

Abstract
The human gut microbiota plays a central role in human well-being and disease. In this study, we present an integrated, iterative approach of computational modeling, in vitro experiments, metabolomics, and genomic analysis to accelerate the identification of metabolic capabilities for poorly characterized (anaerobic) microorganisms. We demonstrate this approach for the beneficial human gut microbe Faecalibacterium prausnitzii strain A2-165. We generated an automated draft reconstruction, which we curated against the limited biochemical data. This reconstruction modeling was used to develop in silico and in vitro a chemically defined medium (CDM), which was validated experimentally. Subsequent metabolomic analysis of the spent medium for growth on CDM was performed. We refined our metabolic reconstruction according to in vitro observed metabolite consumption and secretion and propose improvements to the current genome annotation of F. prausnitzii A2-165. We then used the reconstruction to systematically characterize its metabolic properties. Novel carbon source utilization capabilities and inabilities were predicted based on metabolic modeling and validated experimentally. This study resulted in a functional metabolic map of F. prausnitzii, which is available for further applications. The presented workflow can be readily extended to other poorly characterized and uncharacterized organisms to yield novel biochemical insights about the target organism.
INTRODUCTION
The human gut microbiome is dominated by four phyla, the Bacteroidetes and Firmicutes, which make up 90 to 99% of the identified phylotypes in metagenomic analysis, as well as the Actinobacteria and the Proteobacteria (1), yet it contains an estimated 1,000 species (2). The Human Microbiome Project (3) catalog currently contains over 700 reference genomes of human gut microbes, most of which are still biochemically uncharacterized.
Among firmicutes, F. prausnitzii is one of the most abundant species in humans, accounting for around 8% of the total colonic microbiota (4), and plays an important role in the healthy gut. Indeed, F. prausnitzii was found to be underrepresented in the gut microbiotas of patients with Crohn's disease, active ulcerative colitis, alternating-type irritable bowel syndrome (IBS-A) (5), and diabetes type II (6, 7). One of its main fermentation products, the short-chain fatty acid butyrate, serves as the main energy source for colonocytes and has anti-inflammatory properties (8). F. prausnitzii has been proposed as a potential probiotic for treatment of gut inflammation, as the microbe stimulates the expression of the anti-inflammatory cytokine interleukin 10 (IL-10) in peripheral blood mononuclear cells in vitro. Furthermore, cell supernatants of F. prausnitzii culture reduced the secretion of proinflammatory IL-8 and strongly inhibited NF-κB activation in cancer cells (9). Butyrate alone did not provoke the observed inhibitory effect (9), demonstrating that F. prausnitzii likely secretes an unknown anti-inflammatory metabolite apart from butyrate.
The fermentation pathways and butyrate-producing mechanisms of F. prausnitzii are well described (10). Under in vitro conditions, F. prausnitzii growth is strongly stimulated in the presence of acetate (11). However, other metabolic pathways have been comparatively poorly studied, and its growth requirements are not known (12). F. prausnitzii is known to utilize a variety of carbohydrates, including the prebiotic inulin, apple pectin, and some host-derived carbon sources, such as d-glucosamine and N-acetyl-d-glucosamine (11, 13, 14).
Genome-scale metabolic reconstructions (GENREs) are frequently used in systems biology. These GENREs are generated in a bottom-up manner and capture the genetic, genomic, and biochemical traits of a given organism. The number of well-curated GENREs is increasing steadily (15). Particularly well represented are microbes colonizing the human body (16). GENREs have been applied successfully to the elucidation of biochemical and metabolic properties of a variety of microorganisms, including the Fe(III) reducer Geobacter sulfurreducens (17), the photosynthetic alga Chlamydomonas reinhardtii (18), and the human pathogen Mycoplasma pneumoniae (19).
Another application of GENREs is the investigation of microbial growth requirements and design of minimal media. For instance, constraint-based modeling was used for minimal medium design for the lactic acid bacterium Lactococcus lactis (20), for the eukaryotic pathogen Leishmania major, for which improvements to gene annotations were also proposed (21), and for the pathogenic bacterium Neisseria meningitidis (22). However, the described approaches targeted moderately well studied to well-studied organisms. For N. meningitidis, the amount of literature on growth requirements is particularly high, which facilitated design of a minimal medium (22). Developing minimal media for poorly studied organisms remains challenging, as the predictive potential of a metabolic reconstruction depends on manual curation of the draft reconstruction derived from genome annotation (23).
The aim of the present study was to combine state-of-the-art computational and experimental techniques to elucidate metabolic capabilities of F. prausnitzii type strain A2-165. We assembled a highly curated and validated metabolic reconstruction from the genome sequence, limited biochemical literature, in vitro culture, and metabolomic measurements. The reconstruction was used to develop a chemically defined medium (CDM). During this process, novel secretion products were discovered, leading to refinement of its genome annotation and metabolic reconstruction. We then used the reconstruction to characterize the metabolic properties of F. prausnitzii. Novel carbon sources were proposed and validated in vitro. Finally, we propose a functional metabolic map of this important beneficial gut microbe.
MATERIALS AND METHODS
Manual curation of Model SEED draft reconstruction.
The Faecalibacterium prausnitzii A2-165 genome, consisting of 3,475 protein-coding genes, was retrieved from the Integrated Microbial Genomes-Human Microbiome Project (24) website and imported into the RAST server (25). A draft reconstruction of the microbe was retrieved from the Model SEED pipeline, which is a web-based resource that constructs analysis-ready genome-scale draft metabolic reconstructions based on the genome sequence (26). The draft reconstruction was exported in SBML format and further analyzed using COBRA Toolbox (27) methods.
The content of the draft reconstruction was subsequently manually inspected, curated, validated, and expanded based on a protocol for curation of automated models (26) and established methods in metabolic network reconstruction (23). Similar to our previously described reconstruction approach (28), we manually curated the Model SEED draft reconstruction based on literature and database mining. To streamline the nomenclature used in the draft reconstruction, reaction and metabolite names were translated to BiGG (29) standard identifiers. All reactions, metabolites, and genes included in the draft reconstruction were subsequently inspected and evaluated manually. Reaction directionalities were inspected, and appropriate directionality changes were made based on the BiGG database (29). Gap filling of central pathways was performed and species-specific pathways were filled in manually based on information from the literature. Model predictions were evaluated based on published experimental data. Carbon source utilization predicted by the model was compared with experimental data (11, 13). Furthermore, it was ensured that experimentally observed secretion products (11) could be produced by the model. At this stage of the reconstruction process, the minimal nutrient requirements of F. prausnitzii were predicted in silico (Fig. 2). A minimal medium composition was defined based on computational predictions and tested experimentally (see the section on laboratory procedures). Furthermore, experiments were performed to test carbon sources for which no experimental data were yet published. Based on the results, a final curation step was performed. The finished, manually curated and validated reconstruction, accounting for 602 genes, was named iFpraus_v1.0.
FIG 2.

Description of the overlap between the chemically defined media utilized in this study. Blue circle, compounds included in CDM1 (initial minimal medium proposed by model); orange circle, compounds included in CDM2 (expanded defined medium enabling growth); purple circle, compounds included in CDM3 (final defined medium based on LC-MS data). Green metabolites are those significantly secreted during growth on CDM2; red metabolites are those significantly consumed during growth on CDM2.
Figure 1 sums up the applied reconstruction curation and validation pipeline. A detailed description of the manual curation and validation process can be found in the supplemental material. Details on the biomass reaction can also be found in the supplemental material. The content of the final reconstruction iFpraus_v1.0 is presented in spreadsheet format in Table S7 in the supplemental material and is available in SBML format at http:/thielelab.eu.
FIG 1.
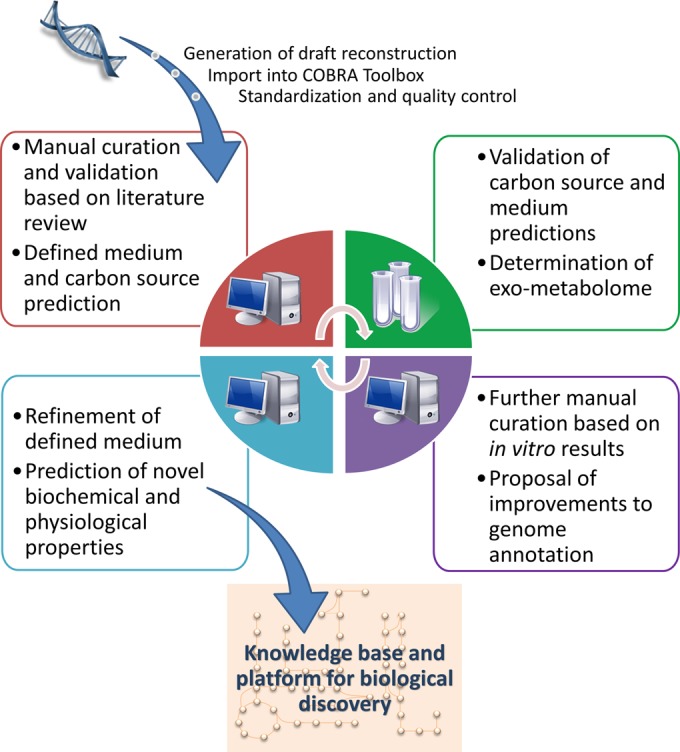
Schematic overview of the reconstruction assembly, curation, and validation pipeline employed in this study.
Genomic analysis.
Comparative genomic identification of transcription factor binding sites and reconstruction of corresponding regulons were performed according to the approach established previously (30), implemented in the RegPredict Web server tool (31). Initial DNA binding site profiles for previously described transcription factors were collected from the RegPrecise database (32). RNA regulatory elements (riboswitches) were identified using the RibEx tool (33).
In silico growth simulations.
Unless stated otherwise, for all simulations with glucose as the carbon source, uptake of glucose was allowed at a rate of 10 mmol g (dry weight)−1 h−1. The uptake rates of other carbon sources were always scaled to the number of carbon atoms with glucose (six carbons) as a reference, e.g., when the glucose uptake rate was 10 mmol g (dry weight)−1 h−1, the uptake rate for lactose (12 carbon atoms) was 5 mmol g (dry weight)−1 h−1.
Gene essentiality in iFpraus_v1.0 was determined using methods implemented in the COBRA Toolbox while simulating growth on glucose minimal medium (see Table S8 in the supplemental material) and rich medium, which consisted of all compounds the model is able to transport. Genes whose deletion resulted in a growth rate of zero were considered essential.
In silico carbon source utilization was tested by simulating minimal medium (see Table S8 in the supplemental material) while omitting glucose and adding the carbon sources one by one.
For simulated YCFAG10 and YCFAG(O2) media (i.e., medium containing yeast extract, Casitone, and fatty acids and supplemented with 25 mM glucose, and the same medium used to determine oxic growth), glucose uptake was allowed up to an uptake rate of 100 mmol g (dry weight)−1 h−1, and oxygen uptake was allowed up to an uptake rate of 100 mmol g (dry weight)−1 h−1. Yeast extract was assumed to consist of all metabolites included in the yeast reconstruction iMM904 (34) that could also be transported by iFpraus_v0.2. Casitone was assumed to consist of all 20 amino acids. Simulation constraints are listed in Table S9 in the supplemental material.
Constraints used to simulate the expanded defined medium CDM2 are listed in Table S10 in the supplemental material.
Phenotypic phase plane analysis.
Phenotypic phase plane analysis was performed as described previously (35). Briefly, fluxes through two exchange reactions representing metabolite uptake or secretion were fixed at different intervals, while biomass production was set as the objective function. For each step, the objective value and the shadow prices for all metabolites in the model were computed and plotted as heat maps. The analysis was performed while minimal medium was simulated (see Table S8 in the supplemental material), with the exception that acetate and carbon source exchange were varied.
Strain used for experimental culture.
A stock of Faecalibacterium prausnitzii A2-165 DSM 17677, isolated by S. H. Duncan (University of Aberdeen, United Kingdom) was donated to and maintained by H. J. M. Harmsen, Department of Medical Microbiology, University of Groningen, Groningen, The Netherlands.
Growth experiments.
Faecalibacterium prausnitzii strain A2-165 was grown under anaerobic conditions at 37°C on CDM1, CDM2, and YCFAG medium (see the supplemental material). One or two colonies of F. prausnitzii A2-165 grown on YCFAG agar were inoculated into the respective growth medium. Carbon source utilizations were performed by adding filter-sterilized carbon source solutions to YCFA medium. The final concentration of carbon sources in the solution was 1 mg/ml. Growth was confirmed by Gram staining as well as measuring acid production. In both minimal medium and carbon source tests, a decrease in pH value of >0.2 was considered indicative of growth.
Growth curve.
A growth curve was plotted for F. prausnitzii A2-165 grown on CDM2. Each growth curve represented the average for three biological replicates. Cell numbers were determined with fluorescence in situ hybridization (FISH) for samples collected at 0 h, 4 h, 8 h, 12 h, 16 h, 20 h, and 24 h. The FISH was performed with the Cy3-labeled probe Fprau645 (36) as described before (37).
Liquid chromatography-mass spectrometry (LC-MS).
Samples for mass spectrometry analysis were taken from the in vitro cultures grown in CDM2 described above for the entire length of the experiment (before inoculation and at 4, 8, 12, 16, 20, and 24 h). The results represented the average of three biological replicates.
An ultra-high-performance liquid chromatography (UPLC) system (UPLC Acquity; Waters, Manchester, United Kingdom) coupled in line with a quadrupole-time of flight hybrid mass spectrometer (Synapt G2; Waters, Manchester, United Kingdom) was used for the analysis of small polar metabolites in spent culture medium as previously reported (38). All materials used in the LC-MS experiments were purchased from Sigma-Aldrich (Germany). All chemicals and solvents were of analytical-grade or higher purity.
A one-way analysis of variance (ANOVA) was used to define measurements presenting a significant change over time (P < 0.05 and P < 0.01).
Growth medium preparation.
The composition of the initial minimal medium, CDM1, was determined by assessing the minimal requirements of the F. prausnitzii reconstruction after manual curation based on databases and literature. To determine the composition of a chemically defined medium allowing F. prausnitzii to grow, the initial minimal medium was enriched with additional vitamins, amino acids, and bases, resulting in CDM2. CDM1 and CDM2 were prepared as follows: components (the composition is presented in Tables S11 and S12 in the supplemental material) were added to double-distilled H2O, the final pH value was set to 6.8, and the solutions were filter sterilized. Cysteine (final concentration 1 mg/ml) was used as a reducing agent, while resazurin was added as a redox indicator.
YCFAG medium was prepared as previously reported (11). Before autoclaving, the pH value of the solution was set to 6.5. YCFA medium consisted of the same components as YCFAG medium, except that glucose was omitted, and was prepared in the same way.
RESULTS
We present an approach combining in silico and in vitro steps to build a high-quality reconstruction of F. prausnitzii A2-165 and gain insight into its metabolic potential. A schematic overview of the workflow is presented in Fig. 1.
Generation of a curated draft metabolic reconstruction.
A draft metabolic reconstruction for any microbial organism with sequence genome can be obtained using the web-based Model SEED resource (26). While this resource greatly accelerates the reconstruction process, substantial manual curation of its content is required to ensure accordance between known biochemical and physiological capabilities of the target organism and its in silico representation.
The F. prausnitzii A2-165 genome, consisting of 3,475 protein-coding genes, was obtained from the Integrated Microbial Genomes-Human Microbiome Project (24) and imported into the RAST server (25). The resulting draft reconstruction, named iFpraus_v0.1, was manually curated, which included inspection of reaction directionalities and gap filling of central pathways and of species-specific pathways based on biochemical information from the available 23 primary research articles. Most importantly, it was ensured that known secretion products (11) and carbon source utilization capabilities (11, 13) were accurately represented. This refined reconstruction was named iFpraus_v0.2 and consisted of 997 reactions across two compartments (extracellular space and cytosol), 818 metabolites, and 585 genes (Table 1). The manual validation and curation process is described in detail in the supplemental material.
TABLE 1.
Comparison of automated (iFpraus_v0.1), curated (iFpraus_v.02), and experimentally validated (iFpraus_v1.0) F. prausnitzii A2-165 reconstruction
| Feature | iFpraus_v0.1 | iFpraus_v0.2 | iFpraus_v1.0 |
|---|---|---|---|
| No. of: | |||
| Total reactions | 820 | 997 | 1,030 |
| Metabolic and transport reactions | 755 | 855 | 873 |
| Exchange and demand reactions | 65 | 142 | 157 |
| Gene-associated reactions | 734 | 793 | 807 |
| Compartment-specific metabolites | 874 | 818 | 833 |
| Genes | 598 | 585 | 602 |
| % reversible reactions | 62.80 | 39.22 | 39.13 |
| % irreversible reactions | 37.20 | 60.78 | 60.87 |
| No. of: | |||
| Blocked reactions | 649 | 220 | 222 |
| Compartments | 2 | 2 | 2 |
| Usable carbohydrates as sole carbon sources | 3 | 17a | 17a |
| Produced published/measured secretion products | 1 | 11 | 17 |
| Ability to produce biomass | No | Yes | Yes |
See Table S1 in the supplemental material.
An established way of assessing the predictive potential of the models derived from the metabolic reconstructions is the comparison of in silico phenotypic traits with experimental data (23). To determine the model's ability to produce known secretion products, biomass production by iFpraus_v0.2 was simulated on YCFAG medium using flux variability analysis. The model predicted that acetate was consumed and butyrate was produced to achieve optimal growth. Formate and d-lactate were produced in some alternate solutions (Table 2). The model thus captured production of F. prausnitzii's major secretion products (11). The calculated growth rate was compared with experimental data collected for F. prausnitzii strain A2-165 grown experimentally in rumen fluid-containing minimal medium with 0.2% glucose (M2G medium) with and without acetate in medium (11). Growth on rumen fluid was approximated by simulating YCFAG medium, which consists of acetate, glucose, Casitone, yeast extract, cysteine, minerals, and vitamins. The predicted growth rate was 0.29 h−1 with acetate in the medium and 0.24 h−1 without acetate. The in vitro growth rate on rumen fluid-containing M2G medium was 0.32 h−1 with acetate and 0.10 h−1 when acetate was omitted (11). While one cannot directly compare in silico and in vitro growth rates, as the medium conditions are different, the model captures F. prausnitzii's known trait that acetate supplementation enhances growth (11). Similarly, qualitative carbon source utilization capabilities predicted by the model were compared with experimental data (11, 13) and found to be in agreement (see Table S1 in the supplemental material).
TABLE 2.
Growth rates and allowed flux spans of secretion products predicted for iFpraus_v0.2 on YCFAG mediuma
| Simulation | Prediction on YCFAG medium |
|---|---|
| Growth rate (h−1) | 0.29 |
| Production (mmol g (dry wt)−1 h−1) of: | |
| Acetate | −16.13 to −8.61 |
| Butyrate | 12.82 to 17.13 |
| Formate | 0 to 5.03 |
| d-Lactate | 0 to 2.51 |
| CO2 | 15.83 to 21.21 |
Constraints for simulated YCFAG medium are listed in Table S9 in the supplemental material. Flux variability analysis (FVA) was carried out with 95% satisfaction of objective required.
Design of a chemically defined medium using an iterative computational, in vitro, and metabolomics approach.
F. prausnitzii A2-165 is routinely cultivated on YCFAG medium but fails to grow when yeast extract is omitted from the growth medium. Furthermore, the absence of acetate significantly retards growth (11). Hitherto, growth requirements for specific vitamins, cofactors, minerals, and amino acids have not been reported. The metabolic model (iFpraus_v0.2) predicted the inability of F. prausnitzii A2-165 to synthesize the amino acids alanine, cysteine, methionine, serine, and tryptophan and the vitamin and cofactors biotin, cobalamin, folic acid, hemin, nicotinic acid or nicotinamide, pantothenic acid, and riboflavin (see Table S2 in the supplemental material). These in silico phenotypes were used to define in vitro a chemically defined medium (named CDM1); however, F. prausnitzii A2-165 was unable to grow on it. CDM1 was supplemented with amino acids, nucleobases, and vitamins (Fig. 2). This modified CDM1, named CDM2, did indeed support the growth of F. prausnitzii in vitro. The experimentally determined growth rate was 0.13 h−1, compared to an in silico growth rate of 0.26 h−1. An explanation of the higher in silico growth rate could be that the applied uptake constraints are higher than the in vitro uptakes fluxes.
To optimize the composition of CDM2, the spent CDM2 of F. prausnitzii was quantified by LC-MS (38) to reveal consumed and secreted metabolite profiles. We found that the concentration of 28 metabolites changed significantly after 20 h growth in CDM2. Of those, 18 were net consumed by F. prausnitzii, while 10 were net secreted (see Table S2 in the supplemental material). Six predicted nonessential amino acids were consumed in significant amounts, namely, arginine, histidine, (iso)leucine, lysine, and phenylalanine (note that isoleucine and leucine cannot be distinguished by our LC-MS approach). Of those, only phenylalanine was included in CDM1, and the remaining five may thus be some of the missing components whose absence prevented growth on CDM1. Significant secretion of glutamine and threonine was observed. Furthermore, five amino acids (alanine, cysteine, proline, serine, and valine) were net secreted, but the observed changes were statistically not significant (see Table S2 in the supplemental material). Of the added metabolites in CDM2, 11, including all four added nucleobases, were taken up (Fig. 2; also, see Table S2 in the supplemental material). These findings reveal that CDM2 represents a defined nonminimal medium that enables growth of F. prausnitzii in silico and in vitro.
F. prausnitzii consumes and secretes a variety of compounds.
We aimed to identify nonessential metabolites through the consumed and secreted metabolite profiles for growth on CDM2. No growth was observed when adenine and xanthine were removed from the medium. The precise mechanism for this phenotype remained ambiguous, as complete biosynthesis pathways for both bases are annotated in the F. prausnitzii A2-165 genome (see Table S3 in the supplemental material). LC-MS analysis revealed a highly significant decrease in adenine (P = 2.363 × 10−7) and a corresponding increase in hypoxanthine (P = 7.034 × 10−7) (see Table S2 in the supplemental material), suggesting that adenine is metabolized via adenine deaminase (FAEPRAA2165_01453 or FAEPRAA2165_01454; EC 3.4.5.2), liberating ammonium ion (Fig. 3). This indicates that adenine serves as an important nitrogen source for these bacteria. Consistently, in silico simulations predicted adenine, unlike other nucleobases, as well as ammonium ion to serve as a sole nitrogen source (see Table S2 in the supplemental material).
FIG 3.

Overview of proposed pathways in F. prausnitzii A2-165 based on genome annotation, the exometabolome, and growth experiments. Putative pathways for which no gene annotation could be identified, as well as substrates known to be transported but for which transporter gene annotations are unknown, are indicated with a question mark. Transporter mechanisms are indicated by icons (see the key).
Furthermore, hemin could be omitted. Hemin biosynthesis is absent (see Table S3 in the supplemental material), and the lack of a requirement suggests that heme-containing proteins may not be encoded by F. prausnitzii A2-165. Indeed, we were unable to identify a cytochrome c oxidase in its genome (see the supplemental material).
We observed a significant uptake of the vitamins biotin, pantothenic acid, and pyridoxal, confirming the model's predictions that they are essential (see Table S2 in the supplemental material). Cobalamin, folic acid, and riboflavin consumption did not reach significance, although they are predicted to be essential. Riboflavin auxotrophy has been previously demonstrated for F. prausnitzii A2-165 (39). Possibly, riboflavin consumption did not reach significance because the vitamin is mainly utilized for an extracellular electron shuttle (39). Thiamine was significantly taken up (see Table S2 in the supplemental material) despite a biosynthesis pathway being annotated in F. prausnitzii A2-165. Surprisingly, pyridoxamine was consumed in significant amounts (see Table S2 in the supplemental material). However, no gene for pyridoxamine 5′-phosphate oxidase (EC 1.4.3.5) could be identified in the F. prausnitzii A2-165 genome, which is a prerequisite for pyridoxamine utilization.
A statistically significant net secretion of p-aminobenzoic acid in the medium was observed between 0 and 4 h, while a decrease occurred between 4 and 20 h, suggesting that F. prausnitzii may be able to utilize or synthesize p-aminobenzoic acid. However, we could not identify the p-aminobenzoic acid biosynthesis pathway in the genome (see Table S2 in the supplemental material), in line with previous results that F. prausnitzii M21/2 does not possess the complete folate biosynthesis pathway (40).
Finally, F. prausnitzii secreted five previously unreported metabolites (see Table S2 in the supplemental material), namely, dihydroorotic acid, N-acetylglutamic acid, N-acetylaspartic acid, and 3-methy2-oxovaleric acid, as well as phenyllactic acid, which has already been shown to be produced by F. prausnitzii strains M21/2 and SL3/3 (41).
We then aimed to refine CDM2. Glutamine and threonine could be omitted from CDM2, in line with model predictions that F. prausnitzii A2-165 is prototrophic for these amino acids. Pyridoxamine and p-aminobenzoic acid could further be omitted, confirming that pyridoxal is a sufficient source of vitamin B6 but pyridoxamine may be unusable. Furthermore, guanine, uracil, and orotic acid could be omitted. Biosynthesis pathways for uracil, guanine, and orotic acid are completely annotated in F. prausnitzii A2-165, confirming that these growth components are nonessential. Deletion of these three compounds from CDM2 resulted in poor and unreliable growth, however. We conclude that F. prausnitzii's growth is unstable in noncomplex media, possibly due to low nutrient concentrations.
The composition of the more refined form of CDM2, named CDM3, which allows growth, though poor and unreliable, of F. prausnitzii in vitro is shown in Fig. 2.
Exometabolomic data guide the refinement of F. prausnitzii's genome annotation.
Uptake of six vitamins was observed, for which the current annotation for F. prausnitzii A2-165 in NCBI Protein, The Seed, IMG, and BioCyc does not include transporters (see Table S3 in the supplemental material). Using comparative genomics analysis, we identified energy-coupling factor (ECF) family transporters for biotin, folic acid, pyridoxine, pantothenic acid, riboflavin, and thiamine in the F. prausnitzii A2-165 genome, supporting the idea that these vitamins can be consumed (Table 3). All transporters require shared ecfAAT energizing components (encoded by FAEPRAA2165_02788-02790), which are energy-coupling modules required for substrate binding that include the components EcfA, EcfA′, and EcfT (42). Furthermore, a currently unannotated pyridoxine-responsive regulon, corresponding to PdxR (FAEPRAA2165_02613), and a niacin-responsive regulon, corresponding to NiaR (FAEPRAA2165_01816), which regulate the associated ECF transporters, were identified. Some of the transporter-encoding genes mentioned are currently mis- or unannotated (Table 3).
TABLE 3.
Proposed improvements and specifications to the genome annotation of F. prausnitzii A2-165
| NCBI Protein ID | Current annotation (NCBI)a | Proposed annotation |
|---|---|---|
| FAEPRAA2165_01205 | Hypothetical protein | Pantothenate transporter gene panT |
| FAEPRAA2165_01298 | Hypothetical protein | Riboflavin transporter gene ribU |
| FAEPRAA2165_01333 | Putative proton-coupled thiamine transporter YuaJ | Thiamine transporter gene thiT |
| FAEPRAA2165_01388 | 4-Phosphoerythronate dehydrogenase | d-Lactate dehydrogenase |
| FAEPRAA2165_01654 | TrpR family protein YerC/YecD | Histidine repressor HisR |
| FAEPRAA2165_01745 | ABC transporter substrate binding protein | Tryptophan ABC transporter gene trpX |
| FAEPRAA2165_01747 | Branched-chain amino acid ABC transporter, permease protein | Tryptophan ABC transporter gene trpY |
| FAEPRAA2165_01748 | ABC transporter, ATP-binding protein | Tryptophan ABC transporter gene trpZ |
| FAEPRAA2165_01816 | 3H domain protein | Niacin-responsive regulator NiaR |
| FAEPRAA2165_01935 | Dehydrogenase, FMN dependent | Hydroxyacid oxidase gene (similar to eukaryotic gene Hao2) |
| FAEPRAA2165_02602 | Hypothetical protein | Niacin transporter gene niaY |
| FAEPRAA2165_02613 | Transcriptional regulator, GntR family | Pyridoxine-responsive regulator PdxR |
| FAEPRAA2165_02615 | Hypothetical protein | ECF-family pyridoxine transporter gene pxdT |
| FAEPRAA2165_02750 | Transporter, major facilitator family protein | MFS-type arginine transporter |
| FAEPRAA2165_02761 | Glycosyl hydrolase family 32 | Beta-fructosidase (levanase/invertase) gene |
| FAEPRAA2165_02762 | Tat pathway signal sequence domain protein | Fructooligosaccharide ABC transporter |
| FAEPRAA2165_02763 | ABC transporter, permease protein | Fructooligosaccharide ABC transporter |
| FAEPRAA2165_02764 | ABC transporter, permease protein | Fructooligosaccharide ABC transporter |
| FAEPRAA2165_02765 | Transcriptional regulator, LacI family | Fructooligosaccharides utilization transcriptional regulator SusR, LacI family |
| FAEPRAA2165_02788 | Cobalt transport protein | EcfT energizing component |
| FAEPRAA2165_02789 | ABC transporter, ATP-binding protein | EcfA′ energizing component |
| FAEPRAA2165_02790 | Cobalt ABC transporter, ATP-binding protein | EcfA energizing component |
| FAEPRAA2165_03014 | Hypothetical protein | Folate transporter gene folT |
| FAEPRAA2165_03033 | NlpA lipoprotein | Predicted methionine ABC transporter gene metQ |
| FAEPRAA2165_03034 | ABC transporter, ATP-binding protein | Predicted methionine ABC transporter gene metP |
| FAEPRAA2165_03035 | ABC transporter, permease protein | Predicted methionine ABC transporter gene metN |
| FAEPRAA2165_03085 | ABC transporter, substrate-binding protein, family 3 | Lysine ABC transporter gene lysX |
| FAEPRAA2165_03087 | ABC transporter, permease protein | Lysine ABC transporter gene lysY |
| FAEPRAA2165_03088 | ABC transporter, permease protein | Lysine ABC transporter gene lysZ |
| FAEPRAA2165_03374 | ABC transporter, ATP-binding protein | Histidine ABC transporter gene hisZ |
| FAEPRAA2165_03375 | ABC transporter, permease protein | Histidine ABC transporter gene hisY |
| FAEPRAA2165_03376 | ABC transporter, substrate-binding protein, family 3 | Histidine ABC transporter gene hisX |
FMN, flavin mononucleotide.
Furthermore, we aimed to confirm the presence of functional biosynthesis pathways and transporters for amino acids not present in CDM1 that were consumed in significant amounts. We newly identified a histidine regulon, corresponding to HisR (FAEPRAA2165_01654), which regulates the histidine biosynthesis operon hisGD1DCBHAFEJ (FAEPRAA2165_00489-480), a T-box regulon, leuABCD, for leucine (FAEPRAA2165_01127-31), and ilvD for isoleucine (FAEPRAA2165_00371) (Table 3). We confirmed the presence of an arginine regulon containing the arginine biosyn-thesis operon argGHCJBDF (FAEPRAA2165_00172-163), argF (FAEPRAA2165_00366), and newly identified a predicted major facilitator superfamily (MFS)-type arginine transporter (FAEPRAA2165_02750). Histidine, arginine, leucine, and isoleucine biosynthesis is thus most likely functional in F. prausnitzii A2-165. We further identified ABC-type transporters for histidine (hisXYZ; FAEPRAA2165_03376-74), lysine (lysXYZ; FAEPRAA2165_03085-88), methionine (metQPN; FAEPRAA2165_03033-35), and tryptophan (trpXYZ; FAEPRAA2165_01745-48) that are controlled by a lysine riboswitch and amino acid-specific T boxes. Finally, we identified a previously misannotated d-lactate dehydrogenase and a putative hydroxyacid oxidase (Table 3; also, see the supplemental material).
In summary, several biomass precursor transporters included in the corresponding biosynthesis regulons were identified, and 33 novel gene annotations for F. prausnitzii A2-165 were proposed. In particular, we identified the transported substrates for transporters with currently generic annotations (Table 3).
Reconstruction content, refinement, and characteristics.
iFpraus_v0.2 was consequently refined. Missing transport reactions for all metabolites observed to be consumed or secreted were added, and the corresponding transporter-including genes (Table 3) were assigned accordingly. Reactions consuming or producing the metabolites were included as necessary to connect the metabolites to the network if supporting gene annotations could be identified (Table 3; also, see the supplemental material). In total, 37 reactions, 15 nonunique metabolites, and 17 genes were added, and six reactions were replaced during the refinement based on exometabolome measurements (for details, see the supplemental material and see Table S2 in the supplemental material). The resulting final reconstruction was named iFpraus_v1.0, accounting for 602 genes, 1,030 reactions, and 833 metabolites, with a final average confidence score of 2.29 (see the supplemental material) and a genome coverage of 17%. Excluding exchange and demand reactions, only 66 of all 1,030 reactions are not supported by genome annotation. iFpraus_v1.0 is able to utilize carbon sources and produce secretion products in good agreement with literature and our in vitro experiments (see Tables S1 and S4 in the supplemental material). The expansion of the reconstruction resulted in only minor changes in growth rate and secretion product predictions (data not shown).
At its end, this integrated systems biology approach yielded a metabolic chart of F. prausnitzii summarizing its metabolic capabilities and connections with the environment (Fig. 2). Our proposed refinements to F. prausnitzii's genome annotation are summed up in Table 3. A comparison of proposed function, NCBI Protein, IMG, BioCyc, and The Seed gene annotations for all genes included in the reconstruction is listed in Table S3 in the supplemental material.
The subsystem participation of reactions in iFpraus_v1.0 was analyzed and compared with that of the previously assembled Bacteroides thetaiotaomicron strain VPI 5482 reconstruction iAH991 (28) (Fig. 4). As expected, the most striking difference between the two reconstructions is the percentage of reactions and genes involved in polysaccharide degradation, which make up 9% and 26%, respectively, in the B. thetaiotaomicron reconstruction but only 2% in F. prausnitzii (Fig. 4). This difference reflects the significantly higher saccharolytic potential of B. thetaiotaomicron, in line with observations that B. thetaiotaomicron possesses 255 glycoside hydrolases and 29 polysaccharide lyases, while the F. prausnitzii genome encodes only 17 glycoside hydrolases and one polysaccharide lyase (43). Moreover, 61% of B. thetaiotaomicron's glycosylhydrolases are noncytosolic (44), while F. prausnitzii possesses mainly cytosolic enzymes. Accordingly, 22% of the genes in the F. prausnitzii reconstruction but only 8% of the genes in the B. thetaiotaomicron reconstruction are involved in transport (Fig. 4b), in line with the fact that firmicutes possesses a number of phosphotransferase systems (PTS) for efficient nutrient uptake while B. thetaiotaomicron has no genes encoding PTS (45).
FIG 4.

Comparison of subsystem participation in the F. prausnitzii A2-165 reconstruction iFpraus_v1.0 and the B. thetaiotaomicron VPI-5482 reconstruction iAH991. (a) Reactions; (b) genes.
Prospective use of the metabolic reconstruction: assessing the metabolic capability of F. prausnitzii in silico and in vitro. (i) Insights into the central metabolism of F. prausnitzii.
Network analysis revealed the central carbon metabolism of F. prausnitzii (Fig. 3). Glucose and most other carbon sources are metabolized via glycolysis. Amino sugars enter metabolism via the amino sugar pathway with fructose-6-phosphate as the product, while uronic acids are converted to pyruvate and glyceraldehyde-3-phosphate via pentose and glucuronate interconversions. Formate is produced via pyruvate formate lyase and can be further converted to CO2 by formate dehydrogenase. The citric acid cycle is incomplete, as malate dehydrogenase (EC 1.1.1.37) and oxoglutarate dehydrogenase (EC 1.2.4.2) are not annotated. However, the steps for conversion of succinate to malate and oxaloacetate to 2-oxoglutarate are present, in agreement with the observation that malate and succinate are formed from fumarate (39). Finally, an incomplete pentose phosphate pathway is annotated in the F. prausnitzii A2-165 genome that does not allow the microbe to generate NADPH via the pentose phosphate pathway. However, the necessary enzymes for synthesis of erythrose-4-phosphate as precursors for synthesis of aromatic amino acids and 5-phosphoribosy1-pyrophosphate (PRPP), which is required for synthesis of purines and pyrimidines, are accounted for (Fig. 3).
(ii) Acetate supplementation increases energy harvest from glucose.
We investigated in silico how acetate consumption enables F. prausnitzii to harvest more energy from carbon sources. Using flux balance analysis (46) while minimizing internal flux, we calculated the predicted ATP yield per mmol glucose, without and with acetate present in the in silico medium. The predicted ATP yields were 2 and 3, respectively. Flux balance analysis revealed that, in the absence of acetate, glucose was fermented to d-lactate and ATP was produced via phosphoenolpyruvate carboxykinase (BiGG ID: PPCKr) (see Fig. S3a in the supplemental material). When acetate uptake was allowed, the model switched from d-lactate to butyrate and CO2 production, leading to proton transport to the extracellular compartment via NADH:ferredoxin oxidoreductase (BiGG ID: FDNADOX_H). This proton motive force enabled ATP production via ATP synthase (BiGG ID: ATPS4) (see Fig. S3a in the supplemental material), thus allowing more efficient utilization of glucose. The predicted flux ratio between ATP synthase and acetate uptake was 1:2; hence, 1 mmol g (dry weight)−1h−1 ATP per 2 mmol g (dry weight)−1h−1 acetate was produced.
(iii) Metabolic modeling reveals novel carbon sources.
Using flux balance analysis, novel carbon sources were predicted. iFpraus_v1.0 has exchange reactions defined for 154 unique metabolites, of which 90 contain carbons. We supplemented the in silico YCFA medium with each of these candidate carbon sources and found that 17 of 90 indeed supported growth in silico, including 14 known sole carbon sources (11, 13) (see Table S1 in the supplemental material) as well as lactose, inosine, and the host-derived monosaccharide N-acetylneuraminic acid. The utilization pathways for these three novel substrates are supported by the genome annotation (for details, see the supplemental material). Furthermore, very poor growth on malate was predicted. Unusable sole carbon sources included all amino acids and dipeptides (see Table S1 in the supplemental material), consistent with reports that F. prausnitzii has little or no ability to utilize peptides (13).
We validated 10 of these predictions in in vitro cell culture (Table 4). As predicted in silico, F. prausnitzii A2-165 grew on lactose and N-acetylneuraminic acid, confirmed by acid production. On inosine, acid production was detectable but weak, and the difference in pH value did not reach 0.2. F. prausnitzii A2-165 is thus able to utilize inosine as a carbon source, but to an extent that may not be sufficient for growth if no other usable carbon source is provided. Furthermore, recent experimental evidence suggested that fumarate can serve as a carbon source (39). The model predicted that F. prausnitzii A2-165 could utilize fumarate as an additional, though not as the sole, carbon source. Our in vitro experiments agreed with this prediction (Table 4). Furthermore, F. prausnitzii A2-165 did not grow on malate, for which poor growth was predicted (Table 4). Both compounds likely fail to sustain growth as sole carbon sources due to the loss of one carbon atom when malate is converted to pyruvate via fumarase (BiGG ID: FUM; FAEPRAA2165_03471-03472) and malic enzyme (BiGG ID: ME1; FAEPRAA2165_03473). Moreover, in agreement with model predictions, F. prausnitzii A2-165 was also unable to grow on arbutin, citrate, mannose, salicin, and succinate (Table 4).
TABLE 4.
Previously undescribed carbon source utilization predicted for iFpraus_v1.0 (F. prausnitzii A2-165) and experimental validation
| Carbon source | Relative in silico growth rate (h−1) | In vivo growth |
|---|---|---|
| Arbutin | No growth | No growth |
| Citrate | No growth | No growth |
| Fumarate | No growth | No growth |
| Inosine | 0.66 | Weak acid production |
| Lactose | 0.78 | Growth |
| Malate | 0.07 | No growth |
| Mannose | No growth | No growth |
| N-Acetylneuraminic acid | 0.98 | Growth |
| Salicin | No growth | No growth |
| Succinate | No growth | No growth |
Relative in silico growth was calculated with the predicted growth rate on glucose as a reference. A pH decrease of >0.2 was considered indicative of growth in vitro.
Taken together, these data indicate that F. prausnitzii's genome encodes numerous transporters and the microorganism is also able to utilize many host- and diet-derived metabolites for energy and biomass production.
(iv) Utilization of oxygen benefits F. prausnitzii.
Recently, it was shown that F. prausnitzii utilizes oxygen or fumarate as electron acceptors for an extracellular electron shuttle involving cysteine or glutathione and riboflavin (39). Using flux balance analysis, we investigated how the addition of oxygen to the in silico YCFAG medium benefits the microbe and changes pathway utilization by F. prausnitzii. Therefore, we modeled two conditions, anoxic growth in YCFAG medium with glucose (YCFAG10 medium) and oxic growth in YCFAG medium with glucose [YCFAG(O2) medium], after experimental conditions (39) and predicted molar yields for secretion products and biomass (see Table S5 in the supplemental material).
We inspected the pathway usage for the two conditions. Flux through the reactions representing the extracellular electron shuttle (BiGG IDs: ESHCYS_FPe, ESHCYS2_FPe, ESHGLU_FPe, and ESHGLU2_FPe) was enabled for YCFAG(O2) but not for YCFAG10 (see Fig. S3b in the supplemental material), confirming that the model correctly captured the recently discovered electron shuttle. Furthermore, oxygen consumption increased the in silico growth rate by 30% (see Table S5 in the supplemental material). Flux balance analysis revealed that flux through a flavin reductase (BiGG ID: FLVRxe; FAEPRAA2165_00362), which regenerates NAD+ from NADH (see Fig. S3b in the supplemental material), increased 15-fold under the YCFAG(O2) condition. In contrast, if no flux through FLVRxe was allowed, oxygen uptake could no longer increase the in silico growth rate (see Table S6 in the supplemental material), demonstrating that this reaction is indeed responsible for the predicted growth stimulation by oxygen. We thus propose that the uptake of oxygen leads to a higher flux through the extracellular electron shuttle and through the stoichiometrically coupled flavin reductase, which subsequently moves the NAD+/NADH ratio in a favorable direction, leading to an increase in growth yield. The model predicted acetate production rather than consumption on YCFAG(O2) medium (see Table S5 in the supplemental material). In contrast, acetate consumption was observed in vitro on YCFAG(O2) medium (39). Due to NAD+ being regenerated mainly via flavin reductase, flux through butyrate fermentation pathway, which also converts NADH to NAD+, was not enforced at optimal growth yield, and acetyl-CoA was mainly converted to acetate. The reason for this discrepancy with in vitro results may be explained by missing regulatory constraints in the model or incomplete knowledge about the molecular composition of the yeast extract present in the in vitro YCFAG medium.
(v) Phenotypic phase plane analysis predicts growth-limiting nutrients.
The effect of secretion product ratio on growth rate was predicted using phenotypic phase plane (PhPP) analysis. Briefly, in the PhPP analysis, two constraining environmental variables (e.g., flux through exchange reactions) are varied, while a third parameter (e.g., biomass production) is optimized. The analysis may reveal different regions, or “phases,” in the resulting phenotypic phase plane representing qualitatively different pathway utilizations. These phases are computed based on shadow prices. A metabolite's shadow price represents the extent to which increasing the flux through the metabolite would increase flux through the particular objective function. By definition, a positive shadow price indicates that increasing the flux through this metabolite would increase the objective function flux, while a negative shadow price represents an associated decrease in objective function flux (35). Using PhPP analysis, we computed the effect of varying carbon source uptake and acetate exchange on growth rate (Fig. 5). Furthermore, the shadow prices for all metabolites, excluding dead-end metabolites, were calculated.
FIG 5.

Phenotypic phase plane analysis performed for the F. prausnitzii A2-165 reconstruction iFpraus_v1.0. Growth rates and shadow prices were plotted as heat maps spanning the feasible solution space. (a) Glucose uptake varied against acetate exchange, plotted in three-dimensional (3D) and 2D format. (b) Galacturonic acid uptake varied against acetate exchange, plotted in 3D and 2D format.
Glucose and galacturonic acid uptake were varied against acetate exchange. The analysis revealed different phenotypic phases. As expected, increasing carbon source uptake flux increased growth rate (Fig. 5). When comparing the point of minimal carbon source uptake rate that still allowed optimal growth (indicated by a star in Fig. 5a and b), it was observed that for glucose, acetate had to be consumed, while for galacturonic acid, acetate had to be produced. The maximally achievable growth rate was equal for both carbon sources. Forcing acetate production thus limited growth on glucose, while on galacturonic acid, the growth yield decreased with increasing acetate consumption. This difference can be explained by their distinct metabolisms. Glucose is fully metabolized via glycolysis, yielding 2 mol of pyruvate per mol. Uronic acids are utilized via pentose and glucuronate interconversions, resulting in 1 mol of glyceraldehyde-3-phosphate and 1 mol of pyruvate per mol (Fig. 3). The reduced flux through glycolysis during growth on uronic acids results in a lower NADH yield per mol of carbon source. Thus, ATP gain via acetate kinase becomes preferable to the NADH-consuming butyrate pathway.
We then examined the shadow prices for the entire phase plane while varying glucose against acetate uptake (see Fig. S4 in the supplemental material). We found that at low glucose uptake rates, the model was carbon limited, as indicated by positive shadow prices for glucose and other usable carbon sources. At optimal growth, however, growth was limited by NAD+, NADP+, ATP, acetyl coenzyme A (acetyl-CoA), amino acids, and cell envelope biosynthesis building blocks. Thus, reducing equivalent availability and energy levels rather than carbon skeleton availability, as well as cost-intensive biomass precursors, is growth limiting in this phase. Furthermore, cost-intensive biomass precursors involved in fatty acid metabolism, e.g., hexadecanoic acid, switched from a positive shadow price at suboptimal growth to a negative shadow price at optimal growth. Increasing the availability of hexadecanoic acid at suboptimal growth would thus save biosynthesis costs, while at optimal growth, the metabolite would be in excess and costly to remove. Shadow price analysis thus predicts how the limited resources need to be distributed in F. prausnitzii to achieve optimal growth.
DISCUSSION
In this work, we manually constructed and validated a metabolic reconstruction of Faecalibacterium prausnitzii A2-165 starting from an automated draft reconstruction. A defined medium enabling growth of the microbe was developed. Previously proposed traits of the microbe, such as generation of a proton motive force, were captured via metabolic modeling. The model was then successfully applied to predict novel carbon source utilization capabilities, which were validated experimentally. Finally, a phenotypic phase plane analysis predicted optimal ratios between acetate exchange and carbon source uptake, as well as growth-limiting metabolites.
We, and others, have previously shown that automatically generated GENREs, based solely on genome annotation, do not adequately capture biochemical characteristics and phenotypic properties of the target organisms (28, 47, 48). However, as poorly characterized organisms have a limited amount of published biochemical data available, a key ingredient for traditional reconstruction of high-quality metabolic reconstructions (23), the genome becomes the main source of metabolic information. Here, we demonstrated that the draft reconstruction in conjunction with in vitro experiments and metabolomic measurements could greatly augment the biochemical knowledge about the target organism. In fact, we propose a comprehensive metabolic map for F. prausnitzii (Fig. 3), which summarizes all its measured, annotated, and predicted metabolic capabilities. While the reconstruction captures qualitative properties well, the predicted formate production rates relative to the butyrate production rates were too low (Table 2). This discrepancy may be due to missing regulatory or kinetic constraints. Butyrate and formate production varies drastically between the different F. prausnitzii strains (11), despite their having the same central fermentation pathways encoded in the genomes. In fact, inclusion of nonmetabolic cellular processes, such as regulation and macromolecular synthesis, with metabolic models has been shown to increase their predictive potential by reducing the set of feasible solutions significantly (49–53).
Guided by the refined metabolic reconstruction, we then developed an initial medium, CDM1, which was further improved through the analysis of metabolomic data. CDM2 enabled growth of F. prausnitzii A2-165, in contrast to CDM1, while not being minimal. The medium should prove useful, as it enables cultivation of F. prausnitzii A2-165 under defined nutrient concentrations. Growth on CDM2 (0.13 h−1) is poorer than on YCFAG medium (0.55 h−1) (62). However, F. prausnitzii is generally difficult to cultivate, and its growth is likely influenced by factors other than medium composition.
It has been proposed that the conversion of acetate to butyrate by colonic bacteria leads to further energy gains for the bacteria in the form of ATP by means of a proton motive force (10). Correspondingly, the model predicted the generation of one additional molecule of ATP per two consumed molecules of acetate via such a proton motive force. This ratio may be validated experimentally in future efforts. Furthermore, it has been shown that extracellular riboflavin drives an extracellular electron shuttle with oxygen as the terminal electron acceptor (54). In line with this observation, we predicted an increase in growth rate in the presence of oxygen due to a riboflavin-utilizing flavin reductase (FAEPRAA2165_00362), which may be a valuable target for further analysis. Moreover, we could not observe significant changes in riboflavin medium concentration, suggesting that the vitamin is utilized extracellularly (see Table S2 in the supplemental material). Riboflavin may be an important factor benefitting F. prausnitzii. It has been proposed that a diet rich in riboflavin may promote the abundance of the beneficial faecalibacteria in the human gut (39, 54).
We furthermore predicted and confirmed lactose and N-acetylneuraminic acid utilization by F. prausnitzii A2-165. N-Acetylneuraminic acid (sialic acid) is an amino sugar found in human mucus and thus extensively present in the human intestine. N-Acetylneuraminic acid utilization confers a competitive advantage to several pathogens and opportunistic pathogens colonizing the gut, including Salmonella enterica, Bacteroides fragilis, and enterohemorrhagic Escherichia coli. F. prausnitzii is one of few human gut commensals possessing the sialic acid utilization gene cluster (55). The microbe's ability to utilize sialic acid may be beneficial for the host if it can successfully outcompete pathogens in mucus-rich environments. The reconstruction further captures the fact that sialic acid can also serve as nitrogen source (55) (see Table S2 in the supplemental material).
Finally, a phenotypic phase plane analysis identified optimal ratios between carbon source uptake and acetate exchange, as well as growth-limiting nutrients that may represent bottlenecks. Maximal growth was limited not by the availability of carbon skeletons but by that of other biomass precursors, such as cost-intensive amino acids and cell envelope building blocks. The model was further predicted to be nitrogen source limited at optimal growth (see the supplemental material). F. prausnitzii might thus benefit from growth on carbon sources that are also nitrogen sources. Correspondingly, increasing the availability of N-acetylglucosamine but not of glucose was predicted to enhance growth at optimal carbon source-to-acetate exchange ratios (see Fig. S4 in the supplemental material). In agreement with this prediction, F. prausnitzii A2-165 grew better on N-acetylglucosamine than on glucose (13). The analysis further suggested that resources need to be distributed optimally to enable optimal growth. Investing resources in metabolites that do not contribute to biomass production was predicted to lower the growth rate.
Another metabolic reconstruction of F. prausnitzii A2-165, iFap484, was recently published (56). However, unlike iFpraus_v1.0, which was experimentally validated and refined based on exo-metabolomic data, iFap484 was constructed solely based on the limited available bibliome for F. prausnitzii A2-165. Furthermore, iFpraus_v1.0 accounts for 118 genes and 317 reactions more and thus has a significantly higher pathway coverage than iFap484.
The potential of systems biology to study both host-microbe and microbe-microbe interactions for microbes relevant to human health is promising (16, 57). An interesting prospective application is the construction of an in silico ex-germfree mouse model. Germfree animals selectively colonized with representative gut microbes are well-established model organisms (58). Experimental data, e.g., in vivo studies on ex-germfree animals colonized with a Bacteroidetes and a Firmicutes representative (59), including a recent study in which germfree rats were colonized selectively with F. prausnitzii and B. thetaiotaomicron (60), could thus be put in context. Furthermore, it has been proposed that increasing the abundance of beneficial faecalibacteria in the gut might have therapeutic potential and contribute to intestinal homeostasis (5). Using F. prausnitzii as a probiotic, however, would require ensuring its survival until its arrival in the colon (5). Using metabolic modeling, formulations that improve the survival of F. prausnitzii in the upper intestinal tract, and prebiotics that increase its abundance in the gut, could be predicted.
Finally, the approach presented here can be readily applied to other uncharacterized gut bacteria. This includes yet-undiscovered potential “keystone” species, which specialize in important functions found only in a selected few microbes (61). The human gut is populated by an estimated 1,000 species (2), most of which have not been characterized. Recent technological advances permit isolation and characterization of microbes from natural microbiota, but their cultivation and phenotypic characterization may be hampered by a lack of appropriate medium availability. Our proposed iterative approach using metabolic reconstruction and metabolomic methods can fill this gap. Performing multiple iterations of computation and experiments could result in the identification of a minimal medium for the target organism.
Supplementary Material
ACKNOWLEDGMENTS
We thank S. Valgeirsdóttir for assisting with the sample preparation for the LC-MS analysis and D. Ravcheev for help with the genomic analysis. We also thank R. M. T. Fleming for valuable discussions.
This work was supported by a Marie Curie International Reintegration grant 19 to I.T. (no. 249261) within the 7th European Community Framework Program and by an ATTRACT program grant to I.T. (FNR/A12/01) from the Luxembourg National Research Fund (FNR).
Footnotes
Published ahead of print 7 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01780-14.
REFERENCES
- 1.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. 2005. Diversity of the human intestinal microbial flora. Science 308:1635–1638. 10.1126/science.1110591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Dore J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Meta HITC, Bork P, Ehrlich SD, Wang J. 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–65. 10.1038/nature08821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.NIH HMP Working Group, Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C, Baker CC, Di Francesco V, Howcroft TK, Karp RW, Lunsford RD, Wellington CR, Belachew T, Wright M, Giblin C, David H, Mills M, Salomon R, Mullins C, Akolkar B, Begg L, Davis C, Grandison L, Humble M, Khalsa J, Little AR, Peavy H, Pontzer C, Portnoy M, Sayre MH, Starke-Reed P, Zakhari S, Read J, Watson B, Guyer M. 2009. The NIH Human Microbiome Project. Genome Res. 19:2317–2323. 10.1101/gr.096651.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze XL, Brown D, Stares MD, Scott P, Bergerat A, Louis P, McIntosh F, Johnstone AM, Lobley GE, Parkhill J, Flint HJ. 2011. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 5:220–230. 10.1038/ismej.2010.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miquel S, Martin R, Rossi O, Bermudez-Humaran L, Chatel J, Sokol H, Thomas M, Wells J, Langella P. 2013. Faecalibacterium prausnitzii and human intestinal health. Curr. Opin. Microbiol. 16:255–261. 10.1016/j.mib.2013.06.003 [DOI] [PubMed] [Google Scholar]
- 6.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S, Li X, Chen W, Xu R, Wang M, Feng Q, Gong M, Yu J, Zhang Y, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, LeChatelier E, Renault P, Pons N, Batto JM, Zhang Z, Chen H, Yang R, Zheng W, Li S, Yang H, Wang J, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K, Wang J. 2012. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490:55–60. 10.1038/nature11450 [DOI] [PubMed] [Google Scholar]
- 7.Karlsson FH, Tremaroli V, Nookaew I, Bergstrom G, Behre CJ, Fagerberg B, Nielsen J, Backhed F. 2013. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 498:99–103. 10.1038/nature12198 [DOI] [PubMed] [Google Scholar]
- 8.Tremaroli V, Backhed F. 2012. Functional interactions between the gut microbiota and host metabolism. Nature 489:242–249. 10.1038/nature11552 [DOI] [PubMed] [Google Scholar]
- 9.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, Grangette C, Vasquez N, Pochart P, Trugnan G, Thomas G, Blottiere HM, Dore J, Marteau P, Seksik P, Langella P. 2008. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. U. S. A. 105:16731–16736. 10.1073/pnas.0804812105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Louis P, Flint HJ. 2009. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 294:1–8. 10.1111/j.1574-6968.2009.01514.x [DOI] [PubMed] [Google Scholar]
- 11.Duncan SH, Hold GL, Harmsen HJ, Stewart CS, Flint HJ. 2002. Growth requirements and fermentation products of Fusobacterium prausnitzii, and a proposal to reclassify it as Faecalibacterium prausnitzii gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 52:2141–2146. 10.1099/ijs.0.02241-0 [DOI] [PubMed] [Google Scholar]
- 12.Miquel S, Martin R, Bridonneau C, Robert V, Sokol H, Bermudez-Humaran LG, Thomas M, Langella P. 2014. Ecology and metabolism of the beneficial intestinal commensal bacterium. Gut Microbes 5:146–151. 10.4161/gmic.27651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopez-Siles M, Khan TM, Duncan SH, Harmsen HJ, Garcia-Gil LJ, Flint HJ. 2012. Cultured representatives of two major phylogroups of human colonic Faecalibacterium prausnitzii can utilize pectin, uronic acids, and host-derived substrates for growth. Appl. Environ. Microbiol. 78:420–428. 10.1128/AEM.06858-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramirez-Farias C, Slezak K, Fuller Z, Duncan A, Holtrop G, Louis P. 2009. Effect of inulin on the human gut microbiota: stimulation of Bifidobacterium adolescentis and Faecalibacterium prausnitzii. Br. J. Nutr. 101:541–550. 10.1017/S0007114508019880 [DOI] [PubMed] [Google Scholar]
- 15.Kim TY, Sohn SB, Kim YB, Kim WJ, Lee SY. 2012. Recent advances in reconstruction and applications of genome-scale metabolic models. Curr. Opin. Biotechnol. 23:617–623. 10.1016/j.copbio.2011.10.007 [DOI] [PubMed] [Google Scholar]
- 16.Thiele I, Heinken A, Fleming RM. 2013. A systems biology approach to studying the role of microbes in human health. Curr. Opin. Biotechnol. 24:4–12. 10.1016/j.copbio.2012.10.001 [DOI] [PubMed] [Google Scholar]
- 17.Mahadevan R, Bond DR, Butler JE, Esteve-Nunez A, Coppi MV, Palsson BO, Schilling CH, Lovley DR. 2006. Characterization of metabolism in the Fe(III)-reducing organism Geobacter sulfurreducens by constraint-based modeling. Appl. Environ. Microbiol. 72:1558–1568. 10.1128/AEM.72.2.1558-1568.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang RL, Ghamsari L, Manichaikul A, Hom EF, Balaji S, Fu W, Shen Y, Hao T, Palsson BO, Salehi-Ashtiani K, Papin JA. 2011. Metabolic network reconstruction of Chlamydomonas offers insight into light-driven algal metabolism. Mol. Syst. Biol. 7:518. 10.1038/msb.2011.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wodke JA, Puchalka J, Lluch-Senar M, Marcos J, Yus E, Godinho M, Gutierrez-Gallego R, dos Santos VA, Serrano L, Klipp E, Maier T. 2013. Dissecting the energy metabolism in Mycoplasma pneumoniae through genome-scale metabolic modeling. Mol. Syst. Biol. 9:653. 10.1038/msb.2013.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oliveira AP, Nielsen J, Forster J. 2005. Modeling Lactococcus lactis using a genome-scale flux model. BMC Microbiol. 5:39. 10.1186/1471-2180-5-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chavali AK, Whittemore JD, Eddy JA, Williams KT, Papin JA. 2008. Systems analysis of metabolism in the pathogenic trypanosomatid Leishmania major. Mol. Syst. Biol. 4:177. 10.1038/msb.2008.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baart GJ, Zomer B, de Haan A, van der Pol LA, Beuvery EC, Tramper J, Martens DE. 2007. Modeling Neisseria meningitidis metabolism: from genome to metabolic fluxes. Genome Biol. 8:R136. 10.1186/gb-2007-8-7-r136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thiele I, Palsson BO. 2010. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 5:93–121. 10.1038/nprot.2009.203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Jacob B, Huang J, Williams P, Huntemann M, Anderson I, Mavromatis K, Ivanova NN, Kyrpides NC. 2012. IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res. 40:D115–D122. 10.1093/nar/gkr1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. 10.1186/1471-2164-9-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, Stevens RL. 2010. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotechnol. 28:977–982. 10.1038/nbt.1672 [DOI] [PubMed] [Google Scholar]
- 27.Schellenberger J, Que R, Fleming RM, Thiele I, Orth JD, Feist AM, Zielinski DC, Bordbar A, Lewis NE, Rahmanian S, Kang J, Hyduke DR, Palsson BO. 2011. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat. Protoc. 6:1290–1307. 10.1038/nprot.2011.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heinken A, Sahoo S, Fleming RM, Thiele I. 2013. Systems-level characterization of a host-microbe metabolic symbiosis in the mammalian gut. Gut Microbes 4:28–40. 10.4161/gmic.22370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schellenberger J, Park JO, Conrad TM, Palsson BO. 2010. BiGG: a Biochemical Genetic and Genomic knowledgebase of large scale metabolic reconstructions. BMC Bioinformatics 11:213. 10.1186/1471-2105-11-213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodionov DA. 2007. Comparative genomic reconstruction of transcriptional regulatory networks in bacteria. Chem. Rev. 107:3467–3497. 10.1021/cr068309+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Novichkov PS, Rodionov DA, Stavrovskaya ED, Novichkova ES, Kazakov AE, Gelfand MS, Arkin AP, Mironov AA, Dubchak I. 2010. RegPredict: an integrated system for regulon inference in prokaryotes by comparative genomics approach. Nucleic Acids Res. 38:W299–W307. 10.1093/nar/gkq531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Novichkov PS, Laikova ON, Novichkova ES, Gelfand MS, Arkin AP, Dubchak I, Rodionov DA. 2010. RegPrecise: a database of curated genomic inferences of transcriptional regulatory interactions in prokaryotes. Nucleic Acids Res. 38:D111–D118. 10.1093/nar/gkp894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abreu-Goodger C, Merino E. 2005. RibEx: a web server for locating riboswitches and other conserved bacterial regulatory elements. Nucleic Acids Res. 33:W690–692. 10.1093/nar/gki445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mo ML, Palsson BO, Herrgard MJ. 2009. Connecting extracellular metabolomic measurements to intracellular flux states in yeast. BMC Syst. Biol. 3:37. 10.1186/1752-0509-3-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edwards JS, Ramakrishna R, Palsson BO. 2002. Characterizing the metabolic phenotype: a phenotype phase plane analysis. Biotechnol. Bioeng. 77:27–36. 10.1002/bit.10047 [DOI] [PubMed] [Google Scholar]
- 36.Suau A, Rochet V, Sghir A, Gramet G, Brewaeys S, Sutren M, Rigottier-Gois L, Dore J. 2001. Fusobacterium prausnitzii and related species represent a dominant group within the human fecal flora. Syst. Appl. Microbiol. 24:139–145. 10.1078/0723-2020-00015 [DOI] [PubMed] [Google Scholar]
- 37.Benus RFJ, van der Werf TS, Welling GW, Judd PA, Taylor MA, Harmsen HJM, Whelan K. 2010. Association between Faecalibacterium prausnitzii and dietary fibre in colonic fermentation in healthy human subjects. Br. J. Nutr. 104:693–700. 10.1017/S0007114510001030 [DOI] [PubMed] [Google Scholar]
- 38.Paglia G, Hrafnsdottir S, Magnusdottir M, Fleming RM, Thorlacius S, Palsson BO, Thiele I. 2012. Monitoring metabolites consumption and secretion in cultured cells using ultra-performance liquid chromatography quadrupole-time of flight mass spectrometry (UPLC-Q-ToF-MS). Anal. Bioanal. Chem. 402:1183–1198. 10.1007/s00216-011-5556-4 [DOI] [PubMed] [Google Scholar]
- 39.Khan MT, Duncan SH, Stams AJ, van Dijl JM, Flint HJ, Harmsen HJ. 2012. The gut anaerobe Faecalibacterium prausnitzii uses an extracellular electron shuttle to grow at oxic-anoxic interphases. ISME J. 6:1578–1585. 10.1038/ismej.2012.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ames TD, Rodionov DA, Weinberg Z, Breaker RR. 2010. A eubacterial riboswitch class that senses the coenzyme tetrahydrofolate. Chem. Biol. 17:681–685. 10.1016/j.chembiol.2010.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Russell WR, Duncan SH, Scobbie L, Duncan G, Cantlay L, Calder AG, Anderson SE, Flint HJ. 2013. Major phenylpropanoid-derived metabolites in the human gut can arise from microbial fermentation of protein. Mol. Nutr. Food Res. 57:523–535. 10.1002/mnfr.201200594 [DOI] [PubMed] [Google Scholar]
- 42.Rodionov DA, Hebbeln P, Eudes A, ter Beek J, Rodionova IA, Erkens GB, Slotboom DJ, Gelfand MS, Osterman AL, Hanson AD, Eitinger T. 2009. A novel class of modular transporters for vitamins in prokaryotes. J. Bacteriol. 191:42–51. 10.1128/JB.01208-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khan MT. 2013. Novel physiological and metabolic insights into the beneficial gut microbe Faecalibacterium prausnitzii—from carbohydrates to current. University Medical Center Groningen, University of Groningen, Groningen, The Netherlands [Google Scholar]
- 44.Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, Hooper LV, Gordon JI. 2003. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science 299:2074–2076. 10.1126/science.1080029 [DOI] [PubMed] [Google Scholar]
- 45.Brigham CJ, Malamy MH. 2005. Characterization of the RokA and HexA broad-substrate-specificity hexokinases from Bacteroides fragilis and their role in hexose and N-acetylglucosamine utilization. J. Bacteriol. 187:890–901. 10.1128/JB.187.3.890-901.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Orth JD, Thiele I, Palsson BO. 2010. What is flux balance analysis? Nat. Biotechnol. 28:245–248. 10.1038/nbt.1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bautista EJ, Zinski J, Szczepanek SM, Johnson EL, Tulman ER, Ching WM, Geary SJ, Srivastava R. 2013. Semi-automated curation of metabolic models via flux balance analysis: a case study with Mycoplasma gallisepticum. Plos Comput. Biol. 9:e1003208. 10.1371/journal.pcbi.1003208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dreyfuss JM, Zucker JD, Hood HM, Ocasio LR, Sachs MS, Galagan JE. 2013. Reconstruction and validation of a genome-scale metabolic model for the filamentous fungus Neurospora crassa using FARM. PLoS Comput. Biol. 9:e1003126. 10.1371/journal.pcbi.1003126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Covert MW, Knight EM, Reed JL, Herrgard MJ, Palsson BO. 2004. Integrating high-throughput and computational data elucidates bacterial networks. Nature 429:92–96. 10.1038/nature02456 [DOI] [PubMed] [Google Scholar]
- 50.Karr JR, Sanghvi JC, Macklin DN, Gutschow MV, Jacobs JM, Bolival B, Jr, Assad-Garcia N, Glass JI, Covert MW. 2012. A whole-cell computational model predicts phenotype from genotype. Cell 150:389–401. 10.1016/j.cell.2012.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thiele I, Fleming RM, Que R, Bordbar A, Diep D, Palsson BO. 2012. Multiscale modeling of metabolism and macromolecular synthesis in E. coli and its application to the evolution of codon usage. PLoS One 7:e45635. 10.1371/journal.pone.0045635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thiele I, Fleming RM, Bordbar A, Schellenberger J, Palsson BO. 2010. Functional characterization of alternate optimal solutions of Escherichia coli's transcriptional and translational machinery. Biophys. J. 98:2072–2081. 10.1016/j.bpj.2010.01.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun Y, Fleming RM, Thiele I, Saunders MA. 2013. Robust flux balance analysis of multiscale biochemical reaction networks. BMC Bioinformatics 14:240. 10.1186/1471-2105-14-240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khan MT, Browne WR, van Dijl JM, Harmsen HJ. 2012. How can Faecalibacterium prausnitzii employ riboflavin for extracellular electron transfer? Antioxid. Redox Signal. 17:1433–1440. 10.1089/ars.2012.4701 [DOI] [PubMed] [Google Scholar]
- 55.Almagro-Moreno S, Boyd EF. 2009. Insights into the evolution of sialic acid catabolism among bacteria. BMC Evol. Biol. 9:118. 10.1186/1471-2148-9-118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.El-Semman IE, Karlsson FH, Shoaie S, Nookaew I, Soliman TH, Nielsen J. 2014. Genome-scale metabolic reconstructions of Bifidobacterium adolescentis L2-32 and Faecalibacterium prausnitzii A2-165 and their interaction. BMC Syst. Biol. 8:41. 10.1186/1752-0509-8-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karlsson FH, Nookaew I, Petranovic D, Nielsen J. 2011. Prospects for systems biology and modeling of the gut microbiome. Trends Biotechnol. 29:251–258. 10.1016/j.tibtech.2011.01.009 [DOI] [PubMed] [Google Scholar]
- 58.Smith K, McCoy KD, Macpherson AJ. 2007. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin. Immunol. 19:59–69. 10.1016/j.smim.2006.10.002 [DOI] [PubMed] [Google Scholar]
- 59.Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A, Shah N, Wang C, Magrini V, Wilson RK, Cantarel BL, Coutinho PM, Henrissat B, Crock LW, Russell A, Verberkmoes NC, Hettich RL, Gordon JI. 2009. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc. Natl. Acad. Sci. U. S. A. 106:5859–5864. 10.1073/pnas.0901529106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wrzosek L, Miquel S, Noordine ML, Bouet S, Chevalier-Curt MJ, Robert V, Philippe C, Bridonneau C, Cherbuy C, Robbe-Masselot C, Langella P, Thomas M. 2013. Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol. 11:61. 10.1186/1741-7007-11-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ze X, Le Mougen F, Duncan SH, Louis P, Flint HJ. 2013. Some are more equal than others: the role of “keystone” species in the degradation of recalcitrant substrates. Gut Microbes 4:236–240. 10.4161/gmic.23998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Duncan SH, Louis P, Thomson JM, Flint HJ. 2009. The role of pH in determining the species composition of the human colonic microbiota. Environ. Microbiol. 11:2112–2122. 10.1111/j.1462-2920.2009.01931.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.