

Abstract
Host defense peptides secreted by colonocytes and Paneth cells play a key role in innate host defenses in the gut. In Crohn's disease, the burden of tissue-associated Escherichia coli commonly increases at epithelial surfaces where host defense peptides concentrate, suggesting that this bacterial population might actively resist this mechanism of bacterial killing. Adherent-invasive E. coli (AIEC) is associated with Crohn's disease; however, the colonization determinants of AIEC in the inflamed gut are undefined. Here, we establish that host defense peptide resistance contributes to host colonization by Crohn's-associated AIEC. We identified a plasmid-encoded genomic island (called PI-6) in AIEC strain NRG857c that confers high-level resistance to α-helical cationic peptides and α- and β-defensins. Deletion of PI-6 sensitized strain NRG857c to these host defense molecules, reduced its competitive fitness in a mouse model of infection, and attenuated its ability to induce cecal pathology. This phenotype is due to two genes in PI-6, arlA, which encodes a Mig-14 family protein implicated in defensin resistance, and arlC, an OmpT family outer membrane protease. Implicit in these findings are new bacterial targets whose inhibition might limit AIEC burden and disease in the gut.
INTRODUCTION
Much experimental and observational data implicate infectious agents in the initiation and maintenance of chronic inflammation in the gastrointestinal tract (1). Escherichia coli is a common Gram-negative species in the mammalian intestine. Through acquisition of well-known virulence factors, such as toxins, adhesins, and secretion systems, E. coli can develop pathogenic traits that participate in intestinal and extraintestinal disease processes (2). Adherent-invasive E. coli (AIEC) is a pathovar associated with Crohn's disease (CD), so named for its ability to adhere to mucosal epithelial cells and invade and survive within epithelial cells and immune cells (3). Several studies have confirmed higher levels of AIEC in adults and children with CD (3–7); AIEC bacteria are approximately six times more likely to be isolated from ileal and colonic samples of adults and children with CD than from healthy controls. Although genetic risk factors in CD are becoming increasingly established (8, 9), research into the interaction between human genes and enteric bacteria that might contribute to the development of disease remains limited.
Paneth cells are a specialized type of secretory cell at the base of small intestinal crypt villi that secrete a number of host defense peptides (sometimes referred to as antimicrobial peptides), including α- and β-defensins, cathelicidins, RegIII family antimicrobial lectins, and lysozyme (10). These host defense molecules, widely used in nature, can kill target microorganisms by disrupting membrane integrity (11). The high local concentration of antimicrobial peptides in the crypt lumen and in the overlying mucous layer likely prevent microbial invasion of the crypt, thereby protecting the gut stem cells at the crypt base. Colonic epithelial cells in the large intestine also release host defense peptides where they provide a similar protective role (10, 12). Enteric pathogens, including Salmonella enterica serovar Typhimurium can partially evade antimicrobial peptides, in part by altering their outer membrane surface charge through lipopolysaccharide (LPS) modifications (13), which contributes to their virulence potential (14).
One of the first human CD susceptibility genes identified was NOD2, in which certain polymorphisms increase the risk of developing disease (15, 16). Early studies suggested that mutant NOD2 alleles could produce defects in the production of some α-defensins from humans (17) and mice (18) and offered a mechanism to account for an altered microbial composition and inflammatory response in CD. However, the precise role of NOD2 in CD remains uncertain based on human (19, 20) and better-controlled mouse studies (21) that show antimicrobial functions of Paneth cells are independent of NOD2 status. Other antimicrobial peptide classes, such as β-defensin 2 and β-defensin 3, are increased in inflamed Crohn's disease lesions (22, 23), while the antimicrobial monokine MIG (monokine induced by gamma interferon) (24) and LL-37 are elevated in the mucosa of ulcerative colitis (UC) patients (25, 26), suggesting that bacteria living at the mucosal surface with an active role in inflammatory bowel disease (IBD) pathogenesis would require mechanisms to resist antimicrobial peptide defenses in the inflamed gut.
Previously we analyzed the genome sequence of AIEC strain LF82 in comparison to AIEC strain NRG857c, both clinical isolates from different patients with ileal CD (27). Although the chromosomes of these two strains are highly similar, NRG857c contains a unique plasmid related to one found in avian-pathogenic E. coli strain APECO1 as well as a plasmid from neonatal meningitis-associated E. coli S88 (28–30). This extrachromosomal plasmid encodes six apparent genomic islands, one of which contains two genes from other bacterial species that are implicated in resistance to host defense peptides. Here, we identify and characterize a genomic island in Crohn's disease-associated strain NRG857c that confers resistance to a vast range of human and synthetic antimicrobial peptides. This genomic island, which we named PI-6, is an important colonization determinant in AIEC, because deletion of this genomic island sensitizes the bacteria to a range of antimicrobial peptides and reduces its ability to infect and colonize murine hosts.
MATERIALS AND METHODS
Ethics statement.
All animal experiments were conducted according to guidelines set by the Canadian Council on Animal Care using protocols approved by the Animal Review Ethics Board at McMaster University.
Bacterial strains.
All bacterial strains and plasmids used in this study are listed in Table 1. AIEC strain NRG857c was isolated from an ileal biopsy sample from a German CD patient (31), and the genome sequence was determined previously (27). Bacteria were routinely grown in LB broth or on LB agar plates. Antibiotic selection was conducted using ampicillin (200 μg/ml), chloramphenicol (34 μg/ml), or kanamycin (50 μg/ml) as required. Deletion of the PI-6 locus, phoP, and the individual arlABC genes was done using the one-step lambda red recombination method (32). Due to extensive intrinsic antibiotic resistance of AIEC NRG857c, we used the modified helper plasmids pKD46-Gm and pCP20-Gm that confer gentamicin resistance (33). All mutants were constructed using the kanamycin resistance gene of pKD4 as the template for construction of the appropriate deletion allele. For competitive infection experiments (see below), the kanamycin resistance cassette was maintained in the infecting strain in order to permit differentiation of the mutant from the unmarked wild-type strain. For in vitro susceptibility testing or for noncompetitive monocolonization of animals, the FLP recombination target (FRT)-flanked resistance gene was removed using pCP20-Gm to generate an unmarked mutant. For construction of the chromosomal ompT mutants, we utilized the allelic exchange plasmid pΔEPompT in an NRG857c background in which the native chloramphenicol acetyltransferase gene (cat) was deleted by lambda red-mediated recombination (34).
TABLE 1.
Bacterial strains and plasmids used in this study
| Bacterial strain or plasmid | Feature(s) | Reference or source |
|---|---|---|
| Bacterial strains | ||
| NRG857c | Clinical AIEC isolate | 31 |
| NRG857ΔPI-6::Kan | Kan-marked deletion of PI-6 operon | This study |
| NRG857ΔPI-6 | Deletion of PI-6 operon | This study |
| NRG857ΔarlA::Kan | Kan-marked deletion of arlA gene | This study |
| NRG857ΔarlA | Deletion of arlA gene | This study |
| NRG857ΔarlB::Kan | Kan-marked deletion of arlB gene | This study |
| NRG857ΔarlB | Deletion of arlB gene | This study |
| NRG857ΔarlC::Kan | Kan-marked deletion of arlC gene | This study |
| NRG857ΔarlC | Deletion of arlC gene | This study |
| NRG857ΔphoP | Deletion of transcriptional regulator gene phoP | This study |
| DK220 | Clinical AIEC isolate | Krause collectiona |
| DK221 | Clinical AIEC isolate | Krause collection |
| DK222 | Clinical AIEC isolate | Krause collection |
| DK223 | Clinical AIEC isolate | Krause collection |
| DK224 | Clinical AIEC isolate | Krause collection |
| UM146 | Clinical AIEC isolate | Krause collection |
| UM147 | Clinical AIEC isolate | Krause collection |
| HM605 | Clinical AIEC isolate | 64 |
| Plasmids | ||
| pKD4 | Template plasmid for lambda red-mediated gene deletion | 32 |
| pKD46-Gm | Lambda red recombinase-expressing vector | 33 |
| pCP20-Gm | FLP-mediated recombination vector | 33 |
| pΔEPompT | Allelic exchange deletion plasmid for chromosomal ompT gene | 34 |
Krause collection, collection of D. O. Krause.
Antimicrobial peptide/defensin susceptibility testing.
The sensitivities of strain NRG857c and derived mutants to the α-helical cationic peptides CP10A, CP28, or LL-37 were determined by using a modified broth microdilution protocol as described by the R. E. W. Hancock laboratory (http://cmdr.ubc.ca/bobh/methods/MODIFIEDMIC.html). Briefly, 10× stocks of the peptide being tested were 2-fold serially diluted (640 μg/ml to 0.06 μg/ml) in a buffer containing 0.02% acetic acid and 0.01% bovine serum albumin. Ten microliters of each of these stocks was added to 85 μl of low-salt LB medium in 96-well polystyrene microtiter plates. The wells were then inoculated with 5-μl portions of bacterial culture containing ∼5 × 104 CFU of the strain being tested. After the bacteria were allowed to grow for 18 to 24 h at 37°C, the wells were visually inspected for inhibition of growth. Values shown represent the modal MICs from 5 to 10 replicates. Time-kill assays were conducted for individual defensin/chemokine molecules. Stationary-phase (16- to 18-h) cultures of the appropriate strains were diluted 1:50 in fresh LB broth and grown for 2 h with shaking at 37°C. One milliliter of bacterial culture was pelleted and washed, and bacteria were resuspended in 10 mM HEPES buffer (pH 7.4). Killing was initiated by adding 50 μg/ml of recombinant human β-defensin 2 (HBD2) or human α-defensin 5 (HD5) or 5 μg/ml of MIG to the washed cultures. Survival was assessed after 2 h by plate count. Controls included bacteria grown in the absence of peptide. The data represent the means of 5 to 10 replicates.
FRET analysis of proteolytic activity.
Bacteria were incubated overnight with shaking at 37°C in LB broth and subcultured into N-minimal medium [50 mM Bis-Tris, 5 mM KCl, 7.5 mM (NH4)2SO4, 0.5 mM K2SO4, 0.5 mM KH2PO4, 38 mM glycerol, 0.1% casamino acids; pH 7.5] supplemented with 0.2% glucose and 1 mM MgCl2 for approximately 3 h to an optical density at 600 nm of approximately 0.5. When required, kanamycin (Kan) (50 μg/ml) was added to the medium. The fluorescence resonance energy transfer (FRET) peptide [2-aminobenzoyl (2-Abz)-SLGRKIQIK2, 4-dinitrophenyl (Dnp)-NH2] containing the omptin dibasic cleavage site RK was purchased from AnaSpec and prepared in phosphate-buffered saline (PBS) as described previously (34). Bacteria were added in triplicate to a black 96-well microtiter plate (Costar) in 75-μl aliquots. Background fluorescence was measured prior to injection of FRET substrate in a BioTek FLx800 multidetection microplate reader with injector. Afterwards, 75 μl of 6 μM FRET peptide was dispensed into the wells, and fluorescence was monitored at an excitation wavelength of 325 nm and an emission wavelength of 430 nm every 2 min for 60 min with shaking between each measurement. Background fluorescence was subtracted from samples for normalization.
Paneth cell isolation and bacterial killing assay.
A section of ileum (∼5 cm) proximal to the ileocecal junction was removed from adult (6- to 10-week-old) C57BL/6 mice (Charles River) and immediately flushed with PBS. The section was opened laterally and then incubated in 2 ml of 30 mM EDTA in PBS. The section was shaken on ice for 5 min, and the section was washed 7 times. Washes were examined microscopically for the presence of Paneth cell-containing ileal crypts in the eluted fractions via staining with 0.25% amido black. Crypts typically eluted in washes 5 to 7. Fractions containing crypts were combined, washed in iPIPES buffer (10 mM PIPES, 150 mM NaCl) and then resuspended in iPIPES buffer to ∼105 crypts/ml. The cells were held on ice (generally less than 1 h) before being used in the killing assay. Stationary-phase (16- to 18-h) bacterial cultures were diluted 1:50 in fresh LB broth and grown for 2 h with shaking at 37°C. One milliliter of bacterial culture was pelleted and washed, and bacteria were resuspended in iPIPES buffer plus 20 μM carbamoyl choline to ∼104 cells ml. To initiate killing, 20 μl of the crypt suspension (∼2,000 crypts) was added to 10 μl of each bacterial culture in a 96-well microtiter plate and incubated at 37°C for 2 h. Control cultures were incubated with iPIPES buffer plus carbamoyl choline only. Surviving bacteria were enumerated by colony counting and normalized to the control culture (iPIPES buffer only). Statistical significance was tested by one-way analysis of variance (ANOVA) with Tukey's posttest. A competitive assay for Paneth cell killing was also developed. For this, individual mutants were competed against the wild-type NRG857c strain during exposure to Paneth cells. The experiment was performed as described above, with the exception that approximately equal numbers of NRG857c and mutant bacteria (marked with a kanamycin resistance cassette) were mixed together before diluting the cultures and initiating killing. Following plating and growth, the ratio of mutant to wild-type bacteria was determined by replica plating on medium containing kanamycin. The competitive index (CI) was determined as follows:
where CFU TS is the number of CFU of the test strain, WT is wild-type strain NRG857c, and the two strains were exposed to Paneth cell secretions (PC) or grown in iPIPES buffer. Statistical significance was tested by one-sample t test against a theoretical competitive index of 1.
Mouse infection studies.
Six- to 8-week-old female CD-1 mice were obtained from Charles River. Twenty-four hours before infection, mice received 20 mg of streptomycin via orogastric gavage as described previously (35). For competitive infections, equal volumes of overnight cultures of wild-type NRG857c strain and the kanamycin-resistant mutant were mixed, and the bacteria were washed in PBS and resuspended to ∼2 × 1010 CFU/ml. Groups of mice received 100 μl of this bacterial suspension by orogastric gavage (total of ∼2 × 109 CFU). Fecal pellets were collected every 2 or 3 days following infection, homogenized in 1 ml of PBS, serially diluted, and plated for colony determination on agar plates. The competitive index was calculated as follows:
where TSoutput is the test strain shed in fecal output and TSinput is the test strain administered to the mice. Mice that cleared the infection or mice with fewer than ∼500 CFU/g feces were excluded from the final analysis. Single-strain infections were carried out as described above using 2 × 109 CFU of the strain being tested.
Histopathological analysis.
At the indicated times (indicated in the figures), the ilea, ceca, and colons of infected mice were removed, fixed in buffered 10% formalin, embedded in paraffin, sectioned into 5-μm slices, and then stained with hematoxylin and eosin (H&E) by Histology Services at McMaster University. Multiple sections from the same tissue from several animals at each time point were scored according to a published pathology scoring matrix (36) with minor modifications (35).
RESULTS
arlABC in pO83 confers resistance to cationic antimicrobial peptides.
Genome sequencing of the CD AIEC isolate NRG857c (27) revealed high sequence conservation and gene synteny with the historical AIEC reference strain, LF82 (37). In spite of this conservation, routine susceptibility testing revealed that strain NRG857c exhibited dramatically increased resistance to the host defense peptide LL-37 compared to strain LF82 (Table 2). Further MIC testing demonstrated that strain NRG857c was also more resistant to two other α-helical cationic antimicrobial peptides, CP10A and CP28 (Table 2). Because the chromosomal gene content of strain NRG857c is highly similar to that of strain LF82, we hypothesized that this phenotypic difference might be related to gene content on the large plasmid in NRG857c that is divergent from that in LF82 (27). Examination of this plasmid, pO83, revealed a genomic island (PI-6) containing two genes whose orthologs in other bacterial species are involved in resistance to cationic antimicrobial peptides (Fig. 1A) (38–40). One of these genes (NRG857_30278 or arlA) is an ortholog to a gene involved in antimicrobial peptide resistance in Salmonella (38) and Pseudomonas aeruginosa (41). The second gene (NRG857_30283 or arlC) is a probable paralog of an ompT family member protease that has emerged as a more widely distributed antimicrobial peptide protease (42, 43). A third gene in PI-6 (NRG857_30279 or arlB) encodes a predicted NAD-dependent epimerase, and its role in peptide resistance was unclear. Deletion of PI-6 sensitized AIEC NRG857c to the α-helical cationic peptides LL-37, CP10A, and CP28 (Table 2). Loss of PI-6 reduced the MIC to LL-37 from 32 to 64 μg/ml to 8 μg/ml, which was similar to that observed in a control phoP mutant as reported previously (40). Similarly, deletion of PI-6 reduced the MIC of both CP10A and CP28. In light of this phenotype, we named the genes encoded within the PI-6 region arlABC for antimicrobial peptide resistance locus ABC. The ΔarlC mutant had identical MICs as the ΔPI-6 mutant to LL-37, CP10A, and CP28. In in vitro susceptibility tests, the ΔarlA and ΔarlB mutants had small, but reproducible MIC reductions to all three peptides tested, demonstrating that these genes also play a role in this type of resistance although to a lesser extent than the putative arlC protease gene. We also tested whether the loss of the PI-6 genes affected resistance to general membrane-disrupting agents. As seen in Table 2, the resistance of the ΔphoP and ΔPI-6 strains to EDTA was reduced ∼2-fold compared to that of strain NRG857c, while only the ΔphoP mutant showed any alteration in susceptibility to SDS. The complementation with the arlABC locus in trans restored growth of the ΔPI-6 strain to wild-type levels. The NRG857c strain also contains a chromosomal ompT gene that we suspected might play a role in resistance to α-helical peptides. However, deletion of ompT alone resulted in only a 2-fold reduction in the MIC to LL-37 compared to the isogenic control (NRG857Δcat) and did not affect the MIC to either CP10A or CP28. The sensitivity profile of the double mutant NRG857ΔcatΔompTΔarlC::aph was similar to that of the single NRG857ΔarlC mutant. Thus, although the chromosomally encoded OmpT protein plays a small role in resistance to cationic antimicrobial peptides, our findings suggest that the plasmid-encoded arlC gene product is more important under the conditions tested.
TABLE 2.
MICs of antimicrobial peptides and membrane-disrupting agents
| Straina | MIC (μg/ml) of antimicrobial peptide: |
MIC of EDTA (mM)b | MIC of SDS (%)b | ||
|---|---|---|---|---|---|
| CP10A | CP28 | LL-37 | |||
| NRG857c* | 16–32 | 8 | 32–64 | 1.25 | >10 |
| NRG857ΔphoP* | 4 | 2 | 8 | 0.625 | 10 |
| NRG857ΔPI-6* | 4–8 | 4 | 8 | 0.312 | >10 |
| NRG857ΔarlA* | 8 | 8 | 16–32 | 1.25 | >10 |
| NRG857ΔarlB* | 8 | 4 | 32 | 1.25 | >10 |
| NRG857ΔarlC* | 4 | 4 | 8 | 1.25 | >10 |
| NRG857Δcat | 16–32 | 8–16 | 64 | ND | ND |
| NRG857ΔcatΔompT | 16–32 | 8 | 32 | ND | ND |
| NRG857ΔcatΔompTΔarlC::aph | 4–8 | 4 | 16 | ND | ND |
| NRG857ΔPI-6(pCR2.1)* | 4 | 2 | 8 | 0.625 | >10 |
| NRG857ΔPI-6(pCRarlABC)* | 64 | 8 | 64 | 1.25 | >10 |
| LF82 | 8 | 4 | 8 | ND | ND |
| LF82(pCR2.1) | 8 | 2 | 16 | ND | ND |
| LF82(pCRarlABC) | 32 | 4 | 64 | ND | ND |
| DK221 | 4 | 4 | 16 | ND | ND |
| DK224 | 8 | 8 | 64 | ND | ND |
| DK234 | 8 | 4 | 8 | ND | ND |
| DK220 | 4 | 2 | 16 | ND | ND |
| DK223 | 4 | 4 | 8 | ND | ND |
| UM146 | 8 | 4 | 16 | ND | ND |
| UM147 | 8 | 16 | 32 | ND | ND |
| HM605 | 16 | 4 | 16 | ND | ND |
The strains with an asterisk were also tested for sensitivity to Tween 20 and Triton X-100, and all showed resistance greater than the highest concentration tested (10% [vol/vol]).
ND, not determined.
FIG 1.
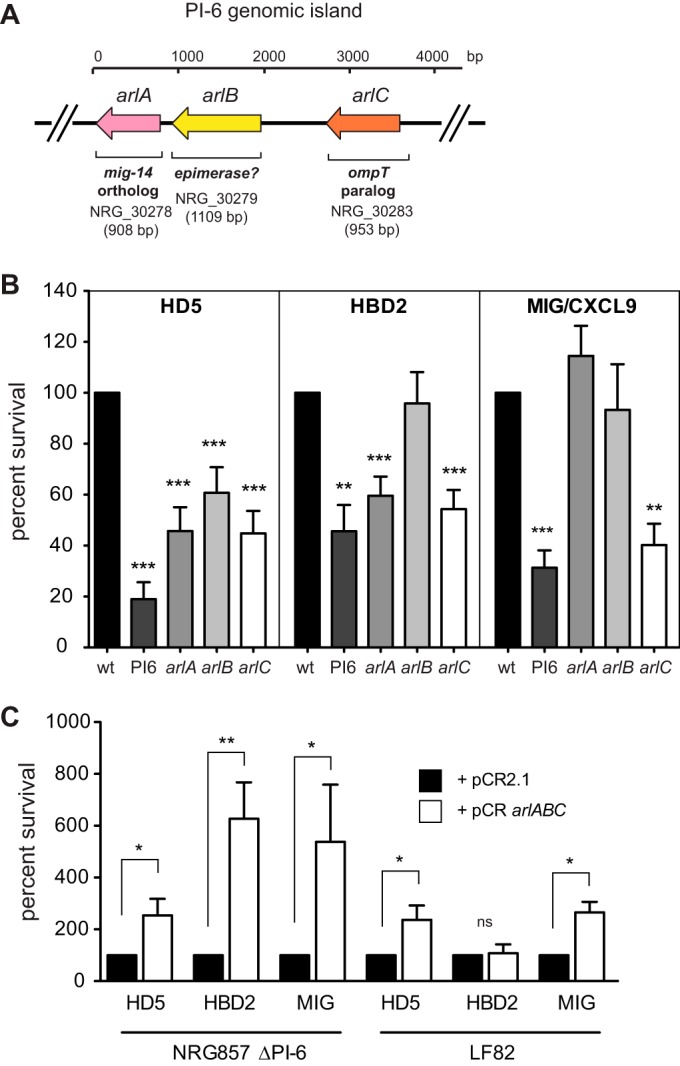
The PI-6 plasmid island encodes three antimicrobial peptide resistance genes. (A) Structure of the PI-6 operon containing the arlA, arlB, and arlC genes. The size of the gene is indicated below the gene. (B) Sensitivity of AIEC NRG857c, ΔPI-6, ΔarlA, ΔarlB, and ΔarlC strains to human α-defensin 5 (HD5), human β-defensin 2 (HBD2), and recombinant murine MIG. Data are expressed as the percent difference in survival following a 2-h exposure to the indicated peptide. Survival in each experiment was normalized to the value for the wild-type (wt) NRG857c strain, and the results shown represent the means plus standard deviations (error bars) of 10 to 15 replicates. Statistical significance was determined by one-way ANOVA. (C) Complementation of NRG857ΔPI-6 and LF82 strains with vector control (pCR2.1) or with a plasmid containing the arlABC locus (pCRarlABC). Data are expressed as the percent difference in survival following a 2-h exposure to the indicated peptide. Survival was normalized to the vector control for each treatment, and statistical significance was determined by Student's t test comparing each peptide treatment independently as indicated. Statistical significance is indicated as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
Complementation of the NRG857ΔPI-6 strain with PI-6 or provision of the AIEC strain LF82 with a plasmid containing the entire PI-6 locus in trans increased resistance to LL-37 by 8-fold (NRG857ΔPI-6) or 4-fold (LF82) relative to a vector control. Increased resistance to CP10A and CP28 was also observed when arlABC was provided in trans. Thus, our data indicate that PI-6 confers resistance to α-helical cationic peptides and that the arlC gene contributes the majority of this activity in vitro.
Screening of human IBD isolates for arlA and arlC.
We designed primers to screen a panel of 97 clinical and control isolates (41 CD, 23 UC, and 33 non-IBD/animal control) described previously (44) for arlA and arlC. In this strain collection, the presence of arlA and arlC was restricted to strains from IBD cases and was not found in strains from non-IBD controls. Five of 96 strains tested positive for arlA (strains NRG857c, HM605, UM147, DK220, and DK224 from 4 CD patients and 1 UC patient) and four were positive for arlC (strains NRG857c, HM605, DK220, and DK224 from 3 CD patients and 1 UC patient). MIC testing of the clinical isolates showed a concordance between the presence of arlA and/or arlC and resistance to LL-37 (Table 2). One discordant strain in this analysis was strain UM146 isolated from a patient with Crohn's disease that was PCR negative for all of the arl genes yet had intermediate resistance to both LL-37 and CP10A. These data suggest that although both arlA and arlC genes of the PI-6 locus are critical for the high-level resistance observed in strain NRG857c, other gene products may also contribute to this phenotype in other AIEC strains.
The arlABC locus increases resistance to human α- and β-defensins and murine MIG.
In addition to the α-helical cathelicidins produced by macrophages and neutrophils, enteric pathogens are exposed to a number of defensin and defensin-like molecules during infection (12). The NRG857ΔPI-6 mutant showed increased susceptibility to both the α-defensin HD5 (19.0% survival relative to strain NRG857c), β-defensin HBD2 (45.6% survival relative to NRG857c), and murine MIG (31.3% survival relative to NRG857c) (Fig. 1B). Interestingly, all of the single-gene deletions of arlA, arlB, or arlC showed significantly increased susceptibility to HD5 relative to the wild-type control (45.7%, 60.7%, and 44.8% survival, respectively), suggesting that the combined action of these gene products might be required for protection from this class of molecules. In contrast, only the arlA and arlC mutants were more sensitive to HBD2 (59.6% and 54.3%, respectively), and only the arlC mutant was significantly sensitized to MIG (40.2% survival) compared to the parental strain. Complementation of the NRG857ΔPI-6 mutant with pCRarlABC containing the entire arlABC locus significantly increased survival following exposure to HD5, HBD2, or MIG compared to the same strain containing empty pCR2.1 (250%, 630%, and 540%, respectively) (Fig. 1C). Similarly, the pCRarlABC plasmid, when introduced into strain LF82, conferred increased resistance to HD5 and MIG (250% and 260%, respectively), demonstrating that these genes are sufficient to provide defensin resistance to a defensin-susceptible strain.
arlC encodes an ompT family protease.
The arlC gene encodes a putative protein of the OmpT family of outer membrane proteases with a high degree of similarity to OmpT and OmpP proteases from E. coli (Fig. 2A). As proteins of the OmpT family have been demonstrated to be important for host defense peptide resistance in enteropathogenic E. coli (EPEC) and enterohemorrhagic E. coli (EHEC) (34), we sought to determine whether the ArlC and OmpT proteins of AIEC strain NRG857c had similar activity. Strain NRG857c exhibited robust cleavage of a synthetic peptide with an internal dibasic recognition site, whereas deletion of the PI-6 locus resulted in a significant reduction in cleavage activity (Fig. 2B). This loss was dependent on the ArlC protein, as the NRG857ΔarlC strain had activity similar to that of the NRG857ΔPI-6 strain. Complementation of the NRGΔPI-6 strain with plasmid-encoded arlABC resulted in high cleavage of the FRET substrate compared to a strain containing the empty vector pCR2.1 (Fig. 2C). These results demonstrate that the ArlC protein is an OmpT family protease and that it functions as such in strain NRG857c.
FIG 2.

ArlC encodes an OmpT family protease. (A) Alignment of ArlC from AIEC strain NRG857c with OmpT and OmpP from Escherichia coli. The red arrows indicate active site residues, and surface-exposed regions are boxed in red. (B) Cleavage of the FRET substrate 2Abz-SLGRKIQI-K(Dnp)-NH2 by strain NRG857c and ΔPI-6 and ΔarlC mutants. Fluorescence is shown in relative fluorescence units (RFU). Data are the means ± standard deviations (error bars) from triplicate determinations and were performed twice. (C) Cleavage of the FRET substrate 2Abz-SLGRKIQI-K(Dnp)-NH2 by NRG857ΔPI-6 mutant containing either pCR2.1 or pCRarlABC.
Deletion of PI-6 or arlA sensitizes AIEC to Paneth cell secretions.
Paneth cells are a specialized secretory cell at the base of ileal crypts that help maintain the barrier function of the intestinal mucosa. They do so by secreting a large and diverse milieu of host defense molecules, including cathelicidins, defensins, lysozyme, antimicrobial C-type lectins, phospholipase, and other proteolytic molecules (10). In an in vitro killing assay, ΔPI-6 cells were highly sensitized to Paneth cells isolated from the ilea of C57BL/6 mice (Fig. 3A). Killing of a ΔphoP mutant, used as a peptide-susceptible control, reached ∼100% after 2 h. The survival of the individual ΔarlA, ΔarlB, or ΔarlC mutants was not different from that of the wild-type control in this assay; however, more-sensitive competition experiments revealed that the ΔarlA mutant showed an ∼50% reduction in survival compared to the wild type (Fig. 3B). These data indicate that PI-6, and in particular ArlA, aids in AIEC resistance to isolated Paneth cells.
FIG 3.
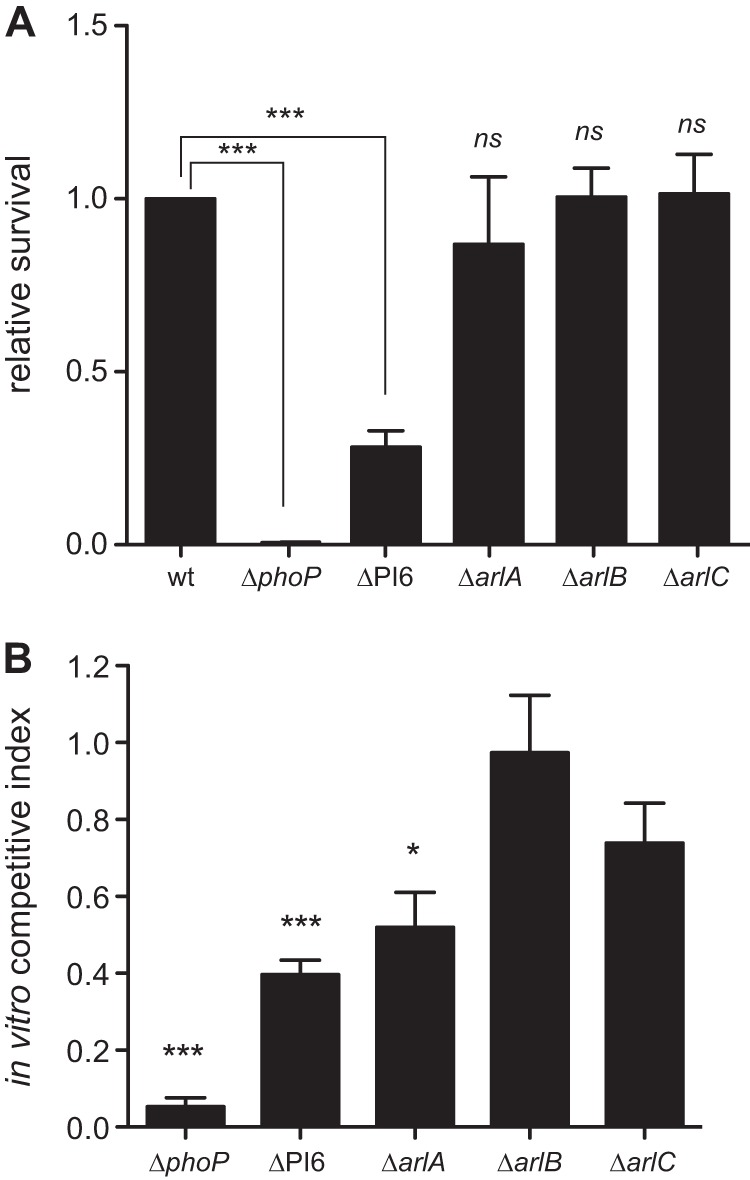
The NRG857ΔPI-6 and NRG857ΔarlA strains exhibit increased sensitivity to Paneth cell secretions. (A) Sensitivity of wild-type NRG857c strain and phoP, PI-6, arlA, arlB and arlC mutant strains to Paneth cell secretions. Values that are significantly different (P < 0.001) by one-way ANOVA are indicated by the bars and three asterisks. Values that are not significantly different (ns) from the value for the wild type are indicated. (B) In vitro competitive index to Paneth cell secretions. Data are the means plus standard errors from 3 separate experiments. Values that are significantly different by one-sample t test are indicated by asterisks as follows: *, P < 0.05; ***, P < 0.001.
The PI-6 locus and the arlA gene are required for competitive fitness in vivo.
In order to quantify the in vivo fitness benefit of ompT, PI-6, and the individual genes therein, we competed mutant strains against the wild-type strain NRG857c using a model of chronic infection in female CD-1 mice (35). The PI-6 mutant was rapidly outcompeted by wild-type NRG857c, beginning on day 2 postinfection and reaching undetectable levels in fecal output by day 7 (Fig. 4A). To determine the gene(s) in PI-6 responsible for this, the individual ΔarlA, ΔarlB, ΔarlC, and ΔompT ΔarlC mutants were competed separately against the wild type in mixed infections of mice. Whereas ΔarlB, ΔarlC, and ΔompT ΔarlC cells competed equally with the wild type over time, the ΔarlA mutant was highly attenuated (Fig. 4B). By day 11 after infection, the arlA mutant was recovered at levels ∼10-fold lower than those of the wild type. These data indicate that under these experimental conditions, the competitive advantage provided by PI-6 during host colonization is driven largely by arlA. This is consistent with the enhanced sensitivity of the arlA mutant to biologically relevant Paneth cell secretions. In in vivo complementation experiments, the ΔPI-6 strain complemented with an arlABC-containing plasmid resulted in ∼100-fold enhanced fitness compared to a ΔPI-6 strain containing empty pCR2.1 vector. These in vivo competitive index experiments demonstrated conclusively that the PI-6 locus, and to a large extent, ArlA, is a critical fitness determinant in a model of chronic AIEC colonization.
FIG 4.

The NRG857ΔPI-6 and NRG857ΔarlA strains exhibit decreased competitive fitness during infection. (A to E) CD-1 mice were infected with equivalent numbers of each strain indicated, and wild-type and mutant AIEC were quantified in fecal output. Data represent the means plus standard errors of the competitive index from 10 to 15 animals from three separate experiments. (F) Complementation of NRG857ΔPI-6 strain with PI-6 in vivo. Data are the means plus standard errors from two separate experiments (five animals per experiment).
The PI-6 locus is required for robust intestinal colonization and induction of cecal pathology in CD-1 mice.
To examine the relationship between AIEC colonization and pathology, we infected groups of CD-1 mice with wild-type strain NRG857c or the ΔPI-6 strain and monitored AIEC burden in feces, tissue-associated bacterial density, and pathology. Carriage of PI-6 allowed AIEC to stabilize at ∼106 CFU/g feces, whereas colonization by the ΔPI-6 mutant was significantly reduced between days 7 to 11 postinfection (Fig. 5A). The level of tissue-associated wild-type AIEC was significantly greater in the colon and ileum compared to the ΔPI-6 mutant at both 7 and 30 days after infection (Fig. 5B).
FIG 5.
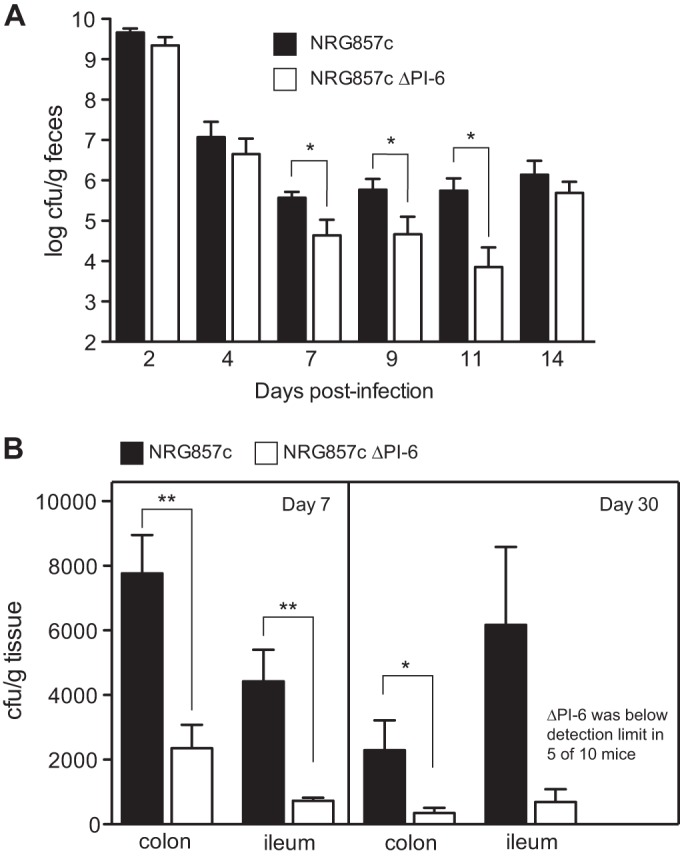
PI-6 contributes to NRG857c colonization of the ileum and colon. Groups of CD-1 mice were colonized with either wild-type NRG857c or the ΔPI-6 mutant, and fecal shedding was measured. At days 7 and 30, the mice were sacrificed, ileal or colonic contents were removed, and the tissue-associated AIEC burden was determined. (A) Fecal output from days 2 to 14 in mice colonized with strain NRG857 or NRG857ΔPI-6. (B) Tissue-associated AIEC on day 7 (n = 5) and day 30 (n = 10). Data are the means plus standard errors from two separate experiments. Values that are significantly different are indicated by bars and asterisks as follows: *, P < 0.05; **, P < 0.01.
We previously showed that wild-type AIEC strain NRG857c elicits chronic inflammation leading to transmural intestinal pathology in both the small and large intestine (35). To quantify the role of PI-6 in this pathology, we infected groups of CD-1 mice with wild-type NRG857c and the ΔPI-6 mutant and quantified ileal and cecal pathology on day 30. The ilea of animals infected with strain NRG857c or with the ΔPI-6 mutant did not show significant differences in pathology, although animal infected with both strains had higher pathology scores than the control animals did (data not shown). In the cecum, strain NRG857c induced transmural inflammation with extensive submucosal edema, hyperplasia, and cellular infiltration in the submucosa, surface epithelium, and mucosa. Crypt loss was also evident in areas of the mucosa where there was increased immune cell infiltration (disease activity index of 14.1) (Fig. 6A to D). The level of histopathology in animals infected with the ΔPI-6 mutant was significantly less than in animals infected with wild-type NRG857c (disease activity index of 9.6) (Fig. 6C and D). This reduction was mainly due to reduced involvement of the surface epithelium and mucosal and submucosal regions of the cecum. The level of histopathology observed in the animals infected with the ΔPI-6 mutant was greater than in the PBS-treated controls, suggesting that although the PI-6 locus contributes to immunopathology, other non-PI-6 bacterial factors are also involved.
FIG 6.

PI-6 contributes to cecal pathology. CD-1 mice were infected for 30 days before the animals were sacrificed and the cecal tips were scored for histopathology. Images are representative of two experiments with five mice per group. (A) PBS-treated, uninfected controls; (B) NRG857c-infected mice; (C) NRG857ΔPI-6-infected mice. The black arrows in panels A to C indicate regions of submucosal edema, cellular infiltration, and hyperplasia. Original magnification, ×200. (D) Pathological scoring of the lumen, surface epithelium, mucosa and submucosa within the ceca of CD-1 mice. (Inset) Cumulative pathology score for the entire section. Values that are significantly different by Student's t test are indicated by bars and asterisks as follows: *, P < 0.05; **, P < 0.01. Scoring for each compartment represents an average of eight views per section, five mice per group, and from two experiments. Data are expressed as the means plus standard errors for each group.
DISCUSSION
The incidence of IBD has been increasing in recent years (45, 46). In spite of this growing disease burden, the cause(s) of IBD remains elusive. Genome-wide association studies have uncovered a large number of genes (∼200) that are associated with IBD (8, 9). Individually, each of these susceptibility loci explains only a small amount of heritability of disease and total disease variance explained by these single nucleotide polymorphisms (SNPs) in IBD is less than 15% (47), indicating that other factors, likely microbial, are involved.
Several studies have now confirmed that the levels of AIEC are higher in humans with Crohn's disease (3, 6). A growing body of work indicates genetic and phenotypic diversity among AIEC isolated from human adults with IBD (7, 27, 35, 44, 48), children with Crohn's disease (4, 5), and companion animals (49, 50). This diversity suggests that host pathways might select for certain AIEC genotypes. Using a model of chronic AIEC infection in mice, we showed that two different clinical isolates of AIEC significantly differed in their ability to colonize the small and large bowel (35). The major genetic differences between these two strains localize to a large extrachromosomal plasmid (27) that we suspected contained genes that impact upon within-host fitness. We prioritized one genomic island, PI-6, because it contained genes that indicated an ability to resist host-derived antimicrobial peptides.
One host pathway implicated in Crohn's disease pathogenesis is the innate defense functions of Paneth cells (51). These cells, located at the base of small intestinal crypts, secrete a large repertoire of antibacterial molecules that are thought to maintain the barrier integrity of the epithelium and help regulate intestinal microbial ecology (10, 52). Infection-induced ablation of Paneth cells leads to outgrowth of members of the family Enterobacteriaceae and increased gut pathology (53), suggesting that Paneth cells create selective pressures on resident and acquired microbes. Some antimicrobial peptide classes, such as β-defensin 2 and β-defensin 3, are increased in inflamed Crohn's lesions (22, 23), suggesting that mucosally associated bacteria with an active role in Crohn's pathogenesis would require mechanisms to resist antimicrobial peptide defenses in the inflamed gut.
Susceptibility tests showed that AIEC NRG857c had higher resistance to α-helical classes of cationic peptides (LL-37 and semisynthetic insect-derived peptides) than did AIEC LF82. This resistance was dependent upon the plasmid-encoded PI-6 island in strain NRG857c, with each of the three genes, particularly arlC, contributing to the observed resistance to α-helical cationic peptides in vitro. The specificity of ArlC for α-helical peptides is consistent with the ArlC-dependent resistance to MIG we observed. MIG, a multifunctional chemokine that also possesses potent antimicrobial activity (24), contains an extended highly cationic C-terminal α-helical domain.
Additional clinical isolates of AIEC that were positive for the arlC gene also showed increased resistance to LL-37. It is important to note that AIEC NRG857c also contains a chromosomal ompT gene that is common to the E. coli lineage and has been shown to be required for cleavage of LL-37 and protamine at the cell surface (34, 54). Our data are consistent with ArlC having combinatorial activity with OmpT toward α-helical cathelicidin molecules like LL-37. This type of enhanced degradation capacity has been seen previously in strains of E. coli carrying the F′ plasmid-encoded OmpP protein as well as chromosomal OmpT (55).
Although there is a significant peptide susceptibility phenotype to α-helical peptides in the ΔarlC mutant in vitro, there is no fitness defect associated with this lesion in outbred CD-1 mice under our experimental conditions. We attempted to potentiate α-helical peptide sensitivity in vivo by competing the ΔarlC ΔompT double mutant against a wild-type strain. Even this strain, which was very sensitive to LL-37 in vitro, exhibited no loss of fitness during chronic intestinal colonization. Although this result was unexpected, it is consistent with the limited importance of the murine cathelicidin peptide, cathelin-related antimicrobial peptide (CRAMP), in intestinal defense against E. coli or Salmonella. For example, Strandberg et al. showed that murine CRAMP was not responsible for selection against Salmonella pmrF mutant with increased in vitro susceptibility to α-helical peptides (56). In a murine model of EHEC-mediated colitis, there was no difference in fecal shedding of EHEC in CRAMP-deficient animals from 3 to 12 days postinfection (57).
Deletion of arlA, which gave only modest susceptibility changes to α-helical peptides, leads to significantly increased sensitivity to HD5 and HBD2 in vitro. In addition, only the ΔarlA mutant showed susceptibility to Paneth cell secretions, and this was the only single gene deletion within PI-6 that was attenuated during chronic mouse infections. The arlA gene is a homolog of the mig-14 gene from Salmonella enterica serovar Typhimurium, a known regulator of cationic peptide resistance (38, 39). The mechanism by which Mig-14 provides resistance to host defense peptides is not fully known. Proteins from this class are associated with the inner membrane where they are thought to decrease the penetrance of active peptide to the inner cytoplasmic membrane (39, 41). Mig-14 family members seem to mediate resistance to a wide range of structural classes of peptides, including colistin, novaspirin G10, CRAMP, and protegrin, which is why ArlA might have the greatest impact on in vivo fitness.
In mice, the α-defensin and β-defensin gene family is greatly expanded and diversified, with C57BL/6 mice containing more than 50 defensin and defensin-like coding genes (58). The repertoire of defensin expression is variable in different mouse strains, and this variability affects microbial gut ecology and resistance to enteric pathogens (59). Transgenic mice expressing the human HD5 α-defensin molecule show greater resistance to infection by Salmonella enterica (60), suggesting that the host defense peptide milieu can affect susceptibility to enteric infection. Although defined genetically, the specific antimicrobial activity of each individual murine defensin (cryptdin) is poorly understood. Of the cryptdins studied biochemically, their antimicrobial activity and selectivity are quite varied (61, 62). While reduced production of ileal α-defensins has been observed in CD patients in some studies (63), other defense molecules such as HBD1 to HBD3 and LL-37 appear to be upregulated in IBD patients, particularly in inflamed regions (22), suggesting that the role of defensin production in the etiology of IBD is complex. Any remodeling and redistribution of the defensin milieu in Crohn's disease would be expected to generate a selective environment for microbes to acquire antimicrobial peptide resistance determinants. Here, we have found that resisting host defense peptides appear to be one mechanism permitting chronic host colonization by AIEC.
ACKNOWLEDGMENTS
This work was supported by grants from the Vertex-Crohn's and Colitis Canada partnership program, the Canadian Institutes of Health Research (CIHR), and the Canada Research Chairs program (to B.K.C.). J.B.M. was supported by a Michael G. DeGroote Postdoctoral Fellowship. C.L.S. was a CIHR-CAG (Canadian Association of Gastroenterology) postdoctoral fellow, and S.A.R.-Y. was supported by an Ontario Graduate Scholarship.
We thank all members of the Coombes lab for helpful discussions. We kindly thank Bob Hancock for the generous gifts of synthetic LL-37, CP10A, CP28, and CP11CN.
Footnotes
Published ahead of print 27 May 2014
REFERENCES
- 1.Sartor RB. 2008. Microbial influences in inflammatory bowel diseases. Gastroenterology 134:577–594. 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 2.Croxen MA, Finlay BB. 2010. Molecular mechanisms of Escherichia coli pathogenicity. Nat. Rev. Microbiol. 8:26–38. 10.1038/nrmicro2265. [DOI] [PubMed] [Google Scholar]
- 3.Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, Gambiez L, Joly B, Cortot A, Colombel JF. 1998. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn's disease. Gastroenterology 115:1405–1413. 10.1016/S0016-5085(98)70019-8. [DOI] [PubMed] [Google Scholar]
- 4.Negroni A, Costanzo M, Vitali R, Superti F, Bertuccini L, Tinari A, Minelli F, Di Nardo G, Nuti F, Pierdomenico M, Cucchiara S, Stronati L. 2012. Characterization of adherent-invasive Escherichia coli isolated from pediatric patients with inflammatory bowel disease. Inflamm. Bowel Dis. 18:913–924. 10.1002/ibd.21899. [DOI] [PubMed] [Google Scholar]
- 5.Schippa S, Iebba V, Totino V, Santangelo F, Lepanto M, Alessandri C, Nuti F, Viola F, Di Nardo G, Cucchiara S, Longhi C, Conte MP. 2012. A potential role of Escherichia coli pathobionts in the pathogenesis of pediatric inflammatory bowel disease. Can. J. Microbiol. 58:426–432. 10.1139/w2012-007. [DOI] [PubMed] [Google Scholar]
- 6.Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, Bringer MA, Swidsinski A, Beaugerie L, Colombel JF. 2004. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn's disease. Gastroenterology 127:412–421. 10.1053/j.gastro.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 7.Baumgart M, Dogan B, Rishniw M, Weitzman G, Bosworth B, Yantiss R, Orsi RH, Wiedmann M, McDonough P, Kim SG, Berg D, Schukken Y, Scherl E, Simpson KW. 2007. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn's disease involving the ileum. ISME J. 1:403–418. 10.1038/ismej.2007.52. [DOI] [PubMed] [Google Scholar]
- 8.Khor B, Gardet A, Xavier RJ. 2011. Genetics and pathogenesis of inflammatory bowel disease. Nature 474:307–317. 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, Anderson CA, Bis JC, Bumpstead S, Ellinghaus D, Festen EM, Georges M, Green T, Haritunians T, Jostins L, Latiano A, Mathew CG, Montgomery GW, Prescott NJ, Raychaudhuri S, Rotter JI, Schumm P, Sharma Y, Simms LA, Taylor KD, Whiteman D, Wijmenga C, Baldassano RN, Barclay M, Bayless TM, Brand S, Buning C, Cohen A, Colombel JF, Cottone M, Stronati L, Denson T, De Vos M, D'Inca R, Dubinsky M, Edwards C, Florin T, Franchimont D, Gearry R, Glas J, Van Gossum A, et al. 2010. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat. Genet. 42:1118–1125. 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bevins CL, Salzman NH. 2011. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Microbiol. 9:356–368. 10.1038/nrmicro2546. [DOI] [PubMed] [Google Scholar]
- 11.Zasloff M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415:389–395. 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 12.Gallo RL, Hooper LV. 2012. Epithelial antimicrobial defence of the skin and intestine. Nat. Rev. Immunol. 12:503–516. 10.1038/nri3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ernst RK, Guina T, Miller SI. 2001. Salmonella typhimurium outer membrane remodeling: role in resistance to host innate immunity. Microbes Infect. 3:1327–1334. 10.1016/S1286-4579(01)01494-0. [DOI] [PubMed] [Google Scholar]
- 14.Groisman EA, Parra-Lopez C, Salcedo M, Lipps CJ, Heffron F. 1992. Resistance to host antimicrobial peptides is necessary for Salmonella virulence. Proc. Natl. Acad. Sci. U. S. A. 89:11939–11943. 10.1073/pnas.89.24.11939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O'Morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent-Puig P, Gower-Rousseau C, Macry J, Colombel JF, Sahbatou M, Thomas G. 2001. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature 411:599–603. 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 16.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nunez G, Cho JH. 2001. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 411:603–606. 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 17.Bevins CL, Stange EF, Wehkamp J. 2009. Decreased Paneth cell defensin expression in ileal Crohn's disease is independent of inflammation, but linked to the NOD2 1007fs genotype. Gut 58:882–883. [PubMed] [Google Scholar]
- 18.Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, Flavell RA. 2005. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307:731–734. 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 19.Simms LA, Doecke JD, Walsh MD, Huang N, Fowler EV, Radford-Smith GL. 2008. Reduced alpha-defensin expression is associated with inflammation and not NOD2 mutation status in ileal Crohn's disease. Gut 57:903–910. 10.1136/gut.2007.142588. [DOI] [PubMed] [Google Scholar]
- 20.Fritz T, Niederreiter L, Tilg H, Blumberg RS, Kaser A. 2010. Controversy over NOD2, inflammation, and defensins. Inflamm. Bowel Dis. 16:1996–1998. 10.1002/ibd.21213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shanahan MT, Carroll IM, Grossniklaus E, White A, von Furstenberg RJ, Barner R, Fodor AA, Henning SJ, Sartor RB, Gulati AS. 19 March 2013. Mouse Paneth cell antimicrobial function is independent of Nod2. Gut 10.1136/gutjnl-2012-304190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aldhous MC, Noble CL, Satsangi J. 2009. Dysregulation of human beta-defensin-2 protein in inflammatory bowel disease. PLoS One 4:e6285. 10.1371/journal.pone.0006285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meisch JP, Nishimura M, Vogel RM, Sung HC, Bednarchik BA, Ghosh SK, Fu P, McCormick T, Weinberg A, Levine AD. 2013. Human beta-defensin 3 peptide is increased and redistributed in Crohn's ileitis. Inflamm. Bowel Dis. 19:942–953. 10.1097/MIB.0b013e318280b11a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cole AM, Ganz T, Liese AM, Burdick MD, Liu L, Strieter RM. 2001. IFN-inducible ELR− CXC chemokines display defensin-like antimicrobial activity. J. Immunol. 167:623–627. 10.4049/jimmunol.167.2.623. [DOI] [PubMed] [Google Scholar]
- 25.Egesten A, Eliasson M, Olin AI, Erjefalt JS, Bjartell A, Sangfelt P, Carlson M. 2007. The proinflammatory CXC-chemokines GRO-alpha/CXCL1 and MIG/CXCL9 are concomitantly expressed in ulcerative colitis and decrease during treatment with topical corticosteroids. Int. J. Colorectal Dis. 22:1421–1427. 10.1007/s00384-007-0370-3. [DOI] [PubMed] [Google Scholar]
- 26.Schauber J, Rieger D, Weiler F, Wehkamp J, Eck M, Fellermann K, Scheppach W, Gallo RL, Stange EF. 2006. Heterogeneous expression of human cathelicidin hCAP18/LL-37 in inflammatory bowel diseases. Eur. J. Gastroenterol. Hepatol. 18:615–621. 10.1097/00042737-200606000-00007. [DOI] [PubMed] [Google Scholar]
- 27.Nash JH, Villegas A, Kropinski AM, Aguilar-Valenzuela R, Konczy P, Mascarenhas M, Ziebell K, Torres AG, Karmali MA, Coombes BK. 2010. Genome sequence of adherent-invasive Escherichia coli and comparative genomic analysis with other E. coli pathotypes. BMC Genomics 11:667. 10.1186/1471-2164-11-667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fricke WF, McDermott PF, Mammel MK, Zhao S, Johnson TJ, Rasko DA, Fedorka-Cray PJ, Pedroso A, Whichard JM, Leclerc JE, White DG, Cebula TA, Ravel J. 2009. Antimicrobial resistance-conferring plasmids with similarity to virulence plasmids from avian pathogenic Escherichia coli strains in Salmonella enterica serovar Kentucky isolates from poultry. Appl. Environ. Microbiol. 75:5963–5971. 10.1128/AEM.00786-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peigne C, Bidet P, Mahjoub-Messai F, Plainvert C, Barbe V, Medigue C, Frapy E, Nassif X, Denamur E, Bingen E, Bonacorsi S. 2009. The plasmid of Escherichia coli strain S88 (O45:K1:H7) that causes neonatal meningitis is closely related to avian pathogenic E. coli plasmids and is associated with high-level bacteremia in a neonatal rat meningitis model. Infect. Immun. 77:2272–2284. 10.1128/IAI.01333-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson TJ, Johnson SJ, Nolan LK. 2006. Complete DNA sequence of a ColBM plasmid from avian pathogenic Escherichia coli suggests that it evolved from closely related ColV virulence plasmids. J. Bacteriol. 188:5975–5983. 10.1128/JB.00204-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eaves-Pyles T, Allen CA, Taormina J, Swidsinski A, Tutt CB, Jezek GE, Islas-Islas M, Torres AG. 2008. Escherichia coli isolated from a Crohn's disease patient adheres, invades, and induces inflammatory responses in polarized intestinal epithelial cells. Int. J. Med. Microbiol. 298:397–409. 10.1016/j.ijmm.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 32.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doublet B, Douard G, Targant H, Meunier D, Madec JY, Cloeckaert A. 2008. Antibiotic marker modifications of lambda Red and FLP helper plasmids, pKD46 and pCP20, for inactivation of chromosomal genes using PCR products in multidrug-resistant strains. J. Microbiol. Methods 75:359–361. 10.1016/j.mimet.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 34.Thomassin JL, Brannon JR, Gibbs BF, Gruenheid S, Le Moual H. 2012. OmpT outer membrane proteases of enterohemorrhagic and enteropathogenic Escherichia coli contribute differently to the degradation of human LL-37. Infect. Immun. 80:483–492. 10.1128/IAI.05674-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Small CL, Reid-Yu SA, McPhee JB, Coombes BK. 2013. Persistent infection with Crohn's disease-associated adherent-invasive Escherichia coli leads to chronic inflammation and intestinal fibrosis. Nat. Commun. 4:1957. 10.1038/ncomms2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coburn B, Li Y, Owen D, Vallance BA, Finlay BB. 2005. Salmonella enterica serovar Typhimurium pathogenicity island 2 is necessary for complete virulence in a mouse model of infectious enterocolitis. Infect. Immun. 73:3219–3227. 10.1128/IAI.73.6.3219-3227.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boudeau J, Glasser AL, Masseret E, Joly B, Darfeuille-Michaud A. 1999. Invasive ability of an Escherichia coli strain isolated from the ileal mucosa of a patient with Crohn's disease. Infect. Immun. 67:4499–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brodsky IE, Ernst RK, Miller SI, Falkow S. 2002. mig-14 is a Salmonella gene that plays a role in bacterial resistance to antimicrobial peptides. J. Bacteriol. 184:3203–3213. 10.1128/JB.184.12.3203-3213.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brodsky IE, Ghori N, Falkow S, Monack D. 2005. Mig-14 is an inner membrane-associated protein that promotes Salmonella typhimurium resistance to CRAMP, survival within activated macrophages and persistent infection. Mol. Microbiol. 55:954–972. 10.1111/j.1365-2958.2004.04444.x. [DOI] [PubMed] [Google Scholar]
- 40.Guina T, Yi EC, Wang H, Hackett M, Miller SI. 2000. A PhoP-regulated outer membrane protease of Salmonella enterica serovar Typhimurium promotes resistance to alpha-helical antimicrobial peptides. J. Bacteriol. 182:4077–4086. 10.1128/JB.182.14.4077-4086.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jochumsen N, Liu Y, Molin S, Folkesson A. 2011. A Mig-14-like protein (PA5003) affects antimicrobial peptide recognition in Pseudomonas aeruginosa. Microbiology 157:2647–2657. 10.1099/mic.0.049445-0. [DOI] [PubMed] [Google Scholar]
- 42.Haiko J, Suomalainen M, Ojala T, Lahteenmaki K, Korhonen TK. 2009. Breaking barriers–attack on innate immune defences by omptin surface proteases of enterobacterial pathogens. Innate Immun. 15:67–80. 10.1177/1753425909102559. [DOI] [PubMed] [Google Scholar]
- 43.Gruenheid S, Le Moual H. 2012. Resistance to antimicrobial peptides in Gram-negative bacteria. FEMS Microbiol. Lett. 330:81–89. 10.1111/j.1574-6968.2012.02528.x. [DOI] [PubMed] [Google Scholar]
- 44.Sepehri S, Khafipour E, Bernstein CN, Coombes BK, Pilar AV, Karmali M, Ziebell K, Krause DO. 2011. Characterization of Escherichia coli isolated from gut biopsies of newly diagnosed patients with inflammatory bowel disease. Inflamm. Bowel Dis. 17:1451–1463. 10.1002/ibd.21509. [DOI] [PubMed] [Google Scholar]
- 45.Martin-de-Carpi J, Rodriguez A, Ramos E, Jimenez S, Martinez-Gomez MJ, Medina E, Navas-Lopez VM, SPIRIT-IBD Working Group of SEGHNP (Sociedad Espanola de Gastroenterologia Hepatologia Nutricion Pediatrica) 23 January 2014. The complete picture of changing pediatric inflammatory bowel disease incidence in Spain in 25 years (1985–2009): the EXPERIENCE registry. J. Crohn's Colitis 10.1016/j.crohns.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 46.Shanahan F, Bernstein CN. 2009. The evolving epidemiology of inflammatory bowel disease. Curr. Opin. Gastroenterol. 25:301–305. 10.1097/MOG.0b013e32832b12ef. [DOI] [PubMed] [Google Scholar]
- 47.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P, Wei Z, Abraham C, Achkar JP, Ahmad T, Amininejad L, Ananthakrishnan AN, Andersen V, Andrews JM, Baidoo L, Balschun T, Bampton PA, Bitton A, Boucher G, Brand S, Buning C, Cohain A, Cichon S, D'Amato M, De Jong D, Devaney KL, Dubinsky M, Edwards C, Ellinghaus D, Ferguson LR, Franchimont D, Fransen K, Gearry R, Georges M, Gieger C, Glas J, Haritunians T, Hart A, Hawkey C, et al. 2012. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491:119–124. 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martinez-Medina M, Aldeguer X, Lopez-Siles M, Gonzalez-Huix F, Lopez-Oliu C, Dahbi G, Blanco JE, Blanco J, Garcia-Gil LJ, Darfeuille-Michaud A. 2009. Molecular diversity of Escherichia coli in the human gut: new ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn's disease. Inflamm. Bowel Dis. 15:872–882. 10.1002/ibd.20860. [DOI] [PubMed] [Google Scholar]
- 49.Martinez-Medina M, Garcia-Gil J, Barnich N, Wieler LH, Ewers C. 2011. Adherent-invasive Escherichia coli phenotype displayed by intestinal pathogenic E. coli strains from cats, dogs, and swine. Appl. Environ. Microbiol. 77:5813–5817. 10.1128/AEM.02614-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simpson KW, Dogan B, Rishniw M, Goldstein RE, Klaessig S, McDonough PL, German AJ, Yates RM, Russell DG, Johnson SE, Berg DE, Harel J, Bruant G, McDonough SP, Schukken YH. 2006. Adherent and invasive Escherichia coli is associated with granulomatous colitis in boxer dogs. Infect. Immun. 74:4778–4792. 10.1128/IAI.00067-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baumgart DC, Sandborn WJ. 2012. Crohn's disease. Lancet 380:1590–1605. 10.1016/S0140-6736(12)60026-9. [DOI] [PubMed] [Google Scholar]
- 52.Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjoberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, Stoel M, Zhou Y, Sodergren E, Weinstock GM, Bevins CL, Williams CB, Bos NA. 2010. Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 11:76–83. 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raetz M, Hwang SH, Wilhelm CL, Kirkland D, Benson A, Sturge CR, Mirpuri J, Vaishnava S, Hou B, Defranco AL, Gilpin CJ, Hooper LV, Yarovinsky F. 2013. Parasite-induced TH1 cells and intestinal dysbiosis cooperate in IFN-gamma-dependent elimination of Paneth cells. Nat. Immunol. 14:136–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stumpe S, Schmid R, Stephens DL, Georgiou G, Bakker EP. 1998. Identification of OmpT as the protease that hydrolyzes the antimicrobial peptide protamine before it enters growing cells of Escherichia coli. J. Bacteriol. 180:4002–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hwang BY, Varadarajan N, Li H, Rodriguez S, Iverson BL, Georgiou G. 2007. Substrate specificity of the Escherichia coli outer membrane protease OmpP. J. Bacteriol. 189:522–530. 10.1128/JB.01493-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Strandberg KL, Richards SM, Tamayo R, Reeves LT, Gunn JS. 2012. An altered immune response, but not individual cationic antimicrobial peptides, is associated with the oral attenuation of Ara4N-deficient Salmonella enterica serovar Typhimurium in mice. PLoS One 7:e49588. 10.1371/journal.pone.0049588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chromek M, Arvidsson I, Karpman D. 2012. The antimicrobial peptide cathelicidin protects mice from Escherichia coli O157:H7-mediated disease. PLoS One 7:e46476. 10.1371/journal.pone.0046476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amid C, Rehaume LM, Brown KL, Gilbert JG, Dougan G, Hancock RE, Harrow JL. 2009. Manual annotation and analysis of the defensin gene cluster in the C57BL/6J mouse reference genome. BMC Genomics 10:606. 10.1186/1471-2164-10-606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gulati AS, Shanahan MT, Arthur JC, Grossniklaus E, von Furstenberg RJ, Kreuk L, Henning SJ, Jobin C, Sartor RB. 2012. Mouse background strain profoundly influences Paneth cell function and intestinal microbial composition. PLoS One 7:e32403. 10.1371/journal.pone.0032403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Salzman NH, Ghosh D, Huttner KM, Paterson Y, Bevins CL. 2003. Protection against enteric salmonellosis in transgenic mice expressing a human intestinal defensin. Nature 422:522–526. 10.1038/nature01520. [DOI] [PubMed] [Google Scholar]
- 61.Masuda K, Sakai N, Nakamura K, Yoshioka S, Ayabe T. 2011. Bactericidal activity of mouse alpha-defensin cryptdin-4 predominantly affects noncommensal bacteria. J. Innate Immun. 3:315–326. 10.1159/000322037. [DOI] [PubMed] [Google Scholar]
- 62.Ouellette AJ, Satchell DP, Hsieh MM, Hagen SJ, Selsted ME. 2000. Characterization of luminal paneth cell alpha-defensins in mouse small intestine. Attenuated antimicrobial activities of peptides with truncated amino termini. J. Biol. Chem. 275:33969–33973. 10.1074/jbc.M004062200. [DOI] [PubMed] [Google Scholar]
- 63.Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, Shen B, Schaeffeler E, Schwab M, Linzmeier R, Feathers RW, Chu H, Lima H, Jr, Fellermann K, Ganz T, Stange EF, Bevins CL. 2005. Reduced Paneth cell alpha-defensins in ileal Crohn's disease. Proc. Natl. Acad. Sci. U. S. A. 102:18129–18134. 10.1073/pnas.0505256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clarke DJ, Chaudhuri RR, Martin HM, Campbell BJ, Rhodes JM, Constantinidou C, Pallen MJ, Loman NJ, Cunningham AF, Browning DF, Henderson IR. 2011. Complete genome sequence of the Crohn's disease-associated adherent-invasive Escherichia coli strain HM605. J. Bacteriol. 193:4540. 10.1128/JB.05374-11. [DOI] [PMC free article] [PubMed] [Google Scholar]