

Abstract
Cytokine production assays have been primarily used in research settings studying novel immunodeficiencies. We sought to determine the diagnostic value of cytokine production assays in patients with recurrent and/or severe infectious diseases (IDs) without known immunodeficiencies and unclassified noninfectious inflammatory disorders (NIIDs). We retrospectively examined cytokine production in whole-blood and peripheral blood mononuclear cell samples from 157 adult patients. A cytokine production rate of <5% of that of healthy controls was considered defective. While monocyte-derived cytokine (tumor necrosis factor alpha [TNF-α], interleukin-1β [IL-1β], and IL-6) production was rarely affected, 30% of all included patients had deficient production of interferon gamma (IFN-γ), IL-17A, or IL-22. Twenty-five percent of the NIID patients displayed defective IFN-γ production, whereas IL-17A production was generally unaffected. In the group of ID patients, defective IFN-γ production was found in 19% and 14% of the patients with viral and bacterial infections, respectively, and in 38%, 24%, and 50% of patients with mycobacterial, mucocutaneous, and invasive fungal infections, respectively. Defective IL-17A and IL-22 production was mainly confined to ID patients with mucocutaneous fungal infections. In conclusion, cytokine production assays frequently detect defective Th1 responses in patients with mycobacterial or fungal infections, in contrast to patients with respiratory tract infections or isolated bacterial infections. Defective IL-17A and IL-22 production was primarily found in patients with fungal infections, while monocyte-derived cytokine production was unaffected. Thus, lymphocyte-derived cytokine production assays are helpful in the diagnostic workup of patients with recurrent infections and suspected immunodeficiencies and have the potential to reveal immune defects that might guide adjunctive immunomodulatory therapy.
INTRODUCTION
Traditionally, primary immunodeficiencies are thought to be represented by rare monogenetic disorders directly affecting immune system development or function, leading to multiple, recurrent, unusually severe, and opportunistic infections in childhood (1, 2). However, in the last decade, this relatively rigid description of immune deficiencies has been challenged (1, 2). The increasing understanding of immune responses and the expansion of the existing investigative tools enabled the deciphering of the genetic basis of many conditions that are accompanied by an increased susceptibility to infections, as well as of various noninfectious inflammatory disorders (NIIDs). Nowadays, it is thought that even individuals suffering from a single unusual infection might be affected by an immune defect, and thus immunodeficiencies should be considered in a wide range of clinical situations (1, 2).
Highly conserved molecular structures of microorganisms, called pathogen-associated molecular patterns (PAMPs) (3), are recognized by pattern recognition receptors (PRR). Defects in PRR and the subsequent signaling pathways are usually accompanied by diminished production of proinflammatory cytokines, and they represent a paradigm shift in the field of primary immunodeficiencies (4, 5). Similarly, various NIIDs are caused by a loss of the regulation of cytokine production and have been coined autoinflammatory disorders (6). In addition to PRR defects, specific immunodeficiencies have been described in patients with defective cytokine or cytokine receptors, such as interleukin-12 (IL-12), IL-12R, interferon gamma (IFN-γ), and IFN-γR defects associated with mycobacterial and Salmonella infections (7, 8), or with IL-17F, and IL-17R deficiencies, which are associated with fungal infections (9).
Until now, an assessment of the integrity of cytokine production in stimulated peripheral blood mononuclear cells (PBMCs) has mainly been used to examine patients with novel primary immunodeficiencies (10, 11). However, cytokine production defects may be much more common in patients with recurrent infectious diseases (IDs) (5) or suspected autoinflammatory NIIDs (6), and an assessment of cytokine production may have diagnostic value in these patients. In the present study, we review cytokine production in a large series of patients suffering from either a recurrent infection, an unusually severe course of infection, an opportunistic infection, or an NIID who presented at our tertiary center for infectious diseases, immunodeficiencies, and autoinflammation.
Case illustration, demonstrating the diagnostic value of cytokine production assessment.
A 28-year-old male known to have diabetes mellitus type I, hypothyroidism, and systemic lupus erythematosus and who was treated with hydroxychloroquine had suffered from five episodes of pneumonia since the age of 25 years. Twice, Streptococcus pneumoniae was isolated from blood cultures. One culture-negative pneumonia was complicated by pancytopenia and autoimmune hemolytic anemia. Furthermore, he had recurrent oropharyngeal candidiasis and persistent dermatomycosis of the right foot. These manifestations were compatible with a large spectrum of diseases, with chronic mucocutaneous candidiasis (CMC) among them. However, Sanger sequencing of exomes 6 to 11 of STAT1, a region in which earlier studies identified mutations (10, 12), revealed no abnormalities. Therefore, the patient was referred for further diagnostic analysis. PBMC stimulation showed normal tumor necrosis factor alpha (TNF-α), IL-1β, and IL-6 production when stimulated with lipopolysaccharide (LPS) and Candida albicans, making it improbable that deficiencies of dectin-1, caspase recruitment domain-containing protein (CARD) 9 mutations, interleukin-1 receptor-associated kinase (IRAK) 4, and myeloid differentiation primary response 88 (MyD88) deficiencies were the cause. However, the production rates of IFN-γ, IL-17A, and IL-22 were <10%, <5%, and <5%, respectively, compared to those of healthy controls, supporting the hypothesized diagnosis of CMC. Further extended genetic analysis by whole-exome sequencing showed a novel missense mutation, replacing aspartic acid with valine at position 24 (D24V) in exome 3 of STAT1, probably with similar effects on STAT1 function as those previously described for the gain-of-function mutations in exons 6 to 11.
MATERIALS AND METHODS
Patients.
All consecutive patients with IDs, recurrent infections, infections of unusual etiology or severity, and so-far unclassified patients suspected for NIID hospitalized or visiting the outpatient clinic of the Department of Internal Medicine, Radboud University Nijmegen Medical Center between 2001 and April 2013 were included in the study. Patients with immunosuppressive drugs, HIV infection, hematological malignancies, or known primary immunodeficiencies were excluded.
Classification and data analysis.
The group of ID patients was classified based on the clinical presentation into: (A) ear-nose-throat or airway infections, (B) pyogenic infections, or (C) opportunistic infections (mainly caused by intracellular bacteria, viruses, and fungi) (13) (Fig. 1). In addition, ID patients were classified based on the main causative microorganism: (a) viral, (b) bacterial, (c) mycobacterial, or (d) fungal infections, with fungal infections further separated into (d1) mucocutaneous fungal infections and (d2) invasive fungal infections (Fig. 2).
FIG 1.

Flow chart of the 157 patients in whom cytokine production assays were performed. WB, whole-blood stimulation; PBMC, peripheral blood mononuclear cell stimulation; A, recurrent respiratory tract infection; B, pyogenic infections; C, opportunistic infections.
FIG 2.
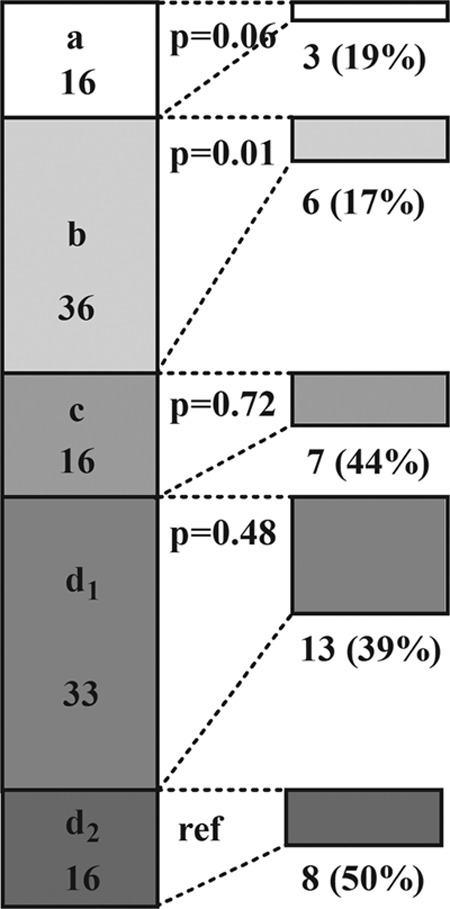
Proportion of patients with infectious diseases (n = 117), stratified according to microbial etiology: viral (a, n = 16), bacterial (b, n = 36), mycobacterial (c, n = 16), mucocutaneous fungal (d1, n = 33), and invasive fungal (d2, n = 16), with low lymphocyte-derived cytokine production as assessed by either whole-blood or peripheral blood mononuclear cell stimulation tests. The number (%) of patients with each kind of infection and a deficient response are shown on the right for each category. P values shown are for a comparison of the proportions between group d2 and the other groups.
Cytokine production assays.
Whole-blood (WB) stimulation assays were performed from 2001 until 2008. Peripheral blood mononuclear cells (PBMCs) stimulation assays were done from 2008 to 2013.
For the WB stimulation assays, venous blood was collected in lithium-heparin tubes, diluted to a final concentration of 1:5 in RPMI 1640 (Dutch modified; Gibco, Invitrogen, Breda, The Netherlands), and plated out at 1 ml/well in 24-well tissue culture plates. Stimulation was performed by adding 100 μl culture medium (negative control) and the following stimuli (final concentrations in parentheses): LPS derived from Salmonella enterica subsp. enterica serovar Typhimurium (10 ng/ml), S. Typhimurium bacteria (heat-killed [HK], 107 microorganisms/ml), Staphylococcus aureus (HK, 107 microorganisms/ml), Aspergillus fumigatus conidia (HK, 107 microorganisms/ml), and C. albicans conidia (HK, 107 microorganisms/ml). Mycobacterium tuberculosis sonicate (HK, 1 μg/ml) or poly(I·C) (50 μg/ml; InvivoGen, San Diego, CA) was also included in case of a mycobacterial or a viral infection, respectively. The microorganisms were killed by heating them for 1 h at 95°C. After incubation for 24 h or 48 h at 37°C, the supernatants were obtained by centrifugation and stored at −20°C until IL-6, IL-1β, tumor necrosis factor alpha (TNF-α) (all 24 h), and IFN-γ (48 h) measurements were performed. The experiments were done in duplicate.
For the PBMC stimulation assay, venous blood samples were collected into EDTA tubes. PBMCs were isolated according to standard protocols, with minor modifications (14). The PBMC fraction was obtained by density centrifugation of blood diluted 1:1 in phosphate-buffered saline (PBS) over Ficoll-Paque (Pharmacia Biotech AB, Uppsala, Sweden). The cells were washed three times in PBS and resuspended in RPMI 1640 (Dutch modified; Gibco, Invitrogen, Breda, The Netherlands) supplemented with 50 mg/liter gentamicin, 2 mM l-glutamine, and 1 mM pyruvate. The cells were counted in a Coulter Counter Z (Beckman Coulter, Mijdrecht, The Netherlands) and adjusted to 5 × 106 cells/ml. Mononuclear cells (5 × 105) in a 100-μl volume were added to round-bottom 96-well plates (Greiner, Alphen aan den Rijn, The Netherlands) and incubated with either 100 μl of culture medium (negative control) or one of the stimuli for 24 h (TNF-α, IL-1β, and IL-6), 48 h (IFN-γ), and/or 7 days (IL-17A and IL-22). Monocyte-derived cytokine production was tested with LPS derived from Escherichia coli, Pam3Cys, muramyl dipeptide, and β-glucan. Both monocyte-derived and lymphocyte-derived cytokine production were tested with HK S. aureus (106 or 107 microorganisms/ml, respectively) and HK C. albicans conidia (105 or 106 microorganisms/ml, respectively). Sonicated M. tuberculosis (1 or 10 μg/ml), HK Mycobacterium abscessus (105 microorganisms/ml), HK Mycobacterium fortuitum (105 microorganisms/ml), or poly(I·C) (5 or 50 μg/ml; InvivoGen, San Diego, CA) was included if the patients suffered from a mycobacterial or viral infection, respectively. αCD3/αCD28 (2.5 × 106 beads/5 × 106 PBMC; Miltenyi Biotec, Bergisch Gladbach, Germany) was used in the lymphocyte-derived cytokine production assays. The microorganisms were killed by heating them for 1 h at 95°C. C. albicans and S. aureus were chosen because of their potency for inducing IFN-γ, since E. coli LPS is a poor inducer of IFN-γ. C. albicans and S. aureus were well suited as strong stimuli for IL-17A and IL-22 release (see Fig. S1 in the supplemental material). Ten percent human pooled serum was added when the PBMCs were incubated for 7 days. After incubation, the supernatants were stored at −20°C until the assay. The experiments were performed in duplicate.
Until 2006, TNF-α was measured using an in-house radioimmunoassay, which has a lower limit of detection of 15 pg/ml. Thereafter, TNF-α was measured with a commercial enzyme-linked immunosorbent assay (ELISA) kit from R&D Systems (Minneapolis, MN), which has a lower detection limit of 78 pg/ml. IL-6 and IFN-γ concentrations were assessed using commercial ELISA kits from Sanquin (Amsterdam, The Netherlands), which has lower detection limits of 15 pg/ml and 8 pg/ml, respectively. IL-1β, IL-17A, and IL-22 were measured using commercial ELISA kits from R&D Systems with 39 pg/ml, 16 to 78 pg/ml, and 78 to 155 pg/ml as the lower limits of detection, respectively.
Cytokine production, i.e., the concentration in the stimulated sample, was compared between patients and healthy volunteers (anonymous blood donors, who gave written informed consent for the use of their blood for scientific purposes as approved by the ethics comittee of the Radboud University Medical Center) tested the same day in the same run. In case of more than one healthy control, the values were pooled and averaged. The cytokine production in these patients was compared with that of healthy volunteers tested in the same experiment. To calculate the proportion of patients with deficient production, cytokine concentrations of <5%, 10%, and 20% compared to healthy volunteers (i.e., patient-to-control ratios, <0.05, <0.10, and <0.20, respectively) were used. For the main analysis, we defined cytokine production to be defective when the production upon exposure to at least one stimulus was <5% compared to that of the healthy controls. This stringent threshold was chosen because healthy controls rarely had cytokine concentrations below the 5th percentile of the concentrations found in the whole population (patients plus controls) (see Table S1 and Fig. S2 in the supplemental material), and the 5% cutoff value was associated with most concordant test results in patients who were tested twice (see Table S2 in the supplemental material). If the experiments were repeated, only concordant results were considered to represent a defective response.
Statistical analysis.
The main outcome measures of the study are descriptive. The proportions of affected patients in the different categories were identified using cross tables. To test the relationship between these categorical variables, the chi-square test was performed, and in case of <5 expected counts, Fisher's exact test was performed. The Mann-Whitney U test or the Kruskal-Wallis with Dunn's posttest were used to compare (absolute) cytokine concentrations between two or more independent groups, respectively. A P value of <0.05 was considered statistically significant.
RESULTS
Patient characteristics.
In total, 157 patients were included: 33 underwent WB stimulation and 124 underwent PBMC stimulation. One hundred forty-six patients were ambulant, and 11 were hospitalized at the time of sampling. Nine patients were receiving antimycobacterial treatment while the tests were performed. The test indications are presented in Fig. 1. In all tested patients, the production of TNF-α, IL-1β, and IL-6 was measured. The production of IFN-γ, IL-17A, and IL-22 was tested in a selection of patients (Fig. 1) due to new developments and insights gained during the implementation of the tests (IL-22 assessment became available in only the last years of the study).
Production of TNF-α, IL-1β, and IL-6 in the WB and PBMC assays.
Only 4 of the 157 tested patients had decreased production of <5% compared to that of the controls for one of the monocyte-derived cytokines, TNF-α, IL-1β, or IL-6 (Table 1). Four patients with an infection had defective TNF-α or IL-6 production. Of the three patients with defective TNF-α production, one had disseminated cryptococcosis, one had tuberculous meningitis, and one had recurrent pneumococcal meningitis with cerebrospinal fluid leakage. The patient with defective IL-6 production and recurrent herpes infections also displayed low IFN-γ production (<10%) upon stimulation with the Toll-like receptor 3 (TLR3) ligand poly(I·C). Except for in the patients with disseminated cryptococcosis, cytokine production was tested after the resolution of infection and showed normal results, precluding a defective result due to downregulated cytokine production during acute infection (15).
TABLE 1.
Infected patients with defective monocyte-derived cytokine production
| Age (yr) | Sexa | Clinical picture | Cytokine | Stimulib | Diagnosis-test interval | Underlying defectc |
|---|---|---|---|---|---|---|
| 34 | M | Disseminated cryptococcosis | TNF-α | 3 | 1 wk | NP |
| 42 | F | Recurrent herpes simplex virus 2 meningitis | IL-6 | 1 | 3 yr | NP |
| 64 | M | Recurrent pneumococcal meningitis | TNF-α | 1 | 4 mo | Cerebrospinal fluid leakage |
| 42 | M | Neurological deterioration after tuberculous meningitis | TNF-α | 3 | 13 mo | NP |
M, male; F, female.
Number of stimuli that resulted in a deficient response.
NP, not performed.
Production of IFN-γ, IL-17A, and IL-22 in the cytokine production assays.
Overall, 56/157 (36%) of the measurements indicated a production rate of <5% compared to that of the healthy controls for at least one of the three cytokines, corresponding to 47 (30%) individual patients (32% ID and 25% NIID) (Fig. 1).
Production of IFN-γ in the WB assay.
IFN-γ production was impaired in 13 patients of the 33 patients tested with the WB assay, 9 of whom had an ID and 4 patients who had an NIDD (Table 2). For patients with an ID, decreased IFN-γ production was found only in those with a pyogenic or opportunistic infection (Fig. 1).
TABLE 2.
Patients with defective IFN-γ production upon whole-blood stimulation
| Age (yr) | Sexa | Groupb | Clinical picture | Stimulic | Diagnosis-test intervald | Lymphocyte count/mm3d | Underlying defecte |
|---|---|---|---|---|---|---|---|
| 33 | F | NIID | Recurrent fever, acne, abdominal pain | 1 | 1 day | 2,110 | PAPA syndrome, Crohn's disease |
| 51 | M | NIID | Secondary sclerosing cholangitis and acute phase response of unknown origin | 2 | Continuously | 2,700 | NP |
| 57 | M | NIID | Urticaria, fever | 5 | 1 day | 1,310 | Schnitzler syndrome |
| 56 | F | NIID | Hidradenitis, recurrent upper respiratory tract infections | 1 | 3 mo | 2,290 | NP |
| 65 | M | Infection | Pneumonia, eczema, asthma | 1 | 10 yr | 2,440 | HIES |
| 33 | M | Infection | Bacterial skin infection, recurrent pneumonia, eczema, onychomycosis | 1 | 10 mo | 1,640 | HIES |
| 51 | F | Infection | M. abscessus pneumonia | 3 | 23 yr | NAf | Cystic fibrosis |
| 45 | F | Infection | Pulmonary aspergillosis S. aureus empyema, skin infection | 4 | 4 mo | 1,610 | HIES |
| 45 | F | Infection | Disseminated cryptococcosis | 3 | 1 wk | NA | NP |
| 46 | M | Infection | Disseminated aspergillosis, pulmonary nocardiosis and Rhodococcus equi infection, Alternaria skin infection | 1 mo/5 mo | 350/850 | CD4 lymphopenia | |
| 45 | M | Infection | Disseminated aspergillosis and candidiasis | 5 | 3 mo | 2,090 | NP |
| 20 | F | Infection | Aspergillus pneumonia, skin infections, eczema | 4 | 8 mo | 1,840 | HIES |
| 63 | M | Infection | Aspergillus pneumonia | 3 | NA | NA | NP |
F, female; M, male.
NIID, noninfectious inflammatory disorder.
Number of stimuli that resulted in a deficient response.
Data from patients tested more than once are shown separated by a slash in the same column.
PAPA, pyogenic sterile arthritis, pyoderma gangrenosum, and acne; NP, not performed, HIES, hyper IgE syndrome.
NA, not available.
Production of IFN-γ, IL-17A, and IL-22 in the PBMC assay.
The results of the assays testing the production of T-lymphocyte-derived cytokines IFN-γ, IL-17A, and IL-22 are summarized in Fig. 1. Overall, 43 (35%) of the tests were indicative of deficient production, corresponding to 35 (28%) individual patients (34% of those with an ID and 19% of those with a NIID). (Tables 3, 4, and 5). When patients were grouped according to microbial etiology, the assay results were most often abnormal in patients with mycobacterial and fungal infections (Table 6 and Fig. 2).
TABLE 3.
Patients with defective IFN-γ production upon PBMC stimulation
| Age (yr) | Sexa | Group | Clinical pictureb | Stimulic,d | Diagnosis-test intervald | Lymphocyte count/mm3d | Underlying defecte |
|---|---|---|---|---|---|---|---|
| 46 | F | NIIDf | Idiopathic erythema nodosum | 1 | 2 yr | 1,790 | NP |
| 36 | M | NIID | Hidradenitis | 1 | Continuously | 2,690 | NP |
| 59 | F | NIID | Hidradenitis, pyoderma gangrenosum, ankylosing spondylitis | 2/2 | Continuously | 1,030/510 | NP |
| 50 | F | NIID | Sterile subareolar abscesses | 2 | 19 mo | 3,040 | NP |
| 26 | F | NIID | Episodic fever, abdominal pain, arthralgia | 1 | 17 mo | 1,880 | NP |
| 18 | F | NIID | EBV-driven cutaneous T-cell lymphoma of childhood | 1 | 9 mo | 950 | 232-kb loss in chromosome 16p11.2 |
| 48 | M | Infection | Progressive multifocal leukoencephalopathy | 2 | 5 mo | NAg | NP |
| 51 | M | Infection | Intraocular varicella-zoster virus infection | 1 | 2 mo | 1,990 | NP |
| 49 | F | Infection | Recurrent varicella-zoster virus infection | 2/2 | 1 mo/NA | 1,680/2,660 | NP |
| 61 | M | Infection | Recurrent bacterial skin infection | 1 | 3 wk/10 mo | 1,220/NA | NP |
| 35 | F | Infection | Recurrent impetigo, RVVC | 1/1/1/1 | 4 mo/6 mo/12 mo/14 mo | 1,820/2,300/2,890/1,820 | NP |
| 56 | F | Infection | Disseminated M. avium infection, Legionella dumoffii pneumonia | 1/1 | 1 mo/2 yr | 1,860/1,810 | NP |
| 68 | F | Infection | M. avium and Mycobacterium intracellulare pneumonia | 1 | 4 mo | 1,680 | NP |
| 54 | M | Infection | Mycobacterium chelonae skin infection | 1 | 9 mo | 2,080 | NP |
| 67 | M | Infection | M. abscessus pneumonia, ABPA | 1/1 | 2 mo/2 mo | 810/800 | NP |
| 67 | M | Infection | M. intracellulare pneumonia | 2 | 3 yr 8 mo | 1,700 | NP |
| 34 | V | Infection | Oral and vaginal candidiasis, furunculosis | 1 | 1 wk | 3,140 | NP |
| 20 | F | Infection | Chronic mucocutaneous candidiasis | 2 | Continuously | NA | NP |
| 48 | F | Infection | Persistent oropharyngeal candidiasis, onychomycosis, asplenia, vitiligo | 1 | Continuously | 1,200 | APECED |
| 28 | F | Infection | RVVC, impetigo, hidradenitis, granulomatous intestinal inflammation | 1 | Continuously | 1,500 | HIES |
| 36 | F | Infection | Chronic mucocutaneous candidiasis | 2 | Continuously | 900 | STAT1 mutation |
| 42 | F | Infection | RVVC | 1 | NA | 1,180 | NP |
| 30 | F | Infection | Recurrent oropharyngeal candidiasis | 1 | 2 mo | 1,940 | NP |
| 49 | M | Infection | Disseminated aspergillosis, pulmonary nocardiosis and R. equi infection, Alternaria skin infection | 1 | 3 yr | 990 | CD4 lymphopenia |
| 57 | M | Infection | Skin abscesses, recurrent pneumonia, eczema | 1 | 2 mo | 2,300 | HIES |
| 63 | F | Infection | Aspergillus skull base osteomyelitis | 1 | 3 yr | NA | NP |
| 66 | F | Infection | Recurrent Candida esophagitis, onychomycosis, hypothyroidism | 1 | Continuously | 900 | CMC, STAT1 negative |
F, female; M, male.
EBV, Epstein-Barr virus; RVVC, recurrent vulvovaginal candidiasis; ABPA, allergic bronchopulmonary aspergillosis.
Number of stimuli that resulted in a deficient response.
Data from patients tested more than once are shown separated by a slash in the same column.
NP, not performed; APECED, autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy; HIES, hyper-IgE syndrome; STAT, signal transducer and activator of transcription; CMC, chronic mucocutaneous candidiasis.
NIID, noninfectious inflammatory disorder.
NA, not available.
TABLE 4.
Infected patients with defective IL-17 production upon PBMC stimulation
| Age (yr) | Sexa | Clinical pictureb | Stimulic | Diagnosis-test interval | Lymphocyte count/mm3 | Underlying defectd |
|---|---|---|---|---|---|---|
| 20 | F | Chronic mucocutaneous candidiasis | 1 | Continuously | NAe | NP |
| 40 | M | Recurrent esophageal candidiasis | 1 | NA | NA | NP |
| 28 | M | Dermatomycosis, oropharyngeal candidiasis, recurrent pneumonia, SLE, DMI, hypothyroidism | 1 | 2 mo (pneumonia) | 1,620 | STAT1 mutation |
| 29 | F | Chronic mucocutaneous candidiasis | 1 | Continuously | NA | NP |
| 36 | F | Chronic mucocutaneous candidiasis | 1 | Continuously | 900 | STAT1 mutation |
| 63 | F | Aspergillus skull base osteomyelitis | 1 | 3 yr | NA | NP |
F, female; M, male.
SLE, systemic lupus erythematosus; DMI, diabetes mellitus type 1.
Number of stimuli that resulted in a deficient response.
NP, not performed; STAT, signal transducer and activator of transcription.
NA, not available.
TABLE 5.
Patients with defective IL-22 production upon PBMC stimulation
| Age (yr) | Sexa | Group | Clinical pictureb | Stimulic | Diagnosis-test interval | Lymphocyte count/mm3 | Underlying defectd |
|---|---|---|---|---|---|---|---|
| 50 | F | NIIDe | Sterile subareolar abscesses | 1 | Continuously | 3,040 | NP |
| 26 | M | Infection | Pyogenic skin infections, acne conglobata | 1 | 9 mo | 2,050 | NP |
| 53 | F | Infection | RVVC, Mycobacterium kansasii pneumonia, viral skin infections | 1 | 2 yr | 1,300 | NP |
| 66 | M | Infection | Recurrent Candida esophagitis | 1 | 5 mo | 1,840 | NP |
| 22 | F | Infection | Chronic mucocutaneous candidiasis | 1 | Continuously | NAf | NP |
| 28 | M | Infection | Dermatomycosis, oropharyngeal candidiasis, recurrent pneumonia, SLE, DMI, hypothyroidism | 1 | 2 mo (pneumonia) | 1,620 | STAT1 mutation |
| 30 | F | Infection | Recurrent oropharyngeal candidiasis | 1 | 2 mo | 1,940 | NP |
| 20 | F | Infection | Chronic mucocutaneous candidiasis | 1 | Continuously | NA | NP |
| 87 | F | Infection | Aspergillus sinusitis | 1 | 2 wk | NA | NP |
| 63 | F | Infection | Aspergillus skull base osteomyelitis | 1 | NA | 3 yr | NP |
F, female; M, male.
RVVC, recurrent vulvovaginal candidiasis; SLE, systemic lupus erythematosus; DMI, diabetes mellitus type 1.
Number of stimuli that resulted in a deficient response.
NP, not performed; STAT, signal transducer and activator of transcription.
NIID, noninfectious inflammatory disorder.
NA, not available.
TABLE 6.
Abnormal tests of patients with infectious diseases, stratified according to microbial etiology, type of assay, and cytokine
| Infection type | No. of WB stimulation assays (n = 25) with IFN-γa |
No. of PBMC stimulation assays (n = 92) total or with low production ofb: |
||||
|---|---|---|---|---|---|---|
| Totalc | Low production | Totald | IFN-γ | IL-17 | IL-22 | |
| Viral | 0 | 0 | 16 | 3 | 0 | 0 |
| Bacterial | 12 | 2 | 24 | 3 | 0 | 1 |
| Mycobacterial | 3 | 1 | 13 | 5 | 0 | 1 |
| Fungal, mucocutaneous | 2 | 0 | 31 | 8 | 5 | 5 |
| Fungal, invasive | 8 | 6 | 8 | 2 | 1 | 2 |
WB, whole blood.
PBMC, peripheral blood mononuclear cells.
Total number of WB stimulation assays performed in patients with infectious diseases.
Total number of PBMC stimulation assays performed in patients with infectious diseases.
Diminished IFN-γ production was found in 21 ID patients and 6 NIDD patients (Fig. 1). Interestingly, three-quarters of the ID patients with low IFN-γ production were patients with mycobacterial and fungal infections (Table 3). One patient with progressive multifocal leukoencephalopathy and two with recurrent herpesvirus infections had low IFN-γ production after stimulation with poly(I·C). Among the 6 NIID patients with defective IFN-γ release, two had hidradenitis suppurativa and one had sterile subareolar abscesses (Zuska's disease) (16), disorders thought to have a shared pathogenesis.
Impaired IL-17A production was observed in 6 ID patients, all of whom were suffering from fungal infection, five of which were refractory mucocutaneous infections and one was an invasive infection (Tables 4 and 6).
Deficient IL-22 production was also predominantly observed in ID patients with mucocutaneous infections, mainly those of fungal etiology (Table 5). The one patient with an NIID and defective IL-22 release had sterile subareolar abscesses.
A final diagnosis of hyper-IgE syndrome (HIES) was reached in 10 patients, five of whom had a STAT3 mutation. Six patients with CMC were included (STAT1 mutations were found in four of them). Deficient IFN-γ production (WB and PBMC combined, C. albicans and S. aureus as stimuli) was a relatively sensitive marker for HIES: 60% of the patients with HIES produced <5% compared to healthy controls, and 80% produced <10% (see Fig. S3 in the supplemental material). All CMC patients displayed deficient production of one of the lymphocyte-derived cytokines upon stimulation with C. albicans and S. aureus: IFN-γ was defective in 67%, IL-17A was defective in 67%, and IL-22 was defective in 75% of the patients (see Fig. S3).
Figure 3 shows the absolute cytokine concentrations of the abnormal tests for controls versus patients, NIID versus ID patients, and the IFN-γ concentrations in patients with mycobacterial, bacterial, and fungal infections.
FIG 3.
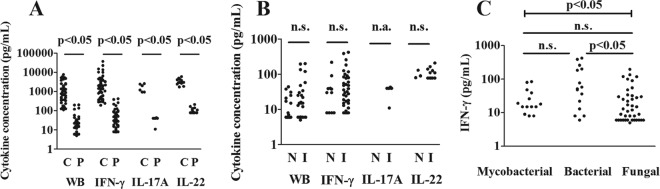
Absolute cytokine concentrations for the abnormal tests (i.e., production of <5% of controls) of patients (P) versus controls (C) (A), noninfectious inflammatory diseases (N) versus inflammatory disorders (I) (B), and mycobacterial versus bacterial and fungal infections (C). The lines show comparisons between two groups (Mann-Whitney U test or Dunn's posttest). The capped line shows the overall result of the Kruskal-Wallis test comparing the three groups. n.s., nonsignificant; n.a., not applicable.
IFN-γ production in patients with mycobacterial infections.
The PBMCs of healthy individuals mount an IFN-γ response when exposed to M. tuberculosis sonicate just by previous exposure or Mycobacterium bovis BCG vaccination (17). Thus, inherent to the reference standard chosen (i.e., <5% of that of healthy controls), a low IFN-γ response in non-BCG-vaccinated ID patients to mycobacterial stimuli, as was found in 6 ID patients, may not be interpreted as defective. Still, 6 other ID patients with mycobacterial infection had deficient IFN-γ production to nonmycobacterial stimuli.
Impact of cutoff values.
Figure 4 illustrates the impact of our stringent cutoff values (<5%) on the diagnostic performance for patients with fungal infections. As also shown in Table S3 in the supplemental material, a higher cutoff value would have identified more patients with low lymphocyte-derived cytokine production, except for patients with viral infections and mycobacterial infections. The increase (44% to 63%) in patients with mycobacterial infections was not significant, probably due to the small sample size. Across all three reference values, mycobacterial and fungal infections were predominantly associated with positive test results. For NIID, with the 5% as reference category (25% [10/40]), the use of 10% and 20% as cutoff values increased the proportion of affected patients to 45% (18/40, P = 0.06) and 50% (20/40, P = 0.02), respectively.
FIG 4.
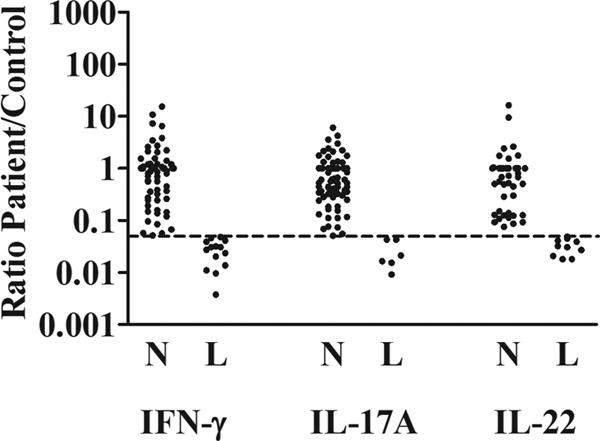
Ratio of IFN-γ, IL-17A, and IL-22 production between patients and healthy volunteers for tests of patients with fungal infections. L, test results considered deficient, i.e., production rate of <5% of that of the controls (ratio, <0.05). N, results considered normal (ratio, >0.05). The horizontal dashed line indicates the reference standard used (ratio, 0.05).
DISCUSSION
Using functional investigations of ex vivo cytokine production, we demonstrate defective cytokine production in one-third of adult patients referred to a tertiary center for infectious diseases, immunodeficiencies, and autoinflammation because of recurrent, severe, or opportunistic infections or suspicion of an NIID.
It is currently unclear to what extent the defects found represent genetic or acquired disorders. Only in a few patients were there family members with similar ailments, making the search for a genetic defect difficult. However, with the advent of powerful genetic techniques, like next-generation sequencing, finding genetic defects guided by functional findings is more in reach. In fact, such an approach was used to detect a STAT1 gene mutation as the cause of autosomal dominant CMC (10).
In our study, deficient production of lymphocyte-derived cytokines was frequently found, especially in patients with opportunistic infections of viral, mycobacterial, or fungal etiology revealing significant abnormalities. Thus, these groups of adult patients should be the targets of investigations on cytokine production defects.
The defective IFN-γ production observed in the samples from many of the patients with mycobacterial infections (mainly nontuberculous mycobacteria [NTM]) reflects the key role of IFN-γ in macrophage activation for mycobacterial killing (7). It is known that patients with mutations in the IL-12/IFN-γ signaling pathway are vulnerable to disseminated infections with mildly virulent mycobacteria. In contrast to our patients, the patients with these defects are often identified at a young age (7, 8), arguing for more subtle defects in our adult patients. Although it has been reported that NTM infection itself may suppress IFN-γ responses (18), in our study, defective cytokine release was also found in patients who had recovered from their NTM infection.
In 7 patients with invasive fungal infections, we found defective IFN-γ production. This underscores the important role of the Th1 response, which in addition to the defense by phagocytic cells, activates the host resistance to fungi (19).
The importance of Th17 responses in mucosal antifungal immunity (9, 10, 20–22) was confirmed by the identification of IL-17A/IL-22 defects in our patients with mucosal fungal infections. In our series, a number of patients were included who turned out to suffer from HIES. These patients did not present with the typical features and/or an informative family history. Based on the functional analysis that yielded defective IFN-γ responses to C. albicans and S. aureus, genetic analysis was performed. The case of the new STAT1 variant also illustrates how functional analysis may guide subsequent genetic search.
Remarkably, two patients with an intracranial Aspergillus infection had impaired IL-22 production, isolated or in combination with IFN-γ and IL-17A deficits. Reportedly, patients with HIES and a defective Th17 response are at risk for invasive fungal infections (19, 23). An impaired mucosal host defense, as demonstrated by defective IL-22 or IL-17A production, may have led to increased fungal colonization of the nasal cavity and paranasal sinuses, impaired epithelial integrity, and invasion of the deeper tissues (24, 25).
Besides the possibility of exploring underlying immunodeficiencies, it is tempting to speculate that a defective IFN-γ functional test may guide the use of adjunctive immunomodulatory therapy, such as recombinant IFN-γ therapy (26). Therapy with IFN-γ has been used successfully for the prevention and treatment of fungal infections (especially aspergillosis) in patients with chronic granulomatous disease (27).
An unexpected finding of the present study was the defective IFN-γ production found in 10 patients with NIID. Of these patients, 4 had hidradenitis suppurativa or sterile subareolar abscesses, in which follicular occlusion was thought to be the initial event in the pathogenesis of the disease. It is widely acknowledged that Th17 cells play a prominent role in autoimmune and autoinflammatory disorders (28). Recently, it has been shown that the IL-23/Th17 pathway is overexpressed in hidradenitis suppurativa (29), and this provides a rationale for the use of biologicals interfering with the Th17 pathway, like ustekinumab (anti-IL-12/IL-23) and anakinra (IL-1Ra) (see http://clinicaltrials.gov/ct2/show/NCT01704534?term=hidradenitis+suppurativa&rank=11 and http://clinicaltrials.gov/ct2/show/NCT01558375?term=hidradenitis+suppurativa&rank=12). Given the known suppressive effect of IFN-γ on the expansion of Th17 cells (28), the absence of IFN-γ production in affected skin (30) and circulating lymphocytes as found here suggests a role of defective Th1 responses for the overproduction of IL-17A.
With regard to the production of monocyte-derived cytokines, we rarely found defects. Disorders characterized by the defective production of monocyte-derived cytokines, such as MyD88 and IRAK-4 deficiency, predispose individuals to recurrent pyogenic bacterial infections during childhood (31). The occurrence of these infections at a young age, the severity of these infections, and the decrease in susceptibility with increasing age likely explain the absence of these functional phenotypes in our cohort of adult patients.
Obviously, our observational explorative study has also some limitations that should be acknowledged. First, our studies are retrospective and were performed over many years; hence, the methodology evolved over time. In addition, as is well-known from previous surveys, ex vivo cytokine production has large interindividual variation (32), hampering the establishment of reference values. We compared cytokine production in infected patients with that of healthy volunteers tested in the same experiment in order to control for procedural variability in the investigations. One limitation of the study is that in most cases, no age- and sex-matched controls were available, and blood samples from anonymous healthy blood donors were used. We applied stringent cutoff values (i.e., <5% of the cytokine production in healthy controls) in order to minimize false-positive results. Hence, we may have missed some mild defects in cytokine production (33). We used fixed concentrations of stimulus, but we are aware that exposure to stimuli in a range of doses might provide extra information. Again, we may have missed some mild defects, as we used relatively high concentrations of ligand. Especially, in autoinflammatory disorders, which are characterized by an exaggerated immune response to relatively mild stimuli, the use of a dose-response curve provides a way to evaluate overproduction. Third, two methods for determining cytokine production were used: a WB stimulation assay and a PBMC stimulation assay. Although the PBMC stimulation assay is more time-consuming, it allows for an assessment of IL-17A and IL-22 production, in addition to that of IFN-γ. Despite differences in the cell types and noncellular components between the two methods, we think the results are generalizable, because the correlation between these methods is good (32). Nevertheless, it is crucial to standardize this methodology in the future in order to make these tests available for larger numbers of patients.
Incorporating measurements of additional cytokines may improve the identification of more immunodeficiencies. For example, assessments of IFN-α, IFN-β, and/or IFN-λ may be of added value for evaluating antiviral responses (34). Circulating leukocytes do not necessarily reflect the entire immune status at the tissue level (35), and the stimulation of nonhematopoietic cells may reveal an immunodeficiency, while PBMCs are normally responsive (34).
In conclusion, a retrospective analysis of ex vivo cytokine release assays in >150 patients with recurrent and/or severe infections or NIID revealed functional defects in Th1 and Th17 responses to be relatively common in patients with mycobacterial, fungal, and viral infections, as well as in patients with NIID. These assays are well suited for detecting defects in immune pathways relating to different infectious phenotypes. These functional findings may guide genetic studies using advanced powerful techniques, thereby establishing a molecular diagnosis. In addition, the findings may guide adjunctive immunomodulatory therapy. Future studies using standardized methodologies to assess cytokine production are warranted to better understand the immunologic defects in patients with infections and NIID.
Supplementary Material
ACKNOWLEDGMENTS
We thank Trees Jansen, Liesbeth Jacobs, and Johanna Jongekrijg for help with the cytokine assessments.
We declare no conflicts of interest.
J.T.O. was supported by the European Regional Development Fund–Province of Gelderland project 2009-010034. F.L.V.D.V. was supported by a Veni grant, and R.V.C. and A.S. were supported by a Vidi grant from the Netherlands Organization for Scientific Research (NWO). M.G.N. was supported by an ERC consolidator grant (310372).
Footnotes
Published ahead of print 28 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/CVI.00152-14.
REFERENCES
- 1.Casanova JL, Abel L. 2007. Primary immunodeficiencies: a field in its infancy. Science 317:617–619. 10.1126/science.1142963 [DOI] [PubMed] [Google Scholar]
- 2.Casanova JL, Fieschi C, Zhang SY, Abel L. 2008. Revisiting human primary immunodeficiencies. J. Intern. Med. 264:115–127. 10.1111/j.1365-2796.2008.01971.x [DOI] [PubMed] [Google Scholar]
- 3.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr 1997. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388:394–397. 10.1038/41131 [DOI] [PubMed] [Google Scholar]
- 4.Netea MG, van der Meer JW. 2011. Immunodeficiency and genetic defects of pattern-recognition receptors. N. Engl. J. Med. 364:60–70. 10.1056/NEJMra1001976 [DOI] [PubMed] [Google Scholar]
- 5.Netea MG, van de Veerdonk FL, van Deuren M, van der Meer JW. 2011. Defects of pattern recognition: primary immunodeficiencies of the innate immune system. Curr. Opin. Pharmacol. 11:412–422. 10.1016/j.coph.2011.03.003 [DOI] [PubMed] [Google Scholar]
- 6.Masters SL, Simon A, Aksentijevich I, Kastner DL. 2009. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu. Rev. Immunol. 27:621–668. 10.1146/annurev.immunol.25.022106.141627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haverkamp MH, van Dissel JT, Holland SM. 2006. Human host genetic factors in nontuberculous mycobacterial infection: lessons from single gene disorders affecting innate and adaptive immunity and lessons from molecular defects in interferon-gamma-dependent signaling. Microbes Infect. 8:1157–1166. 10.1016/j.micinf.2005.10.029 [DOI] [PubMed] [Google Scholar]
- 8.Al-Muhsen S, Casanova JL. 2008. The genetic heterogeneity of Mendelian susceptibility to mycobacterial diseases. J. Allergy Clin. Immunol. 122:1043–1051, quiz 1052–1043. 10.1016/j.jaci.2008.10.037 [DOI] [PubMed] [Google Scholar]
- 9.Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, Migaud M, Israel L, Chrabieh M, Audry M, Gumbleton M, Toulon A, Bodemer C, El-Baghdadi J, Whitters M, Paradis T, Brooks J, Collins M, Wolfman NM, Al-Muhsen S, Galicchio M, Abel L, Picard C, Casanova JL. 2011. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 332:65–68. 10.1126/science.1200439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, Arts P, Rosentul DC, Carmichael AJ, Smits-van der Graaf CA, Kullberg BJ, van der Meer JW, Lilic D, Veltman JA, Netea MG. 2011. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N. Engl. J. Med. 365:54–61. 10.1056/NEJMoa1100102 [DOI] [PubMed] [Google Scholar]
- 11.Netea MG, van der Leij F, Drenth JP, Joosten LA, te Morsche R, Verweij P, de Jong D, Kullberg BJ, van der Meer JW. 2010. Chronic yersiniosis due to defects in the TLR5 and NOD2 recognition pathways. Neth. J. Med. 68:310–315 [PubMed] [Google Scholar]
- 12.Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, Toubiana J, Itan Y, Audry M, Nitschke P, Masson C, Toth B, Flatot J, Migaud M, Chrabieh M, Kochetkov T, Bolze A, Borghesi A, Toulon A, Hiller J, Eyerich S, Eyerich K, Gulácsy V, Chernyshova L, Chernyshov V, Bondarenko A, Grimaldo RM, Blancas-Galicia L, Beas IM, Roesler J, Magdorf K, Engelhard D, Thumerelle C, Burgel PR, Hoernes M, Drexel B, Seger R, Kusuma T, Jansson AF, Sawalle-Belohradsky J, Belohradsky B, Jouanguy E, Bustamante J, Bué M, Karin N, Wildbaum G, Bodemer C, Lortholary O, Fischer A, Blanche S, et al. 2011. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med. 208:1635–1648. 10.1084/jem.20110958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Vries E, European Society for Immunodeficiencies (ESID) members 2012. Patient-centred screening for primary immunodeficiency, a multi-stage diagnostic protocol designed for non-immunologists: 2011 update. Clin. Exp. Immunol. 167:108–119. 10.1111/j.1365-2249.2011.04461.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Endres S, Ghorbani R, Lonnemann G, van der Meer JW, Dinarello CA. 1988. Measurement of immunoreactive interleukin-1 beta from human mononuclear cells: optimization of recovery, intrasubject consistency, and comparison with interleukin-1 alpha and tumor necrosis factor. Clin. Immunol. Immunopathol. 49:424–438. 10.1016/0090-1229(88)90130-4 [DOI] [PubMed] [Google Scholar]
- 15.van Deuren M, Netea MG, Hijmans A, Demacker PN, Neeleman C, Sauerwein RW, Bartelink AK, van der Meer JW. 1998. Posttranscriptional down-regulation of tumor necrosis factor-alpha and interleukin-1beta production in acute meningococcal infections. J. Infect. Dis. 177:1401–1405. 10.1086/517824 [DOI] [PubMed] [Google Scholar]
- 16.Berná-Serna JD, Berná-Mestre JD. 2010. Follicular occlusion due to hyperkeratosis: a new hypothesis on the pathogenesis of mammillary fistula. Med. Hypotheses 75:553–554. 10.1016/j.mehy.2010.07.027 [DOI] [PubMed] [Google Scholar]
- 17.Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Ifrim DC, Saeed S, Jacobs C, van Loenhout J, de Jong D, Stunnenberg HG, Xavier RJ, van der Meer JW, van Crevel R, Netea MG. 2012. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc. Natl. Acad. Sci. U. S. A. 109:17537–17542. 10.1073/pnas.1202870109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeboah-Manu D, Peduzzi E, Mensah-Quainoo E, Asante-Poku A, Ofori-Adjei D, Pluschke G, Daubenberger CA. 2006. Systemic suppression of interferon-gamma responses in Buruli ulcer patients resolves after surgical excision of the lesions caused by the extracellular pathogen Mycobacterium ulcerans. J. Leukoc. Biol. 79:1150–1156. 10.1189/jlb.1005581 [DOI] [PubMed] [Google Scholar]
- 19.Vinh DC. 2011. Insights into human antifungal immunity from primary immunodeficiencies. Lancet Infect. Dis. 11:780–792. 10.1016/S1473-3099(11)70217-1 [DOI] [PubMed] [Google Scholar]
- 20.Maródi L, Cypowyj S, Tóth B, Chernyshova L, Puel A, Casanova JL. 2012. Molecular mechanisms of mucocutaneous immunity against Candida and Staphylococcus species. J. Allergy Clin. Immunol. 130:1019–1027. 10.1016/j.jaci.2012.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, Davis J, Hsu A, Asher AI, O'Shea J, Holland SM, Paul WE, Douek DC. 2008. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452:773–776. 10.1038/nature06764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Netea MG, Kullberg BJ, van der Meer JW. 2005. Severely impaired IL-12/IL-18/IFNgamma axis in patients with hyper IgE syndrome. Eur. J. Clin. Invest. 35:718–721. 10.1111/j.1365-2362.2005.01564.x [DOI] [PubMed] [Google Scholar]
- 23.Vinh DC, Sugui JA, Hsu AP, Freeman AF, Holland SM. 2010. Invasive fungal disease in autosomal-dominant hyper-IgE syndrome. J. Allergy Clin. Immunol. 125:1389–1390. 10.1016/j.jaci.2010.01.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knipping S, Holzhausen HJ, Koesling S, Bloching M. 2007. Invasive aspergillosis of the paranasal sinuses and the skull base. Eur. Arch. Otorhinolaryngol. 264:1163–1169. 10.1007/s00405-007-0336-7 [DOI] [PubMed] [Google Scholar]
- 25.Plantinga TS, van der Velden WJ, Ferwerda B, van Spriel AB, Adema G, Feuth T, Donnelly JP, Brown GD, Kullberg BJ, Blijlevens NM, Netea MG. 2009. Early stop polymorphism in human DECTIN-1 is associated with increased Candida colonization in hematopoietic stem cell transplant recipients. Clin. Infect. Dis. 49:724–732. 10.1086/604714 [DOI] [PubMed] [Google Scholar]
- 26.Netea MG, Brouwer AE, Hoogendoorn EH, Van der Meer JW, Koolen M, Verweij PE, Kullberg BJ. 2004. Two patients with cryptococcal meningitis and idiopathic CD4 lymphopenia: defective cytokine production and reversal by recombinant interferon-gamma therapy. Clin. Infect. Dis. 39:e83–87. 10.1086/425121 [DOI] [PubMed] [Google Scholar]
- 27.Marciano BE, Wesley R, De Carlo ES, Anderson VL, Barnhart LA, Darnell D, Malech HL, Gallin JI, Holland SM. 2004. Long-term interferon-gamma therapy for patients with chronic granulomatous disease. Clin. Infect. Dis. 39:692–699. 10.1086/422993 [DOI] [PubMed] [Google Scholar]
- 28.Bettelli E, Korn T, Oukka M, Kuchroo VK. 2008. Induction and effector functions of TH17 cells. Nature 453:1051–1057. 10.1038/nature07036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schlapbach C, Hänni T, Yawalkar N, Hunger RE. 2011. Expression of the IL-23/Th17 pathway in lesions of hidradenitis suppurativa. J. Am. Acad. Dermatol. 65:790–798. 10.1016/j.jaad.2010.07.010 [DOI] [PubMed] [Google Scholar]
- 30.van der Zee HH, de Ruiter L, van den Broecke DG, Dik WA, Laman JD, Prens EP. 2011. Elevated levels of tumour necrosis factor (TNF)-α, interleukin (IL)-1β and IL-10 in hidradenitis suppurativa skin: a rationale for targeting TNF-α and IL-1β. Br. J. Dermatol. 164:1292–1298. 10.1111/j.1365-2133.2011.10254.x [DOI] [PubMed] [Google Scholar]
- 31.Picard C, Casanova JL, Puel A. 2011. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IκBα deficiency. Clin. Microbiol. Rev. 24:490–497. 10.1128/CMR.00001-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yaqoob P, Newsholme EA, Calder PC. 1999. Comparison of cytokine production in cultures of whole human blood and purified mononuclear cells. Cytokine 11:600–605. 10.1006/cyto.1998.0471 [DOI] [PubMed] [Google Scholar]
- 33.van de Veerdonk FL, Marijnissen RJ, Joosten LA, Kullberg BJ, Drenth JP, Netea MG, van der Meer JW. 2010. Milder clinical hyperimmunoglobulin E syndrome phenotype is associated with partial interleukin-17 deficiency. Clin. Exp. Immunol. 159:57–64. 10.1111/j.1365-2249.2009.04043.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, Alcais A, Picard C, Mahfoufi N, Nicolas N, Lorenzo L, Plancoulaine S, Sénéchal B, Geissmann F, Tabeta K, Hoebe K, Du X, Miller RL, Héron B, Mignot C, de Villemeur TB, Lebon P, Dulac O, Rozenberg F, Beutler B, Tardieu M, Abel L, Casanova JL. 2006. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 314:308–312. 10.1126/science.1128346 [DOI] [PubMed] [Google Scholar]
- 35.Kox M, de Kleijn S, Pompe JC, Ramakers BP, Netea MG, van der Hoeven JG, Hoedemaekers CW, Pickkers P. 2011. Differential ex vivo and in vivo endotoxin tolerance kinetics following human endotoxemia. Crit. Care Med. 39:1866–1870. 10.1097/CCM.0b013e3182190d5d [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.