

Abstract
Hydrosalpinx is a pathological hallmark of tubal infertility associated with chlamydial infection. However, the mechanisms of hydrosalpinx remain unknown. Here, we report that complement factor 5 (C5) contributes significantly to chlamydial induction of hydrosalpinx. Mice lacking C5 (C5−/−) failed to develop any hydrosalpinx, while ∼42% of the corresponding wild-type mice (C5+/+) did so following intravaginal infection with Chlamydia muridarum. Surprisingly, deficiency in C3 (C3−/−), an upstream component of the complement system, did not affect mouse susceptibility to chlamydial induction of hydrosalpinx. Interestingly, C5 activation was induced by chlamydial infection in oviducts of C3−/− mice, explaining why the C3−/− mice remained susceptible to chlamydial induction of hydrosalpinx. Similar levels of live chlamydial organisms were recovered from oviduct tissues of both C5−/− and C5+/+ mice, suggesting that C5 deficiency did not affect C. muridarum ascending infection. Furthermore, C5−/− mice were still more resistant to hydrosalpinx induction than C5+/+ mice, even when live C. muridarum organisms were directly delivered into the upper genital tract, both confirming the role of C5 in promoting hydrosalpinx and indicating that the C5-facilitated hydrosalpinx was not due to enhancement of ascending infection. The C5−/− mice displayed significantly reduced lumenal inflammatory infiltration and cytokine production in oviduct tissue, suggesting that C5 may contribute to chlamydial induction of hydrosalpinx by enhancing inflammatory responses.
INTRODUCTION
Tubal factor infertility caused by sexually transmitted infection with Chlamydia trachomatis is often accompanied by upper genital tract (UGT) pathologies, such as hydrosalpinx (1). The pathogenic mechanisms of C. trachomatis-induced hydrosalpinx in women remain unknown. Intravaginal inoculation with Chlamydia muridarum in mice can induce visible hydrosalpinges that closely mimic those induced by C. trachomatis in women, which has led to the extensive use of the murine model to study mechanisms of C. trachomatis pathogenesis and immunity (2–6). For example, the animal model studies have led to the discovery of a CD4+ T cell-dependent and gamma interferon (IFN-γ)-mediated immunity as a major protective mechanism for mice to control chlamydial infection (7).
Many studies have been carried out to understand the mechanisms of C. muridarum induction of hydrosalpinx. The results are not always consistent. The conflicting data might be due to not only the method used to score hydrosalpinx (microscopic scoring for oviduct dilation versus naked eye or visual observation of swollen oviducts filled with clear fluids), but also the time following infection when hydrosalpinx is assessed (5 weeks versus 8 weeks postinfection). For example, a robust acute inflammatory response was correlated with the development of long-lasting hydrosalpinx observed with the naked eye 8 weeks after infection based on a comparison between C57BL/6J and C3H/HeN mice (8). These long-lasting hydrosalpinges were further correlated with oviduct infection (9, 10). Knockout mice have been used to map host determinants required for chlamydial induction of hydrosalpinx. Mice deficient in Toll-like receptor 2 (TLR2) failed to develop robust acute inflammatory responses and oviduct dilation when detected under microscopy 5 weeks after infection (11). However, it is not clear whether the TLR2-mediated signaling pathway is sufficient for C. muridarum induction of long-lasting hydrosalpinx, since the TLR2-deficient mice did develop chronic inflammation in the oviduct similar to that in wild-type mice (11). Furthermore, mice deficient in MyD88, a critical adaptor molecule of the TLR2-mediated signaling pathway, developed severe, long-lasting hydrosalpinx (12), which further calls into question the role of the TLR2-mediated signaling pathway in chlamydial induction of hydrosalpinx. Indeed, it was recently shown that TLR2-deficient mice developed as severe hydrosalpinx as wild-type mice (13). The same study also revealed that tumor necrosis factor receptor 1 (TNFR1) was critical for hydrosalpinx development following C. muridarum infection, which is consistent with an earlier observation showing an important role of tumor necrosis factor alpha (TNF-α) in chlamydial induction of hydrosalpinx (14). Many other host molecules have also been shown to contribute to chlamydial pathogenicity in the upper genital tract, including matrix metalloproteinases (15), inducible nitric oxide synthase (16), interleukin 1 (IL-1) receptor (17), caspase 1 (18, 19), IL-17 (20), CD28 (21), and CXCR2 (22). However, none of these previous studies have sufficiently addressed whether the reduced pathology was due to reduced oviduct infection, reduced inflammatory responses in the oviduct, or both.
The complement system plays a central role in both host defense response against infection and inflammatory pathology (23). Both microbial components, such as lipopolysaccharide (LPS), and host factors, such as C-reactive protein and immune complexes, can activate complement via one of the three major pathways—the alternative, mannan-binding-lectin, and classical pathways—all of which merge at the level of complement factor 3 (C3). On activation, C3 is cleaved into C3a and C3b. C3b can covalently bind to surfaces of pathogens to enhance phagocytosis. C3b also combines with C3 and C5 convertases to trigger the formation of the lytic membrane attack complex to attack pathogens. The cleavage products C5a and C3a can both promote inflammation and modulate immune responses by binding to C5a receptor (C5aR, or CD88) and C3a receptor (C3aR), respectively (for a review, see reference 24). Because of the important roles of C5a in inflammatory pathologies, various strategies targeting C5a are being developed to reduce pathogenic inflammation (25–28). It appears that targeting C5 rather than C3 to prevent tissue damage caused by complement-enhanced inflammation is a wise strategy. This is because the unwanted side effects of the blockade of C5a/C5aR are much smaller than those of a general blockade, e.g., at the level of C3, and C5a can also be generated in the absence of C3 (29). The role of the complement system in chlamydial infection has also been investigated. Chlamydial elementary bodies (EBs) were found to be able to activate complement (30, 31), and the complement activation seemed to reduce chlamydial infection in cell culture. Interestingly, the complement antichlamydial activity seemed to depend on the early complement components, such as C3, but not late components, including C5 (32). Consistent with these earlier observations are the recent findings that mice deficient in C3 or C3aR, but not C5, displayed drastically increased susceptibility to airway infection with Chlamydia psittaci (33, 34). However, the role of the complement system or C5a in chlamydial induction of hydrosalpinx remains unknown. In the current study, we evaluated the role of the complement system in chlamydial pathogenesis and found that C5, but not C3, contributes significantly to chlamydial induction of hydrosalpinx. C5−/− mice failed to develop significant hydrosalpinx following C. muridarum infection. The decreased susceptibility of C5−/− mice to C. muridarum induction of hydrosalpinx was correlated with reduced inflammation in oviducts but not alteration in chlamydial ascension to the oviduct.
MATERIALS AND METHODS
Chlamydial organisms and infection.
The C. muridarum organisms (strain Nigg) used in the current study were propagated in HeLa cells (human cervical carcinoma epithelial cells; ATCC catalog number CCL2), purified, aliquoted, and stored as described previously (18, 35). Female C5-deficient, or C5−/− (B10.D2-Hc0 H2d H2-T18c/oSnJ; stock number 000461); C5-competent, or C5+/+ (B10.D2-Hc1 H2d H2-T18c/nSnJ; stock number 000463); C3−/− (B6.129S4-C3tm1Crr/J; stock number 003641); and C3+/+ (C57BL/6J; stock number 000664) mice were all purchased at the age of 5 to 6 weeks from Jackson Laboratories (Bar Harbor, ME). It is important to note that there is a 2-base (TA) deletion at positions 62 and 63 of an 83-bp exon near the 5′ end of the c5 gene in B10.D2/oSnJ mice. This deletion creates a stop codon starting four bases after the deletion. A truncated product of 216 amino acids is predicted, although a larger pro-C5 protein may be synthesized. Nevertheless, macrophages from mouse strains carrying this allele do not secrete C5. For infection experiments, each mouse was inoculated intravaginally with 2 × 105 inclusion-forming units (IFU) of live C. muridarum organisms as described previously (16). The animal experiments were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Laboratory Animal Experiments of the University of Texas Health Science Center at San Antonio. For in vitro infection of HeLa cells, HeLa cells grown on coverslips in 24-well plates containing Dulbecco's modified Eagle's medium (DMEM) (Gibco BRL, Rockville, MD) with 10% fetal calf serum (FCS) (Gibco BRL) at 37°C in an incubator supplied with 5% CO2 were inoculated with C. muridarum organisms as described previously (18, 35). The infected cultures were processed for immunofluorescence assay as described below.
Monitoring live C. muridarum organism recovery from swabs and genital tract tissues.
To monitor live organism shedding, vaginal swabs were taken on different days after intravaginal infection. Each swab was suspended in 500 μl of ice-cold sucrose-phosphate-glutamic acid (SPG), followed by vortexing with glass beads, and the released organisms were titrated on HeLa cell monolayers in duplicate, as described previously (12). To monitor upper genital tract infection, the genital tract tissue was harvested sterilely from each mouse on different days after infection, as indicated for individual experiments. Each tissue was cut into 3 segments, including the vagina/cervix (lower genital tract [LGT]), uterus/uterine horn (both sides from the same mouse were combined as a single segment), and oviducts/ovaries (both sides from the same mouse were pooled as a single tissue sample; both belong to the UGT). Each segment sample was homogenized in 500 μl of SPG using a 2-ml mini-tissue grinder (Fisher Scientific, Pittsburgh, PA). After brief sonication, the released live organisms were titrated as described above. The total number of IFU per swab/tissue was calculated based on the number of IFU per field, the number of fields per coverslip, the dilution factors, and inoculation and total sample volumes. An average was taken of serially diluted duplicate samples for any given swab/tissue. The calculated total number of IFU/swab or tissue was converted into log10, and the log10 IFU were used to calculate means and standard deviations for each group at each time point.
Evaluating mouse genital tract tissue pathology and histological scoring.
Mice were sacrificed on day 60 after infection, and the mouse urogenital tract tissues were isolated. Before the tissues were removed from the mouse body, an in situ gross examination was performed for evidence of oviduct hydrosalpinx or any other related abnormalities of the oviducts. The severity of oviduct hydrosalpinx was scored based on the following criteria: no hydrosalpinx (0); hydrosalpinx detectable only after amplification (1); and hydrosalpinx clearly visible with the naked eye, but the size is smaller than (2), equal to (3), or larger than (4) that of the ovary on the same side. The oviducts from the left and right sides of the same mouse were scored separately, and the two scores were added together as the score for the mouse. The excised tissues, after photographing, were fixed in 10% neutral formalin, embedded in paraffin, and serially sectioned longitudinally (at 5 μm/section). Efforts were made to include the cervix, both uterine horns, and oviducts, as well as lumenal structures of each tissue in each section. The sections were stained with hematoxylin and eosin (H&E) as described previously (8). The H&E-stained sections were scored for severity of inflammation and pathologies based on modified schemes established previously (8, 12). Scoring for dilation of the oviduct was as follows: 0, no significant dilation; 1, mild dilation of a single cross section; 2, one to three dilated cross sections; 3, more than three dilated cross sections; and 4, confluent pronounced dilation. Scoring for inflammatory cell infiltrates (at the chronic stage of infection, the infiltrates mainly contain mononuclear cells) was as follows: 0, no significant infiltration; 1, infiltration at a single focus; 2, infiltration at two to four foci; 3, infiltration at more than four foci; and 4, confluent infiltration. The oviducts from the left and right sides of the same mouse were scored separately, and the two scores were added together as the score for the mouse. Scores assigned to individual mice were calculated as means ± standard errors for each group of animals.
Immunofluorescence assay.
HeLa cells grown on coverslips with or without chlamydial infection were fixed and permeabilized for immunostaining as described previously (36–38). Hoechst stain (blue; Sigma) was used to visualize nuclear DNA. For titrating IFU from mouse vaginal swab and oviduct tissue homogenate samples, a mouse anti-chlamydial LPS antibody (clone number MB5H9) (unpublished observations) plus a goat anti-mouse IgG conjugated with Cy3 (red; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) were used to visualize chlamydial inclusions. All immunofluorescence-labeled samples were observed under an Olympus AX-70 fluorescence microscope equipped with multiple filter sets (Olympus, Melville, NY).
Multiplex array for profiling cytokines in oviduct tissue and enzyme-linked immunosorbent assay (ELISA) for detecting C5a.
Oviduct and ovary tissues were harvested from C5+/+ and C5−/− mice, respectively (n = 5 for each group) on day 10 after intrauterine inoculation with C. muridarum to make homogenates, as described previously (10, 12). The homogenates were used for simultaneous measurements of 32 mouse cytokines (23-plex group I [catalog number M60-009RDPD] plus 9-plex group II [MD0-00000EL]) using a multiplex bead array assay (Bio-Plex 200 System; all from Bio-Rad, Hercules, CA) by following the manufacturer's instructions. All cytokines were expressed in pg/ml as means and standard deviations. The means from the two mouse strains were used to calculate ratios and for statistical analysis (Student t test).
To detect C5a in oviduct tissues and sera of mice, oviduct tissues and blood were collected 14 days after infection from C3+/+ and C3−/− mice, respectively (n = 10 for each group). These samples were serially diluted for standard sandwich ELISA detection as described previously (39–41). The capture antibody that specifically recognizes mouse C5a/C5adesArg (see below) while it does not cross-react with C5 (rat IgG1; clone number 152-1486; catalog number 558027; BD Pharmingen, San Jose, CA) was used to coat the ELISA plates. C5a from mouse samples or standard C5a (catalog number 2150-C5-025/CF; R&D Systems, Minneapolis, MN) captured on the plates was detected with a C5a-specific detection antibody conjugated with biotin (rat IgG2a; clone number 152-278; catalog number 558028; BD Pharmingen). The immobilized biotin was quantitated using horseradish peroxidase-conjugated goat anti-biotin (catalog number SP-3010; Vector Laboratories, Burlingame, CA) and a TMB substrate reagent set (catalog number 555214; BD Biosciences, San Jose, CA). The absorbance at 450 nm was used to calculate C5a concentrations (ng per tissue or ml of serum).
Statistical analyses.
A Kruskal-Wallis test was used to analyze the differences in numbers of IFU recovered from mouse swabs and tissue homogenate samples. The pathology score data were analyzed with a Wilcoxon rank sum test. Fisher's exact test was used to analyze category data, including the percentage of mice with oviduct hydrosalpinx. The cytokine concentrations were analyzed using a two-tailed Student t test.
RESULTS
Mice deficient in C5 fail to develop hydrosalpinx following intravaginal infection with C. muridarum.
To probe the role of C5 in chlamydial pathogenesis, we compared the susceptibilities of C5−/− (n = 23) and C5+/+ (n = 24) mice to C. muridarum induction of hydrosalpinx (Fig. 1). We found that following intravaginal infection with C. muridarum, C5−/− mice failed to develop any significant hydrosalpinx, while ∼42% of C5+/+ mice developed significant hydrosalpinx (P < 0.01; Fisher's exact test). The C5+/+ mice achieved a median hydrosalpinx score of greater than 1 (P < 0.05; Wilcoxon rank sum test). These differences in gross pathology between C5−/− and C5+/+ mice were further validated under a microscope, which revealed a highly significant reduction in both oviduct lumenal dilation and inflammatory infiltration (P < 0.01 for both; Wilcoxon rank sum test) in C5−/− mice. Since the data were from a large number of mice in each group and the experiments were carried out independently 3 or 4 times, we are confident that the results are reproducible. Nevertheless, when these two groups of mice were monitored for live organism shedding from the lower genital tract during the infection course prior to sacrifice, there was no difference in either the level or length of live organism shedding or the number of mice positive for shedding between the two groups, suggesting that C5 deficiency increased the mouse resistance to C. muridarum induction of hydrosalpinx in the upper genital tract without affecting C. muridarum infection in the lower genital tract. The latter is consistent with a previous report that no difference in susceptibility to C. muridarum infection was found between mice with or without complement deficiency (42).
FIG 1.

Effect of C5 deficiency on C. muridarum induction of hydrosalpinx and live organism shedding from the lower genital tract. (A) Mice without (a) (C5+/+, or B10.D2-Hc1 H2d H2-T18c/nSnJ; n = 24) or with (b) (C5−/−, or B10.D2-Hc0 H2d H2-T18c/oSnJ; n = 23) deficiency in complement factor C5 were infected intravaginally with C. muridarum. Sixty days after infection, mouse genital tracts were harvested for visually identifying (arrow) and scoring (white numbers) hydrosalpinges. Mice with hydrosalpinx on either side of the oviducts were defined as positive for hydrosalpinx. Hydrosalpinx severity was scored for each oviduct independently, and the scores from both oviducts of the same mouse were added together as the score for that mouse. The hydrosalpinx incidence rate and severity score for each group are listed under the corresponding images. *, P < 0.05; **, P < 0.01 (Fisher's exact test for comparing incidence rates and Wilcoxon rank sum test for severity scores). Note that C5−/− mice failed to develop any significant hydrosalpinx while ∼42% of C5+/+ mice developed hydrosalpinx with a severity score of >1. (B) The same genital tract tissues after scoring for gross pathology were subjected to microscopic examination. Representative images taken under 10× (left, a and b) or 100× (right, a1, a2, b1, and b2) objective lenses are shown. (C) Sections were semiquantitatively scored for both oviduct lumenal dilation (Dila.; squares) and inflammatory infiltration (Infla.; triangles), as described in Materials and Methods. **, P < 0.01 (Wilcoxon rank sum test for comparing both lumenal dilation and inflammatory scores). Note that C5+/+ mice developed significantly more severe lumenal dilation and inflammatory infiltration than C5−/− mice. (D) Mice were also monitored for live organism shedding from the lower genital tract during the infection course prior to sacrifice. The numbers of live organisms recovered from the swabs (a) and of mice remaining positive for shedding live organisms (b) from the C5+/+ and C5−/− groups are plotted along the y axis and against the infection time course (x axis). Note that there was no difference in either the levels or durations of live organism shedding or the numbers of mice positive for shedding between C5+/+ and C5−/− mice. The error bars indicate standard deviations.
Induction of hydrosalpinx in mice deficient in C3 by C. muridarum infection.
Since C3 is an upstream component of C5 in the complement activation cascade, we further evaluated the effect of C3 deficiency on mouse susceptibility to C. muridarum induction of hydrosalpinx (Fig. 2). To our surprise, we found that both C3−/− and C3+/+ mice developed significant hydrosalpinx, with hydrosalpinx incidence rates of ∼88% and median severity scores of >3 (P > 0.05 for both). In terms of lower genital tract infection, there was no difference in either the level or length of live organism shedding or the number of mice positive for shedding between the two groups. These observations demonstrated that C3 did not contribute to mouse susceptibility to either upper genital tract pathology or lower genital tract infection with C. muridarum.
FIG 2.
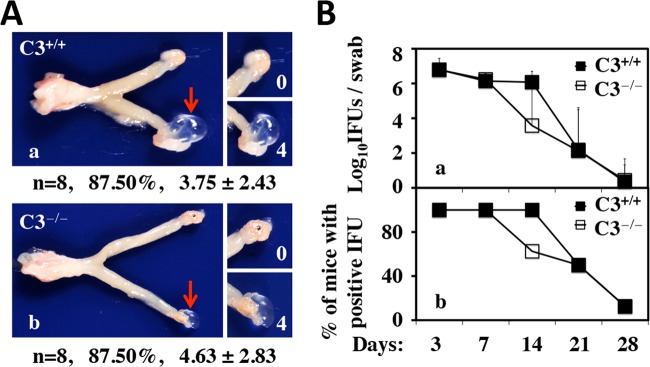
Effects of C3 deficiency on hydrosalpinx development and live organism shedding from the lower genital tract following C. muridarum infection. (A) Mice without (C3+/+, or C57BL/6J wild type; n = 8) (a) or with (C3−/−, or B6.129S4-C3tm1Crr/J; n = 8) (b) deficiency in complement factor C3 were infected intravaginally with C. muridarum. Sixty days after infection, mouse genital tracts were harvested for visually identifying (arrow) and scoring (white numbers) hydrosalpinges and analyzed as described in the legend to Fig. 1. Note that there is no significant difference in either hydrosalpinx incidence rates or hydrosalpinx severity scores between C3+/+ and C3−/− mice. The same genital tract tissues after scoring for gross pathology were subjected to microscopic examination (data not shown). (B) Mice were also monitored for live organism shedding from the lower genital tract during the infection course prior to sacrifice. The numbers of live organisms recovered from swabs (a) and of mice remaining positive for shedding live organisms (b) from the C3+/+ and C3−/− groups are plotted along the y axis. Note that there was no difference in either the levels or durations of live organism shedding or the numbers of mice positive for shedding between C3+/+ and C3−/− groups. The error bars indicate standard deviations.
Activation of C5 in the upper genital tract of mice deficient in C3 following C. muridarum infection.
Since C. muridarum induced hydrosalpinx in C3-deficient mice, we tested whether C. muridarum infection can activate C5 in C3-deficient mice. C5a is a C5 activation product, and antibodies specifically recognizing C5a neoepitopes are available. We used a C5a-specific antibody-based ELISA to monitor C5a in oviduct tissues and blood of mice 14 days after C. muridarum infection. Both C3+/+ and C3−/− mice produced significantly higher levels of C5a in oviducts but not blood after C. muridarum infection (Fig. 3). The level of C5a in the blood of both C3+/+ and C3−/− mice was <4 ng/ml regardless of C. muridarum infection. The C5a level in oviducts was increased from ∼2 ng to 8 ng (per tissue) by C. muridarum infection regardless of C3 deficiency. These observations demonstrated that lower genital tract infection with C. muridarum led to the activation of C5 in the upper genital tract. However, the systemic level of C5 activation is limited.
FIG 3.
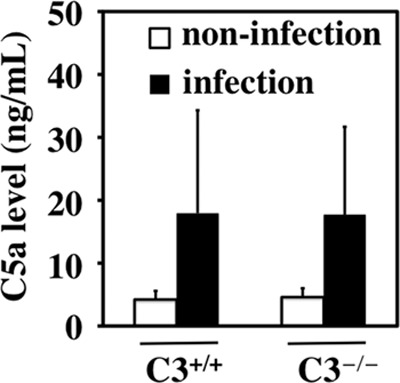
Detection of C5a in oviduct tissues and sera of C3−/− mice following C. muridarum infection. Mice without (C3+/+; n = 10) or with (C3−/−; n = 10) deficiency in complement factor C3 were infected intravaginally with (infection) or without (noninfection) C. muridarum. 14 days after infection, mouse oviduct tissues were collected for measuring C5a using an ELISA. Note that both C3+/+ and C3−/− mice produced higher levels of C5a in oviducts after C. muridarum infection. The error bars indicate standard deviations.
Chlamydial ascending infection is not altered in C5−/− mice.
To understand how C5 activation contributes to hydrosalpinx, we compared C. muridarum ascending infection between C5−/− and C5+/+ mice by monitoring live organism recovery from different segments of genital tracts on days 7, 10, and 14 following intravaginal infection with C. muridarum (Fig. 4). There were no significant differences in the numbers of live organisms recovered from different segments of the genital tract, including the oviduct, between C5+/+ and C5−/− mice. This experiment indicated that C5 did not affect C. muridarum ascending infection. It has been known that direct delivery of live C. muridarum organisms into the upper genital tract via an intrauterine inoculation, which circumvents the requirement for ascending infection, can enhance chlamydial pathogenicity in the upper genital tract (10). We then tested whether intrauterine inoculation of C. muridarum can induce hydrosalpinx in C5−/− mice (Fig. 5). We found that only a minimal level of hydrosalpinx was induced in C5−/− mice, with an ∼27% incidence rate and a median severity score of <1, while the C5+/+ control mice developed significant hydrosalpinx, with an incidence of ∼71% and a severity score of ∼3 (P < 0.05 for both). Clearly, the C5−/− mice were still more resistant to hydrosalpinx induction, even following intrauterine inoculation, both confirming the role of C5 in enhancing mouse susceptibility to hydrosalpinx induction and validating that C5-facilitated hydrosalpinx was probably not due to the enhancement of ascending infection.
FIG 4.
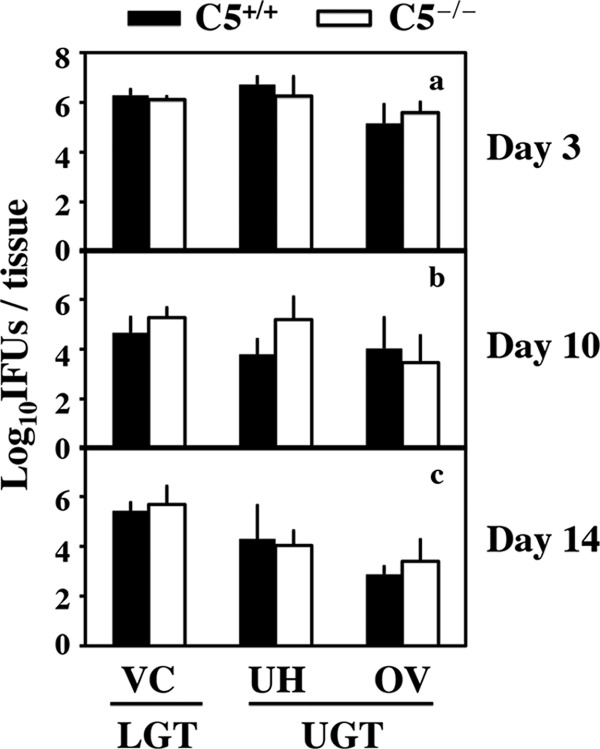
Effect of C5 deficiency on live organism recovery from mouse genital tracts following C. muridarum infection. Mice without (C5+/+; n = 5 for each time point) or with (C5−/−; n = 5 for each time point) deficiency in C5 were infected intravaginally with C. muridarum. Seven (a), 10 (b), or 14 (c) days after infection, mouse genital tract tissues were harvested and separated into vagina/cervix (VC) (representing the LGT) and uterine/uterine horn (UH) and oviduct/ovary (OV) (representing the UGT) segments. These segments were homogenized to measure live chlamydial organisms expressed as log10 IFU. Note that similar numbers of live organisms were recovered from both C5+/+ and C5−/− mice in all tissue segments and at all time points. The error bars indicate standard deviations.
FIG 5.
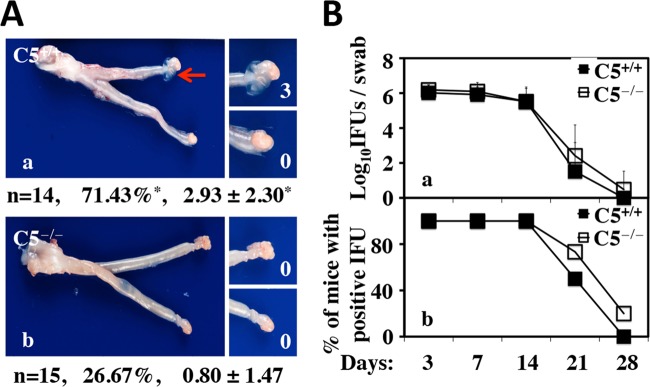
Effect of C5 deficiency on hydrosalpinx development and live organism shedding from the lower genital tract following intrauterine infection with C. muridarum. (A) Mice without (C5+/+; n = 14) (a) or with (C5−/−; n = 15) (b) deficiency in complement factor 5 were infected intrauterinally with C. muridarum. Sixty days after infection, mouse genital tracts were harvested for visually identifying (arrow) and scoring (white numbers) hydrosalpinges, as described in Materials and Methods, and analyzed as described in the legend to Fig. 1. *, P < 0.05 (Fisher's exact test for comparing incidence rates and Wilcoxon rank sum test for severity scores). Note that C5−/− mice developed significantly less hydrosalpinx in terms of both the incidence rate and the severity score. (B) Mice were also monitored for live organism shedding from the lower genital tract during the infection course prior to sacrifice. The numbers of live organisms recovered from swabs (a) and of mice remaining positive for shedding live organisms (b) from the C5+/+ and C5−/− groups are plotted along the y axis. The error bars indicate standard deviations.
Reduced inflammation in the oviducts of C5−/− mice after C. muridarum infection.
We first used microscopy to compare inflammatory responses in the oviducts between C5+/+ and C5−/− mice on days 10, 35, and 60 following intrauterine infection with C. muridarum (Fig. 6). Both oviduct lumenal dilation and inflammatory infiltration were semiquantitated based on criteria established previously (9, 10, 13). We found that C5−/− mice displayed significantly reduced lumenal dilation on days 35 and 60. On day 10 after infection, there was no lumenal dilation in either C5−/− or C5+/+ mice. However, the oviduct lumen was full of inflammatory infiltrates in C5+/+ mice, while the lumenal infiltration was significantly less intensive in C5−/− mice (P < 0.05). C5−/− mice also developed less inflammatory infiltration in oviduct tissue on days 35 and 60 postinfection (P < 001). We then compared the cytokine profiles in oviduct tissues between C5+/+ and C5−/− mice using a multiplex bead array assay (Table 1). We found that levels of 16 of the 32 cytokines were significantly higher in oviducts of C5+/+ than those of C5−/− mice, 6 of which displayed more than 3-fold difference, including IL-1α, IL-1β, IL-2, KC, MIP-1α, and IL-15.
FIG 6.

Effect of C5 deficiency on oviduct inflammatory responses following C. muridarum infection. (A) Mice without (a, c, and e) (C5+/+; n = 5, 6, and 14 for each time point) or with (b, d, and f) (C5−/−; n = 5, 7, and 15 for each time point) deficiency in complement factor 5 were infected intrauterinally with C. muridarum. Ten (a and b), 35 (c and d), or 60 (e and f) days after infection, mouse genital tract tissues were harvested for microscopic examination of both oviduct lumenal dilation and inflammatory infiltration. Representative images of H&E-stained oviduct tissue sections taken under 10× (a to f) or 100× (a1 to f1 and a2 to f2) objective lens are shown. (B) Both oviduct lumenal dilation (squares) and inflammatory infiltration from oviduct tissues (Tissue Infla.; upright triangles) or oviduct lumen (Lumen Infla., inverted triangles) were semiquantitated as described in Materials and Methods. *, P < 0.05; **, P < 0.01 (Wilcoxon rank sum test for comparing both lumenal dilation and inflammatory scores). Note that compared to the C5+/+ mice, C5−/− mice displayed significantly reduced lumenal dilation on days 35 (b) and 60 (c) and less oviduct lumenal inflammatory infiltration on day 10 (a), as well as less oviduct tissue inflammatory infiltration on days 35 (b) and 60 (c), postinfection.
TABLE 1.
Cytokines from oviduct tissue harvested 10 days after intrauterine infectiona
| Cytokine | Level (pg/ml)b |
Ratio (C5+/+/C5−/−) | P value (t test) | |
|---|---|---|---|---|
| C5+/+ (n = 5) | C5−/− (n = 5) | |||
| IL-1α | 579.1 ± 432.2 | 106.1 ± 68.0 | 5.5 | 0.04 |
| IL-1β | 3,884.3 ± 1987.6 | 710.8 ± 554.1 | 5.5 | 0.01 |
| IL-2 | 14.4 ± 7.9 | 3.2 ± 3.2 | 4.5 | 0.02 |
| IL-3 | 7.1 ± 1.8 | 4.0 ± 1.2 | 1.8 | 0.01 |
| IL-4 | 17.7 ± 5.9 | 7.5 ± 3.6 | 2.4 | 0.01 |
| IL-5 | 0 | 0 | ||
| IL-6 | 127.6 ± 224.1 | 4.0 ± 2.4 | 31.8 | 0.25 |
| IL-9 | 316.2 ± 89.8 | 224.2 ± 56.0 | 1.4 | 0.09 |
| IL-10 | 43.5 ± 14.8 | 21.1 ± 9.0 | 2.1 | 0.02 |
| IL-12(p40) | 490.4 ± 155.9 | 322.0 ± 87.9 | 1.5 | 0.07 |
| IL-12(p70) | 61.4 ± 30.0 | 21.0 ± 8.1 | 2.9 | 0.02 |
| IL-13 | 1,124.4 ± 241.9 | 701.5 ± 182.7 | 1.6 | 0.01 |
| IL-17 | 28.5 ± 6.0 | 19.6 ± 5.4 | 1.5 | 0.04 |
| Eotaxin | 1,717.9 ± 551.1 | 672.0 ± 442.8 | 2.6 | 0.01 |
| G-CSF | 2,228.7 ± 2803.9 | 3,684.6 ± 5,086.2 | 0.6 | 0.59 |
| GM-CSF | 113.3 ± 20.5 | 70.2 ± 18.5 | 1.6 | 0.01 |
| IFN-γ | 138.8 ± 72.1 | 49.9 ± 21.5 | 2.8 | 0.03 |
| KC | 195.8 ± 100.5 | 35.5 ± 18.2 | 5.5 | 0.01 |
| MCP-1 | 2,989.1 ± 1,445.5 | 1,406.8 ± 647.4 | 2.1 | 0.06 |
| MIP-1α | 1,089.7 ± 836.9 | 237.0 ± 157.1 | 4.6 | 0.06 |
| MIP-1β | 364.0 ± 142.6 | 121.8 ± 59.4 | 3 | 0.01 |
| RANTES | 2,942.1 ± 918.7 | 1,527.9 ± 1,174.2 | 1.9 | 0.07 |
| TNF-α | 64.7 ± 55.1 | 21.6 ± 7.7 | 3 | 0.12 |
| IL-15 | 308.6 ± 178.8 | 65.1 ± 66.9 | 4.7 | 0.02 |
| IL-18 | 53.6 ± 22.0 | 97.6 ± 128.5 | 0.5 | 0.47 |
| FGF-basic | 1,758.5 ± 429.0 | 1,392.5 ± 464.2 | 1.3 | 0.23 |
| LIF | 56.6 ± 36.5 | 21.1 ± 18.4 | 2.7 | 0.09 |
| M-CSF | 125.5 ± 33.4 | 104.6 ± 44.5 | 1.2 | 0.42 |
| MIG | 36,067.0 ± 8,549.2 | 17,670.5 ± 7,290.0 | 2 | 0.01 |
| MIP-2 | 5,980.6 ± 6,656.1 | 245.1 ± 248.9 | 24.4 | 0.09 |
| PDGF-BB | 0 | 0 | ||
| VEGF | 6,854.4 ± 2,746.5 | 5,148.8 ± 3,497.5 | 1.3 | 0.42 |
Oviduct cytokine profiles in C5+/+ versus C5−/− mice 10 days after intrauterine infection with C. muridarum. Oviduct tissue homogenates were made from C5+/+ (n = 5) and C5−/− (n = 5) mice on day 10 after intrauterine inoculation for simultaneous measurement of 32 cytokines using a multiplex bead array assay.
All cytokines are expressed as means ± standard deviations. The means from the two mouse strains were used for calculating ratios and for Student t test analyses. Note that levels16 of the 32 cytokines were significantly higher in oviducts of C5+/+ than those of C5−/− mice, 6 of which displayed more than 3-fold difference.
DISCUSSION
In the current study, we used the C. muridarum genital tract infection model to investigate the mechanism of hydrosalpinx by probing the role of the complement system in chlamydial pathogenesis. We found that C5 plays a critical role in C. muridarum induction of hydrosalpinx. First, following intravaginal infection with C. muridarum, 10 of the 24 C5+/+ B10.D2/nSnJ mice developed significant hydrosalpinx. The overall median severity score of the 24 mice was 1.33. However, none of the 23 C5−/− B10.D2/oSnJ mice developed any hydrosalpinx when examined by either the naked eye or microscopy. This observation demonstrated that C5−/− mice are highly resistant to C. muridarum induction of hydrosalpinx. The data obtained from the large numbers of mice came from 3 or 4 independent experiments. We are confident that these results are reproducible. Second, when live C. muridarum organisms were directly inoculated into the upper genital tract via intrauterine infection, the C5−/− mice still displayed significant resistance to hydrosalpinx induction. Ten of the 14 C5+/+ mice developed significant hydrosalpinx, with an overall median severity score of 2.93. However, only 4 of the 15 C5−/− mice developed hydrosalpinx, with a median severity score of 0.8. The data came from 2 independent experiments and support the result obtained with intravaginal infection. Intrauterine infection has been known to enhance C. muridarum pathogenicity in the upper genital tract (10), which led to the increase in pathology in both C5+/+ and C5−/− mice. However, the C5−/− mice maintained significant resistance compared to the C5+/+ mice. Third, both C3+/+ and C3−/− mice were induced to develop equally severe hydrosalpinx, demonstrating the specificity of the resistance phenotype observed in C5−/− mice. Finally, although both C5+/+ and C5−/− mice developed similar levels of oviduct infection, the C5−/− mice displayed significantly reduced cytokines in the oviduct tissues. Since C5 is activated by chlamydial infection in the oviduct, the reduced inflammatory cytokines in the oviducts of C5−/− mice suggest that the chlamydia-activated C5a in C5+/+ mice must play a significant role in promoting the development of hydrosalpinx. Altogether, we have presented convincing experimental data supporting a significant role of C5 in chlamydial pathogenicity in the upper genital tract.
Despite the significant resistance to hydrosalpinx induction in C5−/− mice, the C3−/− and C3+/+ mice displayed similar levels of susceptibility to hydrosalpinx induction. This finding was somewhat surprising to us at first, since C3 is an essential upstream component for C5 activation in all three major complement activation pathways. Interestingly, we detected significant levels of C5a in the oviducts of C3−/− mice infected with C. muridarum, suggesting that C. muridarum infection can activate C5 in the absence of C3, which is consistent with previous reports that C5 can be directly activated in C3−/− mice via “intercommunication of the complement system with the coagulation system,” including thrombin and human coagulation factors (F) XIa, Xa, and IXa, as serine proteases, which can effectively cleave (C3 and) C5 (29, 43). C3-independent activation of C5 is said to occur only under unusual circumstances, such as severe tissue damage (and might involve cross talk with additional systems and proteases participating in inflammation and damage besides those of the coagulation cascade). Active infection of oviduct epithelial cells can lead to destruction of the oviduct epithelium and may represent an overwhelming danger signal, at least in the oviduct, leading to local C3-independent bypass activation of C5 in the oviduct tissues. We also found no elevated C5a levels in serum due to C. muridarum infection. These observations are consistent with the concept that genital tract infection with chlamydia may represent a significant danger signal to the productive function but by no means a danger to the livelihood of the host. Thus, the C3-independent bypass activation of C5 was only restricted to the genital tract. The next question is how C. muridarum infection activates C5. Chlamydial EBs were shown to activate complement mainly via the alternative pathway (30, 31). It is not known whether EBs can activate C5 directly or via the coagulation-dependent pathway. Testing of these hypotheses is under way.
The inflammatory role of C5 can be exerted via both C5b and C5a (44). Cell lysis and/or tissue damage caused by C5b-9 membrane attack complexes may amplify inflammatory responses by releasing powerful danger signal ligands that are normally sequestered. In addition, even sublytic C5b-9 complex deposition can enhance inflammatory cytokine production, contributing to the development of nephritis (45). The C5b-9 complexes can activate many different inflammatory pathways and have been known to play significant roles in various kidney pathologies (46). The role of C5a in inflammatory pathologies has been even more extensively illustrated. Because of the important roles of C5a in inflammatory pathologies, strategies targeting C5a are being developed for reducing pathogenic inflammation (25–28). C5a can bind to both C5aR (also called CD88) and C5aR-like 2 (C5L2). Both receptors bind C5a (and its less active derivate C5adesArg) and act in concert to balance immunometabolism in adipose tissue (47) and in other contexts. However, only C5aR, and not C5L2, is coupled to G proteins and is usually regarded as the major receptor for C5a to exert the proinflammatory property (24). As a result, efforts at targeting C5aR to attenuate inflammatory damage have been most successful (48, 49). We have presented convincing evidence that C5 plays a critical role in chlamydial induction of hydrosalpinx. We also found that C5−/− mice displayed significantly reduced levels of cytokines in the oviduct, suggesting that the role of C5 in chlamydial pathogenesis may depend on its proinflammatory property. We recently showed that A/J mice that are deficient in C5 failed to develop significant hydrosalpinx following C. muridarum infection (50). The lack of hydrosalpinx further correlated well with reduced oviduct inflammatory responses (51). These previous observations, together with the finding made in the current study, are consistent with the roles of the complement system in chronic inflammation, such as hepatitis B-induced liver fibrosis (52), and in the pathogenesis of cerebral and placental malaria (53). The next step is to further uncover the pathways C5 uses to exert its proinflammatory property in the oviduct after chlamydial infection, including further confirming the critical role of C5a in C5aR−/− mice, identifying the cells that respond to C5a in the presence of chlamydial infection, and mapping the signaling pathways involved in exacerbating inflammatory pathology. Moreover, the immunomodulatory functions of C5a might also participate in the observed chronic inflammatory responses. Besides the classical functions, such as chemotaxis and promotion of neutrophil granular release in acute inflammation, immunomodulatory functions of C3a and C5a have also been recognized, especially during chronic inflammation (54, 55). C5a augments antigen presentation by dendritic cells (DCs) and prolongs T cell survival and thus can improve adaptive responses. In addition, C5a influences the development of regulatory T cells (24, 56). In the case of an intracellular infection, dysregulated and even improved T cell responses could be deleterious. We found that levels of 16 cytokines were significantly higher in the oviducts of C5+/+ mice, 6 of which (IL-1α, IL-1β, IL-2, KC, MIP-1α, and IL-15) displayed >3-fold difference. These cytokines can serve as the starting points for tracking the signaling pathways and identifying chlamydial ligands involved in C5-mediated hydrosalpinx development.
Clearly, just like the processes of many other pathologies, hydrosalpinx development induced by C. muridarum is multifactorial, despite the fact that we have demonstrated that C5 activation is a significant contributor to chlamydial induction of hydrosalpinx in both B10.D2 and B6 mice. The multifactorial hypothesis is not only supported by the cytokine data discussed above, but also reflected in the varied incidence rates of hydrosalpinx in the different strains of mice. For example, B10.D2 mice displayed a hydrosalpinx incidence rate of ∼42%, while B6 mice had a rate of ∼88%. However, after intrauterine infection (in which the C. muridarum organisms were directly delivered into uterine epithelia), the hydrosalpinx incidence dramatically increased to ∼71% in the B10.D2 mice, suggesting that prevention of ascending infection into the uterine epithelia contributes significantly to the low incidence rate of hydrosalpinx induced by intravaginal infection in this strain of mice. Also significantly, via intrauterine infection, B10.D2 mice deficient in C5 developed hydrosalpinx with an incidence rate of ∼27%, suggesting that C5 may represent just one of many host factors involved in chlamydial induction of hydrosalpinx. On the other hand, the significantly reduced rate of hydrosalpinx in the absence of C5 (27% versus 71%), even following an intrauterine infection, suggests that C5 is a dominant contributor to hydrosalpinx development in this model.
Although we have convincingly demonstrated a significant role of C5 in C. muridarum induction of hydrosalpinx using the murine genital tract infection model, there is no direct evidence for a role of C5 in C. trachomatis induction of hydrosalpinx in women. Given the roles of C5 in chronic inflammation, such as hepatitis B-induced liver fibrosis (52), and in the pathogenesis of cerebral and placental malaria (53), we propose that the complement system also plays a significant role in C. trachomatis induction of hydrosalpinx in women. This hypothesis is consistent with the finding of significant association of the TRAF1/C5 A allele with rheumatoid arthritis (57). Nevertheless, we should be cautious in interpreting the data from the C. muridarum murine genital tract infection model, because it has been recently shown that genital tract infection with C. trachomatis in mice can be controlled by innate immunity alone, while adaptive immunity is required to control C. muridarum genital tract infection (58), highlighting the difference in invasiveness between C. trachomatis and C. muridarum.
ACKNOWLEDGMENT
This work was supported in part by grants from the U.S. National Institutes of Health (G.Z.).
Footnotes
Published ahead of print 19 May 2014
REFERENCES
- 1.Sherman KJ, Daling JR, Stergachis A, Weiss NS, Foy HM, Wang SP, Grayston JT. 1990. Sexually transmitted diseases and tubal pregnancy. Sex. Transm. Dis. 17:115–121. 10.1097/00007435-199007000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Cotter TW, Meng Q, Shen ZL, Zhang YX, Su H, Caldwell HD. 1995. Protective efficacy of major outer membrane protein-specific immunoglobulin A (IgA) and IgG monoclonal antibodies in a murine model of Chlamydia trachomatis genital tract infection. Infect. Immun. 63:4704–4714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pal S, Peterson EM, de la Maza LM. 2005. Vaccination with the Chlamydia trachomatis major outer membrane protein can elicit an immune response as protective as that resulting from inoculation with live bacteria. Infect. Immun. 73:8153–8160. 10.1128/IAI.73.12.8153-8160.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu H, Zhong G. 1999. Interleukin-12 production is required for chlamydial antigen-pulsed dendritic cells to induce protection against live Chlamydia trachomatis infection. Infect. Immun. 67:1763–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murthy AK, Sharma J, Coalson JJ, Zhong G, Arulanandam BP. 2004. Chlamydia trachomatis pulmonary infection induces greater inflammatory pathology in immunoglobulin A deficient mice. Cell Immunol. 230:56–64. 10.1016/j.cellimm.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 6.Morrison SG, Morrison RP. 2005. A predominant role for antibody in acquired immunity to chlamydial genital tract reinfection. J. Immunol. 175:7536–7542. 10.4049/jimmunol.175.11.7536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morrison RP, Caldwell HD. 2002. Immunity to murine chlamydial genital infection. Infect. Immun. 70:2741–2751. 10.1128/IAI.70.6.2741-2751.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah AA, Schripsema JH, Imtiaz MT, Sigar IM, Kasimos J, Matos PG, Inouye S, Ramsey KH. 2005. Histopathologic changes related to fibrotic oviduct occlusion after genital tract infection of mice with Chlamydia muridarum. Sex. Transm. Dis. 32:49–56. 10.1097/01.olq.0000148299.14513.11. [DOI] [PubMed] [Google Scholar]
- 9.Lei L, Chen J, Hou S, Ding Y, Yang Z, Zeng H, Baseman J, Zhong G. 2014. Reduced live organism recovery and lack of hydrosalpinx in mice infected with plasmid-free Chlamydia muridarum. Infect. Immun. 82:983–992. 10.1128/IAI.01543-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang L, Zhang H, Lei L, Gong S, Zhou Z, Baseman J, Zhong G. 2013. Oviduct infection and hydrosalpinx in DBA1/j mice is induced by intracervical but not intravaginal inoculation with Chlamydia muridarum. PLoS One 8:e71649. 10.1371/journal.pone.0071649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Darville T, O'Neill JM, Andrews CW, Jr, Nagarajan UM, Stahl L, Ojcius DM. 2003. Toll-like receptor-2, but not Toll-like receptor-4, is essential for development of oviduct pathology in chlamydial genital tract infection. J. Immunol. 171:6187–6197. 10.4049/jimmunol.171.11.6187. [DOI] [PubMed] [Google Scholar]
- 12.Chen L, Lei L, Chang X, Li Z, Lu C, Zhang X, Wu Y, Yeh IT, Zhong G. 2010. Mice deficient in MyD88 develop a Th2-dominant response and severe pathology in the upper genital tract following Chlamydia muridarum infection. J. Immunol. 184:2602–2610. 10.4049/jimmunol.0901593. [DOI] [PubMed] [Google Scholar]
- 13.Dong X, Liu Y, Chang X, Lei L, Zhong G. 2014. Signaling via TNFR1 but not TLR2 contributes significantly to hydrosalpinx development following Chlamydia muridarum infection. Infect. Immun. 82:1833–1839. 10.1128/IAI.01668-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murthy AK, Li W, Chaganty BK, Kamalakaran S, Guentzel MN, Seshu J, Forsthuber TG, Zhong G, Arulanandam BP. 2011. Tumor necrosis factor alpha production from CD8+ T cells mediates oviduct pathological sequelae following primary genital Chlamydia muridarum infection. Infect. Immun. 79:2928–2935. 10.1128/IAI.05022-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imtiaz MT, Schripsema JH, Sigar IM, Kasimos JN, Ramsey KH. 2006. Inhibition of matrix metalloproteinases protects mice from ascending infection and chronic disease manifestations resulting from urogenital Chlamydia muridarum infection. Infect. Immun. 74:5513–5521. 10.1128/IAI.00730-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramsey KH, Sigar IM, Rana SV, Gupta J, Holland SM, Byrne GI. 2001. Role for inducible nitric oxide synthase in protection from chronic Chlamydia trachomatis urogenital disease in mice and its regulation by oxygen free radicals. Infect. Immun. 69:7374–7379. 10.1128/IAI.69.12.7374-7379.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagarajan UM, Sikes JD, Yeruva L, Prantner D. 2012. Significant role of IL-1 signaling, but limited role of inflammasome activation, in oviduct pathology during Chlamydia muridarum genital infection. J. Immunol. 188:2866–2875. 10.4049/jimmunol.1103461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng W, Shivshankar P, Li Z, Chen L, Yeh IT, Zhong G. 2008. Caspase-1 contributes to Chlamydia trachomatis-induced upper urogenital tract inflammatory pathologies without affecting the course of infection. Infect. Immun. 76:515–522. 10.1128/IAI.01064-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Igietseme JU, Omosun Y, Partin J, Goldstein J, He Q, Joseph K, Ellerson D, Ansari U, Eko FO, Bandea C, Zhong G, Black CM. 2013. Prevention of Chlamydia-induced infertility by inhibition of local caspase activity. J. Infect. Dis. 207:1095–1104. 10.1093/infdis/jit009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andrew DW, Cochrane M, Schripsema JH, Ramsey KH, Dando SJ, O'Meara CP, Timms P, Beagley KW. 2013. The duration of Chlamydia muridarum genital tract infection and associated chronic pathological changes are reduced in IL-17 knockout mice but protection is not increased further by immunization. PLoS One 8:e76664. 10.1371/journal.pone.0076664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen L, Cheng W, Shivshankar P, Lei L, Zhang X, Wu Y, Yeh IT, Zhong G. 2009. Distinct roles of CD28- and CD40 ligand-mediated costimulation in the development of protective immunity and pathology during Chlamydia muridarum urogenital infection in mice. Infect. Immun. 77:3080–3089. 10.1128/IAI.00611-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee HY, Schripsema JH, Sigar IM, Lacy SR, Kasimos JN, Murray CM, Ramsey KH. 2010. A role for CXC chemokine receptor-2 in the pathogenesis of urogenital Chlamydia muridarum infection in mice. FEMS Immunol. Med. Microbiol. 60:49–56. 10.1111/j.1574-695X.2010.00715.x. [DOI] [PubMed] [Google Scholar]
- 23.Sarma JV, Ward PA. 2011. The complement system. Cell Tissue Res. 343:227–235. 10.1007/s00441-010-1034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klos A, Wende E, Wareham KJ, Monk PN. 2013. International Union of Basic and Clinical Pharmacology. LXXXVII. Complement peptide C5a, C4a, and C3a receptors. Pharmacol. Rev. 65:500–543. 10.1124/pr.111.005223. [DOI] [PubMed] [Google Scholar]
- 25.Monk PN, Scola AM, Madala P, Fairlie DP. 2007. Function, structure and therapeutic potential of complement C5a receptors. Br. J. Pharmacol. 152:429–448. 10.1038/sj.bjp.0707332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bao L, Osawe I, Puri T, Lambris JD, Haas M, Quigg RJ. 2005. C5a promotes development of experimental lupus nephritis which can be blocked with a specific receptor antagonist. Eur. J. Immunol. 35:2496–2506. 10.1002/eji.200526327. [DOI] [PubMed] [Google Scholar]
- 27.Fujita E, Farkas I, Campbell W, Baranyi L, Okada H, Okada N. 2004. Inactivation of C5a anaphylatoxin by a peptide that is complementary to a region of C5a. J. Immunol. 172:6382–6387. 10.4049/jimmunol.172.10.6382. [DOI] [PubMed] [Google Scholar]
- 28.Otto M, Hawlisch H, Monk PN, Muller M, Klos A, Karp CL, Kohl J. 2004. C5a mutants are potent antagonists of the C5a receptor (CD88) and of C5L2: position 69 is the locus that determines agonism or antagonism. J. Biol. Chem. 279:142–151. 10.1074/jbc.M310078200. [DOI] [PubMed] [Google Scholar]
- 29.Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, Ward PA. 2006. Generation of C5a in the absence of C3: a new complement activation pathway. Nat. Med. 12:682–687. 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 30.Hall RT, Strugnell T, Wu X, Devine DV, Stiver HG. 1993. Characterization of kinetics and target proteins for binding of human complement component C3 to the surface-exposed outer membrane of Chlamydia trachomatis serovar L2. Infect. Immun. 61:1829–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Megran DW, Stiver HG, Bowie WR. 1985. Complement activation and stimulation of chemotaxis by Chlamydia trachomatis. Infect. Immun. 49:670–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin JS, Yan LL, Ho Y, Rice PA. 1992. Early complement components enhance neutralization of Chlamydia trachomatis infectivity by human sera. Infect. Immun. 60:2547–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bode J, Dutow P, Sommer K, Janik K, Glage S, Tummler B, Munder A, Laudeley R, Sachse KW, Klos A. 2012. A new role of the complement system: C3 provides protection in a mouse model of lung infection with intracellular Chlamydia psittaci. PLoS One 7:e50327. 10.1371/journal.pone.0050327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dutow P, Fehlhaber B, Bode J, Laudeley R, Rheinheimer C, Glage S, Wetsel RA, Pabst O, Klos A. 2014. The complement C3a receptor is critical in defense against Chlamydia psittaci in mouse lung infection and required for antibody and optimal T cell response. J. Infect. Dis. 209:1269–1278. 10.1093/infdis/jit640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen C, Chen D, Sharma J, Cheng W, Zhong Y, Liu K, Jensen J, Shain R, Arulanandam B, Zhong G. 2006. The hypothetical protein CT813 is localized in the Chlamydia trachomatis inclusion membrane and is immunogenic in women urogenitally infected with C. trachomatis. Infect. Immun. 74:4826–4840. 10.1128/IAI.00081-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Z, Lu C, Peng B, Zeng H, Zhou Z, Wu Y, Zhong G. 2012. Induction of protective immunity against Chlamydia muridarum intravaginal infection with a chlamydial glycogen phosphorylase. PLoS One 7:e32997. 10.1371/journal.pone.0032997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhong G, Fan P, Ji H, Dong F, Huang Y. 2001. Identification of a chlamydial protease-like activity factor responsible for the degradation of host transcription factors. J. Exp. Med. 193:935–942. 10.1084/jem.193.8.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhong G, Reis e Sousa C, Germain RN. 1997. Production, specificity, and functionality of monoclonal antibodies to specific peptide-major histocompatibility complex class II complexes formed by processing of exogenous protein. Proc. Natl. Acad. Sci. U. S. A. 94:13856–13861. 10.1073/pnas.94.25.13856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhong G, Toth I, Reid R, Brunham RC. 1993. Immunogenicity evaluation of a lipidic amino acid-based synthetic peptide vaccine for Chlamydia trachomatis. J. Immunol. 151:3728–3736. [PubMed] [Google Scholar]
- 40.Zhong GM, Brunham RC. 1990. Immunoaccessible peptide sequences of the major outer membrane protein from Chlamydia trachomatis serovar C. Infect. Immun. 58:3438–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma J, Bosnic AM, Piper JM, Zhong G. 2004. Human antibody responses to a Chlamydia-secreted protease factor. Infect. Immun. 72:7164–7171. 10.1128/IAI.72.12.7164-7171.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams DM, Schachter J, Weiner MH, Grubbs B. 1984. Antibody in host defense against mouse pneumonitis agent (murine Chlamydia trachomatis). Infect. Immun. 45:674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, Bruckner UB, Nilsson B, Gebhard F, Lambris JD, Huber-Lang M. 2010. Molecular intercommunication between the complement and coagulation systems. J. Immunol. 185:5628–5636. 10.4049/jimmunol.0903678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laursen NS, Magnani F, Gottfredsen RH, Petersen SV, Andersen GR. 2012. Structure, function and control of complement C5 and its proteolytic fragments. Curr. Mol. Med. 12:1083–1097. 10.2174/156652412802480925. [DOI] [PubMed] [Google Scholar]
- 45.Zhang J, Li Y, Shan K, Wang L, Qiu W, Lu Y, Zhao D, Zhu G, He F, Wang Y. 2014. Sublytic C5b-9 induces IL-6 and TGF-beta1 production by glomerular mesangial cells in rat Thy-1 nephritis through p300-mediated C/EBPbeta acetylation. FASEB J. 28:1511–1525. 10.1096/fj.13-242693. [DOI] [PubMed] [Google Scholar]
- 46.Takano T, Elimam H, Cybulsky AV. 2013. Complement-mediated cellular injury. Semin. Nephrol. 33:586–601. 10.1016/j.semnephrol.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 47.Poursharifi P, Lapointe M, Fisette A, Lu H, Roy C, Munkonda MN, Fairlie DP, Cianflone K. 2014. C5aR and C5L2 act in concert to balance immunometabolism in adipose tissue. Mol. Cell. Endocrinol. 382:325–333. 10.1016/j.mce.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 48.Kim H, Erdman LK, Lu Z, Serghides L, Zhong K, Dhabangi A, Musoke C, Gerard C, Cserti-Gazdewich C, Liles WC, Kain KC. 2014. Functional roles for C5a and C5aR but not C5L2 in the pathogenesis of human and experimental cerebral malaria. Infect. Immun. 82:371–379. 10.1128/IAI.01246-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiao H, Dairaghi DJ, Powers JP, Ertl LS, Baumgart T, Wang Y, Seitz LC, Penfold ME, Gan L, Hu P, Lu B, Gerard NP, Gerard C, Schall TJ, Jaen JC, Falk RJ, Jennette JC. 2014. C5a receptor (CD88) blockade protects against MPO-ANCA GN. J. Am. Soc. Nephrol. 25:225–231. 10.1681/ASN.2013020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang H, Zhou Z, Chen J, Wu G, Yang Z, Baseman J, Zhang J, Reddick RL, Zhong G. 7 April 2014. Lack of long lasting hydrosalpinx in A/J mice correlates with rapid but transient chlamydial ascension and neutrophil recruitment in the oviduct following intravaginal inoculation with Chlamydia muridarum. Infect. Immun. 10.1128/IAI.00055-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen J, Zhang H, Zhou Z, Yang Z, Ding Y, Zhong E, Arulanandam B, Baseman J, Zhong G. 2014. Chlamydial induction of hydrosalpinx in 11 strains of mice reveals multiple host mechanisms for preventing upper genital tract pathology. PLoS One 9:e95076. 10.1371/journal.pone.0095076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu R, Lin F, He J, Jin L, Zhang JY, Fu J, Liu H, Wang S, Zhang Z, Wang FS. 2013. Complement 5a stimulates hepatic stellate cells in vitro, and is increased in the plasma of patients with chronic hepatitis B. Immunology 138:228–234. 10.1111/imm.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Biryukov S, Stoute JA. 2014. Complement activation in malaria: friend or foe? Trends Mol. Med. 20:293–301. 10.1016/j.molmed.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 54.Le Friec G, Kohl J, Kemper C. 2013. A complement a day keeps the Fox(p3) away. Nat. Immunol. 14:110–112. 10.1038/ni.2515. [DOI] [PubMed] [Google Scholar]
- 55.Wende E, Laudeley R, Bleich A, Bleich E, Wetsel RA, Glage S, Klos A. 2013. The complement anaphylatoxin C3a receptor (C3aR) contributes to the inflammatory response in dextran sulfate sodium (DSS)-induced colitis in mice. PLoS One 8:e62257. 10.1371/journal.pone.0062257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kemper C, Kohl J. 2013. Novel roles for complement receptors in T cell regulation and beyond. Mol. Immunol. 56:181–190. 10.1016/j.molimm.2013.05.223. [DOI] [PubMed] [Google Scholar]
- 57.Mohamed RH, Pasha HF, El-Shahawy EE. 2012. Influence of TRAF1/C5 and STAT4 genes polymorphisms on susceptibility and severity of rheumatoid arthritis in Egyptian population. Cell Immunol. 273:67–72. 10.1016/j.cellimm.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 58.Sturdevant GL, Caldwell HD. 2 March 2014. Innate immunity is sufficient for the clearance of Chlamydia trachomatis from the female mouse genital tract. Pathog. Dis. 10.1111/2049-632X.12164. [DOI] [PMC free article] [PubMed] [Google Scholar]