

ABSTRACT
Porcine epidemic diarrhea virus (PEDV), a porcine enteropathogenic coronavirus, causes lethal watery diarrhea in piglets and results in large economic losses in many Asian and European countries. A large-scale outbreak of porcine epidemic diarrhea occurred in China in 2010, and the virus emerged in the United States in 2013 and spread rapidly, posing significant economic and public health concerns. Previous studies have shown that PEDV infection inhibits the synthesis of type I interferon (IFN), and viral papain-like protease 2 has been identified as an IFN antagonist. In this study, we found that the PEDV-encoded nucleocapsid (N) protein also inhibits Sendai virus-induced IFN-β production, IFN-stimulated gene expression, and activation of the transcription factors IFN regulatory factor 3 (IRF3) and NF-κB. We also found that N protein significantly impedes the activation of the IFN-β promoter stimulated by TBK1 or its upstream molecules (RIG-I, MDA5, IPS-1, and TRAF3) but does not counteract its activation by IRF3. A detailed analysis revealed that the PEDV N protein targets TBK1 by direct interaction and that this binding sequesters the association between TBK1 and IRF3, which in turn inhibits both IRF3 activation and type I IFN production. Together, our findings demonstrate a new mechanism evolved by PEDV to circumvent the host's antiviral immunity.
IMPORTANCE PEDV has received increasing attention since the emergence of a PEDV variant in China and the United States. Here, we identify nucleocapsid (N) protein as a novel PEDV-encoded interferon (IFN) antagonist and demonstrate that N protein antagonizes IFN production by sequestering the interaction between IRF3 and TBK1, a critical step in type I IFN signaling. This adds another layer of complexity to the immune evasion strategies evolved by this economically important viral pathogen. An understanding of its immune evasion mechanism may direct us to novel therapeutic targets and more effective vaccines against PEDV infection.
INTRODUCTION
Porcine epidemic diarrhea (PED) is an acute, highly contagious, and devastating viral enteric disease with a high mortality rate in suckling pigs. The causal agent, PED virus (PEDV), is an enveloped virus with a single-stranded positive-sense RNA genome of approximately 28 kb (1). At least seven open reading frames (ORFs) have been identified in the PEDV genome, arranged in the order 5′-ORF1a/1b-S-ORF3-E-M-N-3′ (2). ORF1a and ORF1b are located downstream of the 5′ untranslated region (UTR) and encode the viral replicase polyproteins 1a and 1b. The S, E, M, and N genes encode the four major structural proteins spike (S), envelope (E), membrane (M), and nucleocapsid (N), respectively, and ORF3 encodes an accessory protein that is thought to be associated with virulence (1, 3–6).
Historically, PED was first recognized in English feeder and fattening swine in 1971 (7). Since then, outbreaks of PED have been reported in many European countries. At present, PED occurs mainly in Asia, and these outbreaks are more acute and severe than those observed in Europe (1). In 2010, a large-scale outbreak of PED, characterized by watery diarrhea, dehydration, and vomiting, with 80% to 100% morbidity and 50% to 90% mortality in suckling piglets, occurred in swine farms in China (8) and was shown to be caused by a PEDV variant (9, 10). This PEDV variant emerged and spread rapidly in the United States in May 2013, posing significant economic and public health concerns (11–14).
Interferon (IFN) and the IFN-induced cellular antiviral response are the primary defense mechanisms against viral infection. During viral infection, host pattern recognition receptors (PRRs) recognize viral components or replication intermediates, known as pathogen-associated molecular patterns (PAMPs), and trigger the IFN response (15). Retinoic acid-induced gene I (RIG-I) and melanoma differentiation gene 5 (MDA5) are important cytoplasmic PRRs that detect viral RNA PAMPs in a wide range of cell types (16, 17). After sensing the cytoplasmic viral RNAs, RIG-I and/or MDA5 interacts with IFN-β promoter stimulator 1 (IPS-1) (also known as MAVS/VISA/Cardif) to activate the downstream IκB kinase (IKK)-related kinases, such as TANK binding kinase 1 (TBK1) and IKKε, leading to the activation of the critical transcription factors interferon requlatory factor 3 (IRF3) and nuclear factor κB (NF-κB). Phosphorylated IRF3 and NF-κB translocate to the nucleus and directly induce the type I IFNs (18–20).
To combat the antiviral effects of IFN, many viruses, including coronavirus, have evolved elaborate mechanisms to antagonize IFN, and multiple virus-encoded proteins are involved in this process (21–31). To our knowledge, at least eight proteins encoded by severe acute respiratory syndrome coronavirus (SARS-CoV) have been identified as IFN antagonists: nsp1, papain-like protease (PLP), nsp7, nsp15, N, M, ORF3b, and ORF6 (21, 32–40). PEDV also belongs to the family Coronaviridae (1). Previous studies revealed that PEDV infection inhibits the host's IFN responses, and PEDV-encoded PLP2 has been identified as a depressor of the RIG-I-mediated signaling pathway by deubiquitinating RIG-I and STING (41). PEDV encodes >20 mature proteins, and whether other viral proteins antagonize the IFN response is unclear. Because coronavirus nucleocapsid protein is the most abundant protein in virus-infected cells (42), and the N proteins of SARS-CoV and mouse hepatitis virus (MHV) have been identified as IFN antagonists (38, 39, 43), here we investigated the role of PEDV N protein in regulating the IFN response. Our results clearly demonstrate that PEDV N protein also inhibits IFN-β production and IFN-stimulated gene (ISG) expression. Mechanistically, the PEDV N protein targets TBK1 to prevent its interaction with IRF3 after stimulation, leading to a blockade of IFN production.
MATERIALS AND METHODS
Viruses, cells, and reagents.
PEDV strain AJ1102 (GenBank accession number JX188454), which was isolated from a suckling piglet with acute diarrhea in China in 2011 (44), was used in this study. Sendai virus (SEV) was obtained from the Centre of Virus Resource and Information, Wuhan Institute of Virology, Chinese Academy of Sciences. HEK-293T cells were cultured and maintained in RPMI 1640 (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS) at 37°C in a humidified 5% CO2 incubator. Vero cells were cultured and maintained in Dulbecco's modified Eagle's medium (Invitrogen) and used to amplify PEDV. Mouse monoclonal antibodies (MAbs) against Flag, hemagglutinin (HA), and β-actin were purchased from Medical and Biological Laboratories (Japan). Rabbit polyclonal antibodies directed against IRF3, phosphorylated IRF3 (p-IRF3), and TBK1 were obtained from ABclone (China) and Cell Signaling Technology. Human IFN-β enzyme-linked immunosorbent assay (ELISA) kits were purchased from TFB (Japan).
Plasmids.
The IFN-β–Luc, 4×PRDIII/I-Luc (referred to as IRF3-Luc), and 4×PRDII-Luc (referred to as NF-κB–Luc) luciferase reporter plasmids were described previously (45). The expression plasmids for wild-type RIG-I (pEF-Flag-RIG-I) and its constitutively active mutant (pEF-Flag-RIG-IN) were kindly provided by T. Fujita (Tokyo Metropolitan Institute of Medical Science, Japan). The TBK1 and IKKε expression vectors were kindly provided by Himanshu Kuma and Shizuo Akira (Immunology Frontier Research Center, Osaka University, Japan). The TRAF3 expression vector was a gift from Edward W. Harhaj (University of Miami School of Medicine, Miami, FL, USA). The DNA expression constructs encoding MDA5, IPS-1, and IRF3 were described previously (45). To construct the DNA expression vector pCAGGS-HA-N encoding HA-tagged PEDV N protein, standard reverse transcription (RT)-PCR was used to amplify the cDNA of the PEDV N gene from the total RNA extracted from Vero cells infected with PEDV strain AJ1102 with the following primers: forward primer 5′-GGGGGTACCATGGCTTCTGTCAGTTTT-3′ and reverse primer 5′-TTTCTCGAGTTAATTTCCTGTATCGAA-3′. The purified amplicon was cloned into vector pCAGGS-HA. All constructs were validated by DNA sequencing.
Luciferase reporter gene assay.
HEK-293T cells grown in 24-well plates were cotransfected with 0.1 μg/well reporter plasmid, 0.02 μg/well plasmid pRL-TK (Promega) (as an internal control for normalization of the transfection efficiency), and the indicated expression plasmid or an empty control plasmid. Where indicated, the cells were also mock infected or infected with SEV (10 hemagglutinating activity units/well) 24 h after cotransfection. The cells were lysed 12 h later, and firefly luciferase and Renilla luciferase activities were determined with the Dual-Luciferase reporter assay system (Promega), according to the manufacturer's protocol. The data are expressed as relative firefly luciferase activities normalized to Renilla luciferase activities and are representative of three independently conducted experiments.
RNA extraction and quantitative real-time RT-PCR.
To determine the effects of N protein on the expressions of ISG56, ISG54, and ISG20, HEK-293T cells in 24-well plates were transfected with 1 μg of the empty vector or a plasmid encoding PEDV N protein. After 24 h, the cells were mock infected or infected with SEV for 12 h. Total RNA was extracted from the cells with TRIzol reagent (Invitrogen, USA), and an aliquot (1 μg) was reverse transcribed to cDNA by using avian myeloblastosis virus (AMV) reverse transcriptase (Toyobo). The cDNA (1 μl of the 20-μl RT reaction mixture) was then used as the template in a SYBR green PCR assay (Applied Biosystems). The abundance of the individual mRNA transcript in each sample was assayed three times and normalized to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA (the internal control). The primers were designed with Primer Express software v.3.0 (Applied Biosystems), and the sequences are listed in Table 1.
TABLE 1.
Primers used for real-time RT-PCR
| GenBank accession no. | mRNA | Forward primer | Reverse primer |
|---|---|---|---|
| NM_002176 | IFN-β | TCTTTCCATGAGCTACAACTTGCT | GCAGTATTCAAGCCTCCCATTC |
| NM_00201 | ISG20 | CCGTGGCCAGGCTAGAGAT | CCGCTCATGTCCTCTTTCAGT |
| NM_001547 | ISG54 | CACCTCTGGACTGGCAATAGC | GTCAGGATTCAGCCGAATGG |
| NM_001270927 | ISG56 | GCTTTCAAATCCCTTCCGCTAT | GCCTTGGCCCGTTCATAAT |
| NM_002046 | GAPDH | TCATGACCACAGTCCATGCC | GGATGACCTTGCCCACAGCC |
ELISA for IFN-β.
To measure the amount of IFN-β secreted, HEK-293T cells were mock transfected or transfected with 1 μg of the expression plasmid encoding PEDV N protein. At 24 h posttransfection, the cells were treated or not treated with SEV for 12 h. The supernatants were then harvested for an ELISA with a human IFN-β detection kit (Techfields Biochem), according to the manufacturer's instructions.
Immunoblot analysis.
Briefly, HEK-293T cells cultured in 60-mm dishes were transfected with the appropriate plasmids. After 28 h, the cells were harvested by the addition of lysis buffer, and the protein concentrations in the whole-cell extracts were measured. Equal amounts of samples were then subjected to SDS-PAGE and analyzed for the expression of IRF3, p-IRF3, and TBK1 proteins by immunoblotting using rabbit anti-IRF3, anti-p-IRF3 (ABclone), and anti-TBK1 (Cell Signaling Technology) antibodies, respectively. To confirm the expression levels of HA-tagged PEDV N protein, an anti-HA antibody (MBL) was used for immunoblotting. The expression of β-actin was detected with a mouse anti-β-actin monoclonal antibody (MBL) to demonstrate equal protein sample loading.
Coimmunoprecipitation and immunoblot analyses.
For the transient-transfection experiments, HEK-293T cells were transfected with the appropriate plasmids for 28 h. The transfected cells were lysed in 200 μl of lysis buffer (4% SDS, 3% dithiothreitol [DTT], 0.065 mM Tris-HCl [pH 6.8], 30% glycerin) supplemented with a protease inhibitor (phenylmethylsulfonyl fluoride [PMSF]). The lysates were boiled at 100°C for 10 min before they were separated by SDS-PAGE and were then electroblotted onto a polyvinylidene difluoride membrane (Bio-Rad) and analyzed by immunoblotting with the indicated antibodies. For the coimmunoprecipitation analysis, the cells were washed with phosphate-buffered saline (PBS) and lysed for 20 min at 4°C in lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40, 10% glycerin, 0.1% SDS, and 2 mM Na2EDTA. The lysates were then cleared by centrifugation, and the proteins were immunoprecipitated overnight at 4°C with affinity antibodies and protein A+G agarose beads (Beyotime, China). The immunoprecipitates were washed three times with 1 ml of lysis buffer and then analyzed by using standard immunoblotting procedures.
Indirect immunofluorescence assay (IFA).
HEK-293T cells seeded onto microscope coverslips and placed into 24-well dishes were transfected with the PEDV N protein expression plasmid when the cells reached approximately 70% to 80% confluence. The cells were mock infected or infected with SEV 24 h after transfection. At 8 h postinfection, the cells were fixed with 4% paraformaldehyde for 10 min and then permeated with 0.1% Triton X-100 for 10 min at room temperature. After three washes with PBS, the cells were sealed with PBS containing 5% bovine serum albumin for 1 h and then incubated separately with rabbit polyclonal antibody directed against IRF3 (1:200) and mouse monoclonal antibody directed against the HA tag (1:200) for 1 h at room temperature. The cells were then treated with fluorescein isothiocyanate-labeled goat anti-mouse or Cy3-labeled goat anti-rabbit antibodies (Invitrogen) for 1 h and then treated with 4′,6-diamidino-2-phenylindole (DAPI) for 15 min at room temperature. After the samples were washed with PBS, fluorescent images were acquired with a confocal laser scanning microscope (Fluoview ver. 3.1; Olympus, Japan).
RESULTS
PEDV N protein blocks SEV-induced IFN-β production and ISG expression.
To explore whether PEDV N antagonizes IFN-β production, HEK-293T cells were transfected with a DNA expression construct encoding the HA-tagged N protein and then infected with SEV. The cells and supernatants were collected separately for real-time RT-PCR and ELISA of IFN-β. The expression of PEDV N protein was detected by immunoblotting with an anti-HA antibody. As shown in Fig. 1A and B, the ectopic expression of PEDV N protein significantly inhibited SEV-induced mRNA expression and the secretion of IFN-β, indicating that N protein antagonizes IFN-β production.
FIG 1.

N protein significantly inhibits SEV-induced expression of IFN-β and ISGs. HEK-293T cells grown in 24-well plates were transfected with 1 μg of a plasmid encoding PEDV N protein (pCAGGS-HA-N) or an empty vector, and 24 h later, the cells were infected with SEV (10 hemagglutinating activity units/well). Twelve hours after infection, the cells and supernatants were collected separately. (A and C to E) Total RNA was extracted from cells, and the expression levels of the IFN-β (A), ISG56 (C), ISG54 (D), ISG20 (E), and GAPDH genes were evaluated by quantitative real-time RT-PCR. The results are expressed as increases in mRNA levels relative to those in cells transfected without SEV infection and were normalized to the expression level of the GAPDH housekeeping gene. (B) The harvested supernatants were used to detect IFN-β by an ELISA. Anti-HA antibody was used to confirm the expression of PEDV N protein, and anti-β-actin antibody was used to detect β-actin, which served as a protein loading control. The results are representative of data from three independent experiments. WB, Western blot.
It is well known that IFNs initiate a series of signaling cascades through the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway, resulting in the expression of a set of ISGs which collaboratively suppress the replication of viruses and contribute to the development of the adaptive immune response (46). To confirm whether PEDV N protein suppresses IFN signaling, we analyzed the expression levels of some ISGs, including ISG56, ISG54, and ISG20, in cells overexpressing PEDV N protein and treated with IFN-α. The results showed that IFN-α-induced ISG expression could not be inhibited by PEDV N protein, suggesting that PEDV N protein does not antagonize IFN signaling (data not shown). In contrast, overexpression of PEDV N protein significantly reduced the expression levels of ISG56 (Fig. 1C), ISG54 (Fig. 1D), and ISG20 (Fig. 1E) induced by SEV. These results further confirm that PEDV N protein antagonizes the production of IFN-β rather than IFN signaling.
PEDV N protein inhibits activation of IRF3 and NF-κB.
The induction of type I IFN requires the coordinated and cooperative actions of the transcription factors IRF3 and NF-κB. To investigate whether PEDV N protein impairs the activation of IRF3 and NF-κB, cells were cotransfected with pCAGGS-HA-N and the IFN-β–Luc, IRF3-Luc, and NF-κB–Luc luciferase reporter plasmids together with the internal control plasmid pRL-TK and were then infected with SEV. As shown in Fig. 2, the overexpression of PEDV N protein blocked the SEV-induced promoter activities of IFN-β (Fig. 2A), IRF3 (Fig. 2B), and NF-κB (Fig. 2C) in a dose-dependent manner.
FIG 2.
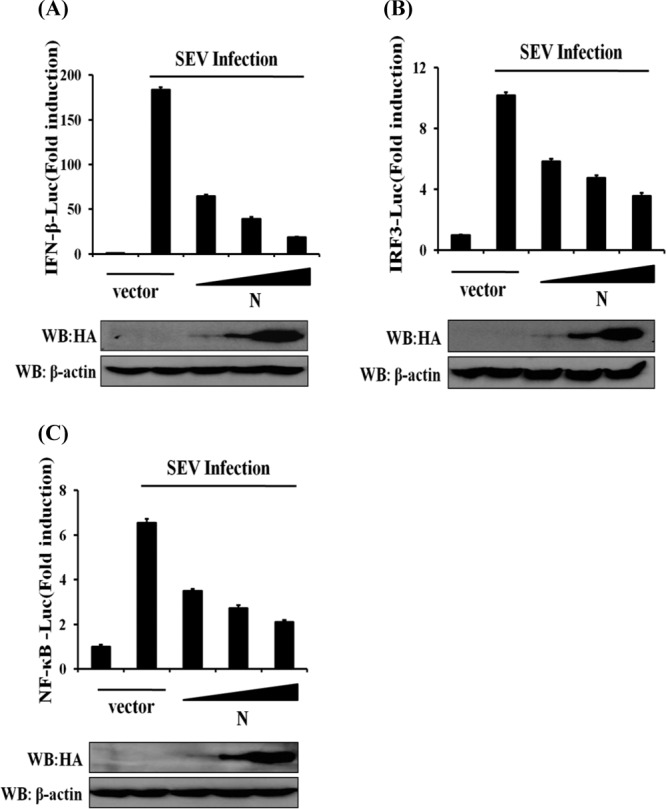
N protein independently inhibits promoter activation of IFN-β, IRF3, and NF-κB. HEK-293T cells were cotransfected with IFN-β–Luc (A), IRF3-Luc (B), or NF-κB–Luc (C) together with the pRL-TK plasmid and increasing quantities (0, 0.2, 0.4, or 0.8 μg) of plasmid pCAGGS-HA-N. Twenty-four hours after the initial transfection, the cells were infected with SEV. Luciferase assays were performed 12 h after infection. The results represent the means and standard deviations of data from three independent experiments. The relative firefly luciferase activity was normalized to the Renilla reniformis luciferase activity, and the untreated empty vector control value was set to 1. The expression of PEDV N protein was confirmed by immunoblotting with an anti-HA antibody.
IRF3 is considered to be the essential transcription factor for type I IFN production, and its phosphorylation and nuclear translocation are the hallmarks of IRF3 activation (18). Because our initial results showed that PEDV N protein blocked SEV-induced IRF3-dependent promoter activity, we investigated the role of PEDV N protein in the phosphorylation and nuclear translocation of IRF3. To this end, HEK-293T cells were transfected with pCAGGS-HA-N and then infected with SEV. The phosphorylation and nuclear translocation of endogenous IRF3 were investigated. As expected, SEV infection markedly enhanced IRF3 phosphorylation, in contrast to that in the mock-treated cells. However, this increase was significantly reduced in the N-protein-expressing cells (Fig. 3A). Consistent with this observation, the nuclear translocation of IRF3 was also blocked by PEDV N protein (Fig. 3B). Collectively, these results further support the notion that PEDV N protein is a potent inhibitor of type I IFN production by impeding the activation of IRF3.
FIG 3.
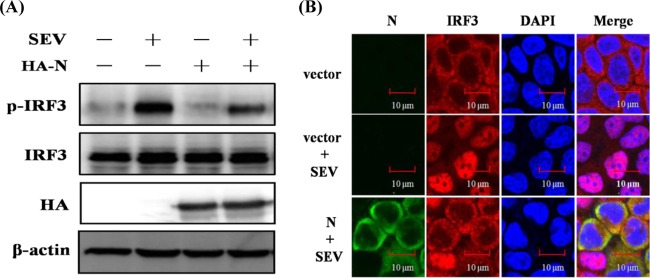
N protein blocks the phosphorylation and nuclear translocation of endogenous IRF3. (A) HEK-293T cells were mock transfected or transfected with an expression plasmid encoding HA-tagged N protein 24 h before being infected with SEV or not for 8 h. Cell lysates were collected for immunoblot analysis with antibodies directed against phosphorylated IRF3 (Ser396), IRF3, HA, or β-actin. (B) HEK-293T cells were transfected with plasmid pCAGGS-HA-N or an empty vector. At 24 h after transfection, the cells were infected with SEV for 8 h. After the fixation and permeation of the cells, an immunofluorescence analysis was performed to detect endogenous IRF3 (red) and N protein (green) with rabbit anti-IRF3 and mouse anti-HA antibodies, respectively. DAPI staining (blue) indicates the locations of the cell nuclei. Fluorescent images were acquired with a confocal laser scanning microscope (Fluoview ver. 3.1; Olympus, Japan). Cells transfected with an empty vector or mock infected with SEV were used as the negative controls.
TBK1 might be the potential target of PEDV N protein.
SEV is a strong inducer of the RIG-I-like receptor (RLR)-mediated IFN signaling pathway (47, 48). The observed blockade of SEV-induced IFN-β production and IRF3 activation by PEDV N protein raises the possibility that the N protein targets one or several components of the RLR signaling pathway. To clarify this, HEK-293T cells were cotransfected with pCAGGS-HA-N and a series of expression constructs encoding the molecules of the RLR signaling pathway, including RIG/RIG-IN, MDA-5, IPS-1, TRAF3, IKKε, TBK1, and IRF3, together with a luciferase reporter plasmid containing the IFN-β promoter and pRL-TK. Luciferase assays were performed 28 h after cotransfection. The overexpression of any molecule of the RLR signaling pathway resulted in a significant activation of the IFN-β promoter (Fig. 4A to F). Interestingly, the activation of the IFN-β promoter driven by TBK1/IKKε or its upstream molecules (RIG-I/RIG-IN, MDA-5, IPS-1, and TRAF3) was inhibited by the PEDV N protein. In contrast, the activation of the IFN-β promoter induced by IRF3 (a molecule downstream of TBK1/IKKε) was not affected by PEDV N protein (Fig. 4F). Consequently, we speculated that PEDV N protein targets TBK1/IKKε or an unknown molecule between TBK1/IKKε and IRF3.
FIG 4.
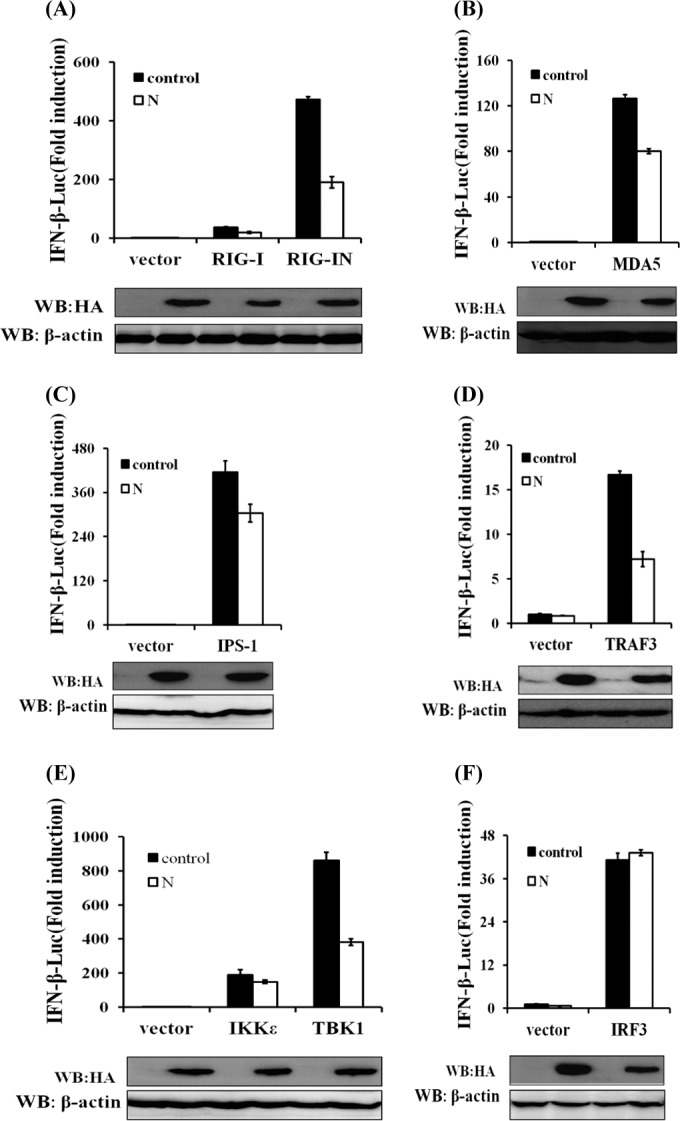
N protein disrupts the TBK1/IKKε-mediated IFN signaling pathway. HEK-293T cells were cotransfected with IFN-β–Luc, the pRL-TK plasmid, and pCAGGS-HA-N together with constructs expressing RIG-I/RIG-IN, MDA-5, IPS-1, TRAF3, IKKε, TBK1, or IRF3. Luciferase assays were performed 28 h after transfection. The results represent the means and standard deviations of data from three independent experiments. The relative firefly luciferase activity was normalized to the Renilla reniformis luciferase activity, and the untreated empty vector control value was set to 1. Western blotting with anti-HA antibody shows expression of PEDV N protein, and Western blotting for β-actin served as a protein loading control.
PEDV N protein interacts with TBK1.
To investigate the possible interaction of N protein and the components of the RLR signaling pathway, HEK-293T cells were transfected with expression constructs encoding HA-tagged PEDV N protein and Flag-tagged RIG-I, MDA-5, IPS-1, TRAF3, IKKε, TBK1, and IRF3. The lysates were then immunoprecipitated with an anti-HA MAb. As shown in Fig. 5A, TBK1 and IKKε were coprecipitated with HA-N, suggesting that there is a direct interaction between PEDV N protein and TBK1/IKKε. Because the PEDV N protein had a more significant blocking effect on TBK1-induced IFN-β promoter activation than on IKKε-induced activation (Fig. 4E), and previous studies suggested that TBK1 has a more important role than IKKε in the induction of IFN in response to double-stranded RNA (dsRNA) or SEV infection (48–51), we concentrated our subsequent investigations on TBK1.
FIG 5.

PEDV N protein interacts with TBK1. (A) HEK-293T cells were transfected with expression constructs encoding HA-tagged PEDV N protein and Flag-tagged RIG-I, MDA-5, IPS-1, TRAF3, IKKε, TBK1, and IRF3. The cells were lysed 28 h after transfection and subjected to immunoprecipitation with anti-HA antibody. The whole-cell lysates (WCL) and immunoprecipitation (IP) complexes were analyzed by immunoblotting (IB) using anti-Flag, anti-HA, or anti-β-actin antibodies. Because three different Flag-tagged control vectors were used to construct the expression plasmids, they were detected in three different gels. (B) Expression plasmids encoding Flag-tagged TBK1 and HA-tagged N protein were cotransfected, and immunoblotting analyses were performed as described above for panel A except for immunoprecipitation with anti-Flag antibody. (C) HEK-293T cells were transfected with expression plasmids encoding HA-tagged N protein and Flag-tagged TBK1. The cells were then fixed for an immunofluorescence assay to detect N protein (red) and TBK1 (green) with anti-HA and anti-Flag antibodies, respectively, 28 h after transfection. DAPI staining (blue) indicates the locations of the cell nuclei. Fluorescent images were acquired with a confocal laser scanning microscope (Fluoview ver. 3.1; Olympus, Japan).
To further confirm the direct interaction between N protein and TBK1, we also tested whether Flag-tagged TBK1 (Flag-TBK1) can pull down the HA-tagged PEDV N protein (HA-N). To this end, HEK-293T cells were transfected with expression constructs encoding Flag-tagged TBK1 and HA-tagged N protein alone or together. The lysates were then immunoprecipitated with an anti-Flag antibody. As shown in Fig. 5B, HA-N coprecipitated with Flag-TBK1, demonstrating that there is a direct interaction between TBK1 and PEDV N protein.
To further test whether PEDV N protein and TBK1 share similar subcellular locations, HEK-293T cells were cotransfected with pCAGGS-HA-N and Flag-tagged TBK1 expression plasmids, and IFA was performed 28 h after their cotransfection. As shown in Fig. 5C, the HA-tagged N protein (detected with anti-HA antibody) and Flag-tagged TBK1 (detected with anti-Flag antibody) colocalized and were distributed mainly in the cytoplasm.
PEDV N protein sequesters the interaction between IRF3 and TBK1.
The production of type I IFN requires the recruitment of TBK1 and the subsequent phosphorylation of IRF3 (18). To determine whether PEDV N protein inhibits TBK1-induced IRF3 phosphorylation, HEK-293T cells were cotransfected with pCAGGS-HA-N and a DNA expression construct encoding TBK1. The phosphorylation of endogenous IRF3 was detected 28 h after transfection. As shown in Fig. 6A, TBK1-induced IRF3 phosphorylation was reduced in cells cotransfected with pCAGGS-HA-N.
FIG 6.
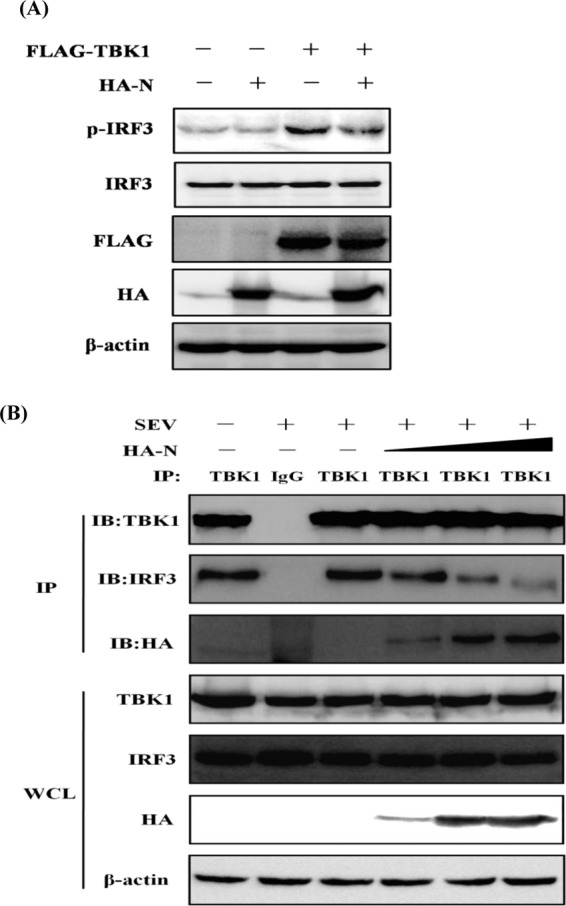
N protein inhibits TBK1-mediated IRF3 phosphorylation and sequesters the interaction between IRF3 and TBK1. (A) HEK-293T cells were cotransfected with an empty vector or expression plasmids encoding HA-tagged N protein and Flag-tagged TBK1. Twenty-four hours after transfection, the cells were mock infected or infected with SEV for 8 h, followed by immunoblotting assays with antibodies directed against phosphorylated IRF3 (Ser396), IRF3, HA, Flag, or β-actin. (B) HEK-293T cells were transfected with increasing amounts (0, 1.5, 3, or 6 μg) of a plasmid expressing HA-tagged N protein. Twenty-four hours after transfection, the cells were mock infected or infected with SEV for 8 h. The cell lysates were immunoprecipitated with rabbit anti-TBK1 or rabbit normal IgG antibody (lane 2). They were then analyzed by immunoblotting (IB) with antibodies directed against TBK1, IRF3, or HA. The whole-cell lysates (WCL) were used to analyze the expression of TBK1, IRF3, and N protein by immunoblotting using anti-TBK1, anti-IRF3, and anti-HA antibodies, respectively.
Previous studies demonstrated a physical interaction between TBK1 and IRF3, which is a critical step in the activation of IRF3 (52). Because our initial results demonstrated that N protein interacts with TBK1, it is reasonable to infer that N protein is an alternative substrate of TBK1, which impairs the recruitment of IRF3 by TBK1. To test this hypothesis, HEK-293T cells were transfected with increasing amounts of pCAGGS-HA-N and then infected with SEV. The PEDV N protein and endogenous IRF3 were immunoprecipitated with a rabbit polyclonal anti-TBK1 antibody, and the coprecipitated IRF3 and N proteins were analyzed by immunoblotting with an anti-IRF3 polyclonal antibody and an anti-HA MAb, respectively. As shown in Fig. 6B, TBK1 efficiently pulled down IRF3. However, the amount of IRF3 that bound to TBK1 gradually decreased as the amount of N protein increased. These results suggest that PEDV N protein physically sequesters the TBK1-IRF3 interaction in a concentration-dependent manner.
DISCUSSION
The IFN system is at the frontline of the host defense against viral invasion. Consequently, many viruses have developed strategies to manipulate IFN signaling with multifunctional viral proteins that target the host innate immune pathway. Previous studies have shown that PLP2 of PEDV antagonizes the IFN response by deubiquitinating RIG-I and STING (41). In the present study, we identified the PEDV-encoded N protein as another IFN antagonist and investigated the underlying molecular mechanism. Our results show that PEDV N protein targets TBK1 by interacting with it, and this binding sequesters the association between TBK1 and IRF3, in turn leading to the inhibition of both IRF3 activation and the production of type I IFN.
TBK1 is a member of the IKK protein kinase family and plays an important role in the regulation of type I IFN signaling (52). It is well known that Toll-like receptors (TLRs) and RLRs are the two main PRRs for RNA viruses. Although TLRs and RLRs recruit different downstream adaptors for IFN signaling, both use TBK1 to phosphorylate IRFs, thus inducing the subsequent transcription of type I IFN (53–55). Thus, TBK1 is at the intersection of the TLR and RLR pathways. Given the importance of TBK1 in the IFN signaling pathway, it is not surprising that viruses have evolved mechanisms that target it to inhibit IFN production. Many studies have demonstrated that viral proteins target TBK1 with diverse mechanisms to negatively modulate the IFN signaling pathway (52). For example, the leader proteinase (Lpro) of foot-and-mouth disease virus deubiquitinates TBK1 to inhibit type I IFN production (45). Herpes simplex virus (HSV) γ134.5 protein, hepatitis C virus (HCV) NS3 protein, vaccinia virus C6 and K7 proteins, SARS-CoV M protein, and the NY-1 hantavirus Gn cytoplasmic tail suppress the formation of the TBK1-containing complex to inhibit IFN-β production (40, 56–60). Poxvirus N1L protein and HCV NS2 protease interact directly with TBK1 to inhibit its activity (61, 62). In this study, we screened several components of the RLR signaling pathway and found that PEDV N protein targets TBK1 by interacting with it, as demonstrated by coimmunoprecipitation and subcellular colocalization experiments. Mechanistically, the interaction between TBK1 and PEDV N protein suggests three possible models. One possibility is that PEDV N protein directly affects the TBK1 kinase activity. The second possibility is that PEDV N protein disrupts the interactions between TBK1 and one or more of the TBK1 adaptors and blocks upstream signaling, as in the mechanism used by the SARS-CoV M protein, which blocks the formation of the TRAF3-TANK-TBK1/IKK complex to disrupt IFN signaling (40). The third possibility is that PEDV N protein prevents the access of a downstream target to the TBK1 complex, as in the mechanism used by HSV γ134.5, which interacts with TBK1 to disrupt the interaction between TBK1 and IRF3 (56). In this study, we noted that TBK1-induced IRF3 phosphorylation was significantly reduced in the presence of PEDV N protein, and IRF3 was dislodged from TBK1 as the level of PEDV N protein increased in the TBK1 immunoprecipitates. These results suggest that PEDV N protein acts as a competitor of IRF3 for TBK1 binding, resulting in the inhibition of both IRF3 activation and subsequent type I IFN production. Although we cannot rule out the other two possible mechanisms by which PEDV N protein might inhibit IFN production, the data in this study demonstrate that the N protein sequesters the interaction between IRF3 and TBK1, shedding light on at least one of the mechanisms underlying the antagonism of IFN by PEDV N protein. The interaction between TBK1 and PEDV N protein might also affect the function of the N protein. A previous study suggested that the Borna disease virus (BDV) P protein interacts with TBK1 to inhibit the TBK1-dependent expression of IFN-β. At the same time, the BDV P protein itself is phosphorylated by TBK1, suggesting that the BDV P protein functions as a viral decoy substrate, preventing the activation of the cellular target proteins of TBK1 (63). Similarly, the VP35 protein of Ebola virus (EBOV) binds to both IKKε and TBK-1 to antagonize IFN, and the VP35 protein is also phosphorylated by IKKε or TBK-1 (64). Previous studies demonstrated that the SARS-CoV N protein is heavily phosphorylated and that phosphorylation may affect nucleocytoplasmic shuttling of N protein (65–67). Although the phosphorylation of PEDV N protein has not been reported, a bioinformatics analysis (http://www.phosphosite.org/) indicated that PEDV N protein might be phosphorylated by IKK protein kinases. If PEDV N protein is really phosphorylated by TBK1, it is possible that the function of PEDV N protein can be modulated by TBK1. The biological implications of this modification for the pathogenesis of PEDV are very interesting, and this issue is under investigation in our laboratory.
As well as TBK1, we also found that PEDV N protein did interact with IKKε. IKKε and TBK1 are structurally and enzymatically similar and share 61% sequence identity. However, the two kinases are not functionally identical. First, the expression patterns of these two kinases are markedly different. TBK1 expression is ubiquitous and constitutive in a wide variety of cells, whereas IKKε expression is restricted to cells of lymphoid origin but is inducible in other cell types (54, 68). Second, the roles of these two kinases in the induction of the IFN response differ, although both kinases directly phosphorylate IRF3 and IRF7 and target identical serine residues. Analyses of the response to viral infection in TBK1- or IKKε-deficient mouse embryonic fibroblasts demonstrated that TBK1 is principally involved in the phosphorylation of IRF3 and IRF7 and subsequent antiviral responses, whereas IKKε has only an accessory role (49–51, 68). In contrast, TBK1 is completely dispensable for the type I IFN responses to viral infection in mouse bone marrow-derived macrophages, in which IKKε is predominant (49–51, 64). Considering these previously reported observations and that the PEDV N protein had a more significant blockade effect on TBK1-induced IFN-β promoter activation than on IKKε-induced activation, we focused on TBK1 in this study. Certainly, the interaction between IKKε and PEDV N protein might play a role in inhibiting both the activation of IRF3 and IFN production. Furthermore, the binding of PEDV N protein to both TBK1 and IKKε may benefit PEDV, which productively infects numerous cells types in vivo.
The PEDV N protein is functionally equivalent to the N proteins of other members of the coronaviruses. The N proteins of SARS-CoV and MHV have been identified as IFN antagonists, but the molecular mechanisms involved are distinct (38, 39, 43). The SARS-CoV N protein blocks the IFN-β response induced by Sendai virus or poly(I · C) but does not inhibit the IFN-β production induced by RIG-IN, IPS-1, TRIF, TBK-1, or IKKε. Furthermore, it does not interact with RIG-I or MDA5, indicating that the SARS-CoV N protein blocks a very early step in IFN-β production, probably at the RNA sensor recognition step (39). The N protein of MHV A59 antagonizes IFN activity by interfering with the RNase L activity associated with the induction of 2′-5′-oligoadenylate synthetase (43), but this has not yet been demonstrated for the SARS-CoV N protein (21). In this study, we have shown that PEDV N protein antagonizes IFN production by sequestering the interaction between IRF3 and TBK1. Furthermore, we also demonstrated that no interaction could be observed between TBK1 and SARS-CoV N protein or MHV N protein (data not shown). Therefore, although all these N proteins of coronaviruses antagonize IFN activity, different mechanisms are used by the nucleocapsid proteins of different coronaviruses.
Compared with our understanding of SARS-CoV and MHV, the molecular mechanism by which PEDV regulates IFN activity remains largely unknown. The identification of virus-encoded IFN antagonists and an understanding of the mechanism of action of each antagonist may direct us toward novel therapeutic targets and more effective vaccines. To date, only PLP2 and N protein (this study) have been identified as IFN antagonists of PEDV. Since the PEDV variant emerged in China and the United States, PEDV has received increasing attention, and more researchers have undertaken the study of PEDV. The identification and characterization of the PEDV-encoded IFN antagonists will broaden our understanding of the pathogenesis of PEDV and accelerate the development of more effective control strategies.
ACKNOWLEDGMENTS
We thank H. Kuma, S. Akira, E. W. Harhaj, D. Guo, and T. Fujita for providing expression constructs.
This work was supported by the National Natural Sciences Foundation of China (31225027 and 31121004) and the Fundamental Research Funds for the Central Universities (2013PY043).
Footnotes
Published ahead of print 28 May 2014
REFERENCES
- 1.Song D, Park B. 2012. Porcine epidemic diarrhoea virus: a comprehensive review of molecular epidemiology, diagnosis, and vaccines. Virus Genes 44:167–175. 10.1007/s11262-012-0713-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duarte M, Gelfi J, Lambert P, Rasschaert D, Laude H. 1993. Genome organization of porcine epidemic diarrhoea virus. Adv. Exp. Med. Biol. 342:55–60 [DOI] [PubMed] [Google Scholar]
- 3.Li C, Li Z, Zou Y, Wicht O, van Kuppeveld FJ, Rottier PJ, Bosch BJ. 2013. Manipulation of the porcine epidemic diarrhea virus genome using targeted RNA recombination. PLoS One 8:e69997. 10.1371/journal.pone.0069997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shirato K, Matsuyama S, Ujike M, Taguchi F. 2011. Role of proteases in the release of porcine epidemic diarrhea virus from infected cells. J. Virol. 85:7872–7880. 10.1128/JVI.00464-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang K, Lu W, Chen J, Xie S, Shi H, Hsu H, Yu W, Xu K, Bian C, Fischer WB, Schwarz W, Feng L, Sun B. 2012. PEDV ORF3 encodes an ion channel protein and regulates virus production. FEBS Lett. 586:384–391. 10.1016/j.febslet.2012.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu X, Zhang H, Zhang Q, Huang Y, Dong J, Liang Y, Liu HJ, Tong D. 2013. Porcine epidemic diarrhea virus N protein prolongs S-phase cell cycle, induces endoplasmic reticulum stress, and up-regulates interleukin-8 expression. Vet. Microbiol. 164:212–221. 10.1016/j.vetmic.2013.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wood EN. 1977. An apparently new syndrome of porcine epidemic diarrhoea. Vet. Rec. 100:243–244. 10.1136/vr.100.12.243 [DOI] [PubMed] [Google Scholar]
- 8.Sun RQ, Cai RJ, Chen YQ, Liang PS, Chen DK, Song CX. 2012. Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerg. Infect. Dis. 18:161–163. 10.3201/eid1801.111259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Liu X, Shi D, Shi H, Zhang X, Feng L. 2012. Complete genome sequence of a porcine epidemic diarrhea virus variant. J. Virol. 86:3408. 10.1128/JVI.07150-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang XM, Niu BB, Yan H, Gao DS, Yang X, Chen L, Chang HT, Zhao J, Wang CQ. 2013. Genetic properties of endemic Chinese porcine epidemic diarrhea virus strains isolated since 2010. Arch. Virol. 158:2487–2494. 10.1007/s00705-013-1767-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Q, Li G, Stasko J, Thomas JT, Stensland WR, Pillatzki AE, Gauger PC, Schwartz KJ, Madson D, Yoon KJ, Stevenson GW, Burrough ER, Harmon KM, Main RG, Zhang J. 2014. Isolation and characterization of porcine epidemic diarrhea viruses associated with the 2013 disease outbreak among swine in the United States. J. Clin. Microbiol. 52:234–243. 10.1128/JCM.02820-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stevenson GW, Hoang H, Schwartz KJ, Burrough ER, Sun D, Madson D, Cooper VL, Pillatzki A, Gauger P, Schmitt BJ, Koster LG, Killian ML, Yoon KJ. 2013. Emergence of porcine epidemic diarrhea virus in the United States: clinical signs, lesions, and viral genomic sequences. J. Vet. Diagn. Invest. 25:649–654. 10.1177/1040638713501675 [DOI] [PubMed] [Google Scholar]
- 13.Huang YW, Dickerman AW, Pineyro P, Li L, Fang L, Kiehne R, Opriessnig T, Meng XJ. 2013. Origin, evolution, and genotyping of emergent porcine epidemic diarrhea virus strains in the United States. mBio 4(5):e00737-13. 10.1128/mBio.00737-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marthaler D, Jiang Y, Otterson T, Goyal S, Rossow K, Collins J. 2013. Complete genome sequence of porcine epidemic diarrhea virus strain USA/Colorado/2013 from the United States. Genome Announc. 1(4):e00555-13. 10.1128/genomeA.00555-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rathinam VA, Fitzgerald KA. 2011. Cytosolic surveillance and antiviral immunity. Curr. Opin. Virol. 1:455–462. 10.1016/j.coviro.2011.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoneyama M, Fujita T. 2009. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 227:54–65. 10.1111/j.1600-065X.2008.00727.x [DOI] [PubMed] [Google Scholar]
- 17.Nakhaei P, Genin P, Civas A, Hiscott J. 2009. RIG-I-like receptors: sensing and responding to RNA virus infection. Semin. Immunol. 21:215–222. 10.1016/j.smim.2009.05.001 [DOI] [PubMed] [Google Scholar]
- 18.Ramos HJ, Gale M., Jr 2011. RIG-I like receptors and their signaling crosstalk in the regulation of antiviral immunity. Curr. Opin. Virol. 1:167–176. 10.1016/j.coviro.2011.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682. 10.1016/j.cell.2005.08.012 [DOI] [PubMed] [Google Scholar]
- 20.Hiscott J, Lin R, Nakhaei P, Paz S. 2006. MasterCARD: a priceless link to innate immunity. Trends Mol. Med. 12:53–56. 10.1016/j.molmed.2005.12.003 [DOI] [PubMed] [Google Scholar]
- 21.Totura AL, Baric RS. 2012. SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Curr. Opin. Virol. 2:264–275. 10.1016/j.coviro.2012.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou H, Perlman S. 2007. Mouse hepatitis virus does not induce beta interferon synthesis and does not inhibit its induction by double-stranded RNA. J. Virol. 81:568–574. 10.1128/JVI.01512-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spiegel M, Pichlmair A, Martinez-Sobrido L, Cros J, Garcia-Sastre A, Haller O, Weber F. 2005. Inhibition of beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J. Virol. 79:2079–2086. 10.1128/JVI.79.4.2079-2086.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thiel V, Weber F. 2008. Interferon and cytokine responses to SARS-coronavirus infection. Cytokine Growth Factor Rev. 19:121–132. 10.1016/j.cytogfr.2008.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roth-Cross JK, Martinez-Sobrido L, Scott EP, Garcia-Sastre A, Weiss SR. 2007. Inhibition of the alpha/beta interferon response by mouse hepatitis virus at multiple levels. J. Virol. 81:7189–7199. 10.1128/JVI.00013-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang R, Jha BK, Ogden KM, Dong B, Zhao L, Elliott R, Patton JT, Silverman RH, Weiss SR. 2013. Homologous 2′,5′-phosphodiesterases from disparate RNA viruses antagonize antiviral innate immunity. Proc. Natl. Acad. Sci. U. S. A. 110:13114–13119. 10.1073/pnas.1306917110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Graham RL, Donaldson EF, Baric RS. 2013. A decade after SARS: strategies for controlling emerging coronaviruses. Nat. Rev. Microbiol. 11:836–848. 10.1038/nrmicro3143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frieman M, Baric R. 2008. Mechanisms of severe acute respiratory syndrome pathogenesis and innate immunomodulation. Microbiol. Mol. Biol. Rev. 72:672–685. 10.1128/MMBR.00015-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frieman M, Heise M, Baric R. 2008. SARS coronavirus and innate immunity. Virus Res. 133:101–112. 10.1016/j.virusres.2007.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Z, Wang Y, Ratia K, Mesecar AD, Wilkinson KD, Baker SC. 2007. Proteolytic processing and deubiquitinating activity of papain-like proteases of human coronavirus NL63. J. Virol. 81:6007–6018. 10.1128/JVI.02747-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng D, Chen G, Guo B, Cheng G, Tang H. 2008. PLP2, a potent deubiquitinase from murine hepatitis virus, strongly inhibits cellular type I interferon production. Cell Res. 18:1105–1113. 10.1038/cr.2008.294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frieman M, Ratia K, Johnston RE, Mesecar AD, Baric RS. 2009. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 83:6689–6705. 10.1128/JVI.02220-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clementz MA, Chen Z, Banach BS, Wang Y, Sun L, Ratia K, Baez-Santos YM, Wang J, Takayama J, Ghosh AK, Li K, Mesecar AD, Baker SC. 2010. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 84:4619–4629. 10.1128/JVI.02406-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Devaraj SG, Wang N, Chen Z, Tseng M, Barretto N, Lin R, Peters CJ, Tseng CT, Baker SC, Li K. 2007. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 282:32208–32221. 10.1074/jbc.M704870200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wathelet MG, Orr M, Frieman MB, Baric RS. 2007. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J. Virol. 81:11620–11633. 10.1128/JVI.00702-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Narayanan K, Huang C, Lokugamage K, Kamitani W, Ikegami T, Tseng CT, Makino S. 2008. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J. Virol. 82:4471–4479. 10.1128/JVI.02472-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zust R, Cervantes-Barragan L, Kuri T, Blakqori G, Weber F, Ludewig B, Thiel V. 2007. Coronavirus non-structural protein 1 is a major pathogenicity factor: implications for the rational design of coronavirus vaccines. PLoS Pathog. 3:e109. 10.1371/journal.ppat.0030109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kopecky-Bromberg SA, Martinez-Sobrido L, Frieman M, Baric RA, Palese P. 2007. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 81:548–557. 10.1128/JVI.01782-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu X, Pan J, Tao J, Guo D. 2011. SARS-CoV nucleocapsid protein antagonizes IFN-beta response by targeting initial step of IFN-beta induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes 42:37–45. 10.1007/s11262-010-0544-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siu KL, Kok KH, Ng MH, Poon VK, Yuen KY, Zheng BJ, Jin DY. 2009. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J. Biol. Chem. 284:16202–16209. 10.1074/jbc.M109.008227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xing Y, Chen J, Tu J, Zhang B, Chen X, Shi H, Baker SC, Feng L, Chen Z. 2013. The papain-like protease of porcine epidemic diarrhea virus negatively regulates type I interferon pathway by acting as a viral deubiquitinase. J. Gen. Virol. 94:1554–1567. 10.1099/vir.0.051169-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zuniga S, Cruz JL, Sola I, Mateos-Gomez PA, Palacio L, Enjuanes L. 2010. Coronavirus nucleocapsid protein facilitates template switching and is required for efficient transcription. J. Virol. 84:2169–2175. 10.1128/JVI.02011-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ye Y, Hauns K, Langland JO, Jacobs BL, Hogue BG. 2007. Mouse hepatitis coronavirus A59 nucleocapsid protein is a type I interferon antagonist. J. Virol. 81:2554–2563. 10.1128/JVI.01634-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bi J, Zeng S, Xiao S, Chen H, Fang L. 2012. Complete genome sequence of porcine epidemic diarrhea virus strain AJ1102 isolated from a suckling piglet with acute diarrhea in China. J. Virol. 86:10910–10911. 10.1128/JVI.01919-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang D, Fang L, Li P, Sun L, Fan J, Zhang Q, Luo R, Liu X, Li K, Chen H, Chen Z, Xiao S. 2011. The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J. Virol. 85:3758–3766. 10.1128/JVI.02589-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Platanias LC. 2005. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5:375–386. 10.1038/nri1604 [DOI] [PubMed] [Google Scholar]
- 47.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172. 10.1038/nature04193 [DOI] [PubMed] [Google Scholar]
- 48.Pythoud C, Rodrigo WW, Pasqual G, Rothenberger S, Martinez-Sobrido L, de la Torre JC, Kunz S. 2012. Arenavirus nucleoprotein targets interferon regulatory factor-activating kinase IKKepsilon. J. Virol. 86:7728–7738. 10.1128/JVI.00187-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, Kawai T, Hoshino K, Takeda K, Akira S. 2004. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 199:1641–1650. 10.1084/jem.20040520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T. 2004. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc. Natl. Acad. Sci. U. S. A. 101:233–238. 10.1073/pnas.2237236100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G. 2004. Differential requirement for TANK-binding kinase-1 in type I interferon responses to Toll-like receptor activation and viral infection. J. Exp. Med. 199:1651–1658. 10.1084/jem.20040528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao W. 2013. Negative regulation of TBK1-mediated antiviral immunity. FEBS Lett. 587:542–548. 10.1016/j.febslet.2013.01.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11:373–384. 10.1038/ni.1863 [DOI] [PubMed] [Google Scholar]
- 54.Clark K, Peggie M, Plater L, Sorcek RJ, Young ER, Madwed JB, Hough J, McIver EG, Cohen P. 2011. Novel cross-talk within the IKK family controls innate immunity. Biochem. J. 434:93–104. 10.1042/BJ20101701 [DOI] [PubMed] [Google Scholar]
- 55.Hacker H, Karin M. 2006. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006:re13. 10.1126/stke.3572006re13 [DOI] [PubMed] [Google Scholar]
- 56.Verpooten D, Ma Y, Hou S, Yan Z, He B. 2009. Control of TANK-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1. J. Biol. Chem. 284:1097–1105. 10.1074/jbc.M805905200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Otsuka M, Kato N, Moriyama M, Taniguchi H, Wang Y, Dharel N, Kawabe T, Omata M. 2005. Interaction between the HCV NS3 protein and the host TBK1 protein leads to inhibition of cellular antiviral responses. Hepatology 41:1004–1012. 10.1002/hep.20666 [DOI] [PubMed] [Google Scholar]
- 58.Unterholzner L, Sumner RP, Baran M, Ren H, Mansur DS, Bourke NM, Randow F, Smith GL, Bowie AG. 2011. Vaccinia virus protein C6 is a virulence factor that binds TBK-1 adaptor proteins and inhibits activation of IRF3 and IRF7. PLoS Pathog. 7:e1002247. 10.1371/journal.ppat.1002247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alff PJ, Gavrilovskaya IN, Gorbunova E, Endriss K, Chong Y, Geimonen E, Sen N, Reich NC, Mackow ER. 2006. The pathogenic NY-1 hantavirus G1 cytoplasmic tail inhibits RIG-I- and TBK-1-directed interferon responses. J. Virol. 80:9676–9686. 10.1128/JVI.00508-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alff PJ, Sen N, Gorbunova E, Gavrilovskaya IN, Mackow ER. 2008. The NY-1 hantavirus Gn cytoplasmic tail coprecipitates TRAF3 and inhibits cellular interferon responses by disrupting TBK1-TRAF3 complex formation. J. Virol. 82:9115–9122. 10.1128/JVI.00290-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.DiPerna G, Stack J, Bowie AG, Boyd A, Kotwal G, Zhang Z, Arvikar S, Latz E, Fitzgerald KA, Marshall WL. 2004. Poxvirus protein N1L targets the I-kappaB kinase complex, inhibits signaling to NF-kappaB by the tumor necrosis factor superfamily of receptors, and inhibits NF-kappaB and IRF3 signaling by Toll-like receptors. J. Biol. Chem. 279:36570–36578. 10.1074/jbc.M400567200 [DOI] [PubMed] [Google Scholar]
- 62.Kaukinen P, Sillanpaa M, Nousiainen L, Melen K, Julkunen I. 2013. Hepatitis C virus NS2 protease inhibits host cell antiviral response by inhibiting IKKepsilon and TBK1 functions. J. Med. Virol. 85:71–82. 10.1002/jmv.23442 [DOI] [PubMed] [Google Scholar]
- 63.Unterstab G, Ludwig S, Anton A, Planz O, Dauber B, Krappmann D, Heins G, Ehrhardt C, Wolff T. 2005. Viral targeting of the interferon-beta-inducing Traf family member-associated NF-kappaB activator (TANK)-binding kinase-1. Proc. Natl. Acad. Sci. U. S. A. 102:13640–13645. 10.1073/pnas.0502883102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Prins KC, Cardenas WB, Basler CF. 2009. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J. Virol. 83:3069–3077. 10.1128/JVI.01875-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peng TY, Lee KR, Tarn WY. 2008. Phosphorylation of the arginine/serine dipeptide-rich motif of the severe acute respiratory syndrome coronavirus nucleocapsid protein modulates its multimerization, translation inhibitory activity and cellular localization. FEBS J. 275:4152–4163. 10.1111/j.1742-4658.2008.06564.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Surjit M, Kumar R, Mishra RN, Reddy MK, Chow VT, Lal SK. 2005. The severe acute respiratory syndrome coronavirus nucleocapsid protein is phosphorylated and localizes in the cytoplasm by 14-3-3-mediated translocation. J. Virol. 79:11476–11486. 10.1128/JVI.79.17.11476-11486.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu CH, Yeh SH, Tsay YG, Shieh YH, Kao CL, Chen YS, Wang SH, Kuo TJ, Chen DS, Chen PJ. 2009. Glycogen synthase kinase-3 regulates the phosphorylation of severe acute respiratory syndrome coronavirus nucleocapsid protein and viral replication. J. Biol. Chem. 284:5229–5239. 10.1074/jbc.M805747200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hiscott J, Nguyen TL, Arguello M, Nakhaei P, Paz S. 2006. Manipulation of the nuclear factor-kappaB pathway and the innate immune response by viruses. Oncogene 25:6844–6867. 10.1038/sj.onc.1209941 [DOI] [PMC free article] [PubMed] [Google Scholar]