

ABSTRACT
Helper T-cell epitope dominance in human immunodeficiency virus type 1 (HIV-1) envelope glycoprotein gp120 is not adequately explained by peptide binding to major histocompatibility complex (MHC) proteins. Antigen processing potentially influences epitope dominance, but few, if any, studies have attempted to reconcile the influences of antigen processing and MHC protein binding for all helper T-cell epitopes of an antigen. Epitopes of gp120 identified in both humans and mice occur on the C-terminal flanks of flexible segments that are likely to be proteolytic cleavage sites. In this study, the influence of gp120 conformation on the dominance pattern in gp120 from HIV strain 89.6 was examined in CBA mice, whose MHC class II protein has one of the most well defined peptide-binding preferences. Only one of six dominant epitopes contained the most conserved element of the I-Ak binding motif, an aspartic acid. Destabilization of the gp120 conformation by deletion of single disulfide bonds preferentially enhanced responses to the cryptic I-Ak motif-containing sequences, as reported by T-cell proliferation or cytokine secretion. Conversely, inclusion of CpG in the adjuvant with gp120 enhanced responses to the dominant CD4+ T-cell epitopes. The gp120 destabilization affected secretion of some cytokines more than others, suggesting that antigen conformation could modulate T-cell functions through mechanisms of antigen processing.
IMPORTANCE CD4+ helper T cells play an essential role in protection against HIV and other pathogens. Thus, the sites of helper T-cell recognition, the dominant epitopes, are targets for vaccine design; and the corresponding T cells may provide markers for monitoring infection and immunity. However, T-cell epitopes are difficult to identify and predict. It is also unclear whether CD4+ T cells specific for one epitope are more protective than T cells specific for other epitopes. This work shows that the three-dimensional (3D) structure of an HIV protein partially determines which epitopes are dominant, most likely by controlling the breakdown of HIV into peptides. Moreover, some types of signals from CD4+ T cells are affected by the HIV protein 3D structure; and thus the protectiveness of a particular peptide vaccine could be related to its location in the 3D structure.
INTRODUCTION
The specificity and phenotype of CD4+ T-helper responses are likely to have a significant influence on the protectiveness of an immune response. In one outstanding example, mucosal immunization of mice with a peptide containing the immunodominant CD4+ T-cell epitope of the rotavirus VP6 protein was sufficient to protect against infection (1). In monkeys, CD4+ T-cell responses to essentially the same epitope were associated with control of natural infection (2). Protection of mice against rotavirus evidently can be mediated solely by CD4+ T cells because neither B cells nor CD8+ T cells are required (3). For simian immunodeficiency virus (SIV) and human immunodeficiency virus (HIV) infections, CD4+ responses are associated with protection from disease or viremia. In monkeys, CD4+ responses correlated with protection against SIV (4, 5), and the vaccinated subjects of a Thai phase III clinical trial (RV144) developed CD4+ responses against HIV Env in addition to nonneutralizing antibodies (6). CD4+ T cells could protect by providing help to B cells or CD8+ T cells and/or by direct action against the virus-infected cells. An association of CD4+ T cells with low viremia in HIV+ persons has been attributed to direct killing (7).
The specificity of CD4+ T-cell responses against a pathogen is often dominated by a small number of epitopes. CD4+ epitope dominance could be a disadvantage against HIV because the breadth of epitopes has been associated with low viremia (8). The breadth of epitopes has also been correlated with the resolution of acute hepatitis C virus infection (9). In the absence of broad responses, the specificity of a dominant CD4+ response could play a crucial role. Whereas low viremia was associated with CD4+ epitopes in HIV Gag, high viremia was associated with a CD4+ epitope in Env (8). CD4+ responses could aggravate disease if the proliferating cells provide targets for HIV infection (10). Although HLA-DRB1-restricted responses were weakly associated with control of HIV, dominant CD4+ epitopes were promiscuously presented by multiple major histocompatibility complex class II (MHC-II) alleles (11). Therefore, strategies for predicting and manipulating CD4+ responses are urgently needed.
Priming and recall of CD4+ T cell epitopes depend on multiple molecular events, including uptake of the antigen into an antigen-presenting cell (APC), proteolytic antigen processing, loading of antigen fragments into the MHC-II antigen-presenting protein, trafficking of the peptide-MHC complex to the cell surface, and recognition of the peptide-MHC complex by the T-cell receptor (TCR) of the CD4+ T cell (12–15). The abundance of specific T cells in the naive or memory populations potentially influences the probability of the recognition step because T cells of particular specificities may be represented at widely different levels (16). Dominance of particular epitopes within an antigen has been long recognized, but mechanisms controlling epitope dominance have not been resolved. Although peptide affinity for the MHC protein is an important factor, it is not well correlated with epitope dominance (17–20).
In HIV gp120, CD4+ epitopes cluster adjacent to the variable loops, a pattern that has been identified in gp120-immunized BALB/c and CBA mice, as well as in HIV-infected persons (21–23). Since this dominance pattern occurs in the context of multiple MHC-II alleles, we attributed it to mechanisms of antigen processing. We have proposed that the proteolytic cleavage of locally disordered antigen segments (typically in variable loops) causes the presentation to T cells of the adjacent epitope-containing segments (22).
The dependence of peptide presentation on proteolytic antigen processing has long been appreciated (24). Several studies have demonstrated that antigen processing at key proteolytic sites can modulate presentation of nearby epitopes (25, 26). However, the responsible protease activities are poorly defined and generally thought to be redundant. In a study on the natural processing of birch allergen Bet v 1, a fragment containing a promiscuous, dominant epitope emerged at early time points of proteolysis with endosomal proteases, suggesting that the initial cleavage at an exposed loop of the intact antigen triggered loading of the C-terminal fragment into the MHC protein (27). These results emphasize the contribution of antigen conformation as it directs the pathway of antigen processing.
Disulfide bonds potentially interfere with antigen processing because they stabilize protein structure and limit the access of proteases and antigen-presenting proteins. Cresswell and coworkers identified an enzyme, gamma interferon (IFN-γ) lysosomal thiol reductase (GILT), that is present in lysosomes and is responsible for cleaving disulfide bonds during antigen processing. GILT knockout mice exhibited a diminished response to two disulfide-rich allergens, hen egg lysozyme and dust mite allergen Der p 1 (28, 29). These results indicate that disulfide bonds can limit T-cell responses, but it remains unclear whether disulfide bonds influence T-cell responses in individuals that have normal GILT function. Following immunization of BALB/c mice, we noted a distinct absence of T-cell responses from regions of HIV gp120 that are enriched in disulfide bonds, but elimination of individual disulfide bonds caused broad reductions in the T-cell response, rather than local increases (30). We attributed the reductions to proteolytic destruction of the antigen, which reduced the amount of antigen fragments available for presentation.
Protease resistance correlates with protein stability because the folded polypeptide does not have sufficient flexibility to conform to the protease active site (31, 32). The contributions by individual disulfide bonds to stability and protease resistance are highly variable, depending on the length of chain between bonded cysteines and the presence of noncovalent interactions that stabilize local conformation. Typically, the elimination of a disulfide bond reduces local or domain-wise conformational stability but does not substantially change the overall conformation (33–36).
Disulfide bond deletions have diverse and complex effects on HIV gp120 processing and CD4+ T-cell epitope dominance. Nine disulfide bonds are almost perfectly conserved in gp120, suggesting that they are essential to the folding or function of the protein. Three disulfides in the outer domain (connecting cysteines 298 to 331, 378 to 441, and 385 to 414) form a nexus from which emerge the V3 loop, V4 loop, and a loop that forms part the bridging sheet. van Anken and coworkers investigated the consequences for folding, assembly, and secretion of HIV envelope trimers caused by elimination of individual disulfide bonds (37). Elimination of the 298-to-331 or 385-to-441 disulfides caused severe defects in folding, assembly, or export of the proteins, whereas elimination of the 378-to-441 disulfide had no consequence for expression or function. We found that elimination of any one of these three disulfides affected the conformation of gp120 and reduced the T-cell responses in BALB/c mice (30). Deletion of the 298-to-331 and 385-to-414 disulfides caused severe and moderate destabilization of gp120, respectively, resulting in globally reduced T-cell responses. Deletion of the 378-to-441 disulfide also destabilized the structure and reduced T-cell responses, but the effect was localized to the gp120 outer domain.
This study continues to probe the causes of CD4+ T-cell epitope dominance by monitoring changes in the T-cell epitope map caused by the disulfide deletions in gp120, except now in CBA mice and with analysis of a suite of T-cell cytokines in addition to proliferation. The standard mucosal immunization of CBA mice with gp120 elicited dominant responses for six epitopes. Although the I-Ak molecule has a highly restricted peptide specificity, all but one immunodominant epitope lacked an aspartic acid for the P1 anchor position of the I-Ak-binding motif. Dominance of the epitopes may be attributed to enhanced yield from antigen processing because each epitope lies adjacent to a highly flexible loop that is likely to serve as an entry point for proteolytic antigen processing. Mutations that eliminated a disulfide bond and destabilized the structure of gp120 increased the intensity of overall T-cell responses. Remarkably, the largest increases in T-cell response were localized to sequences predicted to bind most strongly to the I-Ak molecule, including some sequences that were essentially nonimmunogenic in the wild-type protein. Thus, the disulfide deletions revealed epitopes that had been concealed by the gp120 structure. In contrast, when the immunization was modified by inclusion of CpG, the increase in IFN-γ response followed the dominance pattern observed for the standard immunization.
MATERIALS AND METHODS
Plasmids, proteins, and peptides.
The gp120 DNA from HIV1 strain 89.6 was cloned into the pFastBac-1 vector as previously described (22). Expression and preparation of His6-tagged gp120 protein and its disulfide variants from HIV-1 strain 89.6 were the same as reported previously (30). The 38 peptides (20-mers overlapping by 10 residues) spanning residues 40 to 109 and 180 to 508 of the HIV 89.6 ENV gp120 sequence were synthesized as the carboxy amidomethyl derivatives and aliquoted onto microtiter plates by JPT Peptide Technologies GmbH. For the T-cell proliferation and cytokine assays, peptides were dissolved in cell culture medium, i.e., RPMI 1640 (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen), 2 mM l-glutamine (Invitrogen), 100 IU/ml penicillin, and 100 μg/ml streptomycin.
Immunization.
Six- to 8-week-old female CBA (H-2k) mice (Jackson Laboratory) were immunized intranasally with 20 μg of wild-type or variant gp120 plus 5 μg of mutant (R192G) heat-labile toxin (mLT; kindly provided by John Clements, Tulane University Health Science Center) and/or 10 μg of type C CpG oligonucleotide–human/murine Toll-like receptor 9 (TLR9) ligand (InvivoGen) as an adjuvant in a total volume of 10 μl. The mice received 2 boosts of the same mixture and by the same route at 2-week intervals. One week after the last boost, the mice were euthanized and cardiac blood and spleen were collected from each mouse. All procedures involving animals were IACUC approved under protocol number 3059R2.
T-cell preparation.
Mouse splenocytes were isolated as previously described (22). Briefly, spleens were homogenized in GentleMacs C tubes (Miltenyi Biotec) and dissociated with a GentleMacs dissociator (Miltenyi Biotec) and then filtered through a 40-μm cell strainer (Falcon BD). The cells were pelleted by centrifugation, and red blood cells were lysed using the RBC lysing buffer (Sigma) as per the manufacturer's instructions. The cells were centrifuged and resuspended in cell culture medium. After resuspension, the cells were immediately plated on a 96-well plate in the absence or presence of 2 μg/ml single gp120 peptide or 10 μg/ml gp120 protein in a final volume of 200 μl. Cell density was fixed at 4 × 105 cells per well. Splenocytes were cultured at 37°C in a 5% CO2 atmosphere.
Cytokine secretion and T-cell proliferation.
For assays of cytokine secretion, 115 μl of supernatant was skimmed from each well of the T-cell culture plates after 3 days of culture. The cytokine concentration in the supernatant was measured using a bead-based cytokine assay with kits from Millipore according to the manufacturer's instructions and reported as the logarithm of the raw fluorescence signal, log(Fcytokine). In order to remove plate-to-plate variation in the analysis, all values for a given mouse were normalized to the lowest value observed for peptide-stimulated assays in that mouse, which was set to a value of 1, the approximate sensitivity of the assay in pg/ml. A response was considered positive if the value from each stimulated condition was greater than 2 standard deviations above the average value from the unstimulated condition for all animals in the same group.
For T-cell proliferation assays, 115 μl of fresh cell culture medium containing 1 μCi of tritiated thymidine was added into each well after skimming off the supernatant. The cells were continuously cultured at 37°C for another 18 h in a 5% CO2 atmosphere and harvested onto FilterMAT glass fiber papers (Skatron Instruments) using a cell harvester. Cell proliferation was measured by the incorporation of tritiated thymidine into cellular DNA using a scintillation counter. A response was considered positive if the value was greater than 2 standard deviations above the value obtained without any peptide or protein stimulation (average of six wells for each mouse).
Titration of serum IgG2a and IgG1 antibodies by ELISA.
Serum IgG2a and IgG1 levels were measured against HIV gp120 by enzyme-linked immunosorbent assay (ELISA). Plates were directly coated with gp120 from HIV-1 strain 89.6 at the final concentration of 2.5 μg/ml in phosphate-buffered saline (PBS) in a final volume of 100 μl and incubated overnight. After washing 5 times, plates were blocked with PBS containing 0.5% (vol/vol) Tween 20, 4% (wt/vol) whey, and 10% (vol/vol) fetal bovine serum (blocking buffer) in a final volume of 200 μl for 30 min. After the blocking buffer was removed, plates were incubated for 1 h with a series of blocking-buffer-diluted sera from immunized mice, followed by washing 5 times. Then, the plates were incubated with alkaline phosphatase-labeled anti-mouse IgG1 or anti-mouse IgG2a (Sigma-Aldrich) at a 1:2,000 dilution in the blocking buffer for 1 h, followed by washing 5 times. Development was initiated by addition of 100 μl of 1-mg/ml p-nitrophenyl phosphate (catalog number N2765; Sigma-Aldrich) in 0.1 M ethanolamine, and the reaction was stopped by addition of 50 μl of 2 M NaOH. Absorbance was measured at 405 nm.
Correlations of epitope frequencies to antigen flexibility and statistical analysis.
In order to model preferential cleavage, we developed a simple measure of conformational stability that provides a means to score the epitope likelihood of a given antigen peptide. Our measure is based on a combination of four sources of structural information: b-factors, solvent-accessible surface area, the COREX residue stability score based on the X-ray structure 3JWO (38), and the sequence entropy based on an alignment of 51 gp120s representing 4 clades of HIV-1. The solvent-accessible surface area was calculated using MOLMOL (39). The COREX residue stability was generated with the Web-based tool (40, 41). The Shannon sequence entropy was calculated using BioEdit (42). To combine these disparate sources of information, we take a statistical approach by computing a normalized Z-score for each source of data. Thus, each amino acid in the antigen sequence has four individual Z-scores that provide a measure of conformational stability at that position in the antigen. To combine these Z-scores, we use Stouffer's formula:
| (1) |
This method interprets each Z-score as a test of a single underlying hypothesis (i.e., that an amino acid is in a stable region); for our data, we used a k value of 4. Stouffer's method is analogous to computing Fisher's test statistic from a set of P values collected for tests of a single hypothesis.
Once we have computed this aggregate measure of stability, we use it to construct our scoring profile for the likelihood that the residue occurs in the protease-resistant region of a proteolytic fragment (Lfrag). First, we apply a threshold to identify stable and unstable regions of the antigen sequence. For the results reported here, we simply characterize any amino acid with a negative aggregate Z-score to be “unstable” and set the initial epitope likelihood to zero. For amino acids scoring above the threshold, we use the aggregate Z-score as the Lfrag.
Once the initial Lfrag is computed, we modify the score to incorporate our hypothesis that cleavage occurs in the middle of unstable regions and that epitopes are more likely to occur near the N-terminal end of a C-terminal proteolytic fragment. Prior to a single endoproteolytic cleavage, the epitope sequence is most probably located between the midpoint of a flexible segment and the midpoint of a stable segment. In a collection of nine antigens/allergens, the average length of stable segments was 48 amino acid residues (43), and thus the midpoint of stability occurs an average of 24 residues from the midpoint of flexibility. The probability that a given residue occurs at the N terminus of a C-terminal fragment (LC-frag) was calculated as follows. First, for each stable region, we multiply Lfrag for the N-terminal 24 residues by a factor of 3 and divide Lfrag for the C-terminal 24 residues by a factor of 3. Then, for each unstable region, we linearly raise the score from zero at the midpoint of the unstable region to the value at residue 12 of the first C-terminal stable segment (whose score has already been scaled up 3-fold).
RESULTS
T-cell epitope immunogenicity was examined following the immunization of groups of nine CBA mice with wild-type or a disulfide variant gp120 from HIV strain 89.6. The three variants containing substitutions of paired cysteines for alanines (Fig. 1A) were previously shown to be more sensitive to proteolysis by trypsin and cathepsin S; and they exhibited reduced binding to CD4 and conformation-dependent monoclonal antibodies (30). The purified protein was administered intranasally three times over 4 weeks with mutant (R192G) heat-labile toxin (mLT) from Escherichia coli as an adjuvant (44). This route and frequency were utilized in order to mimic HIV infection, which induces both cellular and humoral responses. One week after the third administration, T-cell responses of individual mice were analyzed by splenocyte restimulation with individual 20-mers that spanned the length of gp120, except for the segment V1-V2 (corresponding to peptides 8 to 15), which contained none of the immunodominant CD4+ epitopes previously observed in CBA mice (22). After 2 days of culture, a sample of the culture medium was tested for the presence of 8 cytokines, and radioactive thymidine was added for analysis of proliferation after another 18 h of culture. Cytokine secretion was analyzed in terms of the logarithm of the bead-based fluorescence signal, log(Fcytokine), in order to minimize data manipulation prior to statistical analysis. Cell proliferation was analyzed as the log incorporation of radioactivity after subtraction of background incorporation. Antibody responses were analyzed for their titer against gp120 and reactivity against individual peptides.
FIG 1.

HIV gp120 structure and epitope dominance. (A) The line graph indicates the number of mice responding to a given peptide. The area graph indicates the likelihood that the peptide contains the N-terminal end of a C-terminal proteolytic fragment (LC-frag). The positions of flexible loops and disulfide bonds are indicated above the graph. Disulfide bonds that were individually deleted in gp120 variants are indicated with heavy brackets, under which the position of the N-terminal cysteine is given. The bar graph indicates the I-Ak peptide-binding motif score (see the text). The six dominant epitopes were positive for an IL-2 response in three or more mice. Each dominant epitope lies on the C-terminal flank of a conformationally unstable loop. Peptide 30 was the only peptide that contained both a dominant epitope and an I-Ak-binding motif. (B) Relationship between conformational stability and LC-frag in the V3 loop and neighboring sequence positions. The aggregate conformational stability score is shown by the thin line; any residue with a score above zero is considered to be part of a stable segment. In the stable segments, LC-frag (thick line) is equal to the aggregate conformational stability with weighting to emphasize epitope probability at the N termini, such that the conformational stability score is upweighted 3-fold for the N-terminal 24 residues of the stable segment and downweighted 3-fold for the C-terminal 24 residues. Additionally, the flexible region preceding the stable segment is increased linearly from zero at the midpoint of the flexible segment to the upweighted peak of the stable segment. (C) Ribbon diagrams of gp120 illustrating positions of the conformationally unstable segments in gp120.
Following immunization with wild-type gp120, six peptides (6, 19, 30, 32, 39, and 47) stimulated a group-wise significant T-cell response, as reported by the median level of at least five cytokines in the group of nine mice using the Wilcoxon signed-rank test. These six peptides also most frequently stimulated a significant response in individual mice, where the stimulated level of cytokine exceeded the average unstimulated level by 2 standard deviations. For example, when interleukin-2 (IL-2) was the analyte, each of the six group-wise significant peptides stimulated a significant response for at least three mice (Fig. 1A). No other peptides stimulated significant responses in as many as three mice. Thus, these six peptides were designated dominant T-cell epitopes for the wild-type gp120. Although the T-cell responses to these peptides cannot be wholly attributed to CD4+ T-helper cells, this cell type is very likely responsible for the bulk of it. Two types of response that are strongly associated with CD4+ T-helper cells, IL-2 secretion and splenocyte proliferation, were well correlated for the dominant peptides (average r, 0.85 in gp120-immunized mice). Any lack of precision in the analysis due to contributions from other cell types was justified by the ability to survey a comprehensive sample of peptides using cells from individual mice.
CD4+ T-cell epitope dominance controlled by gp120 structure.
In order to understand the origin of the dominance pattern, epitopes were predicted on the basis of MHC-II protein (I-Ak) binding and on the basis of conformationally flexible antigen segments that may serve as proteolytic processing sites.
Existing software tools (e.g., NetMHCII) utilize machine learning methods for predicting peptide binding affinity (45). However, none of these tools provides a prediction for the I-Ak allele because the available experimental data for training the prediction algorithms are limited. For example, the Immune Epitope Database (IEDB) currently lists only 513 measurements of T-cell response and 305 measurements of peptide binding for I-Ak, compared to 3,827 measurements of T-cell response and 1,034 measurements of peptide binding for I-Ab (46). Nevertheless, the selectivity of peptide binding to the I-Ak molecule has been thoroughly analyzed on the basis of the X-ray crystal structure of an I-Ak-peptide complex (47). Pockets in the peptide-binding groove for P1, P4, P6, and P9 residues of the peptide interact most favorably with certain types of amino acid residue (48). The P1 residue is the most restricted, almost always being aspartic acid. For the present work, a peptide was identified as containing a minimal I-Ak-binding motif if an Asp residue appeared at least nine residues from the C terminus of the peptide. The minimal I-Ak-binding motif was validated by scoring a nonredundant set of peptides designated I-Ak binding (116 peptides) and I-Ak nonbinding (107 peptides) in the IEDB. Peptides found to have the minimal I-Ak-binding motif were associated with observed I-Ak binding (odds ratio, 3.2; P < 0.0001).
Fourteen gp120 peptides were identified as I-Ak+ peptides because they contained a minimal I-Ak-binding motif (see Table S1 in the supplemental material). Of the six immunodominant peptides in CBA mice, only peptide 30 was among the I-Ak+ peptides (Fig. 1A). Although this method of predicting MHC binding is simplistic, we hypothesized that the strong preference for Asp at P1 would fail to identify dominant epitopes only if mechanisms of antigen processing or presentation interfered with binding or availability of the peptide.
Posttranslational modifications cannot account for the failure to detect CD4+ T-cell responses to I-Ak+ peptides. Some of the I-Ak motifs in gp120 are covalently modified by glycosylation or disulfide bond formation. If the modifications remained present on the processed peptides, the primed T cells would recognize only the modified peptides. However, only four of the I-Ak motifs in gp120 peptides contain asparagine residues that are glycosylated (49). Glycans are added to asparagine residues within the I-Ak motifs of peptides 16, 25, 30, and 43. Moreover, T cells could be restimulated by two of the unmodified peptides. Peptide 30 was dominant, and peptide 16 was immunogenic in one of the variants. Neither can the presence of cysteine in the I-Ak-binding motif account for a failure to immunize. Only peptides 30 and 38 contain cysteine within the I-Ak motif. Peptide 30 was dominant, and peptide 38 raised a response detected by IFN-γ.
The potential influence of the gp120 conformation on peptide immunogenicity was quantified as the likelihood of the peptide occurring at the N-terminal end of a C-terminal proteolytic fragment (LC-frag). We reasoned that conformationally unstable, protease-sensitive sites in the soluble antigen would be most faithfully identified by a combination of parameters that report conformational fluctuations on the basis of different physical principles. A similar approach was previously implemented for prediction of proteolytic sensitivity, although that tool used a somewhat different set of parameters for conformational flexibility (32). In the present work, an instability score was generated for each residue by a combination of the crystallographic b-factor, solvent-exposed surface area, COREX residue stability, and Shannon sequence entropy (see Materials and Methods). Peptides were then assigned zero LC-frag at the middle of a flexible segment and maximum LC-frag within the first 24 amino acid residues of the C-terminally adjacent conformationally stable segment (Fig. 1B).
As previously reported, immunodominant peptides tended to occur on the C-terminal flanks of unstable segments, characterized by high values of LC-frag (Fig. 1A and C). When evaluated by the area under the curve (AUC) of the receiver-operator characteristic, the accuracy of LC-frag for prediction of epitopes was 0.65. The contribution to accuracy by the individual flexibility parameters was examined by determination of AUC values: b-factor, 0.69; solvent-accessible area, 0.75; COREX residue stability, 0.75; and sequence entropy, 0.37. Five of six dominant peptides were associated with a peak of LC-frag, and four of five peaks of LC-frag were associated with a dominant peptide. Thus, LC-frag correlated with CD4+ epitope immunogenicity in gp120. A general implementation of this approach to epitope prediction is under development and will be reported elsewhere.
Disulfide deletions in gp120 enhanced T-cell responses for I-Ak+ peptides.
Disulfide deletion variants of gp120 generally elicited stronger T-cell responses than the wild-type gp120 in CBA mice. For gp120dss298 and gp120dss385, average secretion of IFN-γ, tumor necrosis factor alpha (TNF-α), IL-4, IL-6, and IL-13 was increased (Fig. 2A). For gp120dss378, secretion of only IL-4 was increased relative to wild-type gp120.
FIG 2.

Enhanced T-cell responses for disulfide variants of gp120. (A) Each symbol is the average response to 38 individually tested peptides for splenocytes of a single mouse. Bars and asterisks indicate significant differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001). (B) Average peptide-specific IL-4 responses following immunization of nine mice with wild-type gp120 and disulfide variants. (C) Differences in average secreted IL-4 for disulfide variants versus wild-type gp120. The dashed horizontal line indicates the average difference for all peptides. Bar graphs indicate the I-Ak motif score. I-Ak+ peptides were associated with above-average differences in IL-4 secretion, i.e., were preferentially affected in gp120dss298 and gp120dss385. For the most destabilized disulfide variant, gp120dss298, 12 of 14 I-Ak+ peptides were preferentially affected, and the two exceptions are peptides 30 and 38, which are located in flexible loops.
For all three disulfide variants, the breadth of responding epitopes was increased. The variants primed group-wise significant responses from 8 to 11 peptides in addition to the six peptides that were dominant for wild-type gp120, whereas the wild-type gp120 elicited significant responses from only 3 additional peptides (Table 1). Newly group-wise significant epitopes were observed most frequently with IL-5 (gp120dss298 and gp120dss378) or TNF-α (gp120dss385). In the case of TNF-α, the new epitopes were associated with a larger average response across many epitopes, whereas in the case of IL-5, the increase in response was smaller and more localized (see Fig. S1 in the supplemental material). Some of the additional peptides were I-Ak+. No new group-wise significant epitopes were distinguishable in the IFN-γ and IL-4 responses despite an overall increase in secretion of these cytokines. Although T-cell responses were broadly enhanced for the disulfide variants, the majority of group-wise significant T-cell responses remained on the six dominant epitopes identified for the wild-type gp120.
TABLE 1.
Group-wise significant cytokine responsesd
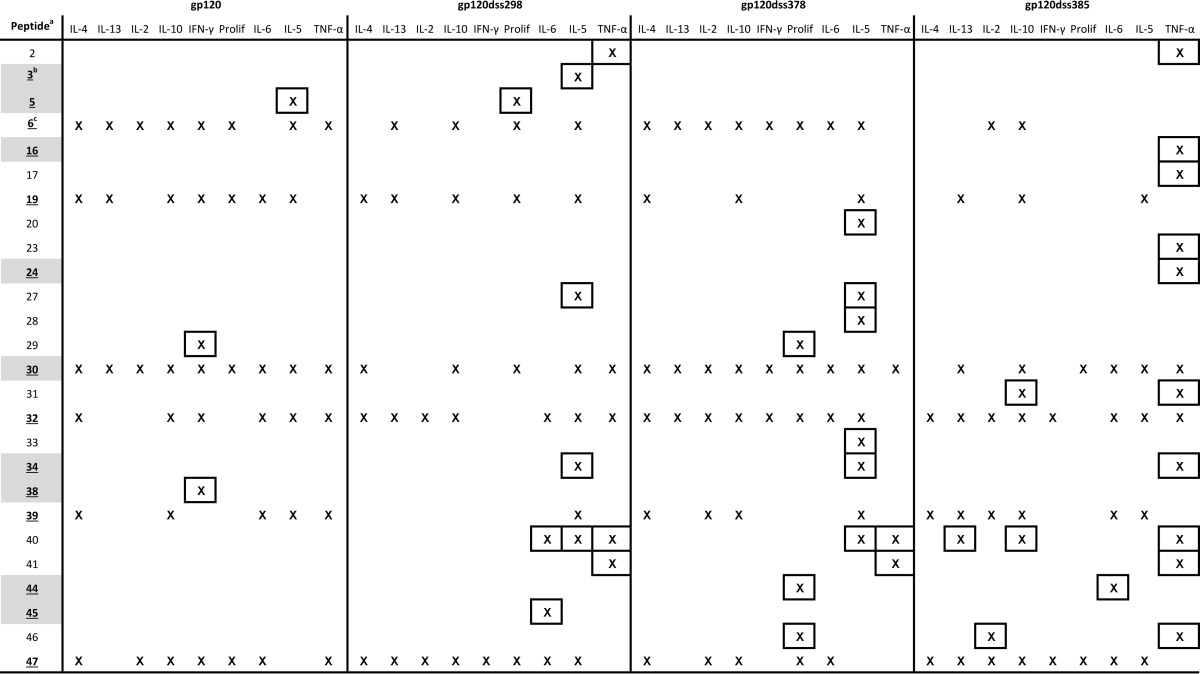
Listed peptides stimulated at least one response. The tested peptides spanned ranges of 2 to 7 and 16 to 47.
Gray highlighting indicates peptides that contain an I-Ak-binding motif.
Underlined numbers and gray highlighting indicate peptides that were dominant in wild-type gp120. Only peptide 30 was both I-Ak+ and dominant.
Boxed responses indicate peptides that were not dominant in wild-type gp120.
Increased T-cell responses for the gp120 disulfide variants were disproportionally localized to I-Ak+ peptides. Changes in the immunogenicity of individual epitopes caused by disulfide deletions were identified by examining the differences in average cytokine secretion between mouse groups. For some cytokines, the average response across all peptides was elevated (Fig. 2A and B; see also Fig. S1 in the supplemental material). This was potentially due to increased immune activation or an enhanced uptake of the antigen, rather than due to a change in antigen processing or presentation. In order to focus on antigen processing or presentation, epitopes that were differentially affected by the disulfide deletions were identified by above-average increases in cytokine secretion. Peptides were scored for differential effect and I-Ak+, and the results were analyzed with a contingency table. I-Ak+ peptides were preferentially associated with above-average increases in cytokine secretion. The strongest association with I-Ak+ status was observed for IL-4 responses by gp120dss298-immunized mice (Table 2; see Table S1 in the supplemental material). The IL-4 response to 12 of the 14 I-Ak+ peptides was elevated (Fig. 2C). The remaining two I-Ak+ peptides (peptides 30 and 38) were located in flexible loops V3 and V4, respectively, where conformational destabilization by the disulfide bond deletion is not expected to have an effect.
TABLE 2.
Number of I-Ak+ peptides (of a total of 14 I-Ak+ peptides) whose responses were differentially affected by disulfide deletiona
| Response | gp120dss298 | gp120dss378 | gp120dss385 |
|---|---|---|---|
| Secretion of: | |||
| IL-4 | 12 | 7 | 8 |
| IL-2 | 10 | 7 | 11 |
| IL-10 | 9 | 6 | 11 |
| IL-6 | 8 | 7 | 8 |
| IL-13 | 7 | 6 | 8 |
| TNF-α | 7 | 4 | 10 |
| IFN-γ | 8 | 5 | 7 |
| IL-5 | 7 | 7 | 4 |
| Cell proliferation | 10 | 6 | 8 |
Numbers in boldface indicate significant association between differential effect and I-Ak+ status: 12 peptides, P < 0.0001; 11 peptides, P < 0.0001; 10 peptides, P = 0.0014; 9 peptides, P = 0.014.
CpG oligonucleotide enhanced IFN-γ responses for dominant peptides.
We sought an alternative method for enhancing T-cell responses in order to test the uniqueness of preferential increases in the responses to I-Ak+ peptides. Inclusion of CpG in the vaccine formulation was expected to cause a Th1 skew in the response, characterized mainly by an increase in IFN-γ response. CpG binds to TLR9 on antigen-presenting cells, resulting in the expression of IL-12 during T-cell priming, which in turn induces primed T cells to produce IFN-γ (50, 51). We hypothesized that the added source of activation would enhance the IFN-γ response and potentially influence antigen processing and epitope dominance.
Three groups of CBA mice were immunized intranasally three times over 4 weeks with gp120 and CpG, mLT, or the combination of mLT and CpG. Splenocytes were then restimulated with individual gp120 peptides as described above. The degree of overall Th1/Th2 polarization was evaluated in terms of the average concentration of cytokine produced in response to all 38 gp120 peptides, measured individually.
T cells from mice immunized with gp120 and CpG proliferated little and secreted low levels of cytokines. T cells from mice immunized with gp120 and mLT proliferated robustly and secreted higher levels of all cytokines. The combination of CpG and mLT skewed cytokine responses toward a Th1 type relative to that for mLT only. The amounts of IFN-γ secreted into the supernatant was significantly larger for the CpG+mLT mice than the amount for the mLT-only mice (Fig. 3A). The CpG+mLT mice also produced smaller amounts of Th2 cytokines (IL-4, IL-5, and IL-6) than those produced by splenocytes from the mLT-only mice. No significant difference was observed in cell proliferation, IL-2, or TNF-α production from the CpG+mLT mice, compared to those from the mLT-only mice. The Th1 polarization effects with CpG were also observed in the amount of serum IgG1 and IgG1a specific for gp120. Ratios of IgG1 to IgG2a for the CpG+mLT mice were significantly lower than those for the mLT-only mice at dilutions up to 8,000-fold (see Fig. S2 in the supplemental material).
FIG 3.

Th1-skewed responses induced by addition of CpG oligonucleotide with mLT as an adjuvant for immunization with gp120. (A) Each symbol is the average response to 38 individually tested peptides for splenocytes of a single mouse. Bars indicate significant differences between mLT and mLT+CpG as in the legend for Fig. 2. (B) Peptide-specific IFN-γ responses following immunization with gp120 and the indicated adjuvant formulation. (C) Difference in secreted IFN-γ for mLT+CpG versus mLT alone. The dashed horizontal line indicates the average difference for all peptides. The bar graph indicates the I-Ak motif score. Immunization of mice with gp120 and mLT or mLT+CpG produced large T-cell responses. Comparing mLT+CpG and mLT, the responses for mLT+CpG were skewed toward Th1, as indicated by the elevated IFN-γ and diminished IL-4, IL-5, and IL-6 amounts. Increases in IFN-γ were not preferentially associated with I-Ak+ peptides; rather, they followed the original dominance pattern.
Peptide-specific changes in T-cell response resulting from CpG were not localized to the I-Ak+ peptides. The largest increases in secreted IFN-γ were associated with peptides that were dominant in the standard immunization, namely, peptides 6 and 32 (Fig. 3B and C). The reduction in IL-4 response was distributed among the epitopes, without preference for either the dominant or I-Ak+ peptides (data not shown). Thus, unlike the results for disulfide variants, changes in the T-cell response caused by CpG in the immunization reflected the dominance hierarchy that was observed with the standard immunization.
Peptide-reactive antibody.
IgGs were tested by ELISA for reactivity to peptides spanning gp120, except for the segment V1-V2. Antibody reactivity indicates the presence of a “linear” epitope because the epitope can be recognized in the context of the protein or the isolated peptide. Typically, linear epitopes are located in conformationally flexible antigen segments. A linear epitope was identified if at least two mice reacted (A450 > 0.1) with the corresponding peptide. The number of linear epitopes identified in each immunogen was as follows: gp120, 9; gp120dss298, 12; gp120dss378, 5; gp120dss385, 6. All four immunogens shared epitopes in peptides 7, 43, and 44. The most remarkable changes in linear epitopes caused by disulfide deletions (difference of two or more mice) were the loss of epitopes at peptides 17, 19, and 20 in all three variants and the appearance of linear epitopes at peptides 21, 22, 24, 30, and 47 in gp120dss298.
DISCUSSION
These studies show that the native gp120 conformation conceals I-Ak-binding sequences that can become immunogenic for CD4+ T cells in a conformationally destabilized gp120 variant. Almost none of the gp120 peptides predicted to bind strongly to I-Ak was immunogenic in the native gp120. It is common for peptides that can bind strongly to MHC proteins to be poorly immunogenic in the context of a protein that contains them (19, 20, 52, 53). The dominance of peptides with relatively low affinity for the MHC protein is likely to be due to mechanisms of antigen processing that increase the availability those peptides.
Earlier studies and the results presented here highlight the dominance of epitopes that lie adjacent to conformationally unstable antigen segments. We have proposed that the unstable segments serve as entry points for proteolytic processing of gp120 and that the adjacent segments on gp120 fragments are preferentially loaded into the MHC proteins (22). This model is consistent with lack of immunogenicity of most I-Ak+ peptides in the native gp120 and enhanced immunogenicity of I-Ak+ peptides in the conformationally destabilized disulfide variants of gp120 (Fig. 4). Destabilization reduces any differences in accessibility of gp120 sequences, whether to processing proteases or the MHC-II protein. The lone example of an immunogenic I-Ak+ peptide in native gp120 (peptide 30) lies next to the highly flexible and protease-sensitive V3 loop (54), where it is likely to be efficiently processed and presented regardless of conformational stability in the gp120.
FIG 4.

Processing and presentation of a dominant epitope and an I-Ak+ epitope that is concealed by a disulfide bond. Proteolytic cleavage makes the dominant epitope available for loading into the MHC protein. In the case of an intact disulfide bond (upper pathway), the I-Ak+ epitope remains unavailable for loading because the disulfide bond stabilizes the partially folded conformation. In the case of a mutated disulfide bond (lower pathway), the I-Ak+ epitope is presented because the protein undergoes additional unfolding.
The differential enhancement of T-cell responses to I-Ak+ epitopes correlated with conformational destabilization in the disulfide variants of gp120. Previous studies using limited proteolysis and the binding of CD4 and monoclonal antibodies indicated that all three disulfide variants were destabilized relative to wild-type gp120 (30). Of the three variants, gp120dss298 was the most destabilized. Destabilization caused by the disulfide deletions was also evident in the increased utilization of linear antibody epitopes. In the present CBA mouse study, gp120dss298 elicited antibodies against a larger number of peptides than did wild-type gp120, suggesting that a larger fraction of gp120dss298 is conformationally disordered. In accord with greater accessibility of I-Ak+ peptides, the differential enhancement of responses to I-Ak+ peptides was most pronounced for gp120dss298.
Selective enhancement of the I-Ak+ epitopes was not obtained by simply generating a stronger immune response. It was possible that the I-Ak+ peptides were preferentially enhanced by disulfide deletions because the response to the dominant epitopes of gp120 (most of which lack an I-Ak motif) was already at its maximum. However, inclusion of CpG in the adjuvant formulation preferentially enhanced responses to peptides that were dominant in the standard immunization. Thus, we conclude that preferential enhancement of responses to the I-Ak+ peptides in the disulfide variants was due to their increased accessibility in the conformationally destabilized disulfide variants, rather than because other epitopes were already at maximum response.
In HIV+ humans, the promiscuous dominance of CD4+ epitopes can be explained by the interaction of gp120 structure with mechanisms of antigen processing. In a sample of seven HIV+ subjects on antiretroviral therapy, the dominant epitopes of gp120 occurred on the C-terminal flanks of conformationally unstable gp120 segments, which we proposed was due to proteolytic cleavage in the unstable segment and preferential loading of the N-terminal segment of the C-terminal cleavage product (21). In a recent study of 93 untreated HIV+ subjects, promiscuously dominant epitopes were identified in Env sequences 30 to 46, 88 to 105, and 207 to 224, which correspond to peptides 1, 6, and 19 in the present study, respectively (11). Peptides 6 and 19 were dominant in CBA mice despite the fact that these peptides lacked an I-Ak motif. Both peptides are located on the C-terminal flanks of conformationally unstable gp120 segments, the β2-α0 loop and the V2 loop, respectively. Peptide 1 may be similarly disposed for presentation in the gp160 envelope glycoprotein because it is located at the protein N terminus. We speculate that peptide 1 was not immunogenic in the context of recombinant gp120 (this study) because the protein was not assembled as the native gp160 trimer, which may protect peptide 1 from destruction.
Dominance of particular epitopes appears to affect the course of infection. In the study of untreated HIV+ subjects, the response corresponding to gp120 peptide 19 was associated with high viremia, whereas a response to any of three Gag epitopes was associated with low viremia (8). Thus, vaccination studies should be concerned with the conformational state of immunogens, not only because it affects the induction of protective antibody but also because it affects the development of particular CD4+ T-cell responses. Although the present work suggests that cryptic CD4+ T-cell epitopes could be targeted by destabilization of HIV ENV, this strategy would most likely reduce the induction of antibodies against conformational neutralizing epitopes, which depend on a stable conformation. It is possible that an immunization protocol could optimize both arms of the immune response by combining stable and unstable immunogens. A similar outcome might be achieved by use of multiple formulations of the same immunogen, as has been observed in recent studies (55, 56).
The gp120 destabilization affected some types of cytokine response more than others. The most general effect was a broad increase in IFN-γ, TNF-α, IL-4, IL-6, and IL-13, which is evident in the average response to gp120dss298 and gp120dss385 (Fig. 2A). For the other types of response (increases in IL-2, IL-5, and IL-10 and proliferation), the average was not significantly affected, and the profiles across epitopes were very similar for all four gp120 proteins (see Fig. S1 in the supplemental material). The mechanistic basis for this difference is unclear, but it is likely to stem from the distinct requirements for differentiation of T-helper cell types (57, 58). We speculate that immunization induces the secretion of key differentiation factors, such as IL-12p70 for Th1 cells, in a time- and tissue-specific manner that overlaps but does not coincide with the wave of antigen presentation. The degree of overlap may depend on the rate of antigen processing and presentation. For example, a wave of rapidly presented epitopes may overlap more closely with the wave of IL-12p70 production, and thus the resulting T cells are skewed to Th1. It is interesting that the average IL-4 and IL-13 were sensitive but the average IL-5 was not sensitive to gp120 stability, in view of the fact that all three cytokines are associated with the Th2 cell type and at least partially regulated by the same transcription factor, GATA3. This result serves to highlight the subtle differences in differentiation requirements for cells that produce particular cytokines, such as IL-5 (59, 60), and suggests that immune functions could be specialized to particular antigens and epitopes by their susceptibility to antigen processing.
In the present study, with CBA mice, the disulfide variants elicited larger T-cell responses than those elicited by wild-type gp120. In C57BL/6 mice, the disulfide variants also elicited larger T-cell responses (data not shown). In contrast, in BALB/c mice, we previously observed that the disulfide variants elicited smaller responses (30). Why the difference between mouse strains? We envision three alternative fates for a potential T-cell epitope: proteolytic destruction, loading into the MHC-II, or remaining unprocessed and not presented. Destabilization of the antigen is expected to reduce the population of unprocessed epitope and increase the amount of epitope available for proteolytic destruction or MHC-II loading. How the increase is distributed between destruction and MHC-II loading depends on mechanisms. It is possible that APCs of BALB/c mice have higher protease activity and therefore destroy a larger fraction of the epitope molecules and reduce the loaded fraction. Alternatively, the protease activities could be similar in the different mouse strains, but the MHC-II of the BALB/c strain (I-Ad) could have lower average peptide-binding affinity than the MHC-II of the other mouse strains; and therefore, I-Ad protects a smaller fraction of epitope molecules from destruction. MHC-II proteins have been shown to protect epitopes from proteolytic destruction (61–63). Further studies in the genetically defined MHC backgrounds of mice will be necessary to generalize mechanisms of antigen processing and then extend them to heterogeneous systems such as in humans.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by NIH R01-AI080367 (to S.J.L.) and NSF CAREER IIS-0643768 (to R.R.M.).
Footnotes
Published ahead of print 11 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00789-14.
REFERENCES
- 1.Choi AH, Basu M, McNeal MM, Flint J, VanCott JL, Clements JD, Ward RL. 2000. Functional mapping of protective domains and epitopes in the rotavirus VP6 protein. J. Virol. 74:11574–11580. 10.1128/JVI.74.24.11574-11580.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao W, Pahar B, Sestak K. 2008. Identification of rotavirus VP6-specific CD4+ T cell epitopes in a G1P[8] human rotavirus-infected rhesus macaque. Virology (Auckl.) 1:9–15 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2855136/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McNeal MM, VanCott JL, Choi AH, Basu M, Flint JA, Stone SC, Clements JD, Ward RL. 2002. CD4 T cells are the only lymphocytes needed to protect mice against rotavirus shedding after intranasal immunization with a chimeric VP6 protein and the adjuvant LT(R192G). J. Virol. 76:560–568. 10.1128/JVI.76.2.560-568.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Letvin NL, Rao SS, Montefiori DC, Seaman MS, Sun Y, Lim SY, Yeh WW, Asmal M, Gelman RS, Shen L, Whitney JB, Seoighe C, Lacerda M, Keating S, Norris PJ, Hudgens MG, Gilbert PB, Buzby AP, Mach LV, Zhang J, Balachandran H, Shaw GM, Schmidt SD, Todd JP, Dodson A, Mascola JR, Nabel GJ. 2011. Immune and genetic correlates of vaccine protection against mucosal infection by SIV in monkeys. Sci. Transl. Med. 3:81ra36. 10.1126/scitranslmed.3002351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pahar B, Gray W, Phelps K, Didier E, deHaro E, Marx P, Traina-Dorge V. 2012. Increased cellular immune responses and CD4+ T-cell proliferation correlate with reduced plasma viral load in SIV challenged recombinant simian varicella virus-simian immunodeficiency virus (rSVV-SIV) vaccinated rhesus macaques. Virology J. 9:160. 10.1186/1743-422X-9-160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Souza MS, Ratto-Kim S, Chuenarom W, Schuetz A, Chantakulkij S, Nuntapinit B, Valencia-Micolta A, Thelian D, Nitayaphan S, Pitisuttithum P, Paris RM, Kaewkungwal J, Michael NL, Rerks-Ngarm S, Mathieson B, Marovich M, Currier JR, Kim JH. 2012. The Thai phase III trial (RV144) vaccine regimen induces T cell responses that preferentially target epitopes within the V2 region of HIV-1 envelope. J. Immunol. 188:5166–5176. 10.4049/jimmunol.1102756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soghoian DZ, Jessen H, Flanders M, Sierra-Davidson K, Cutler S, Pertel T, Ranasinghe S, Lindqvist M, Davis I, Lane K, Rychert J, Rosenberg ES, Piechocka-Trocha A, Brass AL, Brenchley JM, Walker BD, Streeck H. 2012. HIV-specific cytolytic CD4 T cell responses during acute HIV infection predict disease outcome. Sci. Transl. Med. 4:123ra25. 10.1126/scitranslmed.3003165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ranasinghe S, Flanders M, Cutler S, Soghoian DZ, Ghebremichael M, Davis I, Lindqvist M, Pereyra F, Walker BD, Heckerman D, Streeck H. 2012. HIV-specific CD4 T cell responses to different viral proteins have discordant associations with viral load and clinical outcome. J. Virol. 86:277–283. 10.1128/JVI.05577-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schulze zur Wiesch J, Lauer GM, Day CL, Kim AY, Ouchi K, Duncan JE, Wurcel AG, Timm J, Jones AM, Mothe B, Allen TM, McGovern B, Lewis-Ximenez L, Sidney J, Sette A, Chung RT, Walker BD. 2005. Broad repertoire of the CD4+ Th cell response in spontaneously controlled hepatitis C virus infection includes dominant and highly promiscuous epitopes. J. Immunol. 175:3603–3613. 10.4049/jimmunol.175.6.3603 [DOI] [PubMed] [Google Scholar]
- 10.Brenchley JM, Silvestri G, Douek DC. 2010. Nonprogressive and progressive primate immunodeficiency lentivirus infections. Immunity 32:737–742. 10.1016/j.immuni.2010.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ranasinghe S, Cutler S, Davis I, Lu R, Soghoian DZ, Qi Y, Sidney J, Kranias G, Flanders MD, Lindqvist M, Kuhl B, Alter G, Deeks SG, Walker BD, Gao X, Sette A, Carrington M, Streeck H. 2013. Association of HLA-DRB1-restricted CD4(+) T cell responses with HIV immune control. Nat. Med. 19:930–933. 10.1038/nm.3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watts C. 2004. The exogenous pathway for antigen presentation on major histocompatibility complex class II and CD1 molecules. Nat. Immunol. 5:685–692. 10.1038/ni1088 [DOI] [PubMed] [Google Scholar]
- 13.Trombetta ES, Mellman I. 2005. Cell biology of antigen processing in vitro and in vivo. Annu. Rev. Immunol. 23:975–1028. 10.1146/annurev.immunol.22.012703.104538 [DOI] [PubMed] [Google Scholar]
- 14.Painter CA, Stern LJ. 2012. Conformational variation in structures of classical and non-classical MHCII proteins and functional implications. Immunol. Rev. 250:144–157. 10.1111/imr.12003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blum JS, Wearsch PA, Cresswell P. 2013. Pathways of antigen processing. Annu. Rev. Immunol. 31:443–473. 10.1146/annurev-immunol-032712-095910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, Jenkins MK. 2007. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity 27:203–213. 10.1016/j.immuni.2007.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang P, Sidney J, Dow C, Mothe B, Sette A, Peters B. 2008. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput. Biol. 4:e1000048. 10.1371/journal.pcbi.1000048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peterson DA, DiPaolo RJ, Kanagawa O, Unanue ER. 1999. Quantitative analysis of the T cell repertoire that escapes negative selection. Immunity 11:453–462. 10.1016/S1074-7613(00)80120-X [DOI] [PubMed] [Google Scholar]
- 19.Gelder C, Davenport M, Barnardo M, Bourne T, Lamb J, Askonas B, Hill A, Welsh K. 1998. Six unrelated HLA-DR-matched adults recognize identical CD4(+) T cell epitopes from influenza A haemagglutinin that are not simply peptides with high HLA-DR binding affinities. Int. Immunol. 10:211–222. 10.1093/intimm/10.2.211 [DOI] [PubMed] [Google Scholar]
- 20.Kim J, Sette A, Rodda S, Southwood S, Sieling PA, Mehra V, Ohmen JD, Oliveros J, Appella E, Higashimoto Y, Rea TH, Bloom BR, Modlin RL. 1997. Determinants of T cell reactivity to the Mycobacterium leprae GroES homologue. J. Immunol. 159:335–343 [PubMed] [Google Scholar]
- 21.Mirano-Bascos D, Tary-Lehmann M, Landry SJ. 2008. Antigen structure influences helper T-cell epitope dominance in the human immune response to HIV envelope glycoprotein gp120. Eur. J. Immunol. 38:1231–1237. 10.1002/eji.200738011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai G, Steede NK, Landry SJ. 2001. Allocation of helper T-cell epitope immunodominance according to three-dimensional structure in the human immunodeficiency virus type I envelope glycoprotein gp120. J. Biol. Chem. 276:41913–41920. 10.1074/jbc.M106018200 [DOI] [PubMed] [Google Scholar]
- 23.Surman S, Lockey TD, Slobod KS, Jones B, Riberdy JM, White SW, Doherty PC, Hurwitz JL. 2001. Localization of CD4+ T cell epitope hotspots to exposed strands of HIV envelope glycoprotein suggests structural influences on antigen processing. Proc. Natl. Acad. Sci. U. S. A. 98:4587–4592. 10.1073/pnas.071063898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ziegler K, Unanue ER. 1981. Identification of a macrophage antigen-processing event required for I-region-restricted antigen presentation to T lymphocytes. J. Immunol. 127:1869–1875 [PubMed] [Google Scholar]
- 25.Antoniou AN, Blackwood SL, Mazzeo D, Watts C. 2000. Control of antigen presentation by a single protease cleavage site. Immunity 12:391–398. 10.1016/S1074-7613(00)80191-0 [DOI] [PubMed] [Google Scholar]
- 26.Zhu H, Liu K, Cerny J, Imoto T, Moudgil KD. 2005. Insertion of the dibasic motif in the flanking region of a cryptic self-determinant leads to activation of the epitope-specific T cells. J. Immunol. 175:2252–2260. 10.4049/jimmunol.175.4.2252 [DOI] [PubMed] [Google Scholar]
- 27.Mutschlechner S, Egger M, Briza P, Wallner M, Lackner P, Karle A, Vogt AB, Fischer GF, Bohle B, Ferreira F. 2010. Naturally processed T cell-activating peptides of the major birch pollen allergen. J. Allergy Clin. Immunol. 125:711–718, 718.e1–718.e2. 10.1016/j.jaci.2009.10.052 [DOI] [PubMed] [Google Scholar]
- 28.West LC, Grotzke JE, Cresswell P. 2013. MHC class II-restricted presentation of the major house dust mite allergen Der p 1 Is GILT-dependent: implications for allergic asthma. PLoS One 8:e51343. 10.1371/journal.pone.0051343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maric M, Arunachalam B, Phan UT, Dong C, Garrett WS, Cannon KS, Alfonso C, Karlsson L, Flavell RA, Cresswell P. 2001. Defective antigen processing in GILT-free mice. Science 294:1361–1365. 10.1126/science.1065500 [DOI] [PubMed] [Google Scholar]
- 30.Mirano-Bascos D, Steede NK, Robinson JE, Landry SJ. 2010. Influence of disulfide-stabilized structure on the specificity of helper T-cell and antibody responses to HIV envelope glycoprotein gp120. J. Virol. 84:3303–3311. 10.1128/JVI.02242-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hubbard SJ. 1998. The structural aspects of limited proteolysis of native proteins. Biochim. Biophys. Acta 1382:191–206. 10.1016/S0167-4838(97)00175-1 [DOI] [PubMed] [Google Scholar]
- 32.Hubbard SJ, Beynon RJ, Thornton JM. 1998. Assessment of conformational parameters as predictors of limited proteolysis sites in native protein structures. Protein Eng. 11:349–359. 10.1093/protein/11.5.349 [DOI] [PubMed] [Google Scholar]
- 33.Chang SG, Choi KD, Jang SH, Shin HC. 2003. Role of disulfide bonds in the structure and activity of human insulin. Mol. Cells 16:323–330 [PubMed] [Google Scholar]
- 34.van den Berg B, Chung EW, Robinson CV, Dobson CM. 1999. Characterisation of the dominant oxidative folding intermediate of hen lysozyme. J. Mol. Biol. 290:781–796. 10.1006/jmbi.1999.2915 [DOI] [PubMed] [Google Scholar]
- 35.Silvers R, Sziegat F, Tachibana H, Segawa S, Whittaker S, Günther UL, Gabel F, Huang JR, Blackledge M, Wirmer-Bartoschek J, Schwalbe H. 2012. Modulation of structure and dynamics by disulfide bond formation in unfolded states. J. Am. Chem. Soc. 134:6846–6854. 10.1021/ja3009506 [DOI] [PubMed] [Google Scholar]
- 36.Li P, Haque MA, Blum JS. 2002. Role of disulfide bonds in regulating antigen processing and epitope selection. J. Immunol. 169:2444–2450. 10.4049/jimmunol.169.5.2444 [DOI] [PubMed] [Google Scholar]
- 37.van Anken E, Sanders RW, Liscaljet IM, Land A, Bontjer I, Tillemans S, Nabatov AA, Paxton WA, Berkhout B, Braakman I. 2008. Only five of 10 strictly conserved disulfide bonds are essential for folding and eight for function of the HIV-1 envelope glycoprotein. Mol. Biol. Cell 19:4298–4309. 10.1091/mbc.E07-12-1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pancera M, Majeed S, Ban YE, Chen L, Huang CC, Kong L, Kwon YD, Stuckey J, Zhou T, Robinson JE, Schief WR, Sodroski J, Wyatt R, Kwong PD. 2010. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc. Natl. Acad. Sci. U. S. A. 107:1166–1171. 10.1073/pnas.0911004107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koradi R, Billeter M, Wuthrich K. 1996. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14:51–55, 29–32. 10.1016/0263-7855(96)00009-4 [DOI] [PubMed] [Google Scholar]
- 40.Vertrees J, Barritt P, Whitten S, Hilser VJ. 2005. COREX/BEST server: a web browser-based program that calculates regional stability variations within protein structures. Bioinformatics 21:3318–3319. 10.1093/bioinformatics/bti520 [DOI] [PubMed] [Google Scholar]
- 41.Hilser VJ, Freire E. 1996. Structure-based calculation of the equilibrium folding pathway of proteins. Correlation with hydrogen exchange protection factors. J. Mol. Biol. 262:756–772 [DOI] [PubMed] [Google Scholar]
- 42.Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41:95–98 [Google Scholar]
- 43.Landry SJ. 2008. Three-dimensional structure determines the pattern of CD4+ T-cell epitope dominance in influenza virus hemagglutinin. J. Virol. 82:1238–1248. 10.1128/JVI.02026-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cardenas-Freytag L, Cheng E, Mayeux P, Domer JE, Clements JD. 1999. Effectiveness of a vaccine composed of heat-killed Candida albicans and a novel mucosal adjuvant, LT(R192G), against systemic candidiasis. Infect. Immun. 67:826–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nielsen M, Lund O. 2009. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics 10:296. 10.1186/1471-2105-10-296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vita R, Zarebski L, Greenbaum JA, Emami H, Hoof I, Salimi N, Damle R, Sette A, Peters B. 2010. The immune epitope database 2.0. Nucleic Acids Res. 38:D854–D862. 10.1093/nar/gkp1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nelson CA, Viner NJ, Young SP, Petzold SJ, Unanue ER. 1996. A negatively charged anchor residue promotes high affinity binding to the MHC class II molecule I-Ak. J. Immunol. 157:755–762 [PubMed] [Google Scholar]
- 48.Fremont DH, Monnaie D, Nelson CA, Hendrickson WA, Unanue ER. 1998. Crystal structure of I-Ak in complex with a dominant epitope of lysozyme. Immunity 8:305–317. 10.1016/S1074-7613(00)80536-1 [DOI] [PubMed] [Google Scholar]
- 49.Raska M, Takahashi K, Czernekova L, Zachova K, Hall S, Moldoveanu Z, Elliott MC, Wilson L, Brown R, Jancova D, Barnes S, Vrbkova J, Tomana M, Smith PD, Mestecky J, Renfrow MB, Novak J. 2010. Glycosylation patterns of HIV-1 gp120 depend on the type of expressing cells and affect antibody recognition. J. Biol. Chem. 285:20860–20869. 10.1074/jbc.M109.085472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chu RS, Targoni OS, Krieg AM, Lehmann PV, Harding CV. 1997. CpG oligodeoxynucleotides act as adjuvants that switch on T helper 1 (Th1) immunity. J. Exp. Med. 186:1623–1631. 10.1084/jem.186.10.1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tudor D, Dubuquoy C, Gaboriau V, Lefevre F, Charley B, Riffault S. 2005. TLR9 pathway is involved in adjuvant effects of plasmid DNA-based vaccines. Vaccine 23:1258–1264. 10.1016/j.vaccine.2004.09.001 [DOI] [PubMed] [Google Scholar]
- 52.Ma C, Whiteley PE, Cameron PM, Freed DC, Pressey A, Chen SL, Garni-Wagner B, Fang C, Zaller DM, Wicker LS, Blum JS. 1999. Role of APC in the selection of immunodominant T cell epitopes. J. Immunol. 163:6413–6423 [PubMed] [Google Scholar]
- 53.Phelps RG, Jones VL, Coughlan M, Turner AN, Rees AJ. 1998. Presentation of the Goodpasture autoantigen to CD4 T cells is influenced more by processing constraints than by HLA class II peptide binding preferences. J. Biol. Chem. 273:11440–11447. 10.1074/jbc.273.19.11440 [DOI] [PubMed] [Google Scholar]
- 54.Pollard SR, Meier W, Chow P, Rosa JJ, Wiley DC. 1991. CD4-binding regions of human immunodeficiency virus envelope glycoprotein gp120 defined by proteolytic digestion. Proc. Natl. Acad. Sci. U. S. A. 88:11320–11324. 10.1073/pnas.88.24.11320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jaworski JP, Krebs SJ, Trovato M, Kovarik DN, Brower Z, Sutton WF, Waagmeester G, Sartorius R, D'Apice L, Caivano A, Doria-Rose NA, Malherbe D, Montefiori DC, Barnett S, De Berardinis P, Haigwood NL. 2012. Co-immunization with multimeric scaffolds and DNA rapidly induces potent autologous HIV-1 neutralizing antibodies and CD8+ T cells. PLoS One 7:e31464. 10.1371/journal.pone.0031464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li J, Valentin A, Kulkarni V, Rosati M, Beach RK, Alicea C, Hannaman D, Reed SG, Felber BK, Pavlakis GN. 2013. HIV/SIV DNA vaccine combined with protein in a co-immunization protocol elicits highest humoral responses to envelope in mice and macaques. Vaccine 31:3747–3755. 10.1016/j.vaccine.2013.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walsh KP, Mills KH. 2013. Dendritic cells and other innate determinants of T helper cell polarisation. Trends Immunol. 34:521–530. 10.1016/j.it.2013.07.006 [DOI] [PubMed] [Google Scholar]
- 58.Yamane H, Paul WE. 2013. Early signaling events that underlie fate decisions of naive CD4(+) T cells toward distinct T-helper cell subsets. Immunol. Rev. 252:12–23. 10.1111/imr.12032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rank MA, Kobayashi T, Kozaki H, Bartemes KR, Squillace DL, Kita H. 2009. IL-33-activated dendritic cells induce an atypical TH2-type response. J. Allergy Clin. Immunol. 123:1047–1054. 10.1016/j.jaci.2009.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hongjia L, Caiqing Z, Degan L, Fen L, Chao W, Jinxiang W, Liang D. 2014. IL-25 promotes Th2 immunity responses in airway inflammation of asthmatic mice via activation of dendritic cells. Inflammation 10.1007/s10753-014-9830-4 [DOI] [PubMed] [Google Scholar]
- 61.Donermeyer DL, Allen PM. 1989. Binding to Ia protects an immunogenic peptide from proteolytic degradation. J. Immunol. 142:1063–1068 [PubMed] [Google Scholar]
- 62.Mouritsen S, Meldal M, Werdelin O, Hansen AS, Buus S. 1992. MHC molecules protect T cell epitopes against proteolytic destruction. J. Immunol. 149:1987–1993 [PubMed] [Google Scholar]
- 63.von Delwig A, Musson JA, Gray J, McKie N, Robinson JH. 2005. Major histocompatibility class II molecules prevent destructive processing of exogenous peptides at the cell surface of macrophages for presentation to CD4 T cells. Immunology 114:194–203. 10.1111/j.1365-2567.2004.02085.x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.