

Abstract
Heterogeneous vancomycin-intermediate Staphylococcus aureus (hVISA) clinical strain Mu3 spontaneously generates VISA strains at an extremely high frequency (≥1 × 10−6). The generated VISA strains usually grow more slowly than does the parent hVISA strain, but they form colonies on vancomycin-containing agar plates before 48 h of incubation. However, we noticed a curious group of VISA strains, designated “slow VISA” (sVISA), whose colonies appear only after 72 h of incubation. They have extremely prolonged doubling times but have vancomycin MICs of 8 to ∼24 mg/liter when determined after 72 to ∼144 h of incubation. We established strain Mu3-6R-P (6R-P), which has a vancomycin MIC of 16 mg/liter (at 72 h), as a representative sVISA strain. Its cell wall was thickened and autolytic activity was decreased compared to the respective qualities of the parent hVISA strain Mu3. Whole-genome sequencing of 6R-P revealed only one mutation, encoded by rpoB (R512P), which replaced the 512th arginine of the RNA polymerase β-subunit with proline. Its VISA phenotype was unstable, and the strain frequently reverted to hVISA with concomitant losses of pinpoint colony morphology and cell wall thickness and reduced autolytic activity. Sequencing of the rpoB genes of the phenotypic revertant strains revealed mutations affecting the 512th codon, where the proline of 6R-P was replaced with leucine, serine, or histidine. Slow VISA generated in the tissues of an infected patient serves as a temporary shelter for hVISA to survive vancomycin therapy. The sVISA strain spontaneously returns to hVISA when the threat of vancomycin is lifted. The rpoB(R512P) mutation may be regarded as a regulatory mutation that switches the reversible phenotype of sVISA on and off.
INTRODUCTION
Vancomycin-intermediate Staphylococcus aureus (VISA) strains appear spontaneously within the cell population of heterogeneously vancomycin-intermediate S. aureus (hVISA) strains at high frequencies of ≥10−6 (1). The hVISA-to-VISA conversion is caused by either a single mutation or some combined mutations in dozens of discrete genes, explaining the highly frequent occurrence of hVISA-to-VISA conversion (2–4). We showed that a single mutation of ≥20 genes of various metabolic pathways can confer hVISA-to-VISA conversion of S. aureus strain Mu3 and its related strains (4). These genes, such as tarA, tarO, cmk, pykA, trpC, ureD, rpoB, rpoC, and gtaB, were among them. The most prevalent of them were cmk and rpoB; five mutations, each localized in different codons of the respective gene, were shown to cause hVISA-to-VISA conversion as a single contributor (4). Thus, the mechanisms of hVISA-to-VISA conversion are extremely diverse (5, 6).
Although some of the VISA-converted mutant strains have significantly prolonged doubling times, they mostly form colonies on the vancomycin agar plates before 48 h of incubation. They are presumed to be the antecedents of clinically observed VISA strains that emerged from hVISA strains during vancomycin therapy for infected patients. In this study, however, we focused on the VISA mutant strains that appear only after 72 h of incubation on vancomycin-containing plates. This group of VISA strains, designated “slow VISA” (sVISA), has not been given attention before, because these strains were considered aberrant variants of hVISA whose clinical significance was trivial or negligible. We studied strain Mu3-6R-P (6R-P) as a representative of the Mu3-derived sVISA strain, which was isolated in 1997 on an agar plate containing 6 mg/liter vancomycin (1). We found that the strain had a vancomycin MIC of 16 mg/liter, as judged at 72 h incubation, and an extremely prolonged doubling time. It grew slowly even in the absence of vancomycin, but large colonies frequently appeared during propagation of the strains without vancomycin. Whole-genome sequencing revealed that the phenotypic conversion correlated with the mutation located in the 512th codon of the rpoB gene encoding the β-subunit of RNA polymerase holoenzyme. The biological properties of the rpoB mutation were investigated, and the clinical significance of the sVISA phenotype is discussed here.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The S. aureus strains and plasmids used in this study are listed in Table 1. The cloning and transformation of Escherichia coli strain DH5α were carried out by standard techniques (TaKaRa Bio, Inc., Shiga, Japan). All S. aureus strains were cultivated in brain heart infusion (BHI) broth or agar (Difco Laboratories, Detroit, MI) with aeration at 37°C, unless indicated otherwise. The antibiotics tetracycline and chloramphenicol (Sigma-Aldrich Co., St. Louis, MO) were used for the selection of the S. aureus transformants. Vancomycin (Sigma-Aldrich) was used for population analysis and selection of 6R-P.
TABLE 1.
Bacterial strains and plasmids used in this study
| S. aureus strain or plasmid | Descriptiona | Reference or source |
|---|---|---|
| Strains | ||
| Mu3 | hVISA clinical isolate from JUH in 1996, carrying vraS(I5N), stably forms large-sized colonies | 1 |
| Mu50 | VISA clinical isolate from JUH in 1996, carrying vraS(I5N) and rpoB(H481Y) | 1 |
| 6R-P | Mutant isolated from Mu3 by selection with 6 mg/liter vancomycin; spontaneously generates LC and SC | This study |
| L1 | Derivative of 6R-P with large colony morphology | This study |
| L2 | Derivative of 6R-P with large colony morphology | This study |
| L3 | Derivative of 6R-P with large colony morphology | This study |
| N315ΔIP (= ΔIP) | mecI-null mutant (mecI::tetL) of N315P, an N315-derivative strain cured of PCase plasmid | 41 |
| ΔIPrpoB(R512P) | ΔIP introduced with rpoB(R512P) mutation by allelic replacement | This study |
| H14 (= ΔIPvraS(S329L) | Mutant derived from ΔIP by selection with 8 mg/liter imipenem, carrying vraS(S329L) mutation | 10 |
| H14rpoB(R512P) | H14 introduced with rpoB(R512P) mutation by allelic replacement | This study |
| Plasmids | ||
| pKOR1 | E. coli-S. aureus shuttle vector for construction of allelic replacement | 8 |
| pKOR1-rpoB(512P) | pKOR1 harboring the 1,884-bp PCR product of the rpoB gene encompassing its rpoB(R512P) mutation | This study |
| pKOR1-vraS(329L) | pKOR1 harboring the 1,000-bp PCR product of the vraS gene encompassing its vraS(S329L) mutation | 10 |
JUH, Juntendo University Hospital. The strains obtained by antibiotic selection are denoted as mutants.
DNA methods.
The standard methods for DNA manipulations were described previously (7). Genomic DNAs and plasmids were prepared with the QIAamp minikit and the miniprep kit (Qiagen, Inc., Valencia, CA). The restriction enzymes were used as recommended by the manufacturer (TaKaRa). Routine PCR amplification was performed using the Expand high-fidelity system (Roche, Mannheim, Germany).
Construction of ΔIPrpoB(R512P) and H14rpoB(R512P) mutant strains.
For the chromosomal allele replacement of rpoB(R512P) in S. aureus strain N315 or N315ΔIP (ΔIP), we used the pKOR1 allele replacement system, as described previously (8, 9). In brief, a 1,884-bp sequence of rpoB insert DNA encompassing a 1-kb flanking sequence of a phage attachment site was generated by PCR from the chromosomal DNA of strain 6R-P using the primers attB1-rpoB (5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTAAATGGATATTCTGTTATAGTTATATAATA-3′) and attB2-rpoB (5′-GGGG ACCACTTTGTACAAGAAAGCTGGGTCCAGAATCACGTGCTGCAACGTGTTCCA-3′). The resulting plasmids, pKO-rpoB(512P) and pKO-vraS(329L) (10), were introduced by electroporation into S. aureus strains ΔIP and ΔIPrpoB(R512P), respectively, generating the transformants ΔIP(pKO-rpoB[512P]) and ΔIPrpoB(R512P) (pKO-vraS[329L]). The replacement of the chromosomal rpoB or vraS gene was carried out by the two-step procedure, as described previously (10). We confirmed the absence of any unwanted mutations in and around the introduced allele by single nucleotide polymorphism (SNP) analysis (see below for description).
Antibiotic susceptibility tests.
The MIC value was examined by Etest (AB Biodisk, Solna, Sweden), in accordance with the manufacturer's recommendation. BHI agar was used instead of Mueller-Hinton (MH) agar, because BHI agar is more supportive of the expression of the VISA phenotype than any other tested agar (11). In addition to the orthodox 2-fold antibiotic dilution, linear sets of vancomycin concentrations with 1 mg per liter increments were used for the range of 1 to 16 mg per liter. The plates were then incubated at 37°C and read after 48 h. The Etest MIC determination for vancomycin (AB Biodisk) and population analysis were performed under the cultivation of BHI agar.
Growth curve and doubling time.
The 105 CFU of preculture was inoculated into 10 ml of BHI broth and incubated at 37°C with shaking at 25 rpm in a TN-2612 incubator (Advantec, Osaka, Japan). Doubling time (DT) was calculated by the slope of a linear line obtained from a semilogarithmic (semi-log) graph of the growth curve. The log2 optical density at 660 nm (OD660) versus time was plotted for each strain in the exponential growth phase. The DT was calculated with the formula [(t2 − t1) × log2]/(log2 OD660 at t2 − log2 OD660 at t1), where t2 and t1 are the times at the end and start of the logarithmic growth phase, respectively.
Transmission electron microscopy.
The preparation and examination of S. aureus cells by transmission electron microscopy were performed as described previously (3, 12). The cells were grown to an OD660 of about 2.0, using a cultivation temperature of 37°C. Morphometric evaluation of cell wall thickness was performed by using photographs of images taken by an electron microscope (model H-7100; Hitachi, Tokyo, Japan) at a final magnification of ×30,000, and the cell wall thickness was measured as previously described (12). At least 100 cells of each strain with nearly equatorial cut surfaces were measured for an evaluation of cell wall thickness, and the results were expressed as the mean values ± standard deviations in nm.
Whole-genome sequencing of 6R-P.
Whole-genome sequencing of 6R-P was performed with a Genome Sequencer 20 system, a recently introduced highly parallel genome sequencer from 454 Life Sciences (Branford, CT). The sequence assembly and gap closing were carried out as described previously (10, 13). Since 6R-P is an in vitro derivative of Mu3, the resulting sequence of the 6R-P genome was compared to that of Mu3. Genome-wide identification of single nucleotide polymorphisms (SNPs) was performed by whole-genome alignments with the MUMmer (version 3.20) software package (http://mummer.sourceforge.net/). PCR amplification and sequence confirmation of 6R-P chromosomal DNA found no additional mutations, except for an rpoB mutation, which causes substitution of the 512th Arg with Pro.
SNP analysis.
To analyze the SNPs of four strains, 6R-P, 6R-P-L1 (L1), 6R-P-L2 (L2), and ΔIPrpoB(R512P), the 90-bp paired-end read sequencing was performed using a HiSeq 2000 sequencing platform (Illumina, Inc., San Diego, CA) at the Infobio company (Tokyo, Japan). Sequences of 90-bp paired-end reads from each bacterial strain were obtained in a single lane of a flow cell. Image analysis was performed with the Illumina Pipeline Analysis software version 1.8 (14). As a de novo DNA analysis, each read was mapped to the reference whole-genome sequence of Mu3 or N315, and then SNPs were extracted using Genome Traveler software (In Silico Biology, Inc., Yokohama, Japan).
Autolysis assay.
Triton X-100-stimulated autolysin activity in Tris-HCl buffer (pH 7.5) was measured as described previously (15). The cells were grown to the mid-exponential phase (OD660, 2.0), using a cultivation temperature of 37°C. The culture was rapidly chilled, and the cells were washed twice with ice-cold distilled water and suspended to an OD660 of 2.0 in 50 mM Tris-HCl buffer supplemented with 0.05% Triton X-100. Autolysis was measured during incubation at 30°C as a decrease in OD660 using a model TN2612 biophotorecorder (Advantec, Osaka, Japan). All data from the autolysis experiments are reported as percentages of the initial turbidity (at the zero time point).
RNA preparation and microarray analysis.
RNA extraction, cDNA labeling, hybridization, and data analysis for microarray analysis were carried out according to protocols described previously (16). The cells were grown midexponentially to an OD660 of about 2.0, using a cultivation temperature of 37°C. A customized high-density synthetic oligonucleotide array of pre-methicillin-resistant S. aureus (MRSA) clinical strain N315 was designed by using the 2,814,816-bp chromosome genome sequence, including 2,628 predicted open reading frame (ORFs), under the GenBank accession no. NC_002745 (NimbleGen Systems, Inc., Madison, WI). Furthermore, the other 1,128 ORFs coming from the following 13 clinical S. aureus strains and a plasmid, COL, JH1, JH9, MRSA252, MSSA476, Mu3, Mu50, MW2, NCTC 8325, Newman, RF122, USA300 FPR, USA300 TCF, and pLW043, which were absent from N315, were also designed on this array (total, 3,846 ORFs per array). Each probe (60-mer) was replicated 3 times on the microarray to allow for intra-array reproducibility measurements. The arrays were scanned using the NimbleGen MS200 microarray scanner (NimbleGen Systems, Inc.), and the data were extracted by NimbleScan software (NimbleGen Systems, Inc.).
To investigate the statistical analysis of the microarray data, normalization of signal intensity was performed by means of robust multichip analysis (RMA) algorithm (17) and global normalization. Using the three normalized signal intensities, the statistical significance of the data was evaluated with Student's t test. n-fold change ratios were calculated by using the average of the normalized signal intensity. Volcano plot analysis was performed as described previously (18) and constructed by plotting the negative log10 of the P value by t test on the y axis and the log2 of n-fold change on the x axis.
Isolation of sVISA mutants from hVISA strain Mu3.
A total of 105 cells of Mu3 were inoculated into 25 tubes containing 4 ml BHI broth. After 18 h of cultivation at 37°C, portions of the 25 cultures containing about 107 CFU were spread onto BHI agar plates containing 6 mg/liter vancomycin. The colonies that grew only after 96, 120, or 144 h of incubation were picked from each vancomycin plate. A total of 34 colonies were isolated, established as strains, and subjected to further study.
Appearance rate of large colonies from sVISA.
A total of 105 CFU of mutant strain with pinpoint (P) or small (S) colony size were inoculated into 10 ml of BHI broth and incubated at 37°C for 24 h (see the results for the day 1 culture shown in Table 5). Next, a portion of the culture containing 108 CFU was inoculated into 4 ml of fresh BHI, and the daily passage was repeated up to 6 times until a large (L) colony was observed. The appearance and proportion of L colonies in the cell culture were evaluated by plating 50 μl of each of the serial 10-fold dilutions of the daily culture, followed by incubation for 18 h to count L colonies and for 48 h to calculate the total number of colonies (the sum of L, S, and P colonies).
TABLE 5.
Introduction of rpoB(R512P) mutation causes thickening of cell wall observed with transmission electron microscopy
| Strain | Phenotype | Cell wall thickness parameters (nm) |
|
|---|---|---|---|
| Drug-free measurement (mean ± SD) | Ratioa | ||
| Mu3 | hVISA | 21.5 ± 2.4 | 1 (ref) |
| Mu50 | VISA | 32.7 ± 3.0 | 1.52 |
| 6R-P | sVISA | 26.1 ± 2.6 | 1.21 |
| L1 | hVISA | 23.8 ± 2.0 | 1.11 |
| L2 | hVISA | 22.8 ± 1.8 | 1.06 |
| L3 | hVISA | 20.7 ± 1.9 | 0.96 |
| ΔIP | VSSA | 16.4 ± 2.9 | 1 (ref) |
| ΔIPrpoB(R512P) | hVISA | 26.8 ± 3.4 | 1.63 |
| H14 [= ΔIPvraS(S329L)] | hVISA | 26.1 ± 4.3 | 1 (ref) |
| H14rpoB(R512P) | sVISA | 27.3 ± 2.4 | 1.05 |
Relative cell wall thickness compared to those of the reference strains Mu3, ΔIP, and H14. ref, reference thickness.
Statistical analysis of data.
The statistical significance of the data was evaluated with Student's t test.
Microarray data accession number.
The gene expression comparisons 6R-P/Mu3, 6R-P/ΔIP, L1/Mu3, L2/Mu3, Mu3/ΔIP, and Mu50/ΔIP were deposited in the NCBI under GEO accession no. GSE50826.
RESULTS
Characteristic colony morphology of sVISA strain 6R-P.
The sVISA strain 6R-P (vancomycin MIC, 16 mg/liter after 72 h) was isolated from hVISA strain Mu3 (vancomycin MIC, 3 mg/liter) by one-step selection with 6 mg/liter vancomycin (Tables 1 and Table 2). It grew extremely slowly. After 2 days, the drug-free subculture strain 6R-P was inoculated on the drug-free BHI agar plate, followed by incubation at 37°C for 30 h. Figure 1 shows the appearance of the formed colonies of 6R-P. The colonies of 6R-P on the plate were heterogeneous in size and contained large colonies (LC) and small colonies (SC) (Fig. 1), indicating the genetic instability of the pinpoint-sized colony (PC) phenotype of the strain. The patterns of the proportion of vancomycin-resistant subpopulations (population analysis curve) of 6R-P as observed at 48 h and 72 h or longer incubation times were significantly different from one another, as illustrated in Fig. 2A. Colonies on the agar plates containing 7 mg/liter to 14 mg/liter vancomycin were observed only after 72 h of incubation, presumably due to the extremely low growth rate of 6R-P (Fig. 2A). However, after 6 days (144 h) incubation, the population curve of 6R-P became almost equivalent to that of VISA strain Mu50, showing its comparable resistance level to Mu50 (Fig. 2A). To ascertain that the PC strain represents the sVISA phenotype, we repeated colony purification of 6R-P five times in order to enrich the PCs. The resultant culture did not contain LC or SC within the 107 CFU of its cell population. We also established 13 LC strains and 4 SC strains from 6R-P culture. The LC and SC strains stably maintained their colony sizes in at least five subsequent subcultures. In contrast, 6R-P was unstable and kept generating LC or SC at frequencies of 1 in 10−7 to 10−8 during subsequent passages.
TABLE 2.
The genotype and vancomycin susceptibilities of the 6R-P-derived colony-size variants
| Strain | Colony sizea | RpoB 512th amino acid | Codon sequenceb | Vancomycin MIC (mg/liter)e |
|---|---|---|---|---|
| Mu3 | LC | Arginined | CGTd | 3 |
| 6R-P | PC | Proline | CCT | 12 |
| L1 | LC | Leucine | CTT | 3 |
| L9-2 | LC | Leucine | CTT | 3 |
| L2 | LC | Serine | TCT | 3 |
| L8 | LC | Serine | TCT | 3 |
| L12-1 | LC | Serine | TCT | 3 |
| L12-2 | LC | Serine | TCT | 3 |
| L3 | LC | Histidine | CAT | 4 |
| L4 | LC | Histidine | CAT | 3 |
| L5-1 | LC | Histidine | CAT | 4 |
| L5-2 | LC | Histidine | CAT | 3 |
| L5-3 | LC | Histidine | CAT | 3 |
| L5-4 | LC | Histidine | CAT | 3 |
| L9-1 | LC | Histidine | CAT | 3 |
| S11 | SC | Proline | CCT | 3 |
| S13 | SC | Proline | CCT | 3 |
| S14-1 | SC | Proline | CCT | 4 |
| S14-2 | SC | Proline | CCT | 3 |
| Mu50 | LC | Argininec | CGT | 12 |
LC, large colony; PC, pinpoint colony; SC, small colony.
The underlined bases are the changed nucleotides.
Mu50 RpoB has a His-to-Tyr amino acid substitution at the 481st codon.
Wild type.
Determined by Etest after 48 h of incubation.
FIG 1.
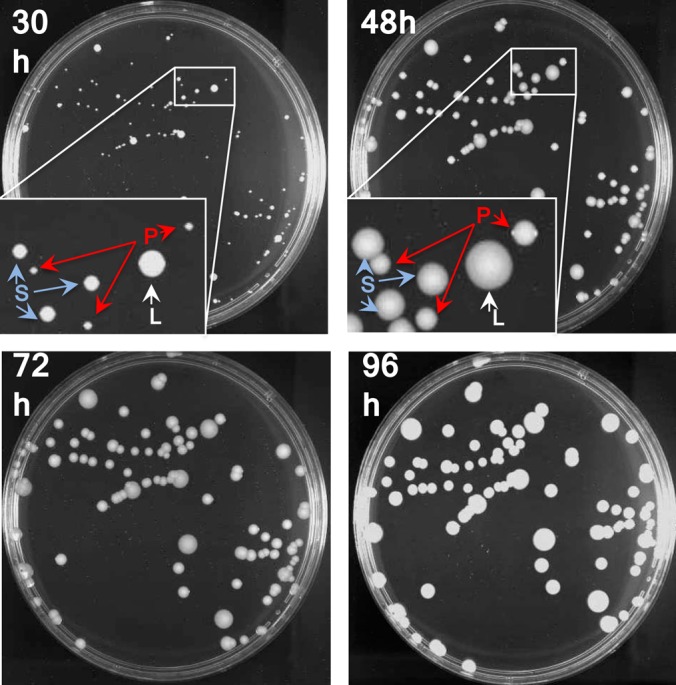
Heterogeneity in the sizes of Mu3-6R-P colonies formed on a drug-free agar plate. About 103 CFU of 6R-P after 2 days of passages in BHI broth were spread on a BHI agar plate. The photos were taken after incubation at 37°C for 30, 48, 72, and 96 h. Inset, magnification of the area in the square. Three sizes of colonies were judged after 30 h of incubation: large (L), small (S) and pinpoint (P). The L and S variants maintained their colony sizes in subsequent passages on the BHI agar plate.
FIG 2.

Analysis of vancomycin-resistant subpopulation profiles (population analysis [PA]). (A) Profiles of Mu3, 6R-P, and Mu50 observed after 48, 72, 96, 120, and 144 h of incubation at 37°C. (B) 6R-P and its phenotypic revertant strains L1, L2, and L3 as observed after 96 h of incubation at 37°C. (C) ΔIP and H14 strains and their derivatives introduced with the rpoB(R512P) mutation. About 107 CFU of the overnight culture of each strain were inoculated on BHI agar plates containing various concentrations of vancomycin. The numbers of colonies formed after incubation of 96 h at 37°C were counted and plotted in a semilogarithmic graph. Note that the shape of the PA curve of H14 is reminiscent of hVISA; however, its resistant subpopulation, as judged by the growth on the plate containing 4 mg/liter vancomycin, is much smaller than that of Mu3, which is >1 × 10−6. We call such an S. aureus strain with reduced susceptibility to vancomycin pre-hVISA.
The vancomycin MICs of the PC strains were 12 and 16 mg/liter, as evaluated by Etest at 48 h and 72 h, respectively, whereas those of the LC and SC strains were 3 to ∼4 mg/liter at both times of incubation. Therefore, the high vancomycin MIC of 6R-P was ascribed to the PC cells.
Identification of rpoB(R512P) mutation as the determinant for the sVISA phenotype and PC morphology of 6R-P.
Whole-genome sequencing identified a single nucleotide difference between 6R-P and the parent strain Mu3. The mutation was found in the rpoB gene, encoding the RNA polymerase β-subunit. The mutation caused an amino acid substitution of the 512th arginine to proline (Table 2). Unlike the rpoB(H481Y) mutation, contributing to the vancomycin resistance in VISA strain Mu50, the rpoB(R512P) mutation of 6R-P did not accompany rifampin resistance (Table 3).
TABLE 3.
Antibiogram and doubling time of Mu3-derived and N315ΔIP-derived rpoB mutant strains
| Straina | RpoB amino acid substitution | Phenotype of vancomycin resistanceb | DT (min)c | Etest MIC (mg/liter) with MH or BHI agar mediumd |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Vancomycin |
Teicoplanin |
Rifampin BHI | Daptomycin BHI | Linezolid BHI | Oxacillin BHI | Imipenem BHI | Bacitracin MH | Gentamicin MH | ||||||
| MH | BHI | MH | BHI | |||||||||||
| Clinical | ||||||||||||||
| Mu50 | H481Y | VISA | 37.1 | 8 | 12 | 12 | 24 | >32 | 4 | 0.38 | >256 | >32 | >256 | >256 |
| Mu3 | WTe | hVISA | 36.9 | 2 | 3 | 12 | 24 | 0.008 | 1.5 | 0.75 | >256 | >32 | 256 | 256 |
| Mu3-derived | ||||||||||||||
| 6R-P | R512P | sVISA | 62.2 | 4 | 12 | 16 | 32 | 0.006 | 3 | 0.5 | >256 | >32 | >256 | >256 |
| L1 | P512L | hVISA | 38.2 | 2 | 3 | 12 | 32 | 0.012 | 2 | 0.75 | >256 | >32 | 256 | 256 |
| L2 | P512S | hVISA | 39.7 | 2 | 3 | 12 | 32 | 0.008 | 2 | 0.75 | >256 | >32 | >256 | 256 |
| L3 | P512H | hVISA | 36.9 | 2 | 4 | 12 | 32 | 0.012 | 2 | 0.75 | >256 | >32 | >256 | >256 |
| N315ΔIP-derived | ||||||||||||||
| ΔIP | WT | VSSA | 26.7 | 0.5 | 1 | 1 | 1.5 | 0.008 | 0.5 | 1 | 8 | 32 | 24 | 1 |
| ΔIPrpoB(R512P) | R512P | hVISA | 41.2 | 1.5 | 4 | 2 | 4 | 0.016 | 3 | 0.75 | >256 | >32 | 256 | 0.25 |
| H14 | WT | hVISA | 28.3 | 1.5 | 3 | 4 | 16 | 0.008 | 2 | 0.75 | >256 | >32 | 64 | 0.75 |
| H14rpoB(R512P) | R512P | sVISA | 45.2 | 4 | 8 | 12 | 24 | 0.012 | 3 | 0.5 | >256 | >32 | >256 | 0.25 |
The 6R-P culture did not include LC or SC at a frequency of >1 × 10−7. H14 is equal to ΔIPvraS(S329L).
sVISA, slow VISA; hVISA, heterogeneous VISA.
DT, doubling time.
MH, Mueller-Hinton broth; BHI, brain heart infusion broth or agar.
WT, wild type.
The strains that had lost their PC morphology and vancomycin resistance were then tested for their rpoB sequences. Remarkably, all the strains that had gained the LC morphology had a second mutation in the 512th codon. The proline at position 512 of 6R-P was changed to leucine, serine, or histidine (Table 2). The strains, L1, L2, and L3, representing three different rpoB512 mutations, were further studied, together with their parent 6R-P, for the phenotypic expression of vancomycin resistance by population analysis (Fig. 2B).
While 6R-P with rpoB(R512P) showed a population curve comparable to that of Mu50 at 96 h, L1, L2, and L3 reverted to the population curves typical of hVISA strains (Fig. 2B). The low growth rate of 6R-P was also lost, together with the raised vancomycin resistance in the three rpoB512 mutant strains (Fig. 3A). These data strongly indicate that the rpoB(R512P) mutation is directly associated with the PC morphology, slow growth, and raised vancomycin resistance of 6R-P.
FIG 3.
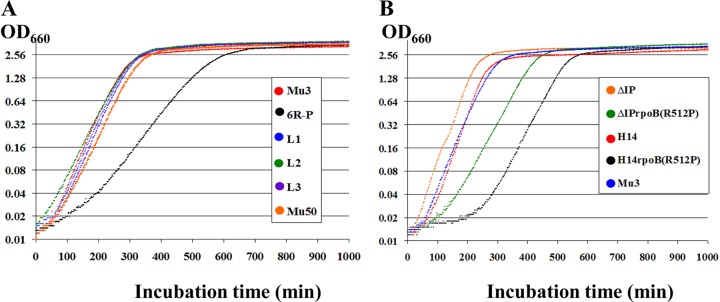
The effect of rpoB(R512P) mutations on the growth of S. aureus strains. (A) Mu3 and Mu3-derived strains in comparison with Mu50; (B) ΔIP-derived strains, and Mu3 as a control. H14 is ΔIPvraS(S329L), having a pre-hVISA phenotype (see Fig. 2C legend). Ten milliliters of BHI broth was inoculated with 107 cells of each strain, followed by incubation with shaking at 37°C for up to 1,000 min. The OD was monitored every 2 min during the cultivation.
The rpoB(R512P) mutation in VSSA strain ΔIP and its derivative strain H14.
To further confirm the effect of the rpoB(R512P) mutation in the cells of different genetic constitutions, the mutated rpoB allele was transferred into a vancomycin-susceptible S. aureus (VSSA) strain, ΔIP, and an in vitro-generated hVISA strain, ΔIP-H14 (H14), an ΔIP derivative in which the vraSR two-component system is derepressed due to the mutation vraS(S329L), to establish ΔIPrpoB(R512P) and H14rpoB(R512P), respectively (Table 1). As shown in Table 3, the vancomycin MICs evaluated by Etest at 48 h for ΔIPrpoB(R512P) and H14rpoB(R512P) were 4 mg/liter and 8 mg/liter, respectively. This was remarkable, since a single mutation was able to convert a VSSA strain into VISA, as judged by the Etest method (Table 3). In the presence of the mutation vraS(S329L), the vancomycin MIC of H14rpoB(R512P) was recorded as 8 mg/liter at 48 h (Table 3), which further increased to 12 mg/liter after 96 h of incubation (see below and Table 4). The MICs of teicoplanin, daptomycin, and bacitracin increased and those of linezolid and gentamicin decreased in both rpoB mutated strains (Table 3). It was also noted that rpoB(R512P) converted the heterogeneous MRSA strain ΔIP into homogeneous MRSA by raising the level of β-lactam resistance (Table 3). The population curves of ΔIP and H14 significantly shifted toward the right upon introduction of the rpoB(R512P) mutation (Fig. 2C). In ΔIP, rpoB(R512P) converted a VSSA strain into hVISA, and in H14, it converted an hVISA strain into VISA (Fig. 2C). A significant delay of growth was observed in the strains into which rpoB(R512P) was introduced (Fig. 3B). The doubling time (DT) of ΔIPrpoB(R512P) was prolonged from 26.7 min for ΔIP to 41.2 min, and that of H14rpoB(R512P) was prolonged to 45.2 min compared to 28.3 min for H14 (Table 3). Therefore, not only the vancomycin resistance and delayed growth rate but the susceptibility patterns to a wide range of antibiotics, such as daptomycin, linezolid, bacitracin, gentamicin, and β-lactams, were also introduced into S. aureus strains ΔIP and H14 by allelic replacement of the rpoB mutation.
TABLE 4.
Properties of sVISA strains compared with those of other MRSA strains with different susceptibilities to vancomycin
| Straina | Category of vancomycin susceptibility | Colony sizeb | Doubling time (min) | Vancomycin MIC (mg/liter) evaluated by Etest after incubation time (h) ofc: |
rpoB genotyped |
No. of LC/total no. of colonies (%) observed after the indicated time (days) of serial passagee |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 48 | 72 | 96 | 120 | 144 | Amino acid change | Nucleotide change | 1f | 2 | 3 | 7 | ||||
| 1-4d | sVISA | PC | 64.8 | 6 | 8 | 8 | 8 | 8 | WT | 32/44 (72.7) | NT | NT | NT | |
| 1-5d | sVISA | PC | 70.7 | 6 | 8 | 8 | 8 | 8 | WT | 488/490 (99.6) | NT | NT | NT | |
| 2-6d | sVISA | PC | 106.6 | 8 | 12 | 12 | 16 | 16 | WT | 6/45 (13.3) | NT | NT | NT | |
| 3-4d | sVISA | PC | 123.8 | 8 | 12 | 12 | 16 | 16 | WT | 6/66 (9.1) | NT | NT | NT | |
| 4-4d | sVISA | PC | 123.8 | 8 | 12 | 12 | 12 | 12 | G744R | GGA→AGA | 217/231 (93.9) | NT | NT | NT |
| 4-5d | sVISA | PC | 68.6 | 8 | 8 | 8 | 12 | 12 | WT | 56/74 (75.7) | NT | NT | NT | |
| 5-4d | sVISA | SC | 60.2 | 8 | 12 | 12 | 12 | 12 | WT | 7/51 (13.7) | NT | NT | NT | |
| 5-5d | sVISA | PC | 58.3 | 8 | 12 | 12 | 12 | 12 | S746F | TCT→TTT | − | 17/6.3 × 104 | NT | NT |
| 6-4d | sVISA | PC | 71.5 | 24 | 24 | 32 | 32 | 32 | WT | − | 2/6.8 × 106 | NT | NT | |
| 11-6d | sVISA | PC | 111.8 | 8 | 16 | 16 | 16 | 16 | WT | 330/335 (98.5) | NT | NT | NT | |
| 12-5d | sVISA | PC | 66 | 8 | 12 | 12 | 12 | 12 | WT | 50/51 (98.0) | NT | NT | NT | |
| 14-4d | sVISA | PC | 64.2 | 8 | 8 | 8 | 8 | 8 | WT | 13/2.3× 104 | NT | NT | NT | |
| 16-4d | sVISA | PC | 61.9 | 6 | 8 | 8 | 8 | 8 | WT | − | − | − | 47/47 (100) | |
| 17-4df | sVISA | PC | 123.8 | 8 | 12 | 12 | 12 | 12 | WT | − | − | − | 4/5.2 × 104 | |
| 17-6d | sVISA | PC | 87.7 | 6 | 8 | 8 | 12 | 12 | S746F | TCT→TTT | 4/43 (9.3) | NT | NT | NT |
| 18-4d | sVISA | PC | 153.9 | 6 | 12 | 12 | 12 | 12 | G977V | GGT→GTG | 25/1,410 (1.8) | NT | NT | NT |
| 18-5d | sVISA | PC | 144.4 | 6 | 12 | 12 | 12 | 12 | WT | 2/1.4 × 104 | NT | NT | NT | |
| 18-6df | sVISA | PC | 182.4 | 4 | 4 | 6 | 6 | 8 | WT | − | − | − | 3/320 (0.9) | |
| 19-4d | sVISA | PC | 141.5 | 8 | 12 | 16 | 24 | 24 | WT | − | 3/1.0× 104 | NT | NT | |
| 19-5d | sVISA | PC | 63 | 8 | 12 | 12 | 12 | 12 | WT | 2/361 (0.5) | NT | NT | NT | |
| 20-4d | sVISA | PC | 64.8 | 12 | 24 | 24 | 24 | 24 | WT | 7/41 (17.0) | NT | NT | NT | |
| 20-6d | sVISA | PC | 77.3 | 8 | 8 | 8 | 8 | 8 | WT | 2/2,600 | NT | NT | NT | |
| 21-4d | sVISA | PC | 83.5 | 6 | 12 | 12 | 12 | 12 | H929T | CAT→TAT | − | 14/2.6× 105 | NT | NT |
| 22-4d | sVISA | SC | 59.2 | 8 | 12 | 12 | 12 | 12 | WT | 3/5,100 | NT | NT | NT | |
| 24-4d | sVISA | SC | 58.2 | 8 | 8 | 8 | 8 | 8 | WT | 2/5,100 | NT | NT | NT | |
| 25-4d | sVISA | PC | 80.6 | 8 | 8 | 12 | 12 | 12 | H929T | CAT→TAT | − | 2/640 (0.3) | NT | NT |
| Mu3 | hVISA | LC | 36.9 | 3 | 3 | 3 | 3 | 3 | WT | NT | NT | NT | NT | |
| Mu50 | VISA | LC | 37.1 | 12 | 12 | 12 | 12 | 12 | H481Y | CAT→TAT | NT | NT | NT | NT |
| 6R-P | sVISA | PC | 62.2 | 12 | 16 | 16 | 16 | 16 | R512P | CGT→CCT | 3/1.0× 107 | 8/93 (8.6) | 8/312 (41.0) | 65/65 (100) |
| L1 | hVISA | LC | 38.2 | 3 | 3 | 3 | 3 | 3 | P512L | CCT→CTT | NT | NT | NT | NT |
| L2 | hVISA | LC | 39.7 | 3 | 4 | 4 | 4 | 4 | P512S | CCT→TCT | NT | NT | NT | NT |
| L3 | VISA | LC | 36.9 | 4 | 6 | 6 | 6 | 6 | P512H | CCT→CAT | NT | NT | NT | NT |
| ΔIP | VSSA | LC | 26.7 | 1 | 1 | 1.5 | 1.5 | 1.5 | WT | NT | NT | NT | NT | |
| ΔIPrpoB(R512P) | hVISA | SC | 41.2 | 4 | 4 | 4 | 4 | 4 | R512P | CGT→CCT | − | − | 2/4.9× 105 | NT |
| H14 | pre-hVISAg | SC | 28.3 | 3 | 3 | 3 | 3 | 3 | WT | − | − | − | NT | |
| H14rpoB(R512P) | VISA | PC | 45.2 | 8 | 8 | 12 | 12 | 12 | R512P | CGT→CCT | − | 2/3.4 × 104 | NT | NT |
The first number in the name of the sVISA strains denotes the culture tube from which the strain was isolated. The suffix denotes the day of appearance of the strain as a visible colony.
Observed at 20 h after streaking of the strain on BHI agar plate without vancomycin. LC, large colony; PC, pinpoint colony; SC, small colony.
If the colony size of the strain was heterogeneous, the smaller colony was picked for the test.
Entire ORF of rpoB gene was sequenced. The Mu3 rpoB sequence was referred to as the wild-type sequence. The underlined base is the changed nucleotide. WT, wild type.
Day 1, the starting culture before propagation; NT, not tested; −, large colonies were not observed (<1 × 108).
The strain seems to have been converted to VISA by a mutation(s). It may not be regarded as sVISA because of its rather stable phenotype throughout the drug-free propagation.
Pre-hVISA, a VSSA strain whose vancomycin-resistant subpopulation does not reach the level of hVISA; i.e., the frequency of the subpopulations of cells that can grow on the plate containing 4 mg/liter of vancomycin is >1 × 10−7 but <1 × 10−6 (see Fig. 2C).
Cell wall thickening with the rpoB(R512P) mutation.
The rpoB(R512P) mutation conferred thickening of the cell wall in S. aureus cells. Thickened cell wall peptidoglycan layers are the mechanistic feature of the VISA phenotype (3, 19). Electron microscopic observation revealed that 6R-P had a thicker cell wall (26.1 ± 2.6 nm) than that of Mu3 (21.5 ± 2.4 nm). This increase in cell wall thickness was lost in the phenotypic revertant strains L1, L2, and L3, whose cell wall thicknesses were comparable to that of Mu3 (Table 5). Therefore, rpoB(R512P) does increase cell wall thickness of Mu3, but its effect was not as significant as in Mu50, whose cell wall thickness was 32.7 ± 3.0 (Table 5). In Mu50, affinity trapping and clogging of the thickened layers of peptidoglycan (PG) constitute the unique resistance mechanism of vancomycin (20). The electron microscopic observation of 6R-P indicated a relatively smaller level of contribution of cell wall thickness to the mechanism of resistance in 6R-P compared to that in Mu50. When introduced into VSSA strain ΔIP, the rpoB(R512P) mutation drastically increased cell wall thickness by 1.63-fold (Table 5). In contrast, almost no increase in cell wall thickness was observed when rpoB(R512P) was introduced into H14 (= ΔIPvraS[S329L]), whose cell wall synthesis is already activated by derepressed expression of the vraSR two-component regulatory system (Table 5). This may signify that the rpoB(R512P) and vraS(S329L) mutations have a redundant or overlapping effect on the cell wall synthesis pathway. In agreement with this view, there were vraSR two-component system regulator genes among the 390 upregulated genes in ΔIPrpoB(R512P) (see below).
The effect of rpoB(R512P) on Triton X-100-stimulated autolytic activity.
Reduced autolytic activity is often associated with VISA strains (21–29). A possible effect of rpoB(R512P) on the Triton X-100-induced autolysis assay was evaluated (Fig. 4). Strains 6R-P and ΔIPrpoB(R512P) showed significantly decreased autolytic activity compared with those of their parent strains, Mu3 and ΔIP, respectively. Moreover, the autolytic activities of L1 and L2 carrying rpoB(R512L) and rpoB(R512S), respectively, reverted to the levels of parent strain Mu3. Thus, the rpoB(R512P) mutation was shown to decrease autolysis.
FIG 4.
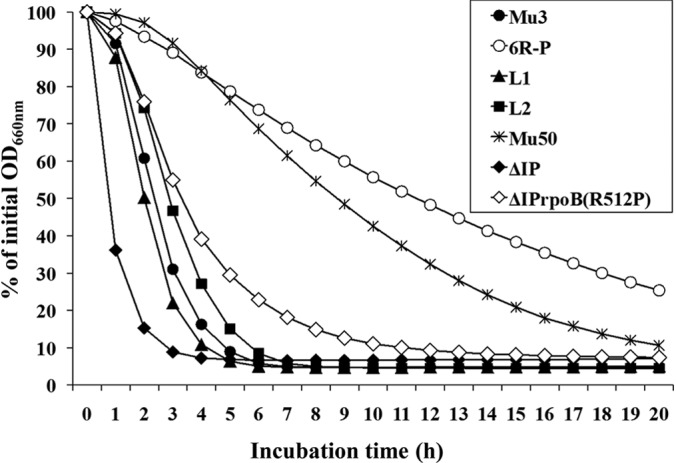
Reduction of autolytic activity in rpoB(R512P) mutant strains. Triton X-100 (0.05%)-stimulated autolysis of two wild-type rpoB strains, Mu3 and ΔIP, and four rpoB mutant strains, 6R-P, L1, L2, and ΔIPrpoB(R512P), and VISA strain Mu50 were tested. Mu50 possesses the rpoB(H481Y) mutation. Autolysis was measured as the decline in optical density versus time and is expressed as the percentage of the initial optical density.
Transcriptional profiles of sVISA strain 6R-P compared to those of VISA strain Mu50.
Microarray analysis of 6R-P versus Mu3 revealed 487 differentially expressed genes (≥2-fold or <0.5-fold; P value < 0.05). Figure 5A illustrates the volcano plots of the microarray data of sVISA strain 6R-P, its phenotypic revertant strains L1 and L2, and VISA strain Mu50 in reference to hVISA strain Mu3. The scatter plots show not only the number of differentially expressed genes but also their degrees of difference from that of Mu3. The widely dispersed pattern of the scatter plots of 6R-P became significantly diminished in the phenotypic revertants L1 and L2 (Fig. 5A). This indicated that the great transcriptional turbulence of 6R-P caused by the rpoB(R512P) mutation was ameliorated by the compensatory mutation(s) in the phenotypic revertant strains (Fig. 5A).
FIG 5.

Volcano plot analysis of transcription profiles. The −log10 (P value for a t test) values are plotted against the log ratios (log2 fold change). (A) Comparison of the level of gene expression of 6R-P, its two LC-sized variants L1 and L2, and Mu50 with that of clinical strain Mu3. (B) Comparison of the transcription profiles of 6R-P and Mu50 with those of Mu3 and ΔIP. The genes which are upregulated (red) or downregulated (blue) more than two times in Mu3 relative to VSSA strain ΔIP were downregulated and upregulated, respectively, in Mu50 relative to Mu3. However, this reversal of gene expression was not seen in 6R-P. As a result, 6R-P showed a much more scattered distribution of gene expression than Mu50 relative to VSSA strain ΔIP. The gray plots show the genes having a P value of ≥0.05 with t test.
When the volcano plot of Mu3/ΔIP was compared with those of Mu50/Mu3 and 6R-P/Mu3, a curious fact was noticeable (Fig. 5B). Most of the downregulated genes in Mu3 relative to those in ΔIP were upregulated in VISA strain Mu50 relative to Mu3, and most of the upregulated genes in Mu3 were downregulated in Mu50. This reversal of the transcriptional profile is considered a sign of relief of the fitness cost by the compensatory mutation(s), which was not observed with 6R-P (Fig. 5B). In Mu3, 499 genes were downregulated (<0.5-fold, P < 0.05), and 255 genes were upregulated (≥2-fold, P < 0.05) relative to those of ΔIP. As a consequence of the reversal of the transcriptional profile, more than half (259 genes [52%]) of the 499 depressed genes in Mu3 were upregulated, and about half (116 genes [45%]) of the 255 enhanced genes in Mu3 were downregulated in Mu50. In contrast, as many as 412 (83%) of the 499 downregulated genes and 184 (72%) of the 255 upregulated genes in Mu3 remained so in 6R-P relative to those in ΔIP. As a result, many of the genes were further depressed or enhanced compared to those in Mu3, and the pattern of the volcano plot of 6R-P/ΔIP became more scattered compared to that of Mu3/ΔIP (Fig. 5B).
Especially notable was the depressive tendency of 6R-P transcription. In 6R-P, a total of 517 and 211 genes were downregulated by >2-fold and >4-fold, respectively, relative to ΔIP. In contrast, the numbers of downregulated genes for Mu50/ΔIP were only 109 and 25, respectively.
Upregulated genes in 6R-P.
Relative to Mu3, only a total of 153 genes were upregulated (≥2-fold, P < 0.05) in 6R-P in contrast to 274 genes upregulated in Mu50/Mu3. Among them, only 24 genes were commonly upregulated. Eight of the commonly upregulated genes were transporters, including two multidrug resistance transporters, SA0650 (norA) and SA1970, and four for ferrichrome transport, SA0603 (fhuB), SA0604 (fhuG), SA1977, and SA1978.
The 129 genes specifically upregulated in 6R-P contained the capsule genes capCDEFGHIKLMNOP, urease-encoding genes ureABCEFGD, and genes involved in molybdopterin biosynthesis, moaA, moaB, mob, modC, and moeA. Three genes, lytM, sceD, and ssaA, associated with autolysis, were upregulated in Mu3/ΔIP. They were further upregulated in 6R-P but not in Mu50. The enhanced expression of all the 153 upregulated genes in 6R-P waned in L1 (L1/Mu3, <2.0-fold), except for the two genes capCD. It was evident that a single compensatory mutation, rpoB(512P) to rpoB(512L), was responsible for this drastic change.
In contrast, a total of 274 genes specifically upregulated in Mu50 contained the genes associated with the raised vancomycin resistance of Mu50, i.e., vraDE, SAS091, vraFG mprF, and dltAD genes, all controlled by the graXRS-vraFG locus (2, 30). The transcription of the genes uppP (bacA) and stgA involved in peptidoglycan synthesis were also enhanced, and the role of fbp in possibly providing more glucose to cell wall synthesis through gluconeogenesis was also enhanced significantly (4.4-fold, P < 0.002). The negative regulators of autolysis, mgrA and sarA, were also upregulated. In contrast to 6R-P (see below), the categories of genes whose activities are required for active cell growth were enhanced in Mu50, e.g., 15 ribosomal protein genes, two aminoacyl-tRNA biosynthesis genes (serS and glyS), and cell division genes (scdA, ftsW, and ftsL).
Downregulated genes in 6R-P.
Relative to Mu3, 334 and 156 genes were downregulated (<0.5, P < 0.05) in 6R-P and Mu50, respectively, of which only 52 genes were commonly downregulated. The genes in the pyrimidine synthesis pathway, pyrRPBCAA,AB,FE, and nitrate/nitrite reductase-associated genes, narG, nasD, nirR, and nasE, were commonly downregulated. The femA gene, encoding cross-bridging of peptidoglycan, was also commonly downregulated. This may contribute to raising vancomycin resistance by creating more vancomycin-binding sites (d-alanyl–d-alanine residues) within the peptidoglycan layers (20). Although downregulated in both strains, pflAB genes, encoding formate acetyltransferase activity, and adh1, encoding alcohol dehydrogenase 1, were much more strongly downregulated in 6R-P; the ratios of the three genes in Mu50/6R-P were 9.8, 14.0, and 4.5, respectively (see Table S1 in the supplemental material). Ten out of the 52 genes, including pyrAA,AB,FE, remained downregulated in L1, while the others, including all the above-mentioned genes, returned to the levels of transcription of Mu3.
As many as 281 genes were specifically downregulated in 6R-P; they were not downregulated >2-fold in Mu50 (i.e., 0.5 ≤ Mu50/Mu3). In the phenotypic revertant strain L1, all the genes returned to the transcription levels of Mu3 (0.5 ≤ L1/Mu3). Among them, five genes were significantly depressed in the teichoic acid biosynthesis pathway, tarI1, tarA, tarJ1, tarG, and tarH (2.1- to ∼3.9-fold depressed), and five genes involved in cell division, mraZ, mraW, ftsW, ftsL, and scdA, were also significantly depressed (2.0- to ∼4.6-fold depressed). Other genes significantly downregulated in 6R-P were two genes in the glycolytic pathway, gapR and gapB (3.7- and 3.5-fold depressed, respectively), two ribosomal protein genes, rpmI and rpsO (both 2.2-fold depressed), and holB, encoding the putative DNA polymerase III delta prime subunit (2.5-fold downregulated). The genes involved in autolysis, cidA, lytS, and lytR, were also downregulated (2.7-, 3.7-, and 3.7-fold depressed, respectively). Depressed expression of these genes may well correspond to the slow growth phenotype of sVISA 6R-P. Curiously, in 6R-P, sgtA, encoding a transglycosylase for peptidoglycan synthesis (31), was 3.1-fold downregulated relative to Mu3, whereas the gene was 2.6-fold upregulated in Mu50 (P < 0.00001), in which peptidoglycan synthesis is activated. There were as many as 22 regulator genes that were >2-fold downregulated: they were (with magnitude of depression) icaR (6.3-fold), lexA (4.3-fold), glnR (4.0-fold), gapR (3.7-fold), lytS and lytR (both 3.7-fold), mepR (3.2-fold), tcaR (2.2-fold), SA1949, encoding putative lytic regulatory protein (2.2-fold), and others. The transcription levels of all of these regulator genes were 0.7- to ∼1.4-fold the range found in L1/Mu3. In contrast, only one putative regulator gene, SA2296, was upregulated in 6R-P relative to Mu3. Among other genes, 2.2-fold depression was observed with SA1919, encoding putative protoporphyrinogen oxidase (hemK), a mutation of which [SAHV_2101(G121R)] was shown to convert Mu3 to VISA (4).
Characterization of other sVISA strains derived from Mu3.
About 2 × 107 CFU of Mu3 were spread on BHI agar plates containing 6 mg/liter vancomycin. After 48 h of incubation, 73 colonies were formed. After 144 h incubation, 155 colonies appeared, which was about a 2-fold greater number of colonies compared to those observed at 48 h of incubation (see Table S1 in the supplemental material). We picked a total of 34 mutually independent colonies (either those derived from different cultures or those that appeared on the same plate on different days of incubation) to establish sVISA strains. After colony purification, the colony sizes were evaluated and classified into three groups (LC, SC, and PC). Most of them (23 strains) were PC, 3 were SC, and 8 were LC strains. The vancomycin MICs of the 26 strains with pinpoint or small colony sizes were determined by Etest. The MIC values of the strains tended to increase with the time of incubation (Table 4). All of them reached MIC values of ≥8 mg/liter before 144 h (6 days) of incubation (Table 4). This was in contrast to Mu50, whose MIC was 12 mg/liter at 48 h and stayed the same up to 144 h of incubation. It was noticeable that 6 strains recorded MIC values greater than that of Mu50: 3 strains with 16 mg/liter, 2 strains with 24 mg/liter, and one strain with 32 mg/liter. The six strains possessed a pinpoint colony morphology comparable with that of 6R-P.
The average ± standard deviation (SD) doubling times of the 3 SC-sized and 23 PC-sized sVISA strains were 59.2 ± 1.0 min and 95.4 ± 35.8 min, respectively, which were much greater than those of hVISA strain Mu3 (36.9 min) and VISA strain Mu50 (37.1 min). Twenty-one out of the 26 sVISA strains grew even more slowly than 6R-P (DT, 62.2 min).
The LC variants were generated from the sVISA strains at surprisingly high frequencies. Out of the 26 sVISA strains, 18 strains (69.2%) yielded LC variants in the day 1 culture (Table 4). LC variants appeared in 23 strains (88.5%) by the third day of passage and in all of the 26 strains by the 7th day of passage (Table 6). However, two strains were much more stable than others in the frequency of appearance of LC revertants (Table 4).
TABLE 6.
Stability of rpoB mutations of sVISA strains
| Strain name | Colony sizea | Doubling time (min) | Vancomycin MIC at 48 h (mg/liter) |
rpoB mutation |
|
|---|---|---|---|---|---|
| Amino acid changeb | Codon sequence | ||||
| 17-6d (parent) | PC | 87.7 | 8 | S746F | TTT |
| 17-6d-L1 | LC | 44.4 | 3 | F746F | TTT |
| 17-6d-L2 | LC | 43.9 | 3 | F746F | TTT |
| 17-6d-L3 | LC | 64.8 | 3 | F746F | TTT |
| 21-4d (parent) | PC | 83.5 | 6 | H929T | TAT |
| 21-4d-L1 | LC | 41 | 3 | T929T | TAT |
| 21-4d-L2 | LC | 38.1 | 3 | T929H | TATGGT→CATGGGc |
| 21-4d-L3 | LC | 37.9 | 2 | T929H | TATGGT→CATGGGc |
| 21-4d-L4 | LC | 37.9 | 3 | T929H | TATGGT→CATGGGc |
| 21-4d-L5 | LC | 46.5 | 3 | H929T | TAT |
Observed at 20 h after streaking of the strain on BHI agar plate without vancomycin.
The rpoB gene sequence encompassing the single mutation was determined.
The sequences of 929th and 930th codons are shown.
Sequencing of the rpoB gene of the 26 sVISA strains revealed four amino acid substitution mutations, rpoB(G744R), rpoB(S746F), rpoB(H929T), and rpoB(G977V), in six strains (23.1%) (Table 6). As a preliminary study of the mechanism of their phenotypic instability, LC variants were isolated from two PC strains, 17-6d (MIC, 8 mg/liter) and 21-4d (MIC, 12 mg/liter), carrying the rpoB(S746F) and rpoB(H929T) mutations, respectively. Three and five LC substrains were established from independent cultures of 17-6d and 21-4d, respectively, and their rpoB sequences, DTs, and vancomycin MICs (by Etest) were determined. Their MIC values returned to 3 mg/liter and DTs to values comparable to that of the parent hVISA strain Mu3. The rpoB mutation rpoB(S746F) remained unchanged in the three LC strains derived from strain 17-6d. On the other hand, three of the five LC strains derived from 21-4d reverted to the wild-type rpoB sequences, with concomitant incorporation of a silent mutation in the next codon (Table 6). The results strongly indicate that a considerable number of sVISA cells are generated and disappear from the cell population of hVISA strain Mu3, in which an rpoB mutation is one of the frequent underlying causative genetic events.
DISCUSSION
sVISA and its clinical significance.
In this study, we introduced a novel VISA phenotype category designated slow VISA (sVISA). The first sVISA strain, 6R-P, was so named because of its extremely low growth rate. The sVISA strains form colonies on agar plates containing relatively high concentrations of vancomycin, such as 6 mg/liter, after 72 h incubation. This colony category has long been neglected for further analysis. The biological properties of sVISA are (i) slow growth, with an average ± SD doubling time of 91.3 ± 35.6 min (58.2 to 182.4 min [n = 26]), (ii) resistance to higher concentrations of vancomycin than extant VISA clinical strains (i.e., ≥6 mg/liter [except for one dubious strain, 18-6d, in Table 4]), (iii) an extremely unstable resistance and growth phenotype, and (iv) rpoB mutation as a frequent cause of the phenotype.
Since sVISA strains grow extremely slowly and are unstable in the resistance phenotype, they have escaped the interest of many clinical microbiologists. However, when observed closely, sVISA has certain clinical significance because of its higher tolerance of vancomycin than that of extant VISA strains. Some sVISA strains are capable of resisting much greater concentrations (16 to ∼32 mg/liter) of vancomycin than a representative clinical VISA strain, like Mu50 (MIC, 12 mg/liter). This gives hVISA a better chance to survive high-dose vancomycin therapy. The frequency of strain Mu3 changing to sVISA from hVISA is as high as or even greater than the frequency of VISA appearing from Mu3. We showed previously that VISA can be obtained from Mu3 and from its related hVISA strains by a single mutation incorporated into any one of 20 different genes (4). Mutations affecting various codons of rpoB were also shown to convert Mu3 and Mu3-related hVISA strains to VISA (3, 4). The fact that such a diverse repertoire of mutations can convert Mu3 to VISA in a single genetic event finally resolved the question of an extremely high rate (≥1 × 10−6) of hVISA-to-VISA conversion (4). In spite of this high rate of hVISA-to-VISA conversion observed in vitro and the frequent isolation of hVISA in clinical samples (32), VISA has rarely been noticed in clinical microbiology laboratories. To explain this paradox, we previously postulated the instability of VISA strains based on the observed decline of the resistance phenotype of VISA during in vitro long-term drug-free passages up to 84 days (19, 33). In this study, we showed that hVISA strain Mu3 generates a novel group of mutant strains with an extremely unstable VISA phenotype, producing phenotypic revertants after only a few days of serial drug-free passages. This would account for the invisibility of VISA in the clinical cases of vancomycin therapeutic failure. After vancomycin treatment, the culture of a patient sample was overgrown by the phenotypic revertants of sVISA strains.
The biological fate of sVISA.
sVISA strains have extremely prolonged doubling times and frequently revert to hVISA by the back mutation or by a compensatory mutation. This property provides hVISA with an important strategy for surviving vancomycin therapy in clinical settings, as the sVISA phenotype serves as a temporary shelter for hVISA against vancomycin chemotherapy. It survives vancomycin therapy by sacrificing vigorous growth until the therapy is lifted, and then it returns to hVISA by a back or compensatory mutation. In this case, there is no trace of sVISA remaining after the course of vancomycin therapy. Another fate of sVISA is the stabilization of the VISA phenotype with improved but a not completely recovered growth rate. One of the three compensatory mutants, L3, has a vancomycin MIC of 4 mg/liter on BHI agar and is much closer to VISA than the other compensated mutants, L1 and L2, as judged from a population analysis (Fig. 2B). Thus, sVISA might be a genetic step that occurs prior to the establishment of a stable class of VISA strains, such as Mu50 and MI (34). Finally, if the VISA phenotype was fixed without any improvement in growth capability, the strain would be clinically unimportant, because it would be diluted out from the body of the patient or from the hospital environment. S. aureus strains 17-4d and 18-6d in Table 4 might represent this category of strains.
Genetic basis for sVISA phenotype in comparison with VISA strain Mu50.
Since rpoB(R512P) was the unique mutation in the genome of 6R-P, we looked for the sequence change in the rpoB gene of its LC variants. In three independently established LC mutants, we identified three distinct sequences in the 512th position of the rpoB gene. This signifies that a great fitness cost accompanies the antibiotic resistance phenotype conferred by the rpoB(R512P) mutation. Another sVISA strain, Mu3-V6-21-4d, carrying the rpoB(H929T) mutation, was also shown to revert to hVISA by back mutation. The great fitness cost of resistance is considered to be the driving force for the back mutation or compensatory mutation observed in Mu3-V6-21-4d and 6R-P. Apparently, the big fitness cost of 6R-P seems to be associated with the drastic change in the transcription profile caused by the introduction of the rpoB(R512P) mutation in Mu3. As shown in Fig. 5B, many of the downregulated genes of Mu3 relative to ΔIP were further downregulated in 6R-P. As a result, genes, such as those associated with cell division and protein synthesis, became severely depressed in 6R-P. This was in great contrast to Mu50, in which those genes were at levels comparable to or even upregulated relative to those of ΔIP. The reversal of transcription profile property observed in Mu50 seems to reflect the compensatory resolution of the fitness cost in the course of the VISA phenotypic development of Mu50. There are nine nonsynonymous SNPs between VISA strain Mu50 and hVISA strain Mu3 (2). It is likely that some of them served as a compensatory mutation(s) to recover homeostasis of the cell physiology disturbed with the stepwise acquisition of the VISA phenotype. In contrast, 6R-P acquired the VISA phenotype through one mutation only, rpoB(R512P). The effect of the mutation was tremendous, causing altered expression of as many as 487 genes when introduced into Mu3. Since a single mutation is enough to convert Mu3 into VISA (4), this great turbulence in the transcription pattern is evidently superfluous and would be hazardous to the cell. If a compensatory mutation occurred to diminish this disturbance and restore an improved growth rate, the mutant would easily overgrow the parent. Although we do not know the genetic mechanism, an extremely high rate of occurrence of back or allelic-replacing mutation was observed. We find it reasonable, because the occurrence of reversal or allelic replacement of the mutated rpoB codon would be much more favorable over any compensatory mutation incorporated in other genes of the chromosome. This is because a mutation in any effector or even in a global regulator gene would not be enough to completely reverse such a large degree of turbulence caused by, e.g., the rpoB(R512P) mutation. Actually, a total of 46 transcription regulator genes, including those of two-component regulatory systems (TCRS), such as lytSR, srrA, arlR, vraSR, saeSR, and arlR, were affected (>1.5-fold, P < 0.05). Therefore, the codon-specific compensatory mutation is the ideal way to resolve the transcriptional upheaval caused by the rpoB(R512P) mutation. The next best compensatory mutation is in the rpoA or rpoC gene encoding the other components of the RNA polymerase (RNAP) holoenzyme together with rpoB (35–37).
Mechanism of resistance of sVISA strain 6R-P compared to that of Mu50.
The precise mechanism for the sVISA phenotype remains to be elucidated. Although 6R-P shares the basic feature of the VISA phenotype, a thickened cell wall, as well as reduced autolytic activity, the genetic mechanism underlying these features may be different from that in Mu50. We found three genes, SAHV_2101, tarA, and tarG, to be among the significantly downregulated group of genes in 6R-P relative to ΔIP: SAHV_2101, 4.8-fold depressed (P = 0.0049); tarA, 3.8-fold depressed (P = 0.0018); and tarG, 9.4-fold depressed (P = 0.00016). In our previous report, the mutations SAHV_2101(G121R) and tarO (llm)(G169R) were able to convert Mu3 into VISA as a single contributory mutation. If we assume that the mutated gene products had decreased biological function, the observed downregulation of the genes in 6R-P may have a similar effect in converting Mu3 into VISA. Although we do not know the function of SAHV_2101, tarO is the first gene of the wall teichoic acid (WTA) synthesis pathway (38). Not only the tarO gene, but also tarA and tarG, present downstream of the WTA synthesis pathway, were significantly depressed in 6R-P, suggesting a depression of the entire WTA synthesis pathway. Since the WTA synthesis and PG synthesis pathways compete for the limited amount of bactoprenol in the cell membrane, the depression of WTA synthesis may lead to an increased use of bactoprenol for synthesizing PG, thus raising the level of vancomycin resistance (39, 40). Evidently, the mutation of rpoB is considered a regulatory mutation that drastically changes the cell physiology by altering dozens of regulator genes, thus affecting the expression of hundreds of genes. It is important to explore the biological significance of the RNAP mutations in the evolution of bacterial species.
In this study, we identified five rpoB mutations associated with sVISA phenotype acquisition: rpoB(R512P), rpoB(S746F), rpoB(G744R), rpoB(G977V), and rpoB(H929T). However, 20 out of 26 sVISA strains obtained from Mu3 had no mutation in rpoB. Therefore, it is possible that other genes cause the sVISA phenotype. We have started to identify those mutations by whole-genome sequencing. This attempt will greatly contribute to the understanding of the genetic mechanisms of vancomycin resistance in S. aureus.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a Grant-in-Aid for Young Scientists B (24791029) and a grant-in-aid (S1201013) from the Ministry of Education, Culture, Sports, Science, and Technology Japan (MEXT) for the Foundation of Strategic Research Projects in Private Universities.
We thank Mitutaka Yoshida (Division of Ultrastructural Research, Juntendo University) for invaluable help in sample preparation and technical support with transmission electron microscopy.
Footnotes
Published ahead of print 19 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02470-13.
REFERENCES
- 1.Hiramatsu K, Aritaka N, Hanaki H, Kawasaki S, Hosoda Y, Hori S, Fukuchi Y, Kobayashi I. 1997. Dissemination in Japanese hospitals of strains of Staphylococcus aureus heterogeneously resistant to vancomycin. Lancet 350:1670–1673. 10.1016/S0140-6736(97)07324-8 [DOI] [PubMed] [Google Scholar]
- 2.Neoh HM, Cui L, Yuzawa H, Takeuchi F, Matsuo M, Hiramatsu K. 2008. Mutated response regulator graR is responsible for phenotypic conversion of Staphylococcus aureus from heterogeneous vancomycin-intermediate resistance to vancomycin-intermediate resistance. Antimicrob. Agents Chemother. 52:45–53. 10.1128/AAC.00534-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsuo M, Hishinuma T, Katayama Y, Cui L, Kapi M, Hiramatsu K. 2011. Mutation of RNA polymerase beta subunit (rpoB) promotes hVISA-to-VISA phenotypic conversion of strain Mu3. Antimicrob. Agents Chemother. 55:4188–4195. 10.1128/AAC.00398-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matsuo M, Cui L, Kim J, Hiramatsu K. 2013. Comprehensive identification of mutations responsible for heterogeneous vancomycin-intermediate Staphylococcus aureus (hVISA)-to-VISA conversion in laboratory-generated VISA strains from hVISA clinical strain Mu3. Antimicrob. Agents Chemother. 57:5843–583. 10.1128/AAC.00425-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Champion MD, Gray V, Eberhard C, Kumar S. 2013. The evolutionary history of amino acid variations mediating increased resistance of S. aureus identifies reversion mutations in metabolic regulators. PLoS One 8:e56466. 10.1371/journal.pone.0056466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Howden BP, McEvoy CR, Allen DL, Chua K, Gao W, Harrison PF, Bell J, Coombs G, Bennett-Wood V, Porter JL, Robins-Browne R, Davies JK, Seemann T, Stinear TP. 2011. Evolution of multidrug resistance during Staphylococcus aureus infection involves mutation of the essential two component regulator WalKR. PLoS Pathog. 7:e1002359. 10.1371/journal.ppat.1002359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katayama Y, Takeuchi F, Ito T, Ma XX, Ui-Mizutani Y, Kobayashi I, Hiramatsu K. 2003. Identification in methicillin-susceptible Staphylococcus hominis of an active primordial mobile genetic element for the staphylococcal cassette chromosome mec of methicillin-resistant Staphylococcus aureus. J. Bacteriol. 185:2711–2722. 10.1128/JB.185.9.2711-2722.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bae T, Schneewind O. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55:58–63. 10.1016/j.plasmid.2005.05.005 [DOI] [PubMed] [Google Scholar]
- 9.Katayama Y, Baba T, Sekine M, Fukuda M, Hiramatsu K. 2013. Beta-hemolysin promotes skin colonization by Staphylococcus aureus. J. Bacteriol. 195:1194–1203. 10.1128/JB.01786-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katayama Y, Murakami-Kuroda H, Cui L, Hiramatsu K. 2009. Selection of heterogeneous vancomycin-intermediate Staphylococcus aureus by imipenem. Antimicrob. Agents Chemother. 53:3190–3196. 10.1128/AAC.00834-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hiramatsu K, Hanaki H. 1998. Glycopeptide resistance in staphylococci. Curr. Opin. Infect. Dis. 11:653–658. 10.1097/00001432-199812000-00002 [DOI] [PubMed] [Google Scholar]
- 12.Cui L, Murakami H, Kuwahara-Arai K, Hanaki H, Hiramatsu K. 2000. Contribution of a thickened cell wall and its glutamine nonamidated component to the vancomycin resistance expressed by Staphylococcus aureus Mu50. Antimicrob. Agents Chemother. 44:2276–2285. 10.1128/AAC.44.9.2276-2285.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui L, Isii T, Fukuda M, Ochiai T, Neoh HM, Camargo IL, Watanabe Y, Shoji M, Hishinuma T, Hiramatsu K. 2010. An RpoB mutation confers dual heteroresistance to daptomycin and vancomycin in Staphylococcus aureus. Antimicrob. Agents Chemother. 54:5222–5233. 10.1128/AAC.00437-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aiba Y, Katayama Y, Hishinuma T, Murakami-Kuroda H, Cui L, Hiramatsu K. 2013. Mutation of RNA polymerase β-subunit gene promotes heterogeneous-to-homogeneous conversion of β-lactam resistance of methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 57:4861–4871. 10.1128/AAC.00720-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gustafson JE, Berger-Bächi B, Strässle A, Wilkinson BJ. 1992. Autolysis of methicillin-resistant and -susceptible Staphylococcus aureus. Antimicrob. Agents Chemother. 36:566–572. 10.1128/AAC.36.3.566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cui L, Lian JQ, Neoh HM, Reyes E, Hiramatsu K. 2005. DNA microarray-based identification of genes associated with glycopeptide resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 49:3404–3413. 10.1128/AAC.49.8.3404-3413.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4:249–264. 10.1093/biostatistics/4.2.249 [DOI] [PubMed] [Google Scholar]
- 18.Cui X, Churchill GA. 2003. Statistical tests for differential expression in cDNA microarray experiments. Genome Biol. 4:210. 10.1186/gb-2003-4-4-210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui L, Ma X, Sato K, Okuma K, Tenover FC, Mamizuka EM, Gemmell CG, Kim MN, Ploy MC, El-Solh N, Ferraz V, Hiramatsu K. 2003. Cell wall thickening is a common feature of vancomycin resistance in Staphylococcus aureus. J. Clin. Microbiol. 41:5–14. 10.1128/JCM.41.1.5-14.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cui L, Neoh HM, Iwamoto A, Hiramatsu K. 2012. Coordinated phenotype switching with large-scale chromosome flip-flop inversion observed in bacteria. Proc. Natl. Acad. Sci. U. S. A. 109:E1647–E1656. 10.1073/pnas.1204307109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boyle-Vavra S, Challapalli M, Daum RS. 2003. Resistance to autolysis in vancomycin-selected Staphylococcus aureus isolates precedes vancomycin-intermediate resistance. Antimicrob. Agents Chemother. 47:2036–2039. 10.1128/AAC.47.6.2036-2039.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hussain FM, Boyle-Vavra S, Shete PB, Daum RS. 2002. Evidence for a continuum of decreased vancomycin susceptibility in unselected Staphylococcus aureus clinical isolates. J. Infect. Dis. 186:661–667. 10.1086/342708 [DOI] [PubMed] [Google Scholar]
- 23.Koehl JL, Muthaiyan A, Jayaswal RK, Ehlert K, Labischinski H, Wilkinson BJ. 2004. Cell wall composition and decreased autolytic activity and lysostaphin susceptibility of glycopeptide-intermediate Staphylococcus aureus. Antimicrob. Agents Chemother. 48:3749–3757. 10.1128/AAC.48.10.3749-3757.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peschel A, Vuong C, Otto M, Götz F. 2000. The d-alanine residues of Staphylococcus aureus teichoic acids alter the susceptibility to vancomycin and the activity of autolytic enzymes. Antimicrob. Agents Chemother. 44:2845–2847. 10.1128/AAC.44.10.2845-2847.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pfeltz RF, Singh VK, Schmidt JL, Batten MA, Baranyk CS, Nadakavukaren MJ, Jayaswal RK, Wilkinson BJ. 2000. Characterization of passage-selected vancomycin-resistant Staphylococcus aureus strains of diverse parental backgrounds. Antimicrob. Agents Chemother. 44:294–303. 10.1128/AAC.44.2.294-303.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakoulas G, Eliopoulos GM, Fowler VG, Jr, Moellering RC, Jr, Novick RP, Lucindo N, Yeaman MR, Bayer AS. 2005. Reduced susceptibility of Staphylococcus aureus to vancomycin and platelet microbicidal protein correlates with defective autolysis and loss of accessory gene regulator (agr) function. Antimicrob. Agents Chemother. 49:2687–2692. 10.1128/AAC.49.7.2687-2692.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sieradzki K, Tomasz A. 1997. Inhibition of cell wall turnover and autolysis by vancomycin in a highly vancomycin-resistant mutant of Staphylococcus aureus. J. Bacteriol. 179:2557–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Utaida S, Pfeltz RF, Jayaswal RK, Wilkinson BJ. 2006. Autolytic properties of glycopeptide-intermediate Staphylococcus aureus Mu50. Antimicrob. Agents Chemother. 50:1541–1545. 10.1128/AAC.50.4.1541-1545.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wootton M, Bennett PM, MacGowan AP, Walsh TR. 2005. Reduced expression of the atl autolysin gene and susceptibility to autolysis in clinical heterogeneous glycopeptide-intermediate Staphylococcus aureus (hGISA) and GISA strains. J. Antimicrob. Chemother. 56:944–947. 10.1093/jac/dki289 [DOI] [PubMed] [Google Scholar]
- 30.Yang SJ, Bayer AS, Mishra NN, Meehl M, Ledala N, Yeaman MR, Xiong YQ, Cheung AL. 2012. The Staphylococcus aureus two-component regulatory system, GraRS, senses and confers resistance to selected cationic antimicrobial peptides. Infect. Immun. 80:74–81. 10.1128/IAI.05669-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reed P, Veiga H, Jorge AM, Terrak M, Pinho MG. 2011. Monofunctional transglycosylases are not essential for Staphylococcus aureus cell wall synthesis. J. Bacteriol. 193:2549–2556. 10.1128/JB.01474-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamakawa J, Aminaka M, Okuzumi K, Kobayashi H, Katayama Y, Kondo S, Nakamura A, Oguri T, Hori S, Cui L, Ito T, Jin J, Kurosawa H, Kaneko K, Hiramatsu K. 2012. Heterogeneously vancomycin-intermediate Staphylococcus aureus (hVISA) emerged before the clinical introduction of vancomycin in Japan: a retrospective study. J. Infect. Chemother. 18:406–409. 10.1007/s10156-011-0330-2 [DOI] [PubMed] [Google Scholar]
- 33.Hiramatsu K. 2001. Vancomycin-resistant Staphylococcus aureus: a new model of antibiotic resistance. Lancet Infect. Dis. 1:147–155. 10.1016/S1473-3099(01)00091-3 [DOI] [PubMed] [Google Scholar]
- 34.Smith TL, Pearson ML, Wilcox KR, Cruz C, Lancaster MV, Robinson-Dunn B, Tenover FC, Zervos MJ, Band JD, White E, Jarvis WR. 1999. Emergence of vancomycin resistance in Staphylococcus aureus. Glycopeptide-Intermediate Staphylococcus aureus Working Group. N. Engl. J. Med. 340:493–501 [DOI] [PubMed] [Google Scholar]
- 35.Comas I, Borrell S, Roetzer A, Rose G, Malla B, Kato-Maeda M, Galagan J, Niemann S, Gagneux S. 2012. Whole-genome sequencing of rifampicin-resistant M. tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat. Genet. 44:106–110. 10.1038/ng.1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reynolds MG. 2000. Compensatory evolution in rifampin-resistant Escherichia coli. Genetics 156:1471–1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brandis G, Wrande M, Liljas L, Hughes D. 2012. Fitness-compensatory mutations in rifampicin-resistant RNA polymerase. Mol. Microbiol. 85:142–151. 10.1111/j.1365-2958.2012.08099.x [DOI] [PubMed] [Google Scholar]
- 38.D'Elia MA, Pereira MP, Chung YS, Zhao W, Chau A, Kenney TJ, Sulavik MC, Black TA, Brown ED. 2006. Lesions in teichoic acid biosynthesis in Staphylococcus aureus lead to a lethal gain of function in the otherwise dispensable pathway. J. Bacteriol. 188:4183–4189. 10.1128/JB.00197-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawai Y, Marles-Wright J, Cleverley RM, Emmins R, Ishikawa S, Kuwano M, Heinz N, Bui NK, Hoyland CN, Ogasawara N, Lewis RJ, Vollmer W, Daniel RA, Errington J. 2011. A widespread family of bacterial cell wall assembly proteins. EMBO J. 30:4931–4941. 10.1038/emboj.2011.358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biswas R, Martinez RE, Göhring N, Schlag M, Josten M, Xia G, Hegler F, Gekeler C, Gleske AK, Götz F, Sahl HG, Kappler A, Peschel A. 2012. Proton-binding capacity of Staphylococcus aureus wall teichoic acid and its role in controlling autolysin activity. PLoS One 7:e41415. 10.1371/journal.pone.0041415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuwahara-Arai K, Kondo N, Hori S, Tateda-Suzuki E, Hiramatsu K. 1996. Suppression of methicillin resistance in a mecA-containing pre-methicillin-resistant Staphylococcus aureus strain is caused by the mecI-mediated repression of PBP 2′ production. Antimicrob. Agents Chemother. 40:2680–2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.