

Abstract
1,2,3-Trichloropropane (TCP) is a toxic compound that is recalcitrant to biodegradation in the environment. Attempts to isolate TCP-degrading organisms using enrichment cultivation have failed. A potential biodegradation pathway starts with hydrolytic dehalogenation to 2,3-dichloro-1-propanol (DCP), followed by oxidative metabolism. To obtain a practically applicable TCP-degrading organism, we introduced an engineered haloalkane dehalogenase with improved TCP degradation activity into the DCP-degrading bacterium Pseudomonas putida MC4. For this purpose, the dehalogenase gene (dhaA31) was cloned behind the constitutive dhlA promoter and was introduced into the genome of strain MC4 using a transposon delivery system. The transposon-located antibiotic resistance marker was subsequently removed using a resolvase step. Growth of the resulting engineered bacterium, P. putida MC4-5222, on TCP was indeed observed, and all organic chlorine was released as chloride. A packed-bed reactor with immobilized cells of strain MC4-5222 degraded >95% of influent TCP (0.33 mM) under continuous-flow conditions, with stoichiometric release of inorganic chloride. The results demonstrate the successful use of a laboratory-evolved dehalogenase and genetic engineering to produce an effective, plasmid-free, and stable whole-cell biocatalyst for the aerobic bioremediation of a recalcitrant chlorinated hydrocarbon.
INTRODUCTION
The chlorinated hydrocarbon 1,2,3-trichloropropane (TCP) is a toxic and carcinogenic nonnatural compound with several useful industrial applications. It is used in the paint industry, as a varnish remover or cleaning agent, and as an intermediate in the production of other chemicals, including polysulfone liquid polymers and hexafluoropropylene (1, 2). Another major use of TCP is the industrial synthesis of epichlorohydrin via propylene chlorination, which generates a waste stream with TCP as the predominant component. TCP is frequently detected as a groundwater pollutant because of improper waste disposal and its recalcitrance to biodegradation. It has a higher density than water, so it moves easily into deeper groundwater layers, leading to widespread contamination as a result of its moderate water solubility. This poses a serious risk to ecosystem quality, as well as to human health when TCP infiltrates drinking water supplies (3). Cleanup of TCP-contaminated sites is problematic because of the physicochemical properties of TCP, including the low sorption capacity of activated carbon (4). TCP can be degraded by reaction with metallic zinc, iron, or zinc oxides (4–6).
Removal of TCP by biodegradation would be an attractive approach for groundwater treatment but is critically dependent on the availability of microorganisms that degrade TCP in a manner that supports cellular maintenance and growth. The relevant physicochemical properties of TCP are similar to those of 1,2-dichloroethane (octanol-water partition coefficient [Kow] values, 1.98 and 1.48 for TCP and 1,2-dichloroethane, respectively; Henry coefficients, 3.4 × 10−4 and 1.18 × 10−4 atm · m3 · mol−1, respectively). A full-scale bioreactor-based treatment process has been used for the removal of 1,2-dichloroethane from contaminated groundwater at a large contaminated site in Lübeck, Germany (7). This process employed the 1,2-dichloroethane degrader Xanthobacter autotrophicus GJ10 (8). The similarity in physical-chemical properties between TCP and 1,2-dichloroethane suggests that biological treatment is feasible once an equally effective microorganism for TCP removal is obtained.
To date, no naturally occurring microbial culture has been described that can degrade TCP and use it as a carbon source under aerobic conditions. Reductive biological dechlorination of TCP and other chloropropanes is possible under anaerobic conditions by reactions that may also occur in the environment (9–13). We previously described oxidative cometabolism of TCP by methanotrophs, but a major drawback of converting TCP by methane monooxygenase is product toxicity (14). Insertion of oxygen preferentially occurs on the terminal carbon, yielding chlorinated carbonyl compounds that undergo an elimination reaction to produce the very toxic compound 2-chloroacrolein (14). Genetic engineering may offer a strategy for producing a strain for the aerobic degradation of TCP (15–17). Theoretical calculations have indicated that a number of transformations, including reductive dechlorination, reductive β-elimination, dehydrochlorination, and nucleophilic substitution by OH−, are thermodynamically favorable (18).
A possible pathway for aerobic TCP mineralization starts with the conversion of TCP to 2,3-dichloro-1-propanol (DCP) by a haloalkane dehalogenase that hydrolyzes a carbon-chlorine bond. Haloalkane dehalogenases are hydrolytic bacterial proteins that belong to the α/β-hydrolase superfamily and cleave carbon-chlorine bonds by hydrolysis via formation of a covalent intermediate. Various haloalkane dehalogenases have been described (19–21), but the only wild-type dehalogenase with significant activity on TCP is a variant called DhaA, which occurs in some strains of Rhodococcus and other bacteria (15, 20, 22). Wild-type DhaA has a poor turnover number and a high Km with TCP. Nevertheless, in view of the widespread occurrence of the dhaA gene (23), it is remarkable that TCP is recalcitrant to aerobic biodegradation, the more so because only 3 to 5 amino acid substitutions are required for better conversion (15, 24). Various attempts by ourselves and others to produce a TCP-degrading culture by classical enrichment have failed, whereas compounds with similar structures and toxicities, such as 1,2-dibromoethane (DBE) and 1,3-dichloropropene, were degraded after prolonged enrichment by organisms using the same DhaA-type haloalkane dehalogenase (22, 23).
Bosma et al. (15), Gray et al. (19), and Pavlova et al. (24) improved the kinetic properties of DhaA for TCP conversion by directed evolution. An engineered dehalogenase mutant (DhaAM2, containing the mutations Cys176Tyr and Tyr273Phe) was 3.5-fold more active with TCP than the wild-type enzyme. In order to obtain a degradation pathway, this improved dehalogenase was expressed with a broad-host-range plasmid in a strain of Agrobacterium radiobacter that metabolizes DCP slowly. The recombinant strain indeed was able to degrade TCP slowly (15), but there were several problems. (i) Degradation was incomplete due to the (R) enantioselectivity of the haloalcohol dehalogenase HheC provided by the host, whereas the enzyme introduced, DhaAM2, produces a racemic mixture of (R)- and (S)-2,3-dichloropropanols (Fig. 1). (ii) HheC prefers vicinal haloalcohols with the halogens on the terminal carbons, i.e., 1,3-dichloro-2-propanol instead of 2,3-dichloro-1-propanol. (iii) The plasmid-based system used for dhaAM2 expression is a broad-host-range cosmid derived from pLAFR3 that carries a tetracycline antibiotic resistance marker and mobilization functions; thus, it is not desirable to use the strain in open applications, such as an immobilized-cell bioreactor. (iv) The plasmid that was used has large segments of DNA that are not necessary for its function and that may cause instability of the strain and loss of the dhaA gene. In accordance with these problems, attempts to degrade TCP in a continuous packed-bed bioreactor inoculated with the recombinant strain and fed TCP-contaminated water failed. Continuous degradation of TCP was obtained with an immobilized-enzyme system composed of the engineered haloalkane dehalogenase DhaA and halohydrin dehalogenase plus epoxide hydrolase from A. radiobacter (25). However, this process requires recombinantly produced immobilized enzymes, imposing a significant cost that may prohibit application.
FIG 1.
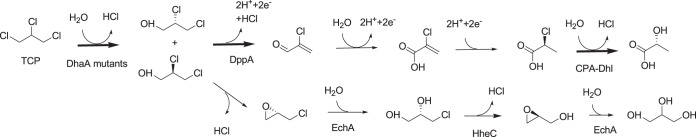
Proposed aerobic degradation pathways for TCP. (Upper route) TCP degradation by the engineered P. putida strain MC4. (Lower route) TCP degradation by the engineered A. radiobacter strain AD1. DhaA, haloalkane dehalogenase; DppA, dichloropropanol dehalogenase/dehydrogenase; CPA, 2-chloropropionic acid dehalogenase; HheC, halohydrin dehalogenase; EchA, epoxide hydrolase. Thick arrows indicate the enzyme activities measured in this study.
The current study aims to remedy the problems mentioned above by construction of a TCP-utilizing recombinant strain that can completely degrade TCP through an improved catabolic pathway that is free of any antibiotic resistance marker and that does not contain a transmissible plasmid. This system is based on a recently described strain of Pseudomonas putida that uses a novel pathway for 2,3-dichloropropanol conversion (26) and on a further improved dehalogenase variant obtained by directed evolution focused on tunnel residues leading to the active site (24, 27).
MATERIALS AND METHODS
Strain and growth conditions.
The host organism used in this study was Pseudomonas putida strain MC4, a 2,3-dichloropropanol-degrading bacterium (26). The organism and its derivatives were grown in LB medium or in a defined medium (MMY) that contained, per liter, 5.4 g of Na2HPO4·12H2O, 1.4 g of KH2PO4, 0.5 g of (NH4)2SO4, 0.2 g of MgSO4·7H2O, 5 ml of a trace element solution, and 5 mg of yeast extract (28). This amount of yeast extract did not produce a significant increase in the optical density at 600 nm (OD600) (less than 0.01). Cells were grown at pH 7.0 and 30°C with shaking (200 to 250 rpm), and when MMY medium was used, 1,2,3-trichloropropane (TCP; 0.5 to 1 mM) or 2,3-dichloro-1-propanol (DCP; 4 mM) was provided as a carbon source. To avoid the loss of TCP by evaporation, bottles were made gas-tight with screw caps that had Viton septa, and samples were collected with a syringe. The gas phase was large enough to provide sufficient oxygen for aerobic mineralization. Inocula consisted of washed cells from a 5-ml overnight culture that was started by inoculating LB medium containing tetracycline (25 μg/ml) with cells from a freshly streaked plate. The MMY cultures were incubated overnight at 30°C for strain MC4. LB medium cultures were incubated at 17°C for 48 h for Escherichia coli TOP10, because the dhaA31 gene was not well expressed in TOP10 at 30°C. In the case of cells containing pIT31, 0.8 mM isopropyl-1-thio-β-d-galactopyranoside (IPTG) was added at the start for the induction of haloalkane dehalogenase synthesis.
Construction of broad-host-range expression plasmids.
The gene encoding improved haloalkane dehalogenase (dhaA31) (24) was placed under the control of the inducible trc promoter (29) or the constitutive dhlA promoter (8). Clones with the dhaA31 gene behind the trc promoter were constructed in the broad-host-range vector pIT2-MCS (28), which is derived from pBBR1MCS2 (30) and carries a tetracycline resistance marker and the trc promoter. Primers PF1 (5′-ACACTATCGGACCATATGTCAGAAATGGGT [NdeI site and start codon underlined]) and PR1 (5′-GATCCGTCGACCTGCAGCCAAGCTTCTAGT [HindIII site underlined; bases in italics include the stop codon and 2 bases of the dhaA31 gene]) were used for the amplification of dhaA31 from plasmid pAQN (24). PCR was carried out with Pfx50 DNA polymerase; the product was purified and ligated at a 1:3 vector-insert ratio in plasmid pBAD; and the mixture was transformed to E. coli TOP10 by chemical transformation or to strain MC4 by electroporation. The resulting plasmid was named pIT31. Transformants were plated on LB agar plates containing tetracycline (25 μg/ml) and were grown at 37°C for E. coli and at 30°C for strain MC4.
In order to place the dhaA31 gene under the control of the strong constitutive dhlA promoter, we used a sequence of X. autotrophicus GJ10 including the ribosome binding site (RBS), but with a variation between the RBS (GGAGG) and the start codon (ATG). In one case, this segment was made one base longer (CTCT → CTCAT) and was designated translation initiation region A. The second version, designated translation initiation region B, contained two mutations between the RBS and the start codon (CTCT → CCAT). Plasmid pIT31 was digested with BamHI and NdeI to remove the trc promoter and was ligated with a 485-bp PCR fragment harboring the dhlA promoter, the dhlA ribosome binding site, and the modified dhlA translation initiation regions. For the isolation of this fragment, X. autotrophicus GJ10 DNA was used as the template in a PCR with primer PF2 (5′-GTCCAGCATCTCTAACGGATCCGGCGATCCCGATAGTACGACTTG [BamHI site underlined]) in combination with the reverse primer PR2a (5′-GTACCTTTCGAATCATATGAGCCTCCGTAAGCGTCTGTCAA [NdeI site and reverse complement start codon underlined; reverse complement RBS italicized]) or PR2b (5′-GTACCTTTCGAATCATATGGCCTCCGTAAGCGTCTGTCAA [NdeI site and reverse complement start codon underlined; inserted G in boldface; reverse complement RBS italicized]). The reverse primers, which differ between the ribosome binding site and the translation start site, gave pIS31A and pIS31B, respectively. In the same way, another improved variant, dhaA27 (24), was cloned under the control of the trc or dhlA promoter to yield pIT27 or pIS27, respectively.
Chromosomal integration of dhaA31B.
The mini-Tn5 system for chromosomal integration consists of a transposon vector (pUT) and a delivery system (31). The plasmid is maintained in E. coli and is transferred to target cells by triparental mating. Plasmid pUT contains two insertion sequences (IS50L and IS50R), which govern transposition in the presence of a transposase, encoded by the tnp gene. A salient feature of the pUT vector is that the transposase gene is positioned outside the transposon itself and consequently is not inserted into the host genome, preventing further mobilization of the transposed DNA fragment (32). Two segments are present between the insertion sequences. One segment carries a unique NotI cloning site, where the gene of interest (dhaA31) can be inserted. The other segment is a marker segment containing a gene for kanamycin resistance and the xylE gene for the detection of an insertion by a chromogenic reaction that occurs when the encoded catechol 2,3-dioxygenase converts catechol (see Fig. S1 in the supplemental material). The marker segment is flanked by two resolution sites (res) and is used to screen exconjugants for the occurrence of transposition events. After transposition, the marker segment can be removed by the introduction of a plasmid carrying the RP4-derived resolvase gene parA, since DNA between the res sequences is excised by the resolvase (33).
To clone the dhaA31 gene into the pUT vector (see Fig. S1 in the supplemental material), a 2-kb dhaA31B fragment containing promoter and terminator sequences was amplified from pIS31B by PCR using primers PF3 (5′-TAGCTAGCGGCCGCGCGAGATCCCGATAGTACGA) and PR3 (5′-TAGCTAGCGGCCGCCAGGGTTATTGTCTCATGAGCG), each of which contained a NotI site (underlined). The PCR product was purified with the QIAquick purification kit for ligation, digested with NotI, and cloned into the unique NotI site of pJMS11 to yield pUT31B. The presence of the 2-kb fragment harboring dhaA31B was confirmed by restriction analysis.
Plasmid pUT31B was used as the delivery plasmid and was mobilized from E. coli CC118(λpir) into target cells by triparental mating using the helper strain E. coli HB101(pRK600), which has an RP4 transfer function (34). The replication of plasmids with the pR6K origin of replication is dependent on the π protein, produced only in E. coli CC118(λpir), and therefore, pRK600 and pUT derivatives are lost in P. putida. Overnight cultures of the required strains were prepared in 2 ml LB medium containing appropriate antibiotics. The recipient strain P. putida MC4 was grown at 30°C; E. coli HB101(pRK600) was grown at 37°C with chloramphenicol (Cm) at 50 μg/ml; and E. coli CC118(λpir)(pUT31B) was grown at 37°C with kanamycin (Km) at 50 μg/ml. Mating was done by mixing 0.02 ml of each culture. After washing and resuspension in 0.02 ml MgSO4 (10 mM), the mixture was spotted onto an LB agar plate and was incubated for 10 to 13 h at 30°C. Then the cells in the spot were resuspended in MgSO4 (10 mM), and the suspension was spread on M9 plates with citrate (10 mM) and Km (50 μg/ml) for selection. Colonies were tested for sensitivity to piperacillin (50 μg/ml) on an LB agar plate to ensure that the whole delivery plasmid was not integrated. Exconjugants obtained after the transposition event were screened in various ways: resistance to Km (75 μg/ml), sensitivity to piperacillin (50 μg/ml), and catechol 1,2-dioxygenase and dehalogenase activities. Catechol 2,3-dioxygenase activity was tested by spraying plates with catechol. The ring cleavage reaction converts catechol to 2-hydroxymuconic semialdehyde, which gives a yellow color. Dehalogenase activities were screened in microtiter plates by suspending cells in water (50 μl) containing TCP (3 mM) or 1,2-dibromoethane (DBE) (5 mM) as the substrate. Plates were kept at 30°C for 4 h with DBE or for 24 h with TCP, and then halide detection reagents were added. Active cells produced a red color.
To remove inverted repeats and antibiotic markers, plasmid pJMSB8, which harbors the RP4 resolvase gene parA (33), was introduced into dehalogenase-positive exconjugants by triparental mating (32, 35), and the desired colonies were selected by testing for the loss of resistance to kanamycin and the retention of haloalkane dehalogenase activity. Plasmid pJMSB8 contains an ampicillin resistance marker for maintenance in E. coli, but once the plasmid is introduced into Pseudomonas, it will be lost due to lack of the π protein required for replication (35, 36).
Southern blotting.
To establish the copy number of the dehalogenase gene after the transposition event, Southern blotting was performed. The experiment was carried out with strains MC4, MC4-52, and MC4-5222. Genomic DNA was isolated with a genomic DNA extraction kit (Sigma) and was digested with SalI restriction endonuclease, which does not cut in the dhaA31 gene sequence. DNA fragments were separated on an 0.8% agarose gel and were transferred to the nylon membrane by diffusion blotting. A 2-kb NotI insert containing dhaA31 was used as a template to generate a probe, which was labeled with digoxigenin using the nonradioactive DIG High Prime kit (Roche). Hybridization was carried out at 60°C, and detection was performed according to the protocol provided by the manufacturer.
DNA sequencing.
Partial genome sequences of P. putida strains MC4-52 and MC4-5222 were obtained by BaseClear (Leiden, The Netherlands) using paired-end Illumina sequencing. Genomic DNA was isolated using the bacterial genomic miniprep kit provided by Sigma-Aldrich according to the accompanying instructions. De novo assembly was carried out with CLC bio software, and the contigs established were analyzed using stand-alone BLAST software to locate the insert carrying the haloalkane dehalogenase gene and to verify the presence of other genes involved in the TCP degradation pathway. Contigs harboring the genes for DppA and DhaA31 were further analyzed by Artemis (http://www.sanger.ac.uk/resources/software/artemis/) (37) and on the RAST server (38).
Enzyme and protein assays.
For the preparation of cell extracts, strain MC4 and its derivatives were cultivated in MMY medium with 4 mM DCP as a carbon source after inoculation with a preculture grown overnight in LB medium at 30°C. Following growth, the cells were harvested by centrifugation, washed with TEMG buffer (10 mM Tris-SO4 [pH 7.5], 1 mM EDTA, 1 mM β-mercaptoethanol, 0.02% sodium azide, 10% glycerol), and resuspended in 1/50 of the original culture volume of TEMG buffer. After ultrasonic disruption of the cells, the lysate was centrifuged (17,000 × g for 30 min). The clear supernatant was used for assays.
Hydrolytic dehalogenase assays were performed by incubating an appropriate amount of cell extract at 30°C in 50 mM Tris-SO4 buffer (pH 8.2) containing TCP (3 mM), 2-chloropropanoic acid (CPA) (5 mM), or DBE (3 mM) as the substrate. Samples were taken at different times (0 to 60 min), and absorbance was measured at 460 nm after the addition of mercuric thiocyanate and ferric ammonium sulfate (39). One unit of enzyme activity is defined as the amount of the enzyme that catalyzes the formation of 1 μmol of a halide per min.
The activity of dichloropropanol dehydrogenase (DppA), a quinohemoprotein that catalyzes a redox reaction, was measured at 30°C by following the reduction of ferricyanide in a spectrophotometer at 420 nm. The reaction mixture (1 ml) consisted of 50 mM Tris-SO4 (pH 8.0), 5 mM DCP, and 1 mM ferricyanide. The extinction coefficient of ferricyanide at 420 nm is 1.02 mM−1 · cm−1. One unit of enzyme activity is defined as the amount of enzyme catalyzing the reduction of 1 μmol of ferricyanide per min. All experiments were performed in duplicate or in triplicate with separately grown cultures. The standard deviation represents the sum of squares of deviations from the mean divided by the number of measurements. Protein concentrations of cell extracts were measured with the Bradford reagent at 595 nm using bovine serum albumin as standard.
Degradation studies in a packed-bed bioreactor.
A lab-scale reactor, consisting of a double-walled cylindrical glass vessel with a volumetric capacity of 1.3 liters and a working volume of 0.7 liter, was designed (see Fig. S2 in the supplemental material). The reactor was filled with 300 g ceramic Raschig rings (10 by 10 by 2 mm; outer diameter, 9 mm; internal diameter, 4.5 mm; length, 9 mm; AceChemPack Tower Packing Co.) as packing material. Sampling ports were present at the top, bottom, and middle of the vessel. TCP-contaminated water was added using a peristaltic pump from a glass supply vessel (20 liters) containing TCP in water. During operation, the inlet concentration decreased slightly due to liquid-gas partitioning of the TCP in the supply vessel. Therefore, the inlet concentration was followed by analyzing samples extracted from the port close to the liquid inlet, which was positioned at the bottom of the reactor, thus achieving an upward liquid flow. Air was introduced as small bubbles from the bottom of the reactor, and the flow was regulated with a mass flow controller. The temperature was kept at 30°C by a water jacket. Water was removed from the top of the reactor for recirculation, analysis, and effluent discharge. Water was recirculated with a peristaltic pump operated at 10-fold the water feed rate. The sampling port for analysis and effluent discharge was at the top of the reactor. The pH was also controlled at the top of the reactor, by using a pH meter with a controller set at 7.0 and adding 0.1 M NaOH when necessary. All pump tubing was from gas-tight Viton material, and sampling ports contained Viton septa to avoid physical loss. Influent air and effluent air were passed through filters to prevent unwanted contamination. The discharge went into a 20-liter glass vessel, and exhaust gas released from that vessel went into a fume hood.
For startup, a 700 ml suspension of cells grown on TCP (OD600, 0.4) was introduced into the reactor, which was left for 4 days with no supply of TCP-contaminated water. During this period, air was introduced at a rate of 0.1 ml/min. Four days after inoculation, a continuous flow of TCP-contaminated water was started, and the concentration was increased slowly from 0.1 to 0.3 mM. From day 35 onward, 0.33 mM TCP was introduced at a rate of 0.1 ml/min (residence time, 116 h; organic loading rate, 0.25 to 0.3 mg TCP · h−1 · liter−1), and the reactor was kept working at that rate. The airflow was maintained at 1 ml/min, which provided more than sufficient oxygen for complete oxidative mineralization of TCP. The effluent concentrations of TCP, DCP, and chloride ions were measured in samples (4.5 ml) taken at different time intervals with a gas-tight syringe through ports closed with Viton septa.
Analytical methods.
Concentrations of halogenated compounds were determined by gas chromatography. Samples (4.5 ml) were extracted with diethyl ether (1.5 ml) containing 0.05 mM mesitylene as an internal standard. The ether layer was analyzed by split injection of 2-μl samples on a gas chromatograph equipped with a flame ionization detector (Agilent) and an HP-5 column (25 m by 0.25 mm by 0.2 μm; Hewlett Packard). The carrier gas was nitrogen (50 kPa), and the temperature program was isothermal for 5 min at 60°C, followed first by an increase to 110°C at a rate of 2°C/min and then by an increase to 130°C at a rate of 15°C/min. Gas chromatography-mass spectrometry was performed on an HP 5890 gas chromatograph equipped with an HP-5 capillary column connected to a type 5971 mass-selective detector (Agilent). Helium was used as a carrier gas (0.9 ml min−1), and the temperature program was the same as that described above.
Halide levels in bioreactor samples were assayed by a colorimetric method as described above.
Nucleotide sequence accession number.
The genome sequences obtained by the Whole Genome Shotgun project carried out in this study have been deposited in DDBJ/EMBL/GenBank under accession number JOJW00000000.
RESULTS AND DISCUSSION
Proposed pathway for TCP degradation.
The construction of a TCP-degrading strain requires an efficient haloalkane dehalogenase variant for conversion to DCP in the first step and a good bacterial host for degrading DCP. Several Rhodococcus haloalkane dehalogenase mutants (e.g., DhaA27, DhaA31) with improved activity toward TCP were obtained by rational design and directed evolution (24). For example, the DhaA31 enzyme shows 36-fold-higher activity (kcat, 1.3 s−1) and 26-fold-higher catalytic efficiency (kcat/Km, 1,050 M−1 · s−1) than the wild-type enzyme (DhaA). As the DCP-mineralizing host, we used P. putida strain MC4, a bacterium that was obtained from the polluted soil of an industrial site by selective enrichment on DCP and grows on a variety of xenobiotic compounds (26). Strain MC4 degrades both stereoisomers of DCP, in agreement with the lack of enantioselectivity of the initial oxidative dehalogenase/dehydrogenase (DppA) that converts DCP to 2-chloroacrolein and 2-chloroacrylic acid. Introduction of the haloalkane dehalogenase in strain MC4 should result in a bacterium with the catabolic pathway shown in Fig. 1.
Two genes encoding haloalkane dehalogenase variants improved by directed evolution were tested in P. putida strain MC4. For this purpose, we constructed the broad-host-range plasmids pIT27, pIT31, pIS31A, and pIS31B, in which the gene encoding haloalkane dehalogenase is expressed under the control of the trc promoter (pIT27, pIT31) (29) or the dhlA promoter region (pIS31A, pIS31B) (8). For the dhlA promoter variants, the short sequence preceding the start codon was mutated in two different ways, since such mutations can strongly influence expression levels (40). In one variant, named A, one extra base (an A) was inserted at the −2 position, leading to the coding strand sequence GGAGGCTCATATG (the ribosome binding site and start codon are underlined). In variant B, the sequence TC at positions −3 and −2 relative to the start codon was replaced by CA, leading to GGAGGCCATATG. The same pBBR1MCS2-derived replicon (30) was used for all broad-host-range expression plasmids, so the copy number was not expected to influence the efficiency of expression.
Plasmids pIT31 and pIS31B, both containing the gene for haloalkane dehalogenase mutant DhaA31 (24), gave slightly higher haloalkane dehalogenase levels in LB medium-grown strain MC4 cells than corresponding plasmids expressing DhaA27 (Table 1). This is in agreement with the published activities of these mutants (24), but in the case of the A variant of the dhlA promoter region, the activities were similar. The activities of the respective dehalogenases were slightly higher with the IPTG-inducible trc promoter than with the strong constitutive dhlA promoters, which possess two −35 consensus sequences (Table 1). Bosma et al. (15) also observed similar expression levels with the trc and dhlA promoters when A. radiobacter AD1 was used as the host. When the two variants of the dhlA promoter region were compared, dehalogenase activity was slightly higher with pIS31B, in agreement with the notion that a C at position −3 relative to the first base of the start codon leads to elevated expression (40). On the basis of these results, the promoter and translation initiation region of pIS31B were selected for further work.
TABLE 1.
Specific activities of haloalkane dehalogenase in P. putida MC4 with various expression constructsa
| Plasmid | DhaA variantb | Promoter | Haloalkane dehalogenase activity (U/mg) |
|---|---|---|---|
| pIT27 | DhaA27 | trc | 0.10 ± 0.02 |
| pIT31 | DhaA31 | trc | 0.16 ± 0.02 |
| pIS27A | DhaA27 | dhlA, A variant | 0.07 ± 0.01 |
| pIS27B | DhaA27 | dhlA, B variant | 0.09 ± 0.01 |
| pIS31A | DhaA31 | dhlA, A variant | 0.06 ± 0.01 |
| pIS31B | DhaA31 | dhlA, B variant | 0.11 ± 0.02 |
Specific activities (U/mg protein) were measured with TCP using extracts from cells grown in LB medium.
Dehalogenase variants described by Pavlova et al. (24).
Chromosomal integration of the mini-Tn5 vector containing dhaA31 into Pseudomonas.
For chromosomal integration of the dhaA31 gene with the constructed constitutive promoter region into P. putida strain MC4, we employed a Tn5-derived transposon vector (pJMS11) developed by de Lorenzo and coworkers (35, 36). After conjugation-mediated introduction of the transposition system (see Materials and Methods), 50 kanamycin (Km)-resistant derivatives of strain MC4 were selected on the basis of growth on M9-Km plates with 10 mM citrate as the carbon source. This selection permitted the Pseudomonas recipient strain to grow when it had acquired the kanamycin resistance gene region but did not allow the E. coli donor strain to grow. The sensitivities of most of the Km-resistant MC4 derivatives to piperacillin indicated the integration of the whole plasmid into the chromosome of strain MC4. Of the 50 Km-resistant MC4 colonies, 45 were sensitive to piperacillin. The Km-resistant colonies also produced catechol 2,3-dioxygenase, as indicated by a yellow color observed after spraying of the colonies with a solution of catechol. A total of 35 Km-resistant colonies that were still able to grow on citrate and DCP as carbon sources were selected, whereas 15 Km-resistant citrate-utilizing colonies lost the capacity to grow on DCP. The cause of the frequent loss of the capacity to degrade DCP is unclear.
The next step was to remove the antibiotic resistance selection markers by subjecting several Km-resistant MC4 derivatives that were still able to grow on DCP to the resolvase protocol (33). After this step, colonies of strain MC4 derivatives were selected on M9 medium containing 5 mM citrate and were tested for Km sensitivity and for the loss of the xylE gene by spraying plates with catechol. We observed that 90% of the colonies that appeared were Km sensitive and had no xylE marker. The presence of a 2-kb fragment carrying the dhaA31 gene was confirmed by PCR analysis with the same primers as those used for cloning. The presence of the dhaA31 gene was also confirmed by testing colonies for haloalkane dehalogenase activity. This activity is lacking in strain MC4, but the derivatives carrying dhaA31 gave a clear positive signal in the halide release assay when tested with DBE or TCP.
To check the stability of the integrated gene on the chromosome, strain MC4-5222 was repeatedly transferred on LB agar plates without TCP and without antibiotics. The strain was examined for the presence of the dehalogenase gene by testing the haloalkane dehalogenase activity of isolated colonies as described above. No colonies that had lost the haloalkane dehalogenase were found when 100 colonies were tested after 10 serial transfers, indicating that strain MC4-5222 is genetically stable, in agreement with previous reports that the chromosomally inserted DNA is stably inherited (34). We estimate the loss at <0.00005 per generation.
Utilization of TCP as a carbon source.
Different derivatives of strain MC4 carrying the dhaA31 gene under the control of promoter region variant B on the chromosome were tested for growth on TCP. For this purpose, we performed batch incubations using MMY medium with TCP (0.5 mM) as the carbon source. Several MC4-derived strains carrying dhaA31 (e.g., MC4-52, MC4-5221, MC4-5222, and MC4-1331) showed growth on TCP. The disappearance of TCP was accompanied by the release of chloride as well as by an increase in optical density when TCP was repeatedly added. In controls containing cells of the wild-type strain MC4, no growth or release of chloride was detected when TCP was the carbon source. Cell extracts prepared from LB medium-grown cultures of the four strains that could use TCP as a carbon source contained haloalkane dehalogenase activity against TCP and 1,2-dibromoethane (Table 2). Strain MC4-5222, which showed the highest growth rate and the highest dehalogenase activity with both TCP and DBE, was selected for further study.
TABLE 2.
Specific haloalkane dehalogenase activity in cell extracts of MC4 derivatives carrying a chromosomal insertion of dhaA31
| Straina | Activity (U/mg) with: |
|
|---|---|---|
| 1,2-Dibromoethane | 1,2,3-Trichloropropane | |
| P. putida MC4 (wild type) | <0.01 | <0.01 |
| MC4-52 | 0.12 | 0.04 |
| MC4-13 | 0.13 | 0.03 |
| MC4-5221 | 0.17 | 0.04 |
| MC4-5222 | 0.27 | 0.10 |
| MC4-1331 | 0.20 | 0.03 |
The MC4 derivatives carry an independent chromosomal insertion of dhaA31 under the control of the B variant of the dhlA translation initiation region.
To determine if a plasmid-carrying strain producing haloalkane dehalogenase [strain MC4(pIS31B)] and strain MC4-5222, carrying the chromosomally integrated dhaA31 gene, expressed all three dehalogenases required for TCP degradation, we measured the activities of DhaA31, haloalcohol dehydrogenase/dehalogenase (DppA), and 2-chloropropionic acid (CPA) dehalogenase in the wild-type strain MC4 and the engineered derivatives (Table 3). DppA is a quinohemoprotein alcohol dehydrogenase that converts 2,3-dichloropropanol to 2-chloroacrolein and 2-chloroacrylic acid in the presence of an artificial electron acceptor such as ferricyanide (1). Assays with cell extracts prepared from cultures grown on DCP (4 mM) showed that DppA and CPA dehalogenase were well expressed in strains MC4 and MC4-5222, whereas in the plasmid-based system MC4(pIS31B), these enzymes showed lower activities (Table 3). Furthermore, the level of DhaA31 was higher in the plasmid-based system MC4(pIS31B) than in the chromosomal integration strain MC4-5222 (Table 3; see also Fig. S3 in the supplemental material). Possibly, the high overexpression of DhaA31 resulting from the strong constitutive dhlA promoter has an effect on the production of DppA and CPA dehalogenase.
TABLE 3.
Specific dehalogenase activities in cell extracts of MC4 and its derivativesa
| Strain | Activity (U/mg)b |
|||
|---|---|---|---|---|
| DhaA31 |
DppA | CPA dehalogenase | ||
| With TCP | With DBE | |||
| MC4 | <0.01 | <0.005 | 0.22 ± 0.01 | 1.05 ± 0.09 |
| MC4(pIS31B) | 0.24 ± 0.06 | 1.3 ± 0.3 | 0.14 ± 0.01 | 0.64 ± 0.06 |
| MC4-5222 | 0.02 ± 0.01 | 0.14 ± 0.01 | 0.35 ± 0.05 | 1.27 ± 0.18 |
Cells were grown in a medium (MMY) with 4 mM DCP as the carbon source.
The activity of DhaA31 was measured with DBE or TCP as the substrate. The activity of DppA was measured with 2,3-dichloropropanol and that of CPA dehalogenase with (S)-2-chloropropionic acid.
The ability of strain MC4-5222 to mineralize TCP was examined in fed-batch culture with repeated addition of TCP. The OD600 and the concentrations of chloride, TCP, and DCP were measured at different times. The culture was inoculated with cells that had been grown overnight in LB medium (starting OD600, 0.05). The initial concentration of TCP was kept low, because when such cultures were started with a small number of cells, TCP added at 1 mM or a higher concentration strongly inhibited growth and caused a decrease in optical density. Even with 0.5 mM TCP, there was an initial reduction in optical density. Growth started after 4 days, as shown by an increase in the OD600. This growth continued almost linearly when TCP was added at regular time intervals. The highest growth rates observed were approximately 0.15 day−1 (note that this may be restricted by the TCP feeding rate) (Fig. 2). Furthermore, after 20 days, the degradation of TCP was rapid, and the accumulated cells of P. putida strain MC4-5222 were able to degrade 1.1 mM TCP quickly. Overall, the total added TCP (3.1 mM) was dechlorinated completely with stoichiometric release of chloride (9.3 mM), as shown in Fig. 2. Thus, the recombinant strain was capable of complete mineralization of TCP. From the optical densities, it was calculated that the growth rate (μ) ranged from 0.0012 to 0.008 h−1. This is similar to the growth rate found by Bosma et al. (15), but strain MC4-5222 has the advantages of complete degradation and no risk of instability of the dehalogenase gene, thanks to its chromosomal insertion.
FIG 2.
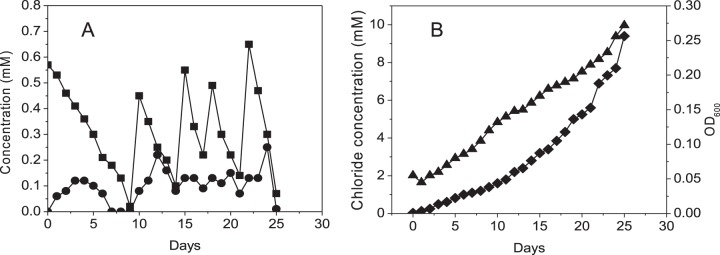
Growth of P. putida MC4-5222 on TCP. Panels A and B represent data from the same culture. TCP was repeatedly added to a total concentration of 3.1 mM. Symbols: ▲, optical density at 600 nm; ◼, TCP; ●, DCP; ◆, chloride.
Higher concentrations of TCP (>1 mM) appeared very toxic to strain MC4-5222, suggesting that further mutations or the introduction of additional genes may be required for improved conversion of high levels of TCP. However, neither extensive ethyl methanesulfonate (EMS) mutagenesis experiments with subsequent selection in batch cultures nor repeated transfer of cultures exposed to TCP at the upper limit of tolerance yielded improved strains in terms of faster growth or higher TCP tolerance. As outlined by Haro and de Lorenzo (41), the rate of metabolic fluxes, the formation of side metabolites, and the physiological control of a degradative pathway may cause a bottleneck that is as important for the function of a catabolic route as the presence of all the enzymes for the pathway itself.
Genetic characterization of a TCP-degrading recombinant.
Southern blotting was used to establish the copy number of the integrated dhaA31 gene. The results indicated that strains MC4-52 and MC4-5222 each contain a single copy of the dhaA31 dehalogenase gene, but at different positions (see Fig. S4 in the supplemental material). The site of integration apparently influences the level of expression of the dhaA31 gene.
The partial genomic sequences of P. putida strains MC4-52 and MC4-5222 were determined by Illumina sequencing, and comparison of the assembled sequences revealed a 26,912-bp contig in MC4-5222 that contained a 2,165-bp fragment insert carrying the dhaA31 gene after position 1305 (see Fig. S5 in the supplemental material). Upstream was a 328-codon open reading frame (ORF) with no similarities to genes of known function and a 308-codon ORF similar to a LysR regulator, both encoded on the complementary strand. Downstream was a 911-codon ORF encoding a putative protein of the P-loop nucleoside triphosphatase (NTPase) superfamily showing 76% identity to a putative transcriptional regulator of Pseudomonas fluorescens Pf0-1. The 27-kb contig into which the dhaA31 gene was integrated also showed similarities to genomic segments of Pseudomonas sp. strain GM78, P. fluorescens Pf0-1, Pseudomonas aeruginosa SBW25, Pseudomonas mendocina YMP, and P. aeruginosa PA7, but further sequence analysis showed that strain MC4 is not highly similar to any bacteria whose genome sequences are deposited at NCBI. By use of BLAST searches, the dppA gene was found on a 34,657-bp contig on which we detected no other genes related to DCP metabolism. Apart from three putative epoxide hydrolase genes, no sequences highly similar to known genes involved in chloropropanol metabolism, such as those for halohydrin dehalogenases or 2-chloroacrylic acid dehalogenase, were detected elsewhere. This suggests that the genes involved in chloroacrolein or chloroacrylic acid metabolism in strain MC4-5222 are not highly similar to known genes.
TCP bioremediation in a fixed-bed bioreactor.
To test the possibility of using strain MC4-5222 for TCP bioremediation, we constructed a fixed-bed reactor and examined the removal of TCP from a contaminated water stream in a continuous process (Fig. 3A). Cells of P. putida MC4-5222 were grown in batch culture and were subsequently used for immobilization on ceramic rings. Attachment of the cells to the support material was allowed to proceed by operating the reactor under fed-batch conditions in buffered medium for 4 days, with repeated dosing of TCP up to 1 mM in total. To prevent stripping of TCP via the exit air, the airflow was kept low throughout all experiments (0.5 to 1.5 ml/min), and the TCP solution (0.33 mM) was supplied at low feed rates (ca. 0.1 ml/min). Air supply remained sufficient, with molar ratios of oxygen to TCP always exceeding 20. The TCP Henry coefficient is 3.2 × 104 to 3.4 × 104 atm · m3 · mol−1; this converts to a dimensionless Henry coefficient (or partitioning coefficient), calculated as the concentration of TCP in the gas phase divided by the concentration of TCP in the liquid phase ([TCP]g/[TCP]l), of 0.012. In view of this Henry coefficient and the airflow rate (Fgas), the TCP that could leave the reactor via the gas phase is calculated from the equation TCPout,g/TCPout,l = (Fgas · [TCP]g)/(Fl · [TCP]l). This suggests that even under the conditions with the strongest aeration (liquid flow rate, 0.1 ml/min; airflow rate, 1.5 ml/min), only 18% of the TCP could leave the reactor via the gas phase. Removal of TCP thus must be due to degradation, not air stripping.
FIG 3.

Degradation of TCP by the genetically engineered P. putida strain MC4-5222 in a fixed-bed reactor. (A) Schematic representation of the reactor, which contained MC4-5222 cells immobilized on ceramic rings and was operated at a rate of 0.008 h−1. (B) Reactor performance as revealed by influent and effluent concentrations. Symbols: ◼, TCP influent; ●, TCP effluent; △, DCP effluent; ◇, chloride effluent.
For a period of 35 days, TCP (0.5 to 1 mM) was supplied continuously, while a high residence time (116 h) was maintained to allow further cell growth and an increase in biofilm size. After 35 days, measurements showed that TCP-degrading cells had been established in the reactor and TCP was being removed. Next, the TCP influent concentration was set at 0.33 to 0.30 mM, and the reactor was operated for another 35 days, over which the concentrations of TCP, DCP, and chloride were monitored by gas chromatography and colorimetric analysis (Fig. 3B). Under these conditions, effective removal of TCP (87 to 93%) with quantitative release of chloride was observed. At day 48, the level of removal of TCP had increased to 95 to 97%. No DCP was detected in the reactor effluent. When cells from the reactor were plated on LB plates and tested for haloalkane dehalogenase, all 90 colonies examined were positive. This demonstrates the possibility of continuous biodegradation of TCP in a bioreactor under aerobic conditions.
Prospects for trichloropropane bioremediation.
The organism described here is probably the best example of a genetically constructed bacterium that grows on an important environmental chemical recalcitrant to biodegradation, for which the availability of a good organism is the bottleneck in the development of a biological treatment process. Several bacterial strains with improved biodegradation potential have been constructed previously (for reviews, see references 42 to 48). Their properties provided important insights into the molecular and physiological causes of recalcitrance and demonstrated that bottlenecks in catabolic pathways can be removed by genetic engineering. On the other hand, successful full-scale application of engineered strains for the bioremediation of xenobiotic compounds has not been reported (42, 49). The only well-documented field trial with a recombinant strain was carried out with Pseudomonas fluorescens HK44, a bacterium that degrades polycyclic aromatic hydrocarbons and harbors a natural catabolic plasmid equipped with a lux bioluminescent reporter (50). A major hurdle in this area is the technical difficulty of constructing organisms to degrade compounds that are really recalcitrant and for which the lack of availability of a suitable organism is the main factor preventing the use of a biological treatment process. This is the case for compounds such as 1,2,3-trichloropropane, 1,1,1-trichloroethane, and trichloroethylene (51). So far, most work has aimed at introducing into a single strain the capacity to degrade a mixture of compounds or it has aimed at expanding the degradation capacity of a single organism (48, 52). The urgency for application is unclear, since the performance of such organisms in a practical bioremediation process may not exceed that of mixed cultures or other natural isolates. Furthermore, there is a chance for successful application of bacteria with a constructed pathway only when the lack of that pathway really is the factor that restricts biodegradation in a practical situation, rather than, for example, substrate bioavailability or oxygen supply.
A serious issue with genetic construction of bioremediation organisms is that engineered organisms do not always utilize the target substrate as well as expected, even if the pathway is well designed biochemically (31, 42). For example, a Pseudomonas strain able to grow on 2-chlorobenzoate was still unable to grow on 2-chlorotoluene as a carbon source, although it possessed all the genes required for degradation in a functional state (41). Similarly, we have observed that the introduction of the 1,2-dichloroethane dehalogenase gene from X. autotrophicus into a 2-chloroethanol-degrading Pseudomonas strain does not allow the resulting recombinant strain to use 1,2-dichloroethane for growth, even though the gene introduced is well expressed and the pathway seems complete (J. R. van der Ploeg and D. B. Janssen, unpublished results). To a lesser extent, the same issue was encountered in the current work. Even though the TCP-degrading Pseudomonas strain MC4-5222 described here can clearly degrade TCP, for unknown reasons the recombinant cells are still quite sensitive to high concentrations of TCP (1 mM), and the growth rate is not as high as desired. One issue may be that side reactions with chlorinated compounds can easily yield reactive by-products (4, 53). High concentrations of TCP can be handled by immobilized enzymes where growth is not required for continuous removal (25).
Notwithstanding these limitations, we found that strain MC4-5222 performed well over a period of a month in a bioreactor for continuous TCP removal, operated with a high water residence time and with TCP as the only organic carbon compound in the influent. An engineered bioremediation organism can likely survive during an in situ or bioreactor treatment process only when the target compound is a predominant pollutant in the groundwater or waste stream to be treated and when its metabolism supports the growth of the organism, e.g., by use as electron donor or acceptor. If the contaminant is a minor component, the engineered strain may have difficulty surviving in situ (31, 49). Due to their application as solvents and intermediates and their formation in specific synthetic chemical processes, compounds such as 1,2-dichloroethane, TCP, and trichloroethene occur as major contaminants at many sites, making them good targets for this approach.
In conclusion, the current paper clearly provides proof of principle for the use of a recombinant TCP-utilizing bacterium for continuous TCP bioremediation. A process using immobilized enzymes was recently reported (25), but the use of recombinant enzymes for bioremediation may suffer from severe cost limitations. For full-scale application, derivatives of strain MC4 with further improved kinetic properties may be needed. The kinetics of the naturally evolved 1,2-dichloroethane-mineralizing bacterium X. autotrophicus GJ10 may serve as a benchmark for successful application. Moreover, we observed that during batch cultivation, the steady-state growth phase was followed by a cell decay phase, which may negatively influence the overall kinetic properties of the strain. If the slow growth and high decay rate are caused by reactive side products (4), a possible improvement could be the use of consortia that contribute to the overall process by removing reactive dead-end metabolites or stimulate the growth of strain MC4 by cross-feeding, as has been observed with strain GJ10 (54).
Supplementary Material
ACKNOWLEDGMENTS
We thank Victor de Lorenzo, Centro Nacional de Biotecnologia, CSIC, Madrid, Spain, for providing the transposon delivery system and advice.
G.S. acknowledges the Higher Education Commission, Government of Pakistan, for financial support. The work of J.D. and M.P. was supported by the Grant Agency of the Czech Republic (P503/12/0572).
Footnotes
Published ahead of print 27 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01620-14.
REFERENCES
- 1.U.S. Environmental Protection Agency. 2009. Toxicological review of 1,2,3-trichloropropane. EPA/635/R-08/010F. U.S. Environmental Protection Agency, Washington, DC [Google Scholar]
- 2.National Toxicology Program. 2002. 1,2,3-Trichloropropane. Rep. Carcinog. 10:248–249 [PubMed] [Google Scholar]
- 3.Kielhorn J, Könnecker G, Pohlenz-Michel C, Schmidt S, Mangelsdorf I. 2003. 1,2,3-Trichloropropane. Concise international chemical assessment document 56. World Health Organization, Geneva, Switzerland [Google Scholar]
- 4.Samin G, Janssen DB. 2012. Transformation and biodegradation of 1,2,3-trichloropropane (TCP). Environ. Sci. Pollut. Res. Int. 19:3067–3078. 10.1007/s11356-012-0859-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salter-Blanc AJ, Tratnyek PG. 2011. Effects of solution chemistry on the dechlorination of 1,2,3-trichloropropane by zero-valent zinc. Environ. Sci. Technol. 45:4073–4079. 10.1021/es104081p [DOI] [PubMed] [Google Scholar]
- 6.Sarathy V, Salter AJ, Nurmi JT, O'Brien Johnson G, Johnson RL, Tratnyek PG. 2010. Degradation of 1,2,3-trichloropropane (TCP): hydrolysis, elimination, and reduction by iron and zinc. Environ. Sci. Technol. 44:787–793. 10.1021/es902595j [DOI] [PubMed] [Google Scholar]
- 7.Stucki G, Thuer M. 1995. Experiences of a large-scale application of 1,2-dichloroethane degrading microorganisms for groundwater treatment. Environ. Sci. Technol. 29:2339–2345. 10.1021/es00009a028 [DOI] [PubMed] [Google Scholar]
- 8.Janssen DB, Pries F, van der Ploeg J, Kazemier B, Terpstra P, Witholt B. 1989. Cloning of 1,2-dichloroethane degradation genes of Xanthobacter autotrophicus GJ10 and expression and sequencing of the dhlA gene. J. Bacteriol. 171:6791–6799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Löffler FE, Champine JE, Ritalahti KM, Sprague SJ, Tiedje JM. 1997. Complete reductive dechlorination of 1,2-dichloropropane by anaerobic bacteria. Appl. Environ. Microbiol. 63:2870–2875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peijnenburg WJGM, Eriksson L, de Groot A, Sjöström M, Verboom HH. 1998. The kinetics of reductive dehalogenation of a set of halogenated aliphatic hydrocarbons in anaerobic sediment slurries. Environ. Sci. Pollut. Res. Int. 5:12–16. 10.1007/BF02986368 [DOI] [PubMed] [Google Scholar]
- 11.Yan J, Rash BA, Rainey FA, Moe WM. 2009. Isolation of novel bacteria within the Chloroflexi capable of reductive dechlorination of 1,2,3-trichloropropane. Environ. Microbiol. 11:833–843. 10.1111/j.1462-2920.2008.01804.x [DOI] [PubMed] [Google Scholar]
- 12.Bowman KS, Nobre MF, da Costa MS, Rainey FA, Moe WM. 2013. Dehalogenimonas alkenigignens sp. nov., a chlorinated-alkane-dehalogenating bacterium isolated from groundwater. Int. J. Syst. Evol. Microbiol. 63:1492–1498. 10.1099/ijs.0.045054-0 [DOI] [PubMed] [Google Scholar]
- 13.Padilla-Crespo E, Yan J, Swift C, Wagner DD, Chourey K, Hettich RL, Ritalahti KM, Löffler FE. 2014. Identification and environmental distribution of dcpA, which encodes the reductive dehalogenase catalyzing the dichloroelimination of 1,2-dichloropropane to propene in organohalide-respiring Chloroflexi. Appl. Environ. Microbiol. 80:808–818. 10.1128/AEM.02927-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bosma T, Janssen DB. 1998. Conversion of chlorinated propanes by Methylosinus trichosporium OB3b expressing soluble methane monooxygenase. Appl. Microbiol. Biotechnol. 50:105–112. 10.1007/s002530051263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bosma T, Kruizinga E, de Bruin EJ, Poelarends GJ, Janssen DB. 1999. Utilization of trihalogenated propanes by Agrobacterium radiobacter AD1 through heterologous expression of the haloalkane dehalogenase from Rhodococcus sp. strain M15-3. Appl. Environ. Microbiol. 65:4575–4581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bosma T, Damborsky J, Stucki G, Janssen DB. 2002. Biodegradation of 1,2,3-trichloropropane through directed evolution and heterologous expression of a haloalkane dehalogenase gene. Appl. Environ. Microbiol. 68:3582–3587. 10.1128/AEM.68.7.3582-3587.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurumbang NP, Dvorak P, Bendl J, Brezovsky J, Prokop Z, Damborsky J. 2014. Computer assisted engineering of the synthetic pathway for biodegradation of a toxic persistent pollutant. ACS Synth. Biol. 3:172–181. 10.1021/sb400147n [DOI] [PubMed] [Google Scholar]
- 18.Bylaska EJ, Glaesemann KR, Felmy AR, Vasiliu M, Dixon DA, Tratnyek PG. 2010. Free energies for degradation reactions of 1,2,3-trichloropropane from ab initio electronic structure theory. J. Phys. Chem. A 114:12269–12282. 10.1021/jp105726u [DOI] [PubMed] [Google Scholar]
- 19.Gray KA, Richardson TH, Kretz K, Short JM, Bartnek F, Knowles R, Kan L, Swanson PE, Robertson DE. 2001. Rapid evolution of reversible denaturation and elevated melting temperature in a microbial haloalkane dehalogenase. Adv. Synth. Catal. 343:607–617. [DOI] [Google Scholar]
- 20.Janssen DB. 2004. Evolving haloalkane dehalogenases. Curr. Opin. Chem. Biol. 8:150–159. 10.1016/j.cbpa.2004.02.012 [DOI] [PubMed] [Google Scholar]
- 21.Koudelakova T, Chovancova E, Brezovsky J, Monincova M, Fortova A, Jarkovsky J, Damborsky J. 2011. Substrate specificity of haloalkane dehalogenases. Biochem. J. 435:345–354. 10.1042/BJ20101405 [DOI] [PubMed] [Google Scholar]
- 22.Poelarends GJ, van Hylckama Vlieg JETV, Marchesi JR, Freitas dos Santos LM, Janssen DB. 1999. Degradation of 1,2-dibromoethane by Mycobacterium sp. strain GP1. J. Bacteriol. 181:2050–2058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poelarends GJ, Zandstra M, Bosma T, Kulakov LA, Larkin MJ, Marchesi JR, Weightman AJ, Janssen DB. 2000. Haloalkane-utilizing Rhodococcus strains isolated from geographically distinct locations possess a highly conserved gene cluster encoding haloalkane catabolism. J. Bacteriol. 182:2725–2731. 10.1128/JB.182.10.2725-2731.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pavlova M, Klvana M, Prokop Z, Chaloupkova R, Banas P, Otyepka M, Wade RC, Tsuda M, Nagata Y, Damborsky J. 2009. Redesigning dehalogenase access tunnels as a strategy for degrading an anthropogenic substrate. Nat. Chem. Biol. 5:727–733. 10.1038/nchembio.205 [DOI] [PubMed] [Google Scholar]
- 25.Dvorak P, Bidmanova S, Damborsky J, Prokop Z. 2014. Immobilized synthetic pathway for biodegradation of toxic recalcitrant pollutant 1,2,3-trichloropropane. Environ. Sci. Technol. 48:6859–6866. 10.1021/es500396r [DOI] [PubMed] [Google Scholar]
- 26.Arif MI, Samin G, van Leeuwen JG, Oppentocht J, Janssen DB. 2012. A novel dehalogenase mechanism for 2,3-dichloro-1-propanol utilization in Pseudomonas putida strain MC4. Appl. Environ. Microbiol. 78:6128–6136. 10.1128/AEM.00760-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lahoda M, Mesters JR, Stsiapanava A, Chaloupkova R, Kuty M, Damborsky J, Kuta Smatanova I. 2014. Crystallographic analysis of 1,2,3-trichloropropane biodegradation by the haloalkane dehalogenase DhaA31. Acta Crystallogr. D Biol. Crystallogr. 70:209–217. 10.1107/S1399004713026254 [DOI] [PubMed] [Google Scholar]
- 28.Schallmey A, den Besten G, Teune IG, Kembaren RF, Janssen DB. 2011. Characterization of cytochrome P450 monooxygenase CYP154H1 from the thermophilic soil bacterium Thermobifida fusca. Appl. Microbiol. Biotechnol. 89:1475–1485. 10.1007/s00253-010-2965-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brosius J, Erfle M, Storella J. 1985. Spacing of the −10 and −35 regions in the Tac promoter. Effect on its in vivo activity. J. Biol. Chem. 260:3539–3541 [PubMed] [Google Scholar]
- 30.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176. 10.1016/0378-1119(95)00584-1 [DOI] [PubMed] [Google Scholar]
- 31.de Lorenzo V. 2009. Recombinant bacteria for environmental release: what went wrong and what we have learnt from it. Clin. Microbiol. Infect. 15:63–65. 10.1111/j.1469-0691.2008.02683.x [DOI] [PubMed] [Google Scholar]
- 32.Panke S, Sanchez-Romero JM, de Lorenzo V. 1998. Engineering of quasi-natural Pseudomonas putida strains for toluene metabolism through an ortho-cleavage degradation pathway. Appl. Environ. Microbiol. 64:748–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kristensen CS, Eberl L, Sanchez-Romero JM, Givskov M, Molin S, de Lorenzo V. 1995. Site-specific deletions of chromosomally located DNA segments with the multimer resolution system of broad-host-range plasmid RP4. J. Bacteriol. 177:52–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kessler B, de Lorenzo V, Timmis KN. 1992. A general system to integrate lacZ fusions into the chromosomes of gram-negative eubacteria: regulation of the Pm promoter of the TOL plasmid studied with all controlling elements in monocopy. Mol. Gen. Genet. 233:293–301. 10.1007/BF00587591 [DOI] [PubMed] [Google Scholar]
- 35.de Lorenzo V, Timmis KN. 1994. Analysis and construction of stable phenotypes in Gram-negative bacteria with Tn5-derived and Tn10-derived minitransposons. Methods Enzymol. 235:386–405. 10.1016/0076-6879(94)35157-0 [DOI] [PubMed] [Google Scholar]
- 36.de Lorenzo V, Herrero M, Sanchez JM, Timmis KN. 1998. Mini-transposons in microbial ecology and environmental biotechnology. FEMS Microbiol. Ecol. 27:211–224. 10.1111/j.1574-6941.1998.tb00538.x [DOI] [Google Scholar]
- 37.Carver T, Berriman M, Tivey A, Patel C, Böhme U, Barrell BG, Parkhill J, Rajandream MA. 2008. Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 24:2672–2676. 10.1093/bioinformatics/btn529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. 10.1186/1471-2164-9-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schanstra JP, Kingma J, Janssen DB. 1996. Specificity and kinetics of haloalkane dehalogenase. J. Biol. Chem. 271:14747–14753. 10.1074/jbc.271.25.14747 [DOI] [PubMed] [Google Scholar]
- 40.Hui A, Hayflick J, Dinkelspiel K, de Boer HA. 1984. Mutagenesis of the 3 bases preceding the start codon of the beta-galactosidase messenger-RNA and its effect on translation in Escherichia coli. EMBO J. 3:623–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haro MA, de Lorenzo V. 2001. Metabolic engineering of bacteria for environmental applications: construction of Pseudomonas strains for biodegradation of 2-chlorotoluene. J. Biotechnol. 85:103–113. 10.1016/S0168-1656(00)00367-9 [DOI] [PubMed] [Google Scholar]
- 42.Cases I, de Lorenzo V. 2005. Genetically modified organisms for the environment: stories of success and failure and what we have learned from them. Int. Microbiol. 8:213–222 [PubMed] [Google Scholar]
- 43.Chen W, Brühlmann F, Richins RD, Mulchandani A. 1999. Engineering of improved microbes and enzymes for bioremediation. Curr. Opin. Biotechnol. 10:137–141. 10.1016/S0958-1669(99)80023-8 [DOI] [PubMed] [Google Scholar]
- 44.Furukawa K. 2003. ‘Super bugs' for bioremediation. Trends Biotechnol. 21:187–190. 10.1016/S0167-7799(03)00054-4 [DOI] [PubMed] [Google Scholar]
- 45.Lau PCK, de Lorenzo V. 1999. Genetic engineering: the frontier of bioremediation. Environ. Sci. Technol. 33:124A–128A. 10.1021/es9926865 [DOI] [PubMed] [Google Scholar]
- 46.Pieper DH, Reineke W. 2000. Engineering bacteria for bioremediation. Curr. Opin. Biotechnol. 11:262–270. 10.1016/S0958-1669(00)00094-X [DOI] [PubMed] [Google Scholar]
- 47.Singh JS, Abhilash PC, Singh HB, Singh RP, Singh DP. 2011. Genetically engineered bacteria: an emerging tool for environmental remediation and future research perspectives. Gene 480:1–9. 10.1016/j.gene.2011.03.001 [DOI] [PubMed] [Google Scholar]
- 48.Timmis KN, Pieper DH. 1999. Bacteria designed for bioremediation. Trends Biotechnol. 5:201–204 [DOI] [PubMed] [Google Scholar]
- 49.Megharaj M, Ramakrishnan B, Venkateswarlu K, Sethunathan N, Naidu R. 2011. Bioremediation approaches for organic pollutants: a critical perspective. Environ. Int. 37:1362–1375. 10.1016/j.envint.2011.06.003 [DOI] [PubMed] [Google Scholar]
- 50.Ripp S, Nivens DE, Ahn Y, Werner C, Jarrell J, Easter JP, Cox CD, Burlage S, Sayler GS. 2000. Controlled field release of a bioluminescent genetically engineered microorganism for bioremediation process monitoring and control. Environ. Sci. Technol. 34:846–853. 10.1021/es9908319 [DOI] [Google Scholar]
- 51.Shields MS, Reagin MJ. 1992. Selection of a Pseudomonas cepacia strain constitutive for the degradation of trichloroethylene. Appl. Environ. Microbiol. 58:3977–3983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rojo F, Pieper D, Engesser K, Knacknuss H, Timmis K. 1987. Assemblage of ortho cleavage route for simultaneous degradation of chloroaromatics and methylaromatics. Science 238:1395–1398. 10.1126/science.3479842 [DOI] [PubMed] [Google Scholar]
- 53.van Hylckama Vlieg J, Poelarends G, Mars A, Janssen DB. 2000. Detoxification of reactive intermediates during microbial metabolism of halogenated compounds. Curr. Opin. Microbiol. 3:257–262. 10.1016/S1369-5274(00)00086-2 [DOI] [PubMed] [Google Scholar]
- 54.van den Wijngaard AJ, van der Kleij RG, Doornweerd RE, Janssen DB. 1993. Influence of organic nutrients and cocultures on the competitive behavior of 1,2-dichloroethane-degrading bacteria. Appl. Environ. Microbiol. 59:3400–3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.