

ABSTRACT
In order to understand and possibly treat B-cell malignancies associated with latent gammaherpesvirus infection, it is vital to understand the factors that control the balance between the two transcriptional states of gammaherpesviruses: latency and lytic replication. We used murine gammaherpesvirus 68 (MHV 68) as a model system to investigate how engagement of endosomal Toll-like receptors (TLRs) impacts reactivation from latency in vitro and establishment of latent infection in vivo. We found that treatment with TLR7 ligand R848 or TLR9 ligand CpG oligodeoxynucleotide (ODN) suppresses reactivation of MHV 68 in vitro. These suppressive effects correlated with the ability to activate cellular transcription factor NF-κB. Downregulation of TLR9 by RNA interference in vitro led to a reduction of nuclear levels of NF-κB p65 and consequently to an increase of spontaneous reactivation in cells latently infected with MHV 68, indicating that the TLR9 pathway suppresses spontaneous reactivation events. In vivo, sustained stimulation of TLR7 by repeated R848 treatment led to an increased frequency of infected splenocytes compared to mock-treated control results. Frequencies of infected splenic B cells in tlr7−/− or tlr9−/− mice after establishment of latency did not differ from those seen with their wild-type counterparts. Nevertheless, MHV 68-infected B cells from tlr9−/− mice showed a higher frequency of reactivation than B cells from wild-type or tlr7−/− mice in ex vivo reactivation assays. Thus, we show a suppressive effect of TLR7 or TLR9 triggering on MHV 68 reactivation that correlates with NF-κB activation and that the mere presence of a functional TLR9 signaling pathway contributes to dampen lytic gammaherpesvirus reactivation in infected cells.
IMPORTANCE A hallmark of gammaherpesviruses is their establishment of latency in B cells that is reversible through lytic reactivation. Latency can result in B-cell malignancies. Activation of the innate immune system is thought to contribute to controlling the switch between the transcriptional states of latency and reactivation. Nevertheless, the mechanisms involved are not clear. Here, we show that engagement of Toll-like receptor 7 (TLR7) and TLR9 suppresses reactivation of murine gammaherpesvirus MHV 68 in vitro and that stimulation of TLR7 in vivo increases the frequency of infected cells. TLR7 and TLR9 are innate immunity sensors of nucleic acids localized in endosomes. Additionally, we demonstrate that impairment of TLR9 signaling in latently infected B cells leads to increased reactivation. Thus, activated endosomal TLR7 and TLR9 pathways play an important role in promoting establishment of latent gammaherpesvirus infection. Counteracting signaling of these pathways allows reactivation and could represent treatment targets in gammaherpesvirus-associated malignancies.
INTRODUCTION
Gammaherpesviruses are double-stranded DNA B-lymphotropic viruses capable of establishing lifelong latent infections. Epstein-Barr virus (EBV) is the most prominent human gammaherpesvirus, with a seroprevalence in the adult population of over 90%. Even though latent infection with EBV is usually asymptomatic, it is associated with several B-cell malignancies, including endemic Burkitt's lymphoma (BL) (1). Notably, the incidence of endemic BL is restricted to areas with holoendemic chronic infection with the malaria parasite Plasmodium falciparum, suggesting a role for coinfections in the etiology of this particular EBV-associated tumor entity (2, 3). The mechanism by which chronic malaria impacts usually asymptomatic gammaherpesvirus infection is not known.
An important hallmark of gammaherpesviruses is sporadic reactivation from latent infection with subsequent production and release of infectious viral particles (4). Sporadic reactivation is essential to ensure transmission to a new host but needs to be tightly controlled since viral gene products represent targets for host immunity (5). Intrinsic and extrinsic factors are thought to control lytic reactivation of gammaherpesviruses. Activation of the innate immune system, in particular, signaling via Toll-like receptors (TLRs), has been shown to be an important factor in the balance between latency and lytic reactivation (6–9). TLRs are a family of pattern-recognition receptors that play a central role in innate immunity (10). How TLR signaling impacts gammaherpesvirus infection is particularly relevant to understanding the potential role of P. falciparum malaria in endemic BL since chronic P. falciparum infection provides constant stimulation of endosomal TLRs (11, 12).
In mammals, four members of the TLR family (TLR3, TLR7, TLR8, and TLR9) are expressed almost exclusively in intracellular compartments (10), where they function as sensors for microbial nucleic acids (13). The natural ligand of TLR3 is double-stranded RNA, while TLR7 senses single-stranded RNA and TLR9 detects unmethylated DNA containing CpG motifs. TLR8 is structurally closely related to TLR7 and independently recognizes single-stranded RNA (14) but is thought to be biologically inactive in mice, having instead a regulatory function in modifying expression and signaling of TLR7 (15). Subsequent to ligation of the receptor, signaling is forwarded via the recruitment of specific Toll/interleukin-1 (IL-1) receptor (TIR)-domain-containing adaptor proteins, including myeloid differentiation primary response protein 88 (MyD88) in the case of TLR7 as well as TLR9 (16), and TIR-domain-containing adaptor-inducing beta interferon (IFN-β) (TRIF) in the case of TLR3 (17). Eventually, signaling via TLR3, TLR7, and TLR9 leads to activation of the nuclear factor-κB (NF-κB) axis, which triggers pro- and anti-inflammatory cytokines (18). Importantly, activation of NF-κB has been demonstrated to be crucial for the establishment and maintenance of latent gammaherpesvirus infection in distinct ways. First, high levels of NF-κB subunit p65 inhibit activation of lytic gene promoters of several gammaherpesviruses (19, 20), and second, recombinant murine gammaherpesvirus 68 (MHV 68) expressing the constitutively active form of the NF-κB inhibitor IκBα is impaired in its ability to establish latent infection in vivo (21). Controversially, triggering of TLR3 or TLR9 but not triggering of TLR7 was found to induce reactivation of MHV 68 (6). This is remarkable since TLR7 and TLR9 share the signaling pathway and TLR3, TLR7, and TLR9 are canonical activators of NF-κB. Thus, additional investigation of this issue is required to precisely assess how activation of TLR signaling pathways impacts gammaherpesvirus reactivation.
Here, we used MHV 68 to study the effect of endosomal TLR triggering and activation of NF-κB on the expression of lytic viral genes and shedding of viral particles in vitro and on the establishment of latent infection in vivo. MHV 68 is commonly accepted as a model system to investigate gammaherpesvirus infection in vivo, since it is highly homologous to the human gammaherpesviruses EBV and Kaposi sarcoma-associated herpesvirus (KSHV) (22). We aimed at advancing the mechanistic understanding of the impact of TLR stimulation on lytic reactivation from latent infection using a cell line harboring latent MHV 68. Further, we tested whether TLR7 or TLR9 signaling contributes to NF-κB activation in latently MHV 68-infected cells and if it impacts viral behavior in vitro and in vivo.
MATERIALS AND METHODS
Ethics statement.
All animal experiments were done according to the guidelines of the Animal Welfare Act provided by the Swiss Federal Veterinary Office (www.bvet.admin.ch), and the veterinarian authorities of the Canton of Zurich, Switzerland, approved all procedures (license 100/2012).
Cell culture and MHV 68 viral production.
MHV 68-infected murine B-cell line S11 (courtesy of Ren Sun, University of California, Los Angeles [UCLA]) was cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum (FCS), 1% penicillin-streptomycin, and 2 mM l-glutamine; baby hamster kidney cells (BHK-21) (23) (courtesy of A. Nash, University of Edinburgh, United Kingdom) were cultured in Glasgow minimal essential medium (GMEM) supplemented with 10% tryptose phosphate broth, 10% heat-inactivated FCS, and 1% penicillin-streptomycin; and NIH3T12 murine fibroblast cells (courtesy of S. Speck, Emory Vaccine Center, Atlanta, GA) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated FCS, 1% penicillin-streptomycin, and 2 mM l-glutamine (all reagents from Invitrogen, Basel, Switzerland).
Wild-type (wt) MHV 68 clone g2.4 (24) was grown in BHK-21 cells (25). Cells and supernatant were harvested at 7 days postinfection (dpi) and centrifuged at 1,400 rpm for 5 min, and the supernatant was passed through a TPP polyethersulfone (PES) 0.22-μm-pore-size filter (Omnilab, Mettmenstetten, Switzerland) and stored at −80°C. MHV 68 titers were determined by a plaque assay on BHK-21 cells.
Mice.
C57BL/6 mice were purchased from Harlan (The Netherlands). Tlr7−/− and tlr9−/− mice on a C57BL/6 background were obtained from the Swiss Immunological Mutant Mouse Repository (SwiMMR, Zurich, Switzerland). All animals were kept in a specific-pathogen-free environment.
Detection of tlr gene expression.
Expression of tlrs was determined by reverse transcription-PCR using gene-specific primers as described in reference 26, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene expression was used as a loading control.
TLR triggering.
S11 cells were resuspended at 1 × 106 cells/ml in supplemented RPMI 1640 and stimulated with 25 μg/ml poly(I·C), 3 μM R848, or 0.5 μM CpG oligodeoxynucleotide (ODN) 1826 (Invivogen, San Diego, CA) 2 h prior to stimulation with 10 ng/ml of 12-O-tetradecanoylphorbol-13-acetate (TPA; Sigma-Aldrich, Buchs, Switzerland). At 24 h after treatment, cell pellets were assayed for MHV 68 lytic gene expression by quantitative PCR (qPCR) and supernatants were tested for MHV 68 production by a plaque assay (see below).
Reverse transcription and qPCR.
Expression of MHV 68 lytic gene ORF50 was determined by qPCR using an ABI 7900HT Fast real-time PCR system (Applied Biosystems, Rotkreuz, Switzerland). Total RNA was extracted from cells using an RNeasy minikit (Qiagen, Hombrechtikon, Switzerland), and contaminating DNA was removed by DNase treatment (DNA-free Ambion; Applied Biosystems, Rotkreuz, Switzerland). cDNA was synthesized using a High Capacity cDNA reverse transcription kit (Applied Biosystems). Amplification of synthesized cDNA was performed using TaqMan Gene Expression master mix (Applied Biosystems). The primers and probe used for MHV 68 ORF50 were as follows: ORF50_fw (5′-CCAACGTGTTCCCAGAAC-3′); ORF50_rev (5′-CGATGAACGCGTCCTCAG-3′); and ORF50_probe (6-carboxyfluorescein [FAM]-TACTCAGGAAGCGTGTCCCGGATCA-black hole quencher 1 [BHQ-1]). Primers and probes for tlr7 (Mm00446590_m1), tlr9 (Mm00446193_m1), ifnb1 (Mm00439552_s1), il-6 (Mm99999064_m1), il-10 (Mm00439614_m1), and the GAPDH gene (Mm99999915_g1) were all from Applied Biosystems.
Plaque assay.
MHV 68 titers in supernatants of S11 cells were determined by a plaque assay as described previously (25). BHK-21 cells were plated onto 6-well plates at 2 × 105 cells/well 1 day prior to infection. Serially diluted viral supernatants were placed onto monolayers and incubated for 1 h at 37°C in 5% CO2. Supernatants were then removed and replaced with complete GMEM containing 1% methyl cellulose (Sigma-Aldrich). After 3 to 4 days, the monolayers were fixed with methanol and stained with neutral red solution (Sigma-Aldrich) and the numbers of plaques were counted.
Preparation of nuclear extracts.
Cells (2 × 106) were washed with Tris-buffered saline (TBS), and the cell pellet was resuspended in 800 μl cold cell lysis buffer (10 mM HEPES [pH 7.9]; 10 mM KCl; 0.1 mM EDTA; 0.1 mM EGTA; 1 mM dithiothreitol [DTT]; 0.5 mM phenylmethylsulfonyl fluoride [PMSF]), kept at 4°C for 15 min, and subjected to a vortex procedure for 10 s after 50 μl of a 10% solution of Nonidet P-40 (Roche, Mannheim, Germany) was added. The homogenate was centrifuged at 10,000 × g for 30 s, and the nuclear pellet was resuspended in 100 μl ice-cold nuclear lysis buffer (20 mM HEPES [pH 7.9]; 0.4 M NaCl; 0.1 mM EDTA; 0.1 mM EGTA; 1 mM DTT; 0.5 mM PMSF) and rocked at 4°C for 15 min on a shaking platform. The nuclear extract was centrifuged at 10,000 × g for 5 min at 4°C, and the supernatant was stored at −80°C.
Western blot analysis.
Nuclear extracts were analyzed by SDS-PAGE on a NuPAGE 10% Bis-Tris gel (Invitrogen). The proteins were transferred onto a Protran nitrocellulose transfer membrane (Whatman, Kent, United Kingdom) and probed with specific antibodies against NF-κB p65 (sc-8008; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), PCNA (sc-25280; Santa Cruz), and beta-actin (catalog no. 4967; Cell Signaling Technology, Beverly, MA).
Detection of a spliced variant of ORF73.
RNA isolation and cDNA synthesis were performed as described above. A 2-μl volume of each cDNA reaction mixture was used as the template in a nested PCR specific for ORF73 spliced transcripts as described previously (27), and products of the reaction were separated on a 1.5% agarose gel. cDNA from S11 cells was used as a positive control, and cDNA generated from uninfected splenocytes was used as a negative control.
Mouse infection, R848 treatment, and limiting-dilution nested PCR.
Mice were infected by injecting 1 × 105 PFU of MHV 68 intraperitoneally (i.p.) in 100 μl phosphate-buffered saline (PBS). Daily treatment was done with 1 mg of R848/kg of body weight in 100 μl PBS based on previous work (28) or with PBS alone by i.p. injection for 19 days. At 20 dpi, mice were sacrificed by inhalation of CO2. Whole spleens were harvested, transferred to ice-cold PBS, and passed through a 70-μm-pore-size nylon mesh cell strainer (BD Falcon; BD Biosciences, Chicago, IL). Erythrocytes were removed by the use of ammonium chloride lysis solution (Stemcell Technologies, Grenoble, France). The frequency of MHV 68 genome-positive cells was determined by a previously published method (29). A nonlinear regression curve was fitted using Prism 6.0b software (GraphPad Software, La Jolla, CA). Frequencies were calculated as the splenocyte number at which 63.2% of reactions were positive based on the Poisson distribution.
TLR downregulation by RNA interference.
S11 cells were transfected with psiRNA-h7SK plasmids expressing short hairpin RNA (shRNA) targeting tlr7, tlr9, or the luciferase gene, as well as green fluorescent protein (GFP) and Zeocin (Invivogen) resistance from a separate promoter (Invivogen). Transfection was performed using a Neon transfection system (Invitrogen) according to the manufacturers' instructions. Transfected cells were selected in complete RPMI 1640 containing 200 μg/ml Zeocin (Invivogen), and successful selection was confirmed by flow cytometry.
Ex vivo MHV 68 reactivation assay.
Mice were sacrificed using a CO2 chamber, and splenocytes were recovered as described above. B cells were isolated by negative selection using a murine B-cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Purified B cells were counted and resuspended in a 2-fold dilution series starting at 1 × 106 cells/ml. Dilutions were plated on monolayers of NIH3T12 murine fibroblasts in a 96-well plate at 100 μl/well, using 12 wells per dilution. As a control, uninfected B cells were plated in 12 wells at 1 × 105 cells per well. To control for preformed virus particles, infected B cells were flash-frozen in liquid nitrogen and thawed in a 37°C water bath to disrupt cells. Serial dilutions of disrupted cells were plated on NIH3T12 monolayers using a method analogous to that used for intact cells. At 14 days later, wells were inspected for cytopathic effect by microscopy and the frequency of reactivation was calculated based on the Poisson distribution.
Statistical analysis.
If not stated otherwise, data were statistically analyzed by the unpaired t test using Prism 6.0b software (GraphPad Software). P values below 0.05 were considered statistically significant.
RESULTS
Stimulation of endosomal TLR7 and TLR9 but not of TLR3 causes nuclear accumulation of NF-κB p65 in S11 cells.
To investigate the effects of TLR triggering on latent gammaherpesvirus infection in vitro, we used the S11 latently MHV 68-infected murine B-cell line. S11 cells originate from a lymphoma harboring MHV 68 in a BALB/c mouse. Each S11 cell carries reactivation-competent MHV 68 (30).
First, we verified that S11 cells express TLR3, TLR7, and TLR9 using reverse transcription-PCR (Fig. 1A). To confirm activation of NF-κB upon TLR triggering, we treated S11 cells with ligands to TLR3, TLR7, TLR9, and NF-κB inhibitor Bay 11-7082 and assessed subsequent nuclear accumulation of NF-κB subunit p65 at different time points by Western blotting (Fig. 1B). To ensure that Bay 11-7082 is able to inhibit activation of NF-κB, we pretreated cells with the inhibitor 2 h prior to stimulation with R848 or CpG ODN.
FIG 1.

Triggering of endosomal TLR7 and TLR9 but not TLR3 activates NF-κB in S11 cells. (A) Expression of TLR3, TLR7, and TLR9 mRNA was detected in S11 cells by reverse transcription-PCR. (B) Triggering of TLR7 and TLR9 but not TLR3 activates NF-κB in S11 cells. NF-κB subunit p65 was detected in nuclear extracts of S11 cells at different time points after mock treatment or treatment with 25 μg/ml poly(I·C), 3 μM R848, 0.5 μM CpG ODN 1826, or 1 μM NF-κB inhibitor Bay 11-7082 by Western blot analysis. PCNA was used as a loading control. (C) Triggering of TLR3 induces a pattern of cytokine expression different from that seen with TLR7 or TLR9. S11 cells were stimulated with TLR ligands as described for panel B and harvested at different time points. Expression of IFN-β, IL-6, and IL-10 was analyzed by qPCR, normalized to GAPDH, and plotted relative to mock-treated control results. Data shown in panel C are means ± standard deviations (SD) of the results of three independent experiments. Some of the error bars are covered by the symbols.
The presence of poly(I·C) did not lead to any accumulation of NF-κB p65 in the nucleus over the course of 48 h. In contrast, the presence of R848, which triggers TLR7, and CpG ODN 1826, which triggers TLR9, led to activation of NF-κB already at 1 h after stimulation. S11 cells treated with R848 or CpG ODN 1826 showed stronger nuclear accumulation of NF-κB p65 than untreated cells for at least 12 h after treatment. In contrast, NF-κB inhibitor Bay 11-7082 reduced nuclear NF-κB p65 to minimal levels 1 h after treatment (Fig. 1B) and for at least 12 h. Bay treatment did not fully inhibit nuclear translocation of NF-κB upon TLR7 or TLR9 triggering (Fig. 1B, lanes 6 and 7) but markedly reduced nuclear NF-κB p65 levels compared to the results seen with stimulated cells (Fig. 1B, lanes 4 and 5). At 24 h after treatment, nuclear levels of NF-κB seemed to have normalized; the weak bands in the first 2 lanes are likely due to suboptimal blotting rather than to reduced activity.
To verify signaling competence, we measured the induction of cytokine expression in response to TLR ligands by qPCR (Fig. 1C). TLR3 ligand poly(I·C) robustly induced expression of IFN-β1 (Fig. 1C, top panel), consistent with activation of the interferon regulatory factor 3 (IRF3) pathway (17), while R848 and CpG ODN 1826 did not. On the other hand, R848 and CpG ODN 1826 induced expression of the cytokines IL-6 (Fig. 1C, middle panel) and IL-10 (Fig. 1C, bottom panel), while poly(I·C) did not, consistent with the observed differences in NF-κB activation.
These results demonstrate that endosomal TLRs activate distinct signaling pathways, resulting in differential levels of induction of transcription factors in the S11 model cells. While stimulation of TLR7 and TLR9 leads to activation of NF-κB and subsequent induction of IL-6 and IL-10, stimulation of TLR3 does not activate NF-κB but rather induces expression of IFN-β, probably via the IRF3 transcription factor.
Stimulation of TLR7 and TLR9 but not TLR3 suppresses both spontaneous and induced MHV 68 lytic reactivation in S11 cells.
A fraction of S11 cells latently infected with MHV 68 constantly undergoes spontaneous lytic reactivation as evidenced by expression of ORF50, the initiator of lytic replication (31), as well as by shedding of infectious MHV 68 particles into the supernatant. The frequency of reactivation of MHV 68 can be increased by treatment with TPA (30).
To monitor the impact of endosomal TLR triggering on MHV 68 reactivation, S11 cells were treated with poly(I·C), R848, and CpG ODN 1826 for 24 h before analyzing mRNA expression levels of immediate early lytic gene ORF50 by qPCR (Fig. 2A, black bars) as well as measuring infectious MHV 68 particles in culture supernatants by a plaque assay (Fig. 2A, white bars). Treatment with 25 μg/ml poly(I·C) did not change expression of ORF50 or the release of infectious particles. Treatment with 3 μM R848 or 0.5 μM CpG ODN 1826, however, decreased ORF50 expression compared to the mock-treated control results. To ensure suppression of the entire MHV 68 lytic transcription program, we also analyzed expression of early lytic gene ORF21 and late lytic gene M7 (32) by qPCR, and the results followed the pattern of ORF50 expression in all samples (data not shown). Consequently, the titer of infectious MHV 68 in the supernatant of cells treated with R848 or CpG ODN was reduced by about 50% compared to the mock-treated control titer. Even though the magnitude of the observed effects was modest, they were reproducible in multiple independent experiments and were statistically significant.
FIG 2.

Triggering of endosomal TLR7 and TLR9 but not TLR3 inhibits expression of the lytic MHV 68 ORF50 gene and reduces viral particle shedding, while TPA or NF-κB inhibitor Bay 11-7082 increases spontaneous reactivation. (A) S11 cells were mock treated or treated with TLR3 ligand poly(I·C), TLR7 ligand R848, TLR9 ligand CpG ODN 1826, or TPA or a combination of TLR ligand followed by TPA 2 h later. At 24 h after the beginning of treatment, expression levels of the MHV 68 lytic ORF50 gene were analyzed by qPCR, normalized to GAPDH, and plotted relative to untreated control results (black bars). Viral titers in the supernatants of treated cells were measured by a plaque assay (white bars). (B) S11 cells were mock treated or treated with TPA, R848, CpG ODN 1826, or NF-κB inhibitor Bay 11-7082 or a combination thereof. At 24 h after treatment, expression levels of the MHV 68 lytic ORF50 gene were analyzed by qPCR (black bars). Viral titers in the supernatants of the treated cells were measured by a plaque assay (white bars). Data shown in panels A and B are means ± SD of the results of three independent experiments. Statistics were calculated using an unpaired t test (*, P < 0.05; **, P < 0.01).
Addition of 10 ng/μl TPA to culture medium for 24 h increased mRNA levels of ORF50 about 4-fold and MHV 68 particle production about 2-fold compared to the levels seen with the untreated control. When the cells were treated with R848 or CpG ODN 1826 2 h before addition of TPA, the prolytic effect was abolished. These results indicate that signaling by TLR7 and TLR9 suppresses spontaneous as well as induced reactivation of MHV 68 while TLR3 signaling does not.
Next, we wanted to see whether the suppressive effect of TLR7 and TLR9 stimulation correlates with their ability to activate NF-κB. To this end, we treated S11 cells with 1 μM NF-κB inhibitor Bay 11-7082 2 h before stimulation with TLR ligands and analyzed lytic reactivation 24 h later (Fig. 2B). We found that pretreatment with Bay 11-7082 completely abolished the suppressive effect of CpG ODN 1826 and partially abolished the effect of R848, indicating that the impact of TLR triggering on lytic replication is at least partially dependent on NF-κB activation. In the absence of TLR stimulation, the treatment with Bay 11-7082 led to a slight increase in lytic reactivation compared to mock-treated control results, in agreement with earlier reports that Bay 11-7082 provokes reactivation of EBV or KSHV from latently infected B cells in vitro (19, 33). Treatment with TPA increased lytic gene expression as well as MHV 68 particle production as seen before.
Stimulation of TLR7 in vivo leads to an increased frequency of infected splenocytes after intraperitoneal injection of MHV 68.
Based on our in vitro data, and since NF-κB activation was shown to be important for the establishment of latent MHV 68 infection in vivo (21), we hypothesized that TLR7-mediated activation of NF-κB would promote the establishment of MHV 68 latency in vivo in a fashion similar to that shown for TLR9 (6).
To test this hypothesis, we infected 10 C57BL/6 mice intraperitoneally (i.p.) with 1 × 105 PFU MHV 68. We chose i.p. infection since this route seeds MHV 68 directly to the spleen without the need of amplification through lytic replication in the respiratory tract (34), therefore allowing the virus to reach the main site of latent infection directly. At 1 day postinoculation (dpi), the mice were segregated into two groups and treated daily with either 1 mg/kg R848–100 μl PBS or PBS alone (mock treatment) i.p. for 19 days. Mice were sacrificed at 20 dpi, a time when lytic MHV 68 infection has been cleared from the spleen and predominantly latent infection can be expected (34).
To investigate whether treatment with R848 following inoculation with MHV 68 facilitates establishment of latent infection, we isolated splenocytes from mice at 20 dpi and subjected them to analysis by limiting-dilution nested PCR targeting MHV 68 gene v-cyclin (ORF72). We found that splenocytes from mice treated daily with R848 showed a frequency of cells positive for MHV 68 genome that was 7-fold higher than that seen with splenocytes from mice subjected to mock treatment with PBS (1 in 100 versus 1 in 750; Fig. 3A).
FIG 3.

Treatment with TLR7 ligand R848 promotes establishment of latent MHV 68 infection in vivo. C57BL/6 mice were infected with MHV 68 by i.p. injection and treated daily i.p. with either 1 mg/kg R848 in 100 μl of PBS or PBS alone (mock) for 19 days. Treated and mock-treated groups contained 5 mice each. (A) Splenocytes isolated from infected mice at 20 dpi were analyzed by limiting-dilution nested PCR targeting ORF72. Data are expressed as mean percentages of positive reactions ± SD. A sigmoidal dose-response curve was fitted by nonlinear regression analysis using GraphPad software, and the frequency of MHV 68-positive splenocytes was calculated based on the Poisson distribution. The resulting curves were compared by the Mann-Whitney test (mock treatment versus R848 treatment P = 0.016). (B) Primary splenocytes from mice infected in vivo with MHV 68 expressed virus latency-associated gene ORF73 (mLANA). Total RNA isolated from bulk splenocytes was reverse transcribed and analyzed for spliced ORF73 transcripts by nested PCR. Amplification of the GAPDH housekeeping gene served as the control for successful reverse transcription. (C) Preformed virus is not detected in disrupted splenocytes from in vivo infection. Splenocytes isolated from mice infected in vivo with MHV 68 were flash-frozen in liquid nitrogen to disrupt cells and plated on NIH3T12 fibroblasts in a limiting-dilution assay. Wells were scored for cytopathic effect (CPE) 14 days after plating. (D) Flash-freezing slightly reduces the infectious titer of cell-free virus. Plaque assays were performed with cell-free virus stocks before and after a freeze-thaw cycle. Data shown are means ± SD of the results of three independent experiments. n.s., not significant.
To confirm latent MHV 68 infection, we tested the splenocytes for expression of a spliced variant of ORF73 (mLANA), a transcript associated with MHV 68 latency (27), by reverse transcription-PCR (Fig. 3B). At 20 dpi, ORF73 spliced transcripts were detected in all samples from mice treated with R848 but inconsistently in samples from mock-treated mice, reflecting the lower frequency of splenocytes harboring latent MHV 68. To ensure that the higher frequency of MHV 68 DNA copies detected by nested PCR (Fig. 3A) represented latent infection and not persistent or secondary lytic replication, we assayed homogenates of disrupted splenocytes from infected mice for the presence of infectious MHV 68. To disrupt splenocytes, the cells were flash-frozen in liquid nitrogen and thawed in a 37°C water bath, a process that kills >99% of cells as judged by a trypan blue exclusion assay. Disrupted splenocytes were plated onto susceptible epithelial cell line NIH3T12 in a limiting-dilution assay, and the plates were scored for cytopathic effect after 14 days. The amount of cytopathic effect observed was negligible in both treated and mock-treated cells (Fig. 3C). To test whether the process of flash-freezing used for cell disruption affected the infectiousness of viral particles, we tested the method with two separate stocks of cell-free virus and analyzed viral titers before and after freezing by a plaque assay (Fig. 3D). Flash-freezing in liquid nitrogen slightly reduced the mean titers of the two separate cell-free virus stocks (by 27% and 23%, respectively) but did not abolish infectiousness completely. We therefore conclude that lytic replication was absent in splenocytes of both R848-treated and control samples and that the viral genome amplified by nested PCR as shown in Fig. 3A stemmed from latent infection. Thus, TLR7 triggering during primary infection promotes the establishment of latency and increases the reservoir size of latent MHV 68.
Silencing of TLR9 but not of TLR7 leads to decreased levels of nuclear NF-κB and enhanced lytic reactivation in S11 cells.
Latently MHV 68-infected S11 cells show a basal level of NF-κB activity (Fig. 1B). Since nucleic acid binding of endosomal TLRs has been implicated in the sensing of various viral infections and since TLR7 and TLR9 both have the ability to activate NF-κB, we aimed at investigating whether the basal activity of NF-κB is mediated by TLR7 or TLR9 signaling in latently MHV 68-infected cells.
To assess the roles of TLR7 and TLR9 in this context, S11 cells were transfected with plasmids expressing shRNA against TLR7, TLR9, or luciferase as a control. Antibiotic selection yielded bulk stable cell lines, and >95% of the cells expressed the GFP cassette included in the vector containing the shRNA as measured by flow cytometry (data not shown). Stably transfected cells showed a reduction in the level of TLR7 mRNA of 70% and a reduction in the level of TLR9 mRNA of 55%, as measured by qPCR (Fig. 4A). S11 cells genetically complemented with shRNA against luciferase (shLuc) did not show any effect on TLR expression and served as a control.
FIG 4.
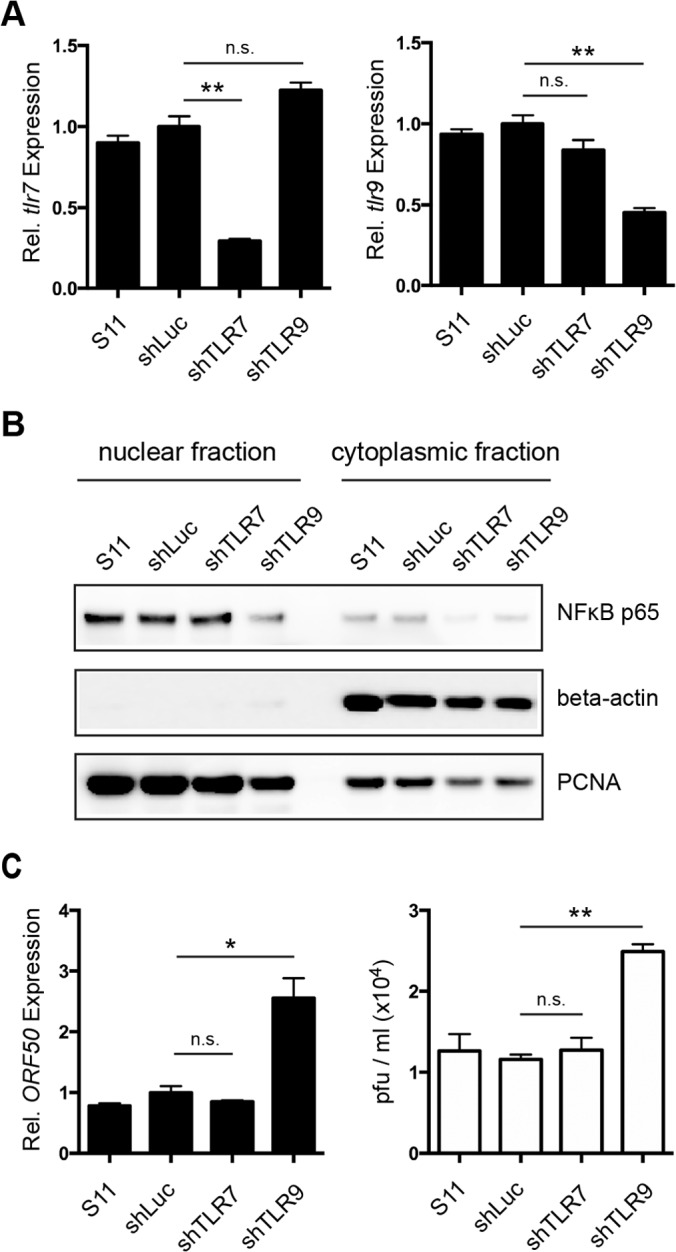
Downregulation of TLR9 but not of TLR7 leads to a decrease in nuclear NF-κB p65 levels and increased spontaneous reactivation in S11 cells. (A) Expression of TLR7 (left side) and TLR9 (right side) was analyzed by qPCR in parental S11 cells and stable cell lines expressing shRNA against luciferase (shLuc), TLR7 (shTLR7), or TLR9 (shTLR9). Expression levels were normalized to GAPDH and plotted relative to shLuc control cells. (B) NF-κB subunit p65, PCNA, and beta-actin were detected in nuclear and cytoplasmic protein fractions from parental S11 and bulk stably transfected cell lines by Western blot analysis. (C) Expression of MHV 68 lytic gene ORF50 was analyzed by qPCR in parental S11 cells and stable cell lines shLuc, shTLR7, and shTLR9 (black bars). Expression levels were normalized to GAPDH and plotted relative to shLuc control cells. Titers of infectious viral particles in the supernatant after 24 h were quantified by a plaque assay (white bars). Data shown in panels A and C are means ± SD of the results of three independent experiments. Statistics were calculated using an unpaired t test (*, P < 0.05; **, P < 0.01; n.s., P > 0.05).
Levels of NF-κB p65 in cells with reduced expression of TLR7 were similar to those in parental S11 cells or in the shLuc control cell line (Fig. 4B). In cells with reduced TLR9 expression, however, lower levels of nuclear NF-κB p65 were detected. We therefore hypothesized that latent MHV 68 infection causes a constant low-level activation of the TLR9 but not the TLR7 signaling pathway culminating in the nuclear translocation of NF-κB p65, thereby contributing to basal NF-κB activation.
Since a reduction in nuclear NF-κB p65 levels induced by the inhibitor Bay 11-7082 in previous experiments resulted in increased reactivation and MHV 68 production (Fig. 2B), we tested whether the bulk stable cell lines silenced for TLR7 or TLR9 differed in their propensities for spontaneous reactivation compared to the parental cell line. Quantification of lytic viral gene expression by qPCR and of infectious MHV 68 production by a plaque assay revealed a 2-fold increase in expression of ORF50 (Fig. 4C, black bars) and a 2-fold increase in the concentration of viral particles in the supernatant (Fig. 4C, white bars) of cell lines silenced for TLR9 but not in the supernatant of cell lines silenced for TLR7 or of the control cells expressing shRNA against luciferase.
These results suggest a contribution of continuous TLR9 signaling to basal NF-κB activation levels and suppression of spontaneous reactivation.
Lack of TLR9 does not impact establishment of latent infection in vivo but favors lytic reactivation of primary B cells ex vivo.
Since activation of the TLR7 or TLR9 pathways supports the establishment and maintenance of latent MHV 68 infection in splenocytes and since MHV 68 infection itself might promote latency by triggering TLR9, we hypothesized that lack of TLR9 would result in a reduced latent reservoir of MHV 68. Thus, we i.p. infected wild-type (wt), tlr7−/−, and tlr9−/− mice (n = 15 for each genotype) with MHV 68 as described in the experiments mentioned above. None of the mice showed signs of distress or illness throughout the experiment. All animals were sacrificed at 34 dpi.
We did not find significant differences between any of the three groups in the frequencies of infected B cells (wt, 1 in 2,600; tlr7−/−, 1 in 2,300; tlr9−/−, 1 in 1,900) by performing limiting-dilution nested PCR on purified B cells (Fig. 5A), indicating that TLR signaling is dispensable for the establishment of latency. Next, we assessed ex vivo reactivation of B cells by performing a limiting-dilution reactivation assay on NIH3T12 fibroblasts and, in parallel, tested for the presence of preformed infectious virus by plating disrupted cells (Fig. 5B). After 14 days, the numbers of reactivation events were minimal in cells from both wt and tlr7−/− mice, indicating the poor reactivation capability of latently infected B cells. In contrast, reactivation was markedly increased in cells from tlr9−/− mice, in line with our in vitro results from silencing TLR7 or TLR9 in S11 cells as well as with the findings reported by Guggemoos et al. (35), who found higher titers of lytic MHV 68 in tlr9−/− mice than in wt mice after i.p. infection. Preformed infectious virus was absent in both wt and tlr7−/− cells and barely detectable in tlr9−/− cells. Thus, it seems that impaired TLR9 signaling renders MHV 68-infected B cells more likely to reactivate whereas impaired TLR7 signaling does not.
FIG 5.

Lack of TLR7 or TLR9 does not impact establishment of infection in B cells upon i.p. injection in vivo, but tlr9−/− B cells show higher reactivation ex vivo. A total of 15 mice each of the wt, tlr7−/−, and tlr9−/− genotypes were infected with MHV 68 and sacrificed on day 34 postinfection. Splenic B cells were isolated by negative selection. (A) Splenic B cells from three mice of the same genotype were pooled and analyzed by limiting-dilution nested PCR (nPCR) as described for Fig. 3. The resulting curves (5 per genotype) were compared by the Mann-Whitney test (wt versus tlr7−/−, P = 0.89; wt versus tlr9−/−, P = 0.1; tlr7−/− versus tlr9−/−, P = 0.1). (B) Splenic B cells were plated in 2-fold dilution series on monolayers of susceptible fibroblasts. In parallel, disrupted samples were plated as controls. At 14 days after seeding, the percentage of wells showing cytopathic effect was determined microscopically and plotted against the number of B cells seeded. A nonlinear regression curve was fitted using GraphPad software.
DISCUSSION
Activation of the innate immune system via TLRs largely impacts gammaherpesvirus latency and reactivation. We here studied the effect of endosomal TLR stimulation on latently MHV 68-infected cells in vitro and on the establishment of gammaherpesvirus infection in vivo using the MHV 68 mouse model. We found that (i) signaling of TLR7 or TLR9 but not of TLR3 inhibits both spontaneous and induced reactivation in the latently MHV 68-infected S11 cells and that this effect is, at least partially, dependent on the induction of NF-κB; (ii) stimulation of TLR7 during infection increases the number of latently infected splenocytes; (iii) downregulation of TLR9 but not of TLR7 reduces basal NF-κB activity and increases spontaneous MHV 68 reactivation in latently infected cells; and (iv) while the lack of TLR7 or TLR9 does not impact the frequency of latently infected B cells in vivo, MHV 68-infected B cells from tlr9−/− mice show an increased propensity to reactivate ex vivo. Our results unprecedentedly show that activation of NF-κB via TLR7 signaling profoundly impacts the establishment and maintenance of latent MHV 68 infection, as is the case for TLR9 signaling. In the absence of external ligands, constant activation of TLR9 caused by persistent MHV 68 infection might contribute to robust activation of NF-κB that is sufficient to enable gammaherpesviral latency.
Our in vitro experiments using S11 cells as a model for established MHV 68 latency in B cells invariably showed that activation of the TLR9 pathway suppresses both spontaneous and induced MHV 68 reactivation (Fig. 2A). It seems likely that this feature is shared by other gammaherpesviruses, as previous studies in our laboratory demonstrated that stimulation of TLR9 similarly leads to a suppression of EBV's master regulator lytic BZLF1 gene in Burkitt's lymphoma cells (8, 9). Our finding that signaling via TLR7 suppresses MHV 68 reactivation is unprecedented. It seems plausible that TLR7 and TLR9 would have comparable effects on MHV68 when stimulated, since they share a signaling pathway via the common MyD88 adaptor molecule. Upon stimulation of either receptor, we observed comparable levels of activation of NF-κB (Fig. 1B). NF-κB was shown to suppress the lytic gene promoters of MHV 68 as well as of the human gammaherpesviruses EBV and KSHV (19, 20), which supports the hypothesis that downregulation of lytic MHV 68 upon TLR7 or TLR9 stimulation in S11 cells is due to activation of NF-κB. Indeed, in our experiments, NF-κB inhibitor Bay counteracted the suppression of viral reactivation by TLR ligands (Fig. 2B). Nevertheless, we cannot exclude the possibility that triggering of endosomal TLRs might activate cellular signaling pathways apart from NF-κB that might impact MHV 68 gene expression. In vitro data of Gargano et al. (6) showed that TLR3 or TLR9 triggering provoked lytic MHV 68 reactivation in the A20HE1 and A20HE2 cell lines whereas TLR7 triggering had no effect. The activation of NF-κB was not tested. Thus, differences between these cell lines in endogenous levels of NF-κB activation or TLR signaling competence might account for the divergent observations. Alternatively, the reason for the apparently contradictory observations may lie in the distinct origin of persistent MHV 68 infection. While S11 cells originate from naturally MHV 68-associated lymphoproliferation, A20HE1 and A20HE2 cell lines derive from murine lymphoma cells secondarily infected in vitro with a recombinant strain of MHV 68 (36). In a different study, increased reactivation of KSHV from primary-effusion lymphoma cells was shown upon treatment with ligands to TLR7 and -8 (7). The critical signaling pathway in those cells was shown to be dependent on the presence of IRF7, however, further demonstrating that the impact of TLR signaling on gammaherpesvirus reactivation may depend on the cellular context.
In vivo, recurrent activation of TLR7 signaling by treatment with R848 following de novo infection of wt mice led to a frequency of latently MHV 68-infected splenocytes higher than that seen with mock treatment (Fig. 3A). This observation is comparable to the findings of Gargano et al. (6), who reported an increased frequency of infected splenocytes in vivo upon stimulation of TLR9 by administration of CpG ODN. A higher frequency of cells positive for MHV 68 DNA indicates an increase in the pool of latently MHV 68-infected cells, and this idea is supported by the detection of MHV 68 latency-associated mLANA transcripts in our experiments (Fig. 3B). Failure to detect mLANA transcripts in two samples from untreated controls reflects the limit of detection of the nested PCR rather than an absence of infection, since splenocytes from all mice were positive for MHV 68 DNA (Fig. 3A) but only 5% to 10% of latently infected cells are expected to express the mLANA gene (27). Based on our observations in vitro, we hypothesize that both TLR7-induced activation and TLR9-induced activation of NF-κB promote maintenance and expansion of the latently infected B-cell pool. Notably, stimulation with TLR ligands has been shown to induce proliferation of murine B cells (37) and is known to synergize with EBV in driving proliferation of EBV-infected human B cells (38), which could explain the finding of elevated frequencies of infected cells without the need for lytic replication. The absence of preformed virus in our samples from R848-treated mice (Fig. 3C) indicates a reactivation-independent mechanism for the expansion of the latent MHV 68 reservoir rather than its occurring, e.g., through de novo infection of B cells by MHV 68 released upon TLR stimulation, as suggested by the results of in vitro experiments by Gargano and colleagues (6).
We observed a constant activation of the NF-κB pathway in the S11 model cell line, as evidenced by high basal levels of NF-κB subunit p65 in the nucleus (Fig. 4B) driving survival and proliferation (39). NF-κB levels in the nucleus were reduced by stable downregulation of TLR9 expression but not of TLR7 expression by RNA interference, which consequently led to an increased rate of spontaneous reactivation (Fig. 4C), implying a constant stimulation of TLR9 in infected S11 cells. Unlike TLR9, TLR7 did not contribute to NF-κB activation in S11 cells in the absence of external stimulation (Fig. 3B). It has been shown by various groups that TLR9 is triggered upon infection by herpesviruses (40, 41), including the gammaherpesviruses EBV (42) and KSHV (43). Many viruses have evolved strategies to benefit from NF-κB activation either directly by having NF-κB binding sites in their promoters or indirectly by using the enhanced proliferation and survival of the host cell due to NF-κB signaling to their advantage (44). For MHV 68, suppression of lytic viral genes mediated by NF-κB induction upon infection might serve as a negative-feedback loop with the potential to limit productive replication and initiate the latent state during primary infection in vivo. The importance of functional NF-κB for the establishment of MHV 68 latency in vivo has been elegantly demonstrated by Krug et al. (21), who reported severe impairment of the establishment of splenic latency by a recombinant strain of MHV 68 that expresses a constitutively active form of the NF-κB inhibitor IκBαM. Furthermore, NF-κB signaling has been shown to be an important factor for the survival of lymphoma cells latently infected with KSHV (45) and of EBV-transformed B cells in vitro (46).
In the context of de novo infection in vivo, our experiments with mice lacking either TLR7 or TLR9 did not show significant differences in the frequencies of infected splenic B cells following i.p. infection. It is possible that more-severe deficiency in TLR signaling impacts the number of latently infected cells since a 10-fold decrease in the level of infected splenocytes was reported in myd88−/− mice that lack functional signaling of most TLRs (47). While the frequencies of infected B cells in mice of different genotypes were comparable in our experiments (Fig. 5A), B cells from tlr9−/− mice showed a greater propensity to reactivate ex vivo whereas the tlr7−/− B cells did not (Fig. 5B). This is in agreement with our results obtained with S11 cells.
Taken together, our results point toward an important role of TLR7- and TLR9-mediated activation of NF-κB in promoting persistent infection of MHV 68 in B cells and show that continuous TLR9 signaling, possibly caused by persistent MHV 68 infection, might contribute to maintaining NF-κB activation during latency. Considering that we did not see a significant difference in the frequencies of infected B cells in vivo, it is conceivable that there are other systems in place contributing to NF-κB activation, for example, the retinoic-acid-inducible-gene I (RIG-I) system, which synergizes with TLR9 to induce the immune response to herpes simplex virus (40). Other gammaherpesviruses use activation of NF-κB in the host cell for their own benefit, as evidenced by EBV's binding to the CD21 cellular receptor, which induces NF-κB expression that in turn mediates activation of the viral latent gene promoter (48) or the sustained moderate activation of NF-κB reported upon infection of endothelial cells by KSHV that regulates viral gene expression (49).
Finally, the latency-promoting effect of endosomal TLR-mediated NF-κB activation reported here suggests a possible mechanism for the role of coinfections that are implicated in the pathogenesis of certain gammaherpesvirus-associated malignancies. Indeed, with respect to EBV-associated B-cell malignancies, P. falciparum malaria is epidemiologically associated with endemic BL, and the parasite was reported to directly stimulate TLR9 (11, 12). Earlier we showed that P. falciparum hemozoin suppresses lytic reactivation of EBV in BL cells (9), further underscoring activation of TLR signaling pathways as a possible mechanistic link between chronic malaria infection and the incidence of endemic BL. The situation in KSHV-associated primary effusion lymphoma, however, may be distinct, as suggested by the lytic reactivation of KSHV following triggering of TLR7/8 (7). Nevertheless, our results are important in the light of TLR ligands being increasingly used as adjuvants in the treatment of infectious diseases, cancer, or autoimmunity because of their potential to promote gammaherpesvirus latency.
ACKNOWLEDGMENTS
This work was supported by grants from the Swiss National Foundation (310040-114118), the Edoardo R., Giovanni, Giuseppe and Chiarina Sassella Foundation, the OPO Foundation, the Wolfermann-Nägeli Foundation, and the Forschungskredit of the University of Zurich, an unrestricted grant from AstraZeneca, a UBS donation by a client, a GSK International Award for Research of the Japanese Society of Immunology & Allergology in Otorhinolaryngology, and Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology and the Japan Society for Promotion of Science (research project 22591915).
Footnotes
Published ahead of print 18 June 2014
REFERENCES
- 1.Young LS, Rickinson AB. 2004. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 4:757–768. 10.1038/nrc1452 [DOI] [PubMed] [Google Scholar]
- 2.Burkitt D. 1962. A children's cancer dependent on climatic factors. Nature 194:232–234. 10.1038/194232a0 [DOI] [PubMed] [Google Scholar]
- 3.Burkitt DP. 1969. Etiology of Burkitt's lymphoma–an alternative hypothesis to a vectored virus. J. Natl. Cancer Inst. 42:19–28 [PubMed] [Google Scholar]
- 4.Speck SH, Ganem D. 2010. Viral latency and its regulation: lessons from the gamma-herpesviruses. Cell Host Microbe 8:100–115. 10.1016/j.chom.2010.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rickinson AB, Moss DJ. 1997. Human cytotoxic T lymphocyte responses to Epstein-Barr virus infection. Annu. Rev. Immunol. 15:405–431. 10.1146/annurev.immunol.15.1.405 [DOI] [PubMed] [Google Scholar]
- 6.Gargano LM, Forrest JC, Speck SH. 2009. Signaling through Toll-like receptors induces murine gammaherpesvirus 68 reactivation in vivo. J. Virol. 83:1474–1482. 10.1128/JVI.01717-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gregory SM, West JA, Dillon PJ, Hilscher C, Dittmer DP, Damania B. 2009. Toll-like receptor signaling controls reactivation of KSHV from latency. Proc. Natl. Acad. Sci. U. S. A. 106:11725–11730. 10.1073/pnas.0905316106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ladell K, Dorner M, Zauner L, Berger C, Zucol F, Bernasconi M, Niggli FK, Speck RF, Nadal D. 2007. Immune activation suppresses initiation of lytic Epstein-Barr virus infection. Cell. Microbiol. 9:2055–2069. 10.1111/j.1462-5822.2007.00937.x [DOI] [PubMed] [Google Scholar]
- 9.Zauner L, Melroe GT, Sigrist JA, Rechsteiner MP, Dorner M, Arnold M, Berger C, Bernasconi M, Schaefer BW, Speck RF, Nadal D. 2010. TLR9 triggering in Burkitt's lymphoma cell lines suppresses the EBV BZLF1 transcription via histone modification. Oncogene 29:4588–4598. 10.1038/onc.2010.203 [DOI] [PubMed] [Google Scholar]
- 10.Takeda K, Akira S. 2005. Toll-like receptors in innate immunity. Int. Immunol. 17:1–14. 10.1093/intimm/dxh186 [DOI] [PubMed] [Google Scholar]
- 11.Pichyangkul S, Yongvanitchit K, Kum-arb U, Hemmi H, Akira S, Krieg AM, Heppner DG, Stewart VA, Hasegawa H, Looareesuwan S, Shanks GD, Miller RS. 2004. Malaria blood stage parasites activate human plasmacytoid dendritic cells and murine dendritic cells through a Toll-like receptor 9-dependent pathway. J. Immunol. 172:4926–4933. 10.4049/jimmunol.172.8.4926 [DOI] [PubMed] [Google Scholar]
- 12.Coban C, Ishii KJ, Kawai T, Hemmi H, Sato S, Uematsu S, Yamamoto M, Takeuchi O, Itagaki S, Kumar N, Horii T, Akira S. 2005. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J. Exp. Med. 201:19–25. 10.1084/jem.20041836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11:373–384. 10.1038/ni.1863 [DOI] [PubMed] [Google Scholar]
- 14.Jurk M, Heil F, Vollmer J, Schetter C, Krieg AM, Wagner H, Lipford G, Bauer S. 2002. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat. Immunol. 3:499. 10.1038/ni0602-499 [DOI] [PubMed] [Google Scholar]
- 15.Demaria O, Pagni PP, Traub S, de Gassart A, Branzk N, Murphy AJ, Valenzuela DM, Yancopoulos GD, Flavell RA, Alexopoulou L. 2010. TLR8 deficiency leads to autoimmunity in mice. J. Clin. Invest. 120:3651–3662. 10.1172/JCI42081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGettrick AF, O'Neill LA. 2004. The expanding family of MyD88-like adaptors in Toll-like receptor signal transduction. Mol. Immunol. 41:577–582. 10.1016/j.molimm.2004.04.006 [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, Akira S. 2002. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J. Immunol. 169:6668–6672. 10.4049/jimmunol.169.12.6668 [DOI] [PubMed] [Google Scholar]
- 18.Bonizzi G, Karin M. 2004. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 25:280–288. 10.1016/j.it.2004.03.008 [DOI] [PubMed] [Google Scholar]
- 19.Brown HJ, Song MJ, Deng H, Wu TT, Cheng G, Sun R. 2003. NF-B inhibits gammaherpesvirus lytic replication. J. Virol. 77:8532–8540. 10.1128/JVI.77.15.8532-8540.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Izumiya Y, Izumiya C, Hsia D, Ellison TJ, Luciw PA, Kung HJ. 2009. NF-kappaB serves as a cellular sensor of Kaposi's sarcoma-associated herpesvirus latency and negatively regulates K-Rta by antagonizing the RBP-Jkappa coactivator. J. Virol. 83:4435–4446. 10.1128/JVI.01999-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krug LT, Moser JM, Dickerson SM, Speck SH. 2007. Inhibition of NF-kappaB activation in vivo impairs establishment of gammaherpesvirus latency. PLoS Pathog. 3:e11. 10.1371/journal.ppat.0030011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Virgin HW, IV, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, Speck SH. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. 71:5894–5904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clarke GD, Stoker MG, Ludlow A, Thornton M. 1970. Requirement of serum for DNA synthesis in BHK 21 cells: effects of density, suspension and virus transformation. Nature 227:798–801. 10.1038/227798a0 [DOI] [PubMed] [Google Scholar]
- 24.Efstathiou S, Ho YM, Minson AC. 1990. Cloning and molecular characterization of the murine herpesvirus 68 genome. J. Gen. Virol. 71(Pt 6):1355–1364. 10.1099/0022-1317-71-6-1355 [DOI] [PubMed] [Google Scholar]
- 25.Sunil-Chandra NP, Efstathiou S, Arno J, Nash AA. 1992. Virological and pathological features of mice infected with murine gamma-herpesvirus 68. J. Gen. Virol. 73(Pt 9):2347–2356. 10.1099/0022-1317-73-9-2347 [DOI] [PubMed] [Google Scholar]
- 26.Edwards AD, Diebold SS, Slack EM, Tomizawa H, Hemmi H, Kaisho T, Akira S, Reis e Sousa C. 2003. Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 alpha+ DC correlates with unresponsiveness to imidazoquinolines. Eur. J. Immunol. 33:827–833. 10.1002/eji.200323797 [DOI] [PubMed] [Google Scholar]
- 27.Allen RD, III, Dickerson S, Speck SH. 2006. Identification of spliced gammaherpesvirus 68 LANA and v-cyclin transcripts and analysis of their expression in vivo during latent infection. J. Virol. 80:2055–2062. 10.1128/JVI.80.4.2055-2062.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baenziger S, Heikenwalder M, Johansen P, Schlaepfer E, Hofer U, Miller RC, Diemand S, Honda K, Kundig TM, Aguzzi A, Speck RF. 2009. Triggering TLR7 in mice induces immune activation and lymphoid system disruption, resembling HIV-mediated pathology. Blood 113:377–388. 10.1182/blood-2008-04-151712 [DOI] [PubMed] [Google Scholar]
- 29.Tibbetts SA, McClellan JS, Gangappa S, Speck SH, Virgin HW., IV 2003. Effective vaccination against long-term gammaherpesvirus latency. J. Virol. 77:2522–2529. 10.1128/JVI.77.4.2522-2529.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Usherwood EJ, Stewart JP, Nash AA. 1996. Characterization of tumor cell lines derived from murine gammaherpesvirus-68-infected mice. J. Virol. 70:6516–6518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu TT, Usherwood EJ, Stewart JP, Nash AA, Sun R. 2000. Rta of murine gammaherpesvirus 68 reactivates the complete lytic cycle from latency. J. Virol. 74:3659–3667. 10.1128/JVI.74.8.3659-3667.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez-Guzman D, Rickabaugh T, Wu TT, Brown H, Cole S, Song MJ, Tong L, Sun R. 2003. Transcription program of murine gammaherpesvirus 68. J. Virol. 77:10488–10503. 10.1128/JVI.77.19.10488-10503.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grossmann C, Ganem D. 2008. Effects of NFkappaB activation on KSHV latency and lytic reactivation are complex and context-dependent. Virology 375:94–102. 10.1016/j.virol.2007.12.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weck KE, Barkon ML, Yoo LI, Speck SH, Virgin HW., IV 1996. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J. Virol. 70:6775–6780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guggemoos S, Hangel D, Hamm S, Heit A, Bauer S, Adler H. 2008. TLR9 contributes to antiviral immunity during gammaherpesvirus infection. J. Immunol. 180:438–443. 10.4049/jimmunol.180.1.438 [DOI] [PubMed] [Google Scholar]
- 36.Forrest JC, Speck SH. 2008. Establishment of B-cell lines latently infected with reactivation-competent murine gammaherpesvirus 68 provides evidence for viral alteration of a DNA damage-signaling cascade. J. Virol. 82:7688–7699. 10.1128/JVI.02689-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gururajan M, Jacob J, Pulendran B. 2007. Toll-like receptor expression and responsiveness of distinct murine splenic and mucosal B-cell subsets. PLoS One 2:e863. 10.1371/journal.pone.0000863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iskra S, Kalla M, Delecluse HJ, Hammerschmidt W, Moosmann A. 2010. Toll-like receptor agonists synergistically increase proliferation and activation of B cells by Epstein-Barr virus. J. Virol. 84:3612–3623. 10.1128/JVI.01400-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karin M, Lin A. 2002. NF-kappaB at the crossroads of life and death. Nat. Immunol. 3:221–227. 10.1038/ni0302-221 [DOI] [PubMed] [Google Scholar]
- 40.Rasmussen SB, Jensen SB, Nielsen C, Quartin E, Kato H, Chen ZJ, Silverman RH, Akira S, Paludan SR. 2009. Herpes simplex virus infection is sensed by both Toll-like receptors and retinoic acid-inducible gene-like receptors, which synergize to induce type I interferon production. J. Gen. Virol. 90:74–78. 10.1099/vir.0.005389-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sato A, Linehan MM, Iwasaki A. 2006. Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc. Natl. Acad. Sci. U. S. A. 103:17343–17348. 10.1073/pnas.0605102103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fiola S, Gosselin D, Takada K, Gosselin J. 2010. TLR9 contributes to the recognition of EBV by primary monocytes and plasmacytoid dendritic cells. J. Immunol. 185:3620–3631. 10.4049/jimmunol.0903736 [DOI] [PubMed] [Google Scholar]
- 43.West JA, Gregory SM, Damania B. 2012. Toll-like receptor sensing of human herpesvirus infection. Front. Cell. Infect. Microbiol. 2:122. 10.3389/fcimb.2012.00122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Santoro MG, Rossi A, Amici C. 2003. NF-kappaB and virus infection: who controls whom. EMBO J. 22:2552–2560. 10.1093/emboj/cdg267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keller SA, Schattner EJ, Cesarman E. 2000. Inhibition of NF-kappaB induces apoptosis of KSHV-infected primary effusion lymphoma cells. Blood 96:2537–2542 [PubMed] [Google Scholar]
- 46.Cahir-McFarland ED, Davidson DM, Schauer SL, Duong J, Kieff E. 2000. NF-kappa B inhibition causes spontaneous apoptosis in Epstein-Barr virus-transformed lymphoblastoid cells. Proc. Natl. Acad. Sci. U. S. A. 97:6055–6060. 10.1073/pnas.100119497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gargano LM, Moser JM, Speck SH. 2008. Role for MyD88 signaling in murine gammaherpesvirus 68 latency. J. Virol. 82:3853–3863. 10.1128/JVI.02577-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sugano N, Chen W, Roberts ML, Cooper NR. 1997. Epstein-Barr virus binding to CD21 activates the initial viral promoter via NF-kappaB induction. J. Exp. Med. 186:731–737. 10.1084/jem.186.5.731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sadagopan S, Sharma-Walia N, Veettil MV, Raghu H, Sivakumar R, Bottero V, Chandran B. 2007. Kaposi's sarcoma-associated herpesvirus induces sustained NF-kappaB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J. Virol. 81:3949–3968. 10.1128/JVI.02333-06 [DOI] [PMC free article] [PubMed] [Google Scholar]