGTGACCAAA | Na+/H+-dicarboxylate symporter | |
| cgR_2222 | dctP1 | 3.49 | 7.77 | AAAGGGATCACCATT | Putative C4-dicarboxylate-binding protein |
| cgR_2896 | gntK | 3.18 | 1.83 | AAAGGTCTGACTAAT | Putative gluconate kinase |
| cgR_2120 | aceE | 3.09 | 0.95 | TTAGGTACGACCAAA | Pyruvate dehydrogenase |
| cgR_0784 | icd | 2.52 | 1.07 | CTTGGTGTGATCTTT | Isocitrate dehydrogenase |
| cgR_2547 | ptsS | 2.29 | 1.69 | AATAGTGCCACCTTT | Enzyme II sucrose protein |
All ChIP-chip peaks were located upstream of the genes listed in the Gene ID column.
Relative peak height ratios of the array data in the ChIP DNA to input DNA are shown in the base 2 logarithm.
Relative ratios of the transcript levels in the gntR1-deleted cells to those in the wild-type strain were determined by DNA microarray analysis.
Binding of GntR1 to the promoter regions of icd, gapB, pckA, and maeB genes.
To explore the more detailed view of the GntR1-mediated regulation, we selected the carbon metabolism genes that were newly identified in the ChIP-chip analysis, and the GntR1 binding motif was found within the 500 bp from the translation initiation codon (icd, gapB, pyk, and maeB). The microarray results showed that gntR1 deletion resulted in the reduction in the gapB and maeB transcripts, whereas transcriptional levels of icd and pyk were not significantly changed by gntR1 deletion. To test the binding of GntR1 to the promoter region of these genes, we conducted EMSA with purified GntR1 protein. We prepared the DNA fragment with or without mutation (see Fig. S2 in the supplemental material) at the putative GntR1 binding motif as a probe used for EMSA (Fig. 2A). Incubation of these probes corresponding to the wild-type upstream regions of icd, gapB, and pyk with GntR1 protein resulted in a DNA-protein complex. EMSA using their mutated DNA probes showed no complex with GntR1. These results indicate that GntR1 specifically binds to the predicted site and also verify the accurate prediction of the GntR1 binding motif (Fig. 1). Upstream of the maeB gene, there are two possible GntR1 binding sites, one located upstream of the transcription start site and the other overlapping the translational start codon. Indeed, addition of GntR1 resulted in the two shifted bands detected in EMSA. Mutation either in the mut1 site or in the mut2 site (see Fig. S2 in the supplemental material) resulted in one shifted band, and the mutation at both sites resulted in the complete disappearance of shifted bands. These results indicate that there are two GntR1 binding sites within the maeB promoter region. Binding affinity of GntR1 to gntK promoter region is diminished in the presence of gluconate or glucono-δ-lactone (13). We then tested whether gluconate affects the binding of GntR1 to the gapB, pyk, icd, and maeB promoters (Fig. 2B). As expected, the GntR1 band shift was diminished in the presence of 50 mM gluconate. However, gluconate did not completely inhibit the GntR1 binding to these promoter DNAs, as observed in the gntK promoter.
FIG 2.

Binding of GntR1 to the promoter regions of the predicted target genes. (A) The DNA fragment (2 nM) with or without mutation at putative GntR1 binding sites was incubated with 0, 15, and 60 nM GntR1. DNA fragments complexed with GntR1 are indicated by closed arrowheads, and free DNA is indicated by an open arrowhead. (B) Effects of gluconate on the binding of GntR1 to target promoters. The promoter DNA fragments of icd, gapB, and pyk were incubated with 15 nM GntR1. The DNA fragment of maeB was incubated with 0, 15, and 60 nM GntR1. DNA binding reactions were performed in the presence or absence of 50 mM gluconate.
Effects of mutation at GntR1 binding site on the promoter activity of icd, gapB, pyk, and maeB.
To investigate the role of GntR1 on the promoter activity of icd, gapB, pyk, and maeB, we constructed the translational promoter-lacZ fusions with or without mutation at the GntR1 binding site and integrated them into the chromosome of C. glutamicum R. The strains were grown in nutrient-rich A medium with 2% glucose or 2% gluconate or 1% glucose plus 1% gluconate, and the promoter activity of the exponentially growing cells was determined by analyzing the β-galactosidase activity (Fig. 3). The activity of the icd promoter carrying the mutation at the GntR1 binding site was less than one-half that of the wild-type promoter both in the presence and in the absence of gluconate. The promoter activity of gapB was also decreased by a mutation of the GntR1 binding site in cells grown in the absence of gluconate. In the gluconate-grown cells, there was no difference between the wild type and the GntR1 binding site mutant. In contrast to icd and gapB, the pyk promoter activity was increased about 3.5-fold by the GntR1 site mutation. A slight increase in wild-type pyk promoter activity was observed in the presence of glucose and gluconate. Mutation at each or both of the two GntR1 sites in the maeB promoter resulted in the marked decrease in the promoter activity both in the presence and in the absence of gluconate. These results suggest that GntR1 activates the promoters of icd, gapB, and maeB, whereas GntR1 represses the pyk promoter. Expression of gapB and maeB were decreased by deletion of gntR1 (Table 3), which supports the idea that GntR1 is an activator of gapB and maeB. Gluconate reduced the GntR1-mediated activation of the gapB promoter. The effect of gluconate on the pyk promoter was modest and was not observed in icd and maeB. These results suggest that gluconate partially inhibited GntR1 activity, which affects target promoter activities to various extents.
FIG 3.
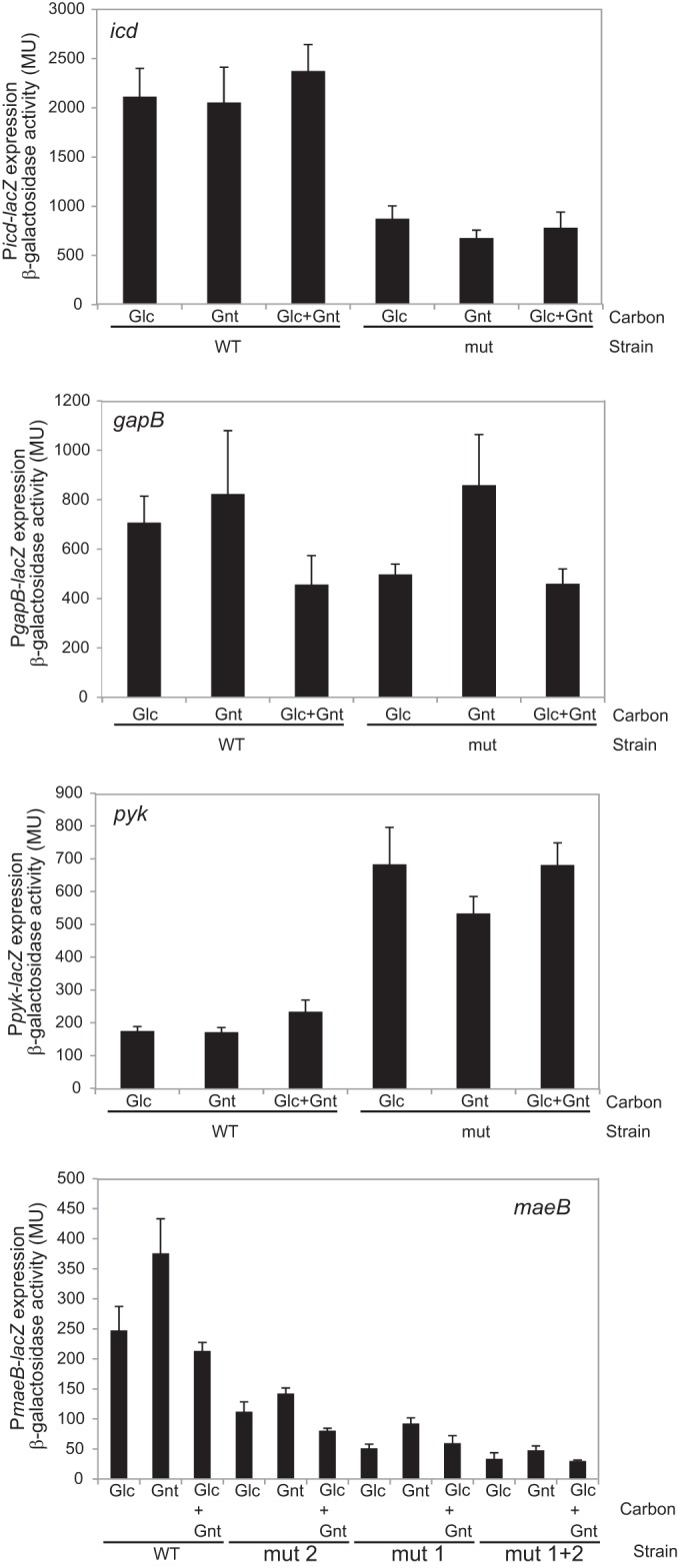
Effects of the mutation at GntR1 binding sites on the promoter activity of the predicted target genes. The strains containing icd, gapB, pyk, or maeB promoter-lacZ fusion with or without mutations at GntR1 binding sites were grown in nutrient-rich A medium supplemented with 2.0% (wt/vol) glucose or 2.0% gluconate or 1.0% glucose plus 1.0% gluconate. Culture samples were taken at an OD610 of 2.0, and β-galactosidase activities were determined. The values are the means of results from three independent experiments, and standard deviations are indicated.
DISCUSSION
GntR1/2 is known to control the expression of genes of gluconate utilization (gntP and gntK), the pentose phosphate pathway (tkt, tal, zwf, opcA, devB, and gnd), the phosphotransferase system (ptsG and ptsS), and gluconeogenesis (pckA) in C. glutamicum ATCC 13032, indicating that GntR1/2 is one of the important transcriptional regulators for carbon metabolism (13–15). In C. glutamicum R, the gntR2 gene is missing and gntR1 is solely responsible for the induction of the gnd gene by gluconate (14). In this study, we conducted ChIP-chip analysis to explore the genome-wide binding target of GntR1 in vivo. In the ChIP-chip analysis, 56 regions, including all the seven previously identified GntR1 binding sites, were enriched by GntR1, indicating that many more genes are under the control of GntR1 than previously thought. The marked increase of the GntR1 target site by ChIP-chip analysis allowed us to predict a more precise consensus sequence of the GntR1 binding sites (Fig. 1). The predicted sequence forms an inverted repeat, which is not obvious in the previous report (13). Mutation at the consensus sequence effectively eliminated the binding of GntR1 (Fig. 2), supporting the accurate prediction of the binding motif.
Transcriptome analysis of the effect of gntR1 deletion showed that expression of 70 genes was increased, whereas expression of 193 genes was decreased (see Table S2 in the supplemental material). As expected, expression of the genes encoding gluconate utilization and the pentose phosphate pathway was increased, and expression of ptsG and pckA was decreased by the deletion of gntR1. However, expression of the ptsS gene, which showed decreased expression in the gntR1- and gntR2-deleted strain in a previous paper (13), showed increased expression in our microarray analysis. This inconsistency may be caused by the different growth media used in the studies and requires more-extensive analysis to determine the role of GntR1 on the regulation of ptsS. Among the genes listed in Table S2 in the supplemental material, 23 genes, including ptsG, pckA, ptsS, tkt, gntP, gnd, and gntV, were also detected as probable direct targets of GntR1 in ChIP-chip analysis. Of the 23 genes, the GntR1 binding peak was located within the intergenic region of four sets of divergently oriented genes, i.e., cgR_1513 (gnd)-cgR1514, cgR_1623 (ctaB)-cgR_1624 (tkt), cgR_6135-cgR_2388 (betT), and cgR_2646-cgR_2647, suggesting that binding of GntR1 to the intergenic region simultaneously controls its downstream genes. Many genes showed different expression by gntR1 deletion in microarray, but their upstream regions showed no significant interaction with GntR1 in ChIP-chip analysis. Thus, it is likely that these genes are indirectly controlled by GntR1. In this context, it is noteworthy that, as shown by ChIP-chip analysis, GntR1 binds upstream of cgR_0453 and cgR_2263, both encoding TetR family transcriptional regulators. Upregulation of cgR_2263 mRNA by gntR1 deletion was also observed in microarray analysis. Although the regulatory target of these transcriptional regulators has not been reported, GntR1 may indirectly affect the expression of a number of genes by controlling the expression of these regulators. Another possible indirect effect by gntR1 deletion may be due to the reduced glucose utilization. The glucose uptake rate of a gntR1 and gntR2 double deletion strain was about one-third that of the wild-type strain (13), which is probably the result of reduced expression of the ptsG gene. Decreased glucose utilization may relieve the catabolite repression by glucose. For example, bglF-bglA-bglG genes, coding for the β-glucoside catabolic genes, are under strict control of glucose repression (20). Therefore, increased expression of the bglF-bglA-bglG genes may be the result of weakened glucose repression.
We selected four carbon metabolism genes (icd, gapB, pyk, and maeB) for further analysis of the regulation by GntR1. These genes are known to be controlled by several transcriptional regulators. For example, expression of gapB and maeB are diminished in a ramA-deleted strain while pyk expression is increased. Binding of RamA to the promoter region of gapB, maeB, and pyk was confirmed by EMSA (21, 22). Expression of pyk was also increased in a sugR mutant, and binding of SugR to the pyk promoter region was confirmed by EMSA (23). Expression of maeB is subject to complex regulation. Promoter activity is downregulated by RamB, the TetR-type regulator MalR, and AmtR, a master regulator of nitrogen regulation (22). However, participation of GntR1 in the regulation of these genes has not been reported. Binding of GntR1 to the promoter region of these genes was observed by EMSA, confirming the ChIP-chip result (Fig. 2). Mutations at the predicted GntR1 binding consensus sequence resulted in the disappearance of the shifted bands, indicating that the binding is specific and the predicted GntR1 consensus sequence is reliable.
Promoter-reporter analysis of the icd, gapB, pyk, and maeB genes with or without mutation at the GntR1 binding site indicates that GntR1 is an activator of icd and gapB while GntR1 is a repressor of pyk. In the maeB promoter, mutation at both of the GntR1 binding sites resulted in reduced promoter activity, consistent with the microarray data, which showed deletion of the gntR1 gene reduced the mRNA level of maeB to about one-half of the level in the wild-type strain. The transcription start sites of icd, gapB, and maeB were determined previously (see Fig. S2 in the supplemental material) (24, 25). The GntR1 binding sites of icd, gapB, and maeB site 1 are located upstream of the transcription start site, which is in accord with the activation of these promoters by GntR1. However, maeB site 2 overlaps the translation start codon (see Fig. S2 in the supplemental material); therefore, caution is needed in interpreting the result of maeB site 2 mutation. Although the translation initiation codon was avoided for mutation, it may affect the recognition of ribosomes to maeB-lacZ mRNA, leading to the defect in translation. Experiments such as in vitro transcription analysis of GntR1 with the site 2-mutated maeB promoter may reveal whether binding of GntR1 to site 2 causes activation of the maeB promoter.
DNA binding affinity of GntR1 to the gntK promoter is decreased by gluconate and glucono-δ-lactone (13). Similarly, the binding of GntR1 to icd, gapB, pyk, and maeB promoter DNA is partially inhibited by gluconate (Fig. 2B). Gluconate eliminated the effect of GntR1 activation for the gapB promoter, partially derepressed the pyk promoter, and showed no obvious effect on icd and maeB promoters (Fig. 3). The different effects of gluconate on these promoters in vivo may be caused by the interactions of other transcriptional regulators as mentioned above. The regulation of carbon metabolism genes by GntR1 is summarized in Fig. 4. The DNA binding activity of GntR1 is diminished by gluconate or glucono-lactone, indicating that GntR1 acts as a sensor of the presence or absence of these sugar molecules (13). In agreement with this, a DNA fragment of gluconate catabolic genes was highly enriched by ChIP-chip analysis, and these genes showed a strong increase in mRNA level in the gntR1-deleted strain (see Table S2 in the supplemental material). In the absence of gluconate, GntR1 represses the gluconate utilization system and the pentose phosphate pathway but activates the glucose and sucrose uptake (13). These regulations are thought to balance the utilization of gluconate with glucose or sucrose in C. glutamicum. What is the role of GntR1 in the regulation of other carbon metabolism genes? pyk is downregulated, while pckA is upregulated by GntR1 (Fig. 4). These regulations, together with the activation of ptsG and ptsS, should support PEP-dependent uptake of glucose and sucrose in the absence of gluconate. Indeed, the consumption of glucose is lowered by gntR1 and gntR2 deletion (13). In addition to the regulation of pyk and pckA, upregulation of gapB and maeB by GntR1 may improve fitness in the presence of carbon sources other than gluconate and PTS-sugar by facilitating gluconeogenesis. It is also noteworthy that icd and maeB positively compensate for the function of the pentose phosphate pathway in the generation of NADPH in C. glutamicum (26–28). Thus, it is conceivable that the GntR1 plays a role in coordinated utilization of gluconate and other carbon sources. Further studies on the involvement of GntR1 in gluconeogenesis will be of interest, because in this pathway the gapB gene product consumes NADPH rather than generating it. The carbon metabolic flux of C. glutamicum has been intensely studied; for example, supply of NADPH, change in the flux of pyruvate node, and on-and-off gluconeogenesis (29–31). Our results imply a GntR1-dependent fine-tuning of these fluxes at the transcriptional level. Advances in understanding of the unique transcriptional regulatory network consisting of various carbon metabolism genes under the control of GntR1/2 along with SugR, RamA, RamB, GlxR, etc., should be valuable for fine-tuning of bioprocesses using this industrially important microorganism.
FIG 4.

Model of the regulation of carbon catabolic genes by GntR1 in C. glutamicum. Metabolic pathways and genes activated by GntR1 are in red, while blue indicates repression. Abbreviations: G6P, glucose 6-phosphate; 6PGL, 6-phospho glucono 1,5-lactone; 6PG, 6-phospho gluconate; Ru5P, ribulose 5-phosphate; Xu5P, xylulose 5-phosphate; R5P, ribose 5-phosphate; GA3P, glyceraldehyde 3-phosphate; S7P, sedoheptulose 7-phosphate; F6P, fructose 6-phosphate; E4P, erythrose 4-phosphate.
Supplementary Material
ACKNOWLEDGMENTS
We thank C. A. Omumasaba (RITE) for critical reading of the manuscript. We thank K. Toyoda for valuable advice on microarray data analysis.
This work was partially supported by a grant from the New Energy and Industrial Technology Development Organization (NEDO).
Footnotes
Published ahead of print 30 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01860-14.
REFERENCES
- 1.Kalinowski J, Bathe B, Bartels D, Bischoff N, Bott M, Burkovski A, Dusch N, Eggeling L, Eikmanns BJ, Gaigalat L, Goesmann A, Hartmann M, Huthmacher K, Krämer R, Linke B, McHardy AC, Meyer F, Möckel B, Pfefferle W, Pühler A, Rey DA, Rückert C, Rupp O, Sahm H, Wendisch VF, Wiegräbe I, Tauch A. 2003. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J. Biotechnol. 104:5–25. 10.1016/S0168-1656(03)00154-8 [DOI] [PubMed] [Google Scholar]
- 2.Liebl W. 2005. Corynebacterium taxonomy, p 9–34 In Eggeling L, Bott M. (ed), Handbook of Corynebacterium glutamicum. CRC Press, Boca Raton, FL [Google Scholar]
- 3.Inui M, Murakami S, Okino S, Kawaguchi H, Vertès AA, Yukawa H. 2004. Metabolic analysis of Corynebacterium glutamicum during lactate and succinate productions under oxygen deprivation conditions. J. Mol. Microbiol. Biotechnol. 7:182–196. 10.1159/000079827 [DOI] [PubMed] [Google Scholar]
- 4.Okino S, Noburyu R, Suda M, Jojima T, Inui M, Yukawa H. 2008. An efficient succinic acid production process in a metabolically engineered Corynebacterium glutamicum strain. Appl. Microbiol. Biotechnol. 81:459–464 [DOI] [PubMed] [Google Scholar]
- 5.Okino S, Suda M, Fujikura K, Inui M, Yukawa H. 2008. Production of D-lactic acid by Corynebacterium glutamicum under oxygen deprivation. Appl. Microbiol. Biotechnol. 78:449–454. 10.1007/s00253-007-1336-7 [DOI] [PubMed] [Google Scholar]
- 6.Ikeda M, Nakagawa S. 2003. The Corynebacterium glutamicum genome: features and impacts on biotechnological processes. Appl. Microbiol. Biotechnol. 62:99–109. 10.1007/s00253-003-1328-1 [DOI] [PubMed] [Google Scholar]
- 7.Yukawa H, Omumasaba CA, Nonaka H, Kós P, Okai N, Suzuki N, Suda M, Tsuge Y, Watanabe J, Ikeda Y, Vertès AA, Inui M. 2007. Comparative analysis of the Corynebacterium glutamicum group and complete genome sequence of strain R. Microbiology 153:1042–1058. 10.1099/mic.0.2006/003657-0 [DOI] [PubMed] [Google Scholar]
- 8.Schröder J, Tauch A. 2010. Transcriptional regulation of gene expression in Corynebacterium glutamicum: the role of global, master and local regulators in the modular and hierarchical gene regulatory network. FEMS Microbiol. Rev. 34:685–737. 10.1111/j.1574-6976.2010.00228.x [DOI] [PubMed] [Google Scholar]
- 9.Teramoto H, Inui M, Yukawa H. 2011. Transcriptional regulators of multiple genes involved in carbon metabolism in Corynebacterium glutamicum. J. Biotechnol. 154:114–125. 10.1016/j.jbiotec.2011.01.016 [DOI] [PubMed] [Google Scholar]
- 10.Teramoto H, Inui M, Yukawa H. 2009. Regulation of expression of genes involved in quinate and shikimate utilization in Corynebacterium glutamicum. Appl. Environ. Microbiol. 75:3461–3468. 10.1128/AEM.00163-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wendisch VF, de Graaf AA, Sahm H, Eikmanns BJ. 2000. Quantitative determination of metabolic fluxes during coutilization of two carbon sources: comparative analyses with Corynebacterium glutamicum during growth on acetate and/or glucose. J. Bacteriol. 182:3088–3096. 10.1128/JB.182.11.3088-3096.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toyoda K, Teramoto H, Inui M, Yukawa H. 2011. Genome-wide identification of in vivo binding sites of GlxR, a cyclic AMP receptor protein-type regulator in Corynebacterium glutamicum. J. Bacteriol. 193:4123–4133. 10.1128/JB.00384-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frunzke J, Engels V, Hasenbein S, Gatgens C, Bott M. 2008. Co-ordinated regulation of gluconate catabolism and glucose uptake in Corynebacterium glutamicum by two functionally equivalent transcriptional regulators, GntR1 and GntR2. Mol. Microbiol. 67:305–322. 10.1111/j.1365-2958.2007.06020.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka Y, Ehira S, Teramoto H, Inui M, Yukawa H. 2012. Coordinated regulation of gnd, which encodes 6-phosphogluconate dehydrogenase, by the two transcriptional regulators GntR1 and RamA in Corynebacterium glutamicum. J. Bacteriol. 194:6527–6536. 10.1128/JB.01635-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klaffl S, Brocker M, Kalinowski J, Eikmanns BJ, Bott M. 2013. Complex regulation of the phosphoenolpyruvate carboxykinase gene pck and characterization of its GntR-type regulator IolR as a repressor of myo-inositol utilization genes in Corynebacterium glutamicum. J. Bacteriol. 195:4283–4296. 10.1128/JB.00265-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inui M, Suda M, Okino S, Nonaka H, Puskás LG, Vertès AA, Yukawa H. 2007. Transcriptional profiling of Corynebacterium glutamicum metabolism during organic acid production under oxygen deprivation conditions. Microbiology 153:2491–2504. 10.1099/mic.0.2006/005587-0 [DOI] [PubMed] [Google Scholar]
- 17.Tanaka Y, Okai N, Teramoto H, Inui M, Yukawa H. 2008. Regulation of the expression of phosphoenolpyruvate: carbohydrate phosphotransferase system (PTS) genes in Corynebacterium glutamicum R. Microbiology 154:264–274. 10.1099/mic.0.2007/008862-0 [DOI] [PubMed] [Google Scholar]
- 18.Inui M, Kawaguchi H, Murakami S, Vertès AA, Yukawa H. 2004. Metabolic engineering of Corynebacterium glutamicum for fuel ethanol production under oxygen-deprivation conditions. J. Mol. Microbiol. Biotechnol. 8:243–254. 10.1159/000086705 [DOI] [PubMed] [Google Scholar]
- 19.Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2:28–36 [PubMed] [Google Scholar]
- 20.Tanaka Y, Teramoto H, Inui M, Yukawa H. 2011. Translation efficiency of antiterminator proteins is a determinant for the difference in glucose repression of two beta-glucoside phosphotransferase system gene clusters in Corynebacterium glutamicum R. J. Bacteriol. 193:349–357. 10.1128/JB.01123-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Auchter M, Cramer A, Hüser A, Rückert C, Emer D, Schwarz P, Arndt A, Lange C, Kalinowski J, Wendisch VF, Eikmanns BJ. 2011. RamA and RamB are global transcriptional regulators in Corynebacterium glutamicum and control genes for enzymes of the central metabolism. J. Biotechnol. 154:126–139. 10.1016/j.jbiotec.2010.07.001 [DOI] [PubMed] [Google Scholar]
- 22.Krause JP, Polen T, Youn JW, Emer D, Eikmanns BJ, Wendisch VF. 2012. Regulation of the malic enzyme gene malE by the transcriptional regulator MalR in Corynebacterium glutamicum. J. Biotechnol. 159:204–215. 10.1016/j.jbiotec.2012.01.003 [DOI] [PubMed] [Google Scholar]
- 23.Engels V, Lindner SN, Wendisch VF. 2008. The global repressor SugR controls expression of genes of glycolysis and of the L-lactate dehydrogenase LdhA in Corynebacterium glutamicum. J. Bacteriol. 190:8033–8044. 10.1128/JB.00705-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han SO, Inui M, Yukawa H. 2008. Transcription of Corynebacterium glutamicum genes involved in tricarboxylic acid cycle and glyoxylate cycle. J. Mol. Microbiol. Biotechnol. 15:264–276. 10.1159/000117614 [DOI] [PubMed] [Google Scholar]
- 25.Han SO, Inui M, Yukawa H. 2007. Expression of Corynebacterium glutamicum glycolytic genes varies with carbon source and growth phase. Microbiology 153:2190–2202. 10.1099/mic.0.2006/004366-0 [DOI] [PubMed] [Google Scholar]
- 26.Gourdon P, Baucher MF, Lindley ND, Guyonvarch A. 2000. Cloning of the malic enzyme gene from Corynebacterium glutamicum and role of the enzyme in lactate metabolism. Appl. Environ. Microbiol. 66:2981–2987. 10.1128/AEM.66.7.2981-2987.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Omumasaba CA, Okai N, Inui M, Yukawa H. 2004. Corynebacterium glutamicum glyceraldehyde-3-phosphate dehydrogenase isoforms with opposite, ATP-dependent regulation. J. Mol. Microbiol. Biotechnol. 8:91–103. 10.1159/000084564 [DOI] [PubMed] [Google Scholar]
- 28.Eikmanns BJ, Rittmann D, Sahm H. 1995. Cloning, sequence analysis, expression, and inactivation of the Corynebacterium glutamicum icd gene encoding isocitrate dehydrogenase and biochemical characterization of the enzyme. J. Bacteriol. 177:774–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kiefer P, Heinzle E, Zelder O, Wittmann C. 2004. Comparative metabolic flux analysis of lysine-producing Corynebacterium glutamicum cultured on glucose or fructose. Appl. Environ. Microbiol. 70:229–239. 10.1128/AEM.70.1.229-239.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoon Yang T, Wittmann C, Heinzle E. 2006. Respirometric 13C flux analysis. Part II. In vivo flux estimation of lysine-producing Corynebacterium glutamicum. Metab. Eng. 8:432–446. 10.1016/j.ymben.2006.03.002 [DOI] [PubMed] [Google Scholar]
- 31.Sahm H, Eggeling L, de Graaf AA. 2000. Pathway analysis and metabolic engineering in Corynebacterium glutamicum. Biol. Chem. 381:899–910 [DOI] [PubMed] [Google Scholar]
- 32.Toyoda K, Teramoto H, Inui M, Yukawa H. 2009. Involvement of the LuxR-type transcriptional regulator RamA in regulation of expression of the gapA gene, encoding glyceraldehyde-3-phosphate dehydrogenase of Corynebacterium glutamicum. J. Bacteriol. 191:968–977. 10.1128/JB.01425-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waldminghaus T, Skarstad K. 2010. ChIP on Chip: surprising results are often artifacts. BMC Genomics 11:414. 10.1186/1471-2164-11-414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Homann OR, Johnson AD. 2010. MochiView: versatile software for genome browsing and DNA motif analysis. BMC Biol. 8:49. 10.1186/1741-7007-8-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Machanick P, Bailey TL. 2011. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27:1696–1697. 10.1093/bioinformatics/btr189 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.