

ABSTRACT
Macrophages must react to a large number of pathogens and their effects. In chronic HIV infection, the microenvironment changes with an influx of microbial products that trigger Toll-like receptors (TLRs). That dynamic nature can be replicated ex vivo by the proinflammatory (M1-polarized) and alternatively activated (M2-polarized) macrophages. Thus, we determined how polarized macrophages primed by various TLR agonists support HIV replication. Triggering of TLR2, -3, -4, -5, and -8 reinforced the low level of permissiveness in polarized macrophages. HIV was inhibited even more in M1-polarized macrophages than in macrophages activated only by TLR agonists. HIV was inhibited before its integration into the host chromosome. Polarization and triggering by various TLR agonists resulted in distinct cytokine profiles, endocytic activity, and distinct upregulation of restriction factors of HIV. Thus, different mechanisms likely contribute to the HIV-inhibitory effects. In chronic HIV infection, macrophages might become less permissive to HIV due to changes in the microenvironment. The high level of reactivity of polarized macrophages to TLR triggering may be exploited for immunotherapeutic strategies.
IMPORTANCE Macrophages are a major target of HIV-1 infection. Different cell types in this very heterogeneous cell population respond differently to stimuli. In vitro, the heterogeneity is mimicked by their polarization into proinflammatory and alternatively activated macrophages. Here we explored the extent to which agonists triggering the TLR family affect HIV replication in polarized macrophages. We found that a number of TLR agonists blocked HIV replication substantially when given before infection. We also report the mechanisms of how TLR agonists exert their inhibitory action. Our findings may advance our understanding of which and how TLR agonists block HIV infection in polarized macrophages and may facilitate the design of novel immunotherapeutic approaches.
INTRODUCTION
Macrophages are part of our defenses against a hostile environment (1). They protect us from infecting pathogens through multiple mechanisms, including phagocytosis, antigen presentation, immunoregulation, and clearing of apoptotic and necrotic cells. For this broad range of functions, they have evolved considerable heterogeneity and plasticity (2) and are equipped with scavenger receptors, sensing receptors (e.g., Toll-like receptors [TLRs], C-type lectins, helicases, NOD- or RIG-like receptor), and opsonic receptors (e.g., Fc and complement) (1) that shape their profiles.
One group of sensing receptors, TLRs, recognizes conserved motifs of all kinds of infecting agents. TLRs are located on the cell membrane (i.e., TLR1, -2, -4, -5, -6, and -10) or in endosomes (i.e., TLR3, -7, -8, and -9). All TLRs (except TLR3) share the same adaptor protein, MyD88 (3). TLR3 uses TRIF, and TLR4 signals through either pathway, depending on the stimulus. Simply put, TLR signaling culminates in activation of cytokines or alpha interferon (IFN-α) by activating the mitogen-activated protein kinase pathway, NF-κB, or various interferon regulatory factors (4). TLR signaling and its downstream effects are cell specific. Indeed, TLR expression patterns/signaling have been well explored in most white blood cells, but we know substantially less about their expression and function in macrophages (5). This knowledge, however, is needed to understand interactions between the first line of defense and invading pathogens and, in particular, to benefit from targeted TLR triggering as a therapeutic modality.
Macrophages are also a target for pathogens, including HIV. In fact, HIV has a preferential cellular tropism for CD4+ T cells, and macrophages are thought to be essential for HIV infection (6). Studies, however, are mostly limited to ex vivo-generated monocyte-derived macrophages (MDMs). During heterosexual transmission, macrophages are one of the first cell types to encounter HIV, and they are important for manufacturing and disseminating virus, as well as for generating an adaptive immune response (6). Productively infected macrophages may also promote bystander killing of T cells. Notably, macrophages from different tissues are thought to be variably permissive to HIV and, importantly, act as a long-living reservoir of HIV. Microglial and alveolar macrophages may live for several weeks to years.
Studies of macrophages have been limited mainly to ex vivo-generated MDMs. MDMs can be polarized ex vivo. Exposure to IFN-γ/tumor necrosis factor alpha (TNF-α) yields proinflammatory (M1-polarized) macrophages, and exposure to interleukin-4 (IL-4) yields alternatively activated (M2-polarized) macrophages (7). M1 has potent microbicidal properties and promotes strong IL-12-dependent Th1 responses. M2 supports Th2-associated effector functions and has a role in resolving inflammation (reviewed in reference 8). Importantly, the action of TLR triggering in polarized macrophages is largely unknown.
HIV replication differs in M1- and M2-polarized macrophages (9). As modeled by Herbein and Varin, M2-polarized monocytes/macrophages predominate before HIV infection. A shift to M1-polarized macrophages occurs during acute HIV infection, and a shift back to M2-polarized macrophages takes place in later stages of HIV infection (10). Cytokine profiles are also distinct throughout the course of HIV disease, with a preponderance of proinflammatory cytokines occurring in the acute phase (11–13). The cytokine profile also depends on the disease progression rate (13).
In addition, vigorous HIV replication and subsequent depletion of the lymphoid cells in acute HIV infection disrupt the integrity of the gastrointestinal tract, with the ensuing translocation of microbial elements (14, 15), and that translocation may contribute to the shaping of macrophage populations. Indeed, we showed that chronic HIV infection in HIV-infected humanized mice results in an increased rate of bacterial translocation, which is most likely at the origin of the malfunctioning of macrophages observed in this model (16).
Here, we explored the interactions of TLR triggering, M1 and M2 polarization, and HIV infection. Specifically, we addressed one major question. Does TLR triggering have an effect on HIV replication in M1- and M2-polarized macrophages? In this context, we also completed a comprehensive and comparative analysis of the effects of TLR triggering on HIV infection in MDMs (i.e., nonpolarized macrophages). Our findings add to the understanding of macrophage function in HIV pathogenesis.
MATERIALS AND METHODS
PBMC isolation, generation of MDMs, reagents, and plasmids.
Buffy coats from HIV-negative individuals were obtained from the local blood donation center in Zurich, Switzerland (http://www.blutspendezurich.ch/). Human peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll (Axis-Shield PoC AS) gradient centrifugation. To generate MDMs, monocytes were isolated using CD14 microbeads (catalog no. 130-050-201; Miltenyi) and cultured in RPMI 1640 medium (BioWhittaker) supplemented with 5% fetal calf serum, 5% human type AB serum (catalog no. H1513; Sigma), 2 mM l-glutamine, and 1% penicillin-streptomycin for 1 week. Monocytes were seeded at a cell count of 1.5 × 105 per cm2. M1 and M2 polarization was induced by adding IFN-γ (20 ng/ml) and TNF-α (2 ng/ml) or IL-4 (20 ng/ml) (catalog numbers 11343536, 113440047, and 11340017, respectively; ImmunoTools), as described previously (9).
We used Pam2CSK4 at 100 ng/ml; poly(I·C) at 20 μg/ml; LPS at 20 ng/ml; flagellin at 100 ng/ml; 3M-001, 3M-002, and R-848 at 3 μM each; and CpG2006 at 5 μg/ml. Pam2CSK4, poly(I·C), and flagellin were purchased from InvivoGen (catalog numbers tlrl-pam2, tlrl-pic, and trl-l-pstfla, respectively), and LPS was purchased from Sigma (catalog no. 62326). TLR agonists 3M-001, 3M-002, and R-848 were kindly provided by 3M (St. Paul, MN). CpG2006 was purchased from Microsynth, and fluorescein isothiocyanate (FITC)-dextran was purchased from Sigma (catalog numbers FD10, FD70, and FD150). Lamivudine was obtained from the AIDS repository and used at a concentration of 100 μM. Antibody to the IFN-α receptor (IFN-αR) was from Alexis (catalog no. PBL21385). Luciferase expression was quantified with a luciferase assay system from Promega (catalog no. E1501). Endotoxin was measured using a Limulus amebocyte lysate chromogenic endotoxin quantification kit (catalog no. 88282; Pierce).
Generating viruses.
We made a stock of replication-competent HIV by transfecting 293T cells with the proviral DNA YU-2. To prepare replication-incompetent viruses packaged by various envelope (Env) proteins, an HIV proviral construct encoding a luciferase reporter gene (pNL4-3.Luc.R_E_) was cotransfected with vesicular stomatitis virus glycoprotein (VSV-G) env, ADA env (HIV subtype B envelope), or a #8 env expression vector in a ratio of 1:1 with polyethylenimine (PEI) in 293T cells, as described previously (17). The #8 env expression vector was from primary CCR5-tropic HIV-1 isolates (i.e., HIV env was PCR amplified and cloned into the retroviral vector pEneo) (18). Supernatants were harvested 48 h later, filtered, and frozen at −80°C until use.
HIV p24 capsid antigen (p24) ELISA.
A twin-site sandwich enzyme-linked immunosorbent assay (ELISA) was performed essentially as described previously (19). Briefly, a polyclonal antibody (Ab) was adsorbed to a solid phase to capture p24 antigen (Ag) from a detergent lysate of virions. Bound p24 was visualized with an alkaline phosphatase-conjugated anti-p24 monoclonal antibody (MAb) and luminescent detection system.
Quantitative PCR for measuring TLR1 to TLR10 and gene profiling.
We quantified TLR1 to TLR10 mRNA and the anti-HIV restriction factors (i.e., APOBEC3G, TREX, PPIA, TRIM22, TRIM5α, BST2, IFN-β1, and SAMHD1) with commercially available primers and probes (assays on demand; Applied Biosystems). Reactions were performed as described previously (20). Hydroxymethylbilane synthase (HMBS) (also from Applied Biosystems) was used as a housekeeping gene.
Data generated by real-time quantitative PCR were analyzed in two steps. First, the mean normalized gene expression (MNE) for every sample, based on the threshold cycle value of the gene of interest in relation to that of the housekeeping gene, HMBS, was determined using the software application Q-Gene (calculation procedure for MNE 2) (21). Second, normalized gene expression in samples of interest relative to that in the controls was assessed by calculating the ratio of the MNE for HIV-infected cells to the MNE for mock-infected cells.
We profiled a selected number of 84 genes with the Toll-like receptor signaling pathway PCR array from SABiosciences Qiagen (catalog no. PAHS-018A), according to the instructions of the manufacturer.
Quantifying proviral DNA with Alu PCR.
Alu PCR was used with primers specific for human Alu sequences and HIV-1-based lentiviral vector sequences. Briefly, DNA was extracted from MDMs infected with YU-2 and pretreated with the various TLR agonists with a QIAamp DNA minikit (Qiagen), according to the manufacturer's instructions. Proviral DNA was amplified from 200 ng of the extracted DNA in a total volume of 50 μl with 1 μM each primer and 2.5 U of JumpStart Taq DNA polymerase (Sigma-Aldrich). PCR was performed in a DNA thermal cycler (Biometra, Goettingen, Germany) with 30 cycles of denaturation at 95°C for 45 s, annealing at 58°C for 60 s, and extension at 72°C for 3 min 30 s; an initial denaturation at 95°C for 5 s; and a final extension at 72°C for 10 min. The primers for the Alu repeats were 5′-GCGCGGTGGCTCACGCCTGTAAT-3′ (sense) and 5′-CTTAATACTGACGCTCTCGCACC-3′ (antisense). Nested PCR was performed using primers specific for the long terminal repeat (LTR) region of HIV-1-based lentiviral vector sequences, to efficiently quantify the provirus. Briefly, DNA amplified in the Alu PCR (25 μl) was added in a total volume of 50 μl in the presence of 1 μM each primer and 2.5 U of JumpStart Taq DNA polymerase (Sigma-Aldrich). PCR was performed in an iQ 5 system (Bio-Rad, Munich, Germany) with 50 cycles of denaturation at 95°C for 10 s, annealing at 55°C for 5 s, and extension at 60°C for 40 s; an initial denaturation at 95°C for 5 s; and a final extension at 72°C for 10 min. The primers for the LTR repeats were 5′-ATAAAGCTTGCCTTGAGTG-3′ (sense) and 5′-TGACTAAAAGGGTCTGAGGGATCTCTAGTTACCAG-3′ (antisense). The probe was 5′-TGTGTGCCCGT-3′. Primers and probes were purchased from Microsynth (Balgach, Switzerland). Results were considered valid only if the same results were obtained in at least two separate experiments.
Cytokine measurements.
Human cytokines were quantified using a multiplexed particle-based flow cytometric cytokine assay (22). Cytokine kits were purchased from R&D. The procedures closely followed those described in the manufacturer's instructions. The analysis was conducted using a conventional flow cytometer (Guava EasyCyte Plus; Millipore, Zug, Switzerland).
Measuring endocytosis.
For assessment of endocytic activity, polarized or TLR-primed MDMs were incubated with 0.1 mg/ml FITC-dextran of 70 kDa for 1 h and harvested to quantify macrophages that had taken up FITC-dextran by flow cytometry.
Flow cytometry.
Macrophages were incubated with phosphate-buffered saline containing 0.1% EDTA for staining of cell surface markers and with 5% trypsin for assessment of endocytosis for 5 min; subsequently, macrophages were mechanically detached with a cell scraper. We used MAbs to CD14, CD4, and CCR5 (all from Becton, Dickinson) and to TLR2, -3, and -4 (Lucerna Chemie AG). Stained cells were acquired on a CyAn ADP analyzer (Beckmann Coulter), and data were analyzed using FlowJo software. We first defined the live cells by the side scatter/forward scatter gate and then quantified the number of cells by the specific marker of interest.
Statistics.
We used the software GraphPad Prism (version 5.04) for statistical analyses. We indicate the statistical test that we used in the text below and in the figure legends and considered a P value of ≤0.5 to be statistically significant.
RESULTS
TLR2, -3, -4, and -8 exposure reinforces the low permissiveness to HIV in polarized macrophages.
We wondered how M1- and M2-polarized macrophages support HIV replication when stimulated by different TLR agonists. HIV permissiveness was much less in M1-polarized macrophages than in M2-polarized macrophages (Fig. 1A). Macrophages (MDMs) treated with TLR agonists yielded a heterogeneous picture: MDMs primed with TLR2, -3, -4, and -8 agonists supported HIV replication poorly, but priming by TLR5, -7, and -9 agonists had no effect (Fig. 1B; also see Fig. S1 in the supplemental material).
FIG 1.

M1 and M2 polarization and triggering of TLR2, -3, -4, and -8 rendered MDMs poorly permissive to HIV infection. We infected various polarized or primed macrophages for 6 h with the replication-competent CCR5-tropic strain YU-2, subsequently washed the cells, and added back culture medium with the corresponding TLR agonists. The culture medium that was added back to M1- and M2-polarized macrophages contained no cytokines. Supernatants were collected at days 4, 7, and 11 to monitor HIV replication by quantifying p24. (A) Permissiveness of matched M1- and M2-polarized macrophages and MDMs (control). The area under the curve (AUC) of the p24 Ag over time was determined for each experiment to consider the replication dynamics and not to focus on one individual time point (n = 8). Each dot represents the data point obtained with one buffy coat. (B) Effects of various TLR agonists on HIV infection in MDMs. (C) Effects of TLR agonists on HIV infection in M1- and M2-polarized macrophages. Taking into account the interindividual permissiveness of macrophages to HIV, the area under the curve of the p24 Ag over time for TLR-treated MDMs was normalized to that for the untreated HIV-infected control (B and C). Statistical analysis was done using a paired t test with a two-tailed P value (A and C) and one-way analysis of variance (ANOVA), followed by Bonferroni's multiple-comparison test (B). *, P < 0.05 compared to MDMs. Pam2CSK4 triggers TLR2, poly(I·C) triggers TLR3, LPS triggers TLR4, flagellin triggers TLR5, 3M-001 triggers TLR7, 3M-002 triggers TLR8, R-848 triggers TLR7/8, and CpG2006 triggers TLR9.
Triggering of TLR2, -3, -4, or -8 on M1- and M2-polarized macrophages led to a further decrease of their HIV permissiveness (Fig. 1C). Remarkably, polarization induced an inhibitory effect by TLR5 triggering, but not in MDMs. TLR7 and -9 triggering had no effect in polarized macrophages, as observed in MDMs. We also wondered whether the extent of permissiveness to HIV differs in MDMs and polarized macrophages after TLR triggering. The inhibitory effect on HIV clearly tended to be reinforced by TLR triggering in M1-polarized but not in M2-polarized macrophages compared to that in MDMs (see Fig. 2 in the supplemental material). TLR agonists did not reduce the viability of macrophages for up to 7 days after adding them (see Fig. 3 in the supplemental material).
FIG 2.

TLR mRNA expression pattern in MDMs and M1- and M2-polarized macrophages. (A) MDMs were generated by culturing highly purified monocytes over 1 week. MDMs (106) were harvested, and RNA was extracted for quantifying the TLRs by real-time PCR. Data are presented as the ratio of the MNE of the distinct TLRs to the MNE of HMBS. (B) TLR expression level of M1- and M2-polarized macrophages in relation to that for matched MDMs. The levels of TLR expression of polarized macrophages were compared to those of unstimulated MDMs by a paired t test with a two-tailed P value.
FIG 3.

TLR2, -3, and -4 protein expression in M1- and M2-polarized macrophages and untreated MDMs. M1- and M2-polarized macrophages and untreated MDMs were harvested and stained with MAbs against TLR2, -3, and -4. Since TLR3 is located endosomally, we performed a permeabilization step before adding the MAb. The results of one representative example of three experiments are shown. Red lines, unstained; green lines, MDMs; blue lines, M1- and M2-polarized macrophages stained with the corresponding MAb. Max, maximum; PE, phycoerythrin; BV, brilliant violet.
M1-polarized macrophages show more TLR expression than MDMs.
We examined the baseline expression of TLR mRNAs in MDMs. TLR1 and -2 levels were very high. TLR3 to TLR8 were easily detectable, but TLR9 and -10 were at the limit of detection (Fig. 2A). Thus, MDMs are equipped with all known TLRs, at least at the level of mRNAs. M1 polarization resulted in the marked upregulation of all TLRs except TLR5, which was downregulated (Fig. 2B). M2 polarization showed less impressive changes with upregulation of TLR4, -5, and -9.
Next, we determined if the mRNA expression levels of TLRs in polarized macrophages corresponded to protein levels. We focused our efforts on TLR2, -3, and -4 because they showed prominent increases in mRNA levels overall and antibodies that reliably bind those TLRs are available. M1-polarized macrophages had higher levels of TLR2 and -4 mRNA expression than MDMs and showed parallel increases in mRNA and protein levels (Fig. 3). On the basis of similar mRNA expression levels, M2-polarized macrophages had protein expression levels similar to those of MDMs. Strikingly, TLR3 protein expression levels did not change, regardless of polarization.
Cytokine secretion and expression profiling of TLR-dependent genes in polarized and TLR-stimulated MDMs.
At the baseline, polarized macrophages expressed amounts of cytokines/chemokines similar to the amounts expressed by MDMs, with one exception: M1-polarized macrophages displayed increased levels of the IFN-induced chemokines CXCL9 and CXCL10 (Fig. 4A). This is explained by the fact that IFN-γ is an essential component of the M1 polarization medium. MDMs responded vigorously to Pam2CSK4, LPS, or 3M-002 by releasing various cytokines and chemokines (Fig. 4A). The response to poly(I·C), flagellin, or 3M-001, even though TLR3, -5, and -7 were expressed at levels similar to those for TLR8, was substantially less or there was no response at all. As expected, on the basis of the TLR9 expression level, no cytokine was released after CpG2006 treatment. The levels of several cytokines, IL-12p70, IFN-α, IL-15, and IL-18, were below the detection limit (the detection limits of the assays for these cytokines were <5, <15, <2, and <4 pg/ml, respectively).
FIG 4.

The most prominent cytokine secretion in response to TLR triggering was in M1-polarized macrophages, followed by MDMs. (A) MDMs were polarized or exposed to Pam2CSK4, poly(I·C), LPS, flagellin, 3M-001, 3M-002, and CpG2006 for 24 h, and supernatants were analyzed for cytokines and chemokines (n ≥ 3). (B) M1- and M2-polarized macrophages were challenged with 3M-002 (TLR8 agonist) for 24 h. Subsequently, supernatants were collected to quantify the amounts of TNF-α, IL-6, and macrophage inflammatory protein 1α (MIP-1α) released (n = 3). MDM (ctrl) → 3M-002, untreated macrophages (control) treated with 3M-002; M1 → 3M-002, M1-polarized macrophages treated with 3M-002; M2 → 3M-002, M2-polarized macrophages treated with 3M-002. The agonists are described in the legend to Fig. 1. The results for MDMs (control [Ctrl]) and either polarized macrophages or macrophages treated with the distinct TLR agonists were compared by the unpaired t test with a one-tailed P value.
We next wanted to know if polarization affects cytokine release in response to TLR triggering, as exemplified by the TLR8 agonist 3M-002. We found that M1-polarized macrophages reacted the most to 3M-002, followed by MDMs and M2-polarized macrophages (Fig. 4B). In addition, M1-polarized macrophages showed a clear trend to a more pronounced response to the TLR4 agonist LPS and the TLR7 agonist 3M-001 than MDMs (see Fig. S4 in the supplemental material). The cytokine data obtained in response to TLR agonists were corroborated by expression profiling of TLR-dependent genes by the Toll-like receptor signaling pathway PCR array (SABiosciences Qiagen). M1 polarization resulted in the upregulation of NF-κB-driven genes, but M2 polarization had the opposite effect (see Fig. S5 in the supplemental material). Triggering of TLR2, -4, -5, and -8 had an even more prominent effect on NF-κB-driven genes than M1 polarization. The signatures of TLR2-, TLR4-, and TLR8-primed macrophages are consistent with TNF-α secretion.
TLR agonists act very early in HIV replication and do not depend primarily on IFN-α secretion.
Using replication-incompetent lentiviral constructs pseudotyped with CCR5-tropic HIV or VSV envelopes with an LTR-driven luciferase reporter gene, we found that the TLR agonists (i.e., TLR2, -3, -4, -5, and -8 agonists) resulted in a marked decrease of luciferase expression (Fig. 5A). 3M-001 (TLR7 agonist) and CpG2006 (TLR9 agonist) had no effect at all. The TLR8 agonist, 3M-002, appeared to act on the very first steps of the HIV replication cycle, since the anti-HIV effects vanished when it was given immediately after infection (Fig. 5B and C). Indeed, using a nested Alu PCR, we found that treatment with the TLR2, -3, -4, and -8 agonists resulted in markedly reduced levels of proviral DNA, corroborating the finding that the TLR agonists blocked HIV replication before HIV integration (Fig. 5C). We excluded the possibility that triggering of TLRs modifies the levels of CD4 and CCR5 expression (see Table S1 in the supplemental material).
FIG 5.

HIV replication is inhibited before translation in M1- and M2-polarized macrophages and in TLR-primed macrophages. (A) Polarized and TLR-primed macrophages were inoculated by spinoculation (centrifugation at 1,200 × g for 2 h at room temperature) with HIV env-pseudotyped reporter (luciferase) viruses and 24 h later were analyzed for luciferase activity (n = 3). ADA is a well-described envelope, and env #8 is the envelope of a primary isolate cloned by our laboratory. Both were obtained using CCR5. (B) 3M-002 (TLR8 agonist) was added to the culture medium either for 24 h before or immediately after HIV challenge with VSV env-pseudotyped reporter (luciferase) (the same inoculation protocol used for HIV env-pseudotyped viruses). After 24 h, the MDMs were analyzed for luciferase activity. n.s., not significant. (C) Analysis of integrated HIV DNA by Alu PCR with primers specific for human Alu sequences and for HIV-1-based lentiviral vector sequences. MDMs were pretreated with the various TLR agonists and infected with the replication-competent HIV strain YU-2, and DNA was extracted 24 h later. The agonists are described in the legend to Fig. 1.
Since TLR agonists are potent inducers of type I IFNs, we determined if IFN is a key player in the anti-HIV effects. Various treatments of the macrophages resulted in no measurable IFN-α secretion (detection limit, <15 pg/ml). However, IFN-β levels were increased in response to poly(I·C) by real-time PCR (see Fig. 8). In any case, blocking the IFN axis with neutralizing antibodies to the IFN receptor (IFN-R) either modestly reversed the HIV-inhibitory effect in response to TLR3 and -4 or had no effect at all in the case of TLR8 triggering (Fig. 6). Notably, HIV replication was massively increased in MDMs when IFN-R was blocked, suggesting that HIV-infected macrophages are modulated by type I IFNs. We verified that the viral stock was free of endotoxin (23). Irrespective of this issue, the production or release of IFN-α does not explain the potent HIV-inhibitory effect of the TLR8 agonist 3M-002. The inhibitory effects obtained with the TLR3 and -4 agonists poly(I·C) and LPS, respectively, can only partially be attributed to the production or release of IFN-α, since we would otherwise expect a massive increase in HIV replication similar to that in the controls when adding neutralizing antibodies to IFN-αR. Inefficient blocking of the IFN pathway by the neutralizing antibody to IFN-αR might be another explanation for the partial reversal of the inhibitory action by the TLR3 and -4 agonists, but we excluded this possibility since at the concentration used, the neutralizing antibody to IFN-αR entirely prevented the upregulation of the IFN-stimulated genes IFIT1 and RSAD2 in MDMs treated with IFN-α, speaking in favor of its efficacy (see Fig. S6A in the supplemental material).
FIG 8.

Triggering of TLR3 and -4 results in the upregulation of a number of restriction factors of HIV. Polarized macrophages and MDMs were harvested after a 24-h treatment with various TLR agonists in concert with untreated MDMs and analyzed for the various anti-HIV restriction factors and PPIA (PPIA, peptidylprolyl isomerase A gene; cyclophilin, protein produced by PPIA) as outlined in the Materials and Methods section. Statistical analysis was done using analysis of variance, followed by Bonferroni's multiple-comparison test. *, P < 0.05 compared to unstimulated MDMs.
FIG 6.
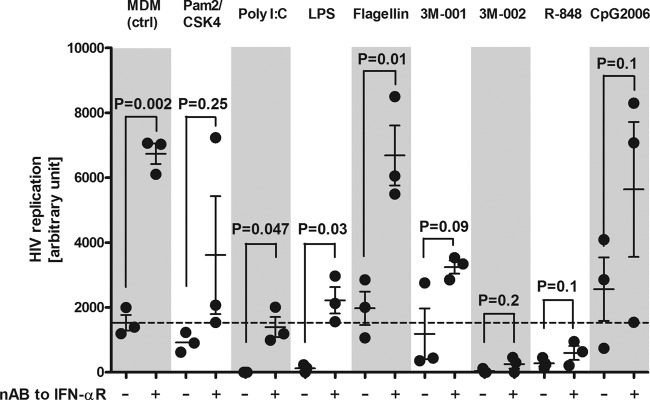
Neutralization of IFN-α with antibodies to the IFN receptor reversed the TLR-dependent HIV-inhibitory effect only to a small extent or not at all. MDMs or TLR-primed macrophages were pretreated with neutralizing Ab (nAB) to IFN-R and subsequently infected with YU-2. HIV replication was monitored over time by quantifying the p24 in the supernatants (n = 3). The area under the curve of the p24 Ag over time is presented for each individual experiment. The agonists are described in the legend to Fig. 1. Statistical analyses were done using the paired t test with a two-tailed P value.
Since HIV enters macrophages by a macropinocytosis-like pathway (24), we also investigated the endocytic capacity by quantifying the uptake of FITC-labeled dextrans of 10, 70, and 150 kDa (25). M1-polarized macrophages and macrophages primed with TLR3, -4, and -8 agonists showed reduced dextran uptake, and M2-polarized macrophages and macrophages primed with the TLR2 agonist showed dextran uptake similar to that of MDMs (Fig. 7). Thus, no uniform pattern subsequent to polarization or priming explains the default antiviral program.
FIG 7.

M1 polarization and triggering of TLR3, -4, and -8 result in substantially less endocytic activity than that from MDMs. For assessing endocytic activity, polarized macrophages or TLR-primed MDMs were incubated with 0.1 mg/ml FITC-dextran of 70 kDa for 1 h and harvested to quantify macrophages that had taken up FITC-dextran by flow cytometry. (A) Mean fluorescence intensity of one representative example. Red lines, macrophages exposed to FITC dextran at 4°C; green lines, MDMs; blue lines, MDMs treated with the various TLRs. (B) Compilation of the results of all the experiments performed. Data from assays with FITC-dextran at either 10 or 150 kDa were similar. We used the paired t test to calculate the statistics. The agonists are described in the legend to Fig. 1. MFI, mean fluorescence intensity.
Triggering of TLR3 and -4 results in the upregulation of several restriction factors of HIV.
We focused the analysis of restriction factors to HIV, based on the work of Cobos Jimenez et al. (26). We observed that triggering of MDMs by the TLR3 and -4 agonists poly(I·C) and LPS, respectively, resulted in the upregulation of all restriction factors examined and in the suppression of cyclophilin, a cellular factor that is incorporated into viral particles. The lack of cyclophilin attenuates viral replication (Fig. 8). In contrast to TLR3 and -4 agonists, polarization and the other TLR agonists resulted in changes to only one of those factors or none at all. Very nicely, adding the neutralizing antibody to IFN-αR reduced substantially but not completely the level of IFN-β when adding poly(I·C) to MDMs, data congruent with a modest direct triggering effect by poly(I·C) and a much more dramatic autocrine effect after a first wave of IFN-β release. The efficacy of the neutralization of the IFN axis by the antibody to IFN-αR is convincingly demonstrated, in that it completely prevented the poly(I·C)-dependent upregulation of all restriction factors examined (see Fig. 6B in the supplemental material).
DISCUSSION
Here, we examined the effects of TLR triggering on the HIV permissiveness of M1- and M2-polarized macrophages and MDMs. We have three major findings. (i) TLR2, -3, -4, and -8 triggering and polarization induced a default program that rendered MDMs poorly permissive to HIV by blocking HIV replication before the integration of HIV into the host chromosome. (ii) TLR2, -3, -4, and -8 triggering reinforced even the low permissive state of polarized macrophages vis-à-vis HIV. (iii) M1-polarized macrophages, in particular, stood out for their high reactivity to TLR agonists, as illustrated by the upregulated TLRs and prominent secretion of cytokines. Thus, polarized macrophages are more responsive to TLR triggering than MDMs. This particular property should be kept in mind when developing immunomodulatory strategies. On the other hand, polarization in concert with TLR triggering may limit viral replication in natural HIV infection.
We found that polarization and triggering of TLR2, -3, -4, and -8 rendered macrophages poorly supportive of HIV replication. Our data agree with those from previous reports of HIV replication in polarized macrophages and macrophages treated with TLR agonists (9, 27–31). Remarkably, TLR triggering reinforced the anti-HIV effects in polarized macrophages but did so more in M1-polarized than in M2-polarized macrophages. In contrast to unpolarized MDMs, polarization made macrophages responsive even to the TLR5 agonist flagellin and, thus, poorly permissive to HIV. In contrast, the TLR7 and -9 agonists had no anti-HIV activity even in polarized macrophages. The TLR-dependent anti-HIV effect in MDMs was not at its peak: the absolute inhibition in M1-polarized macrophages was even more pronounced.
We hypothesized that the higher reactivity of the TLR axis in polarized macrophages explains most of their non-HIV permissive state. M1 polarization resulted in the marked upregulation of all TLRs except TLR5, and M2 polarization resulted in the rather modest upregulation of TLR4, -5, and -9. Other TLRs were not affected by M2 polarization. We selected TLR2, -3, and -4 to determine if mRNA levels reflect protein expression levels. We focused on these three TLRs because they showed prominent increases in mRNA expression levels and reliable antibodies for staining them are available. mRNA and protein levels were congruent in M1-polarized macrophages, with parallel increases being found for TLR2 and -4, but in M2-polarized macrophages, there were no changes overall. TLR3 behaved differently: it showed no change in protein levels, irrespective of increases in mRNA levels. Thus, our data must be interpreted cautiously. So far, increases in TLR expression might contribute to the reinforced anti-HIV effect in M1-polarized macrophages after TLR triggering. However, other mechanisms must be involved in the increased inhibitory effects in M2-polarized macrophages and most likely also in M1-polarized macrophages exposed to the various TLR agonists; for example, in M1-polarized macrophages, the TLR5 agonist flagellin had an anti-HIV effect despite decreased levels of TLR5 expression.
MDMs and polarized macrophages had similar baseline levels of cytokines and chemokines, except for the levels of CXCL9 and CXCL10 in M1 polarization. MDMs responded to TLR2, -4, and -8 agonists with vigorous cytokine secretion and to TLR3 and -5 agonists with less pronounced secretion. Over time, M1-polarized macrophages secreted substantially more TNF-α than M2-polarized macrophages or MDMs in response to the TLR8 agonist 3M-002, corroborating the finding of the higher reactivity of M1-polarized macrophages. Sensitization was also observed when M1-polarized macrophages were triggered by LPS or 3M-001 (TLR7 agonist). While these data are mostly consistent with the data reported by Cassol et al. (9), that the levels of IL-10 in M2-polarized macrophages did not increase in our hands was, at first glance, surprising. However, the differences in cytokine secretion patterns can largely be explained by analysis of the cytokines; we assessed cytokines in the supernatant 1 day after polarization, and Cassol et al. (9) focused on days 3 and 7.
PCR profiling of TLR-dependent genes supports the data obtained with cytokines: a low to moderate increase of TLR-dependent genes in M1-polarized macrophages may be optimal for a TLR agonist to display a maximal response. The minor changes in M2 macrophages in the PCR array data and cytokine secretion are consistent with a previous report (32) on transcriptome analysis of various macrophage types. Besides, we observed no or a very low increase in the expression of TLR-dependent genes in poly(I·C)- or 3M-001 (TLR7 agonist)-treated MDMs and a very strong increase in expression in MDMs treated with Pam2CSK4, LPS, flagellin, or 3M-002. The higher reactivity of M1-polarized macrophages might explain the reinforced nonpermissive HIV state in response to the various TLR agonists. However, M2 macrophages clearly behaved differently than M1 macrophages, and only minor changes from the behavior of MDMs were observed.
We also investigated the level at which polarization and triggering of TLRs interfere with HIV replication and potential mechanisms in the poorly HIV-permissive state of macrophages. We focused on polarization and TLR triggering. The ultimate mechanism(s) for either is still controversial, and a comparison for obtaining detailed insight would be straightforward. We used a one-replication-round-pseudotyped HIV strain encoding a luciferase reporter gene. HIV replication was clearly inhibited in polarized MDMs or MDMs primed with TLR2, -3, -4, and -8. We complemented the data obtained with the luciferase assay by quantifying the proviral DNA in MDMs treated with the various TLR agonists. We found that the TLR2, -3, -4, and -8 agonists resulted in markedly reduced proviral DNA, indicating that HIV replication was inhibited before integration. The rather discordant effects of the TLR5 agonist—a lack of any effect on proviral integration but a substantial decrease in luciferase activity—may point to a TLR5 agonist-mediated mechanism acting after the integration of HIV. Strikingly, this TLR5 agonist-mediated anti-HIV activity is no longer apparent in the spread of infection.
As an example, priming was effective against HIV only when the TLR8 agonist was given before the HIV challenge in a one-round replication assay. This finding suggests that TLRs' inhibitory activity is effective at a very early stage (e.g., when they interfere with HIV trafficking or via a restriction factor). The expression level of the CD4 and CCR5 HIV receptor complex was not affected by the different treatments, and so HIV entry does not appear to be a restrictive step.
Not everyone agrees with us. For example, we and Cassol et al. (9) detected a clear reduction in HIV replication in polarized macrophages, but they found a decrease in the number of M1- and M2-polarized macrophages expressing CD4 that might contribute to the anti-HIV effects and a restriction in later steps of the HIV replication cycle in M2 macrophages. Indeed, they observed the synthesis and accumulation of viral proteins in M2-infected macrophages. We saw no differences in CD4 expression and a block in HIV replication. Long before the concept of M1 and M2 polarization was suggested, Schuitemaker et al. reported that HIV was inhibited in a dose-dependent manner when macrophages were cultured with IL-4 for 5 days (27). They found no downmodulation of CD4, similar to our findings. Irrespective of this open question, TLR agonists unequivocally had a reinforcing anti-HIV effect in polarized macrophages.
HIV is thought to enter macrophages by endocytosis (33). Indeed, the different treatments affected endocytosis substantially: M1-polarized macrophages, as well as MDMs treated with poly(I·C), LPS, and 3M-002, displayed a significant reduction of endocytosis. To what extent reduced endocytosis or even a TLR-triggered autophagic process contributes to the anti-HIV effect is unknown. The TLR-dependent reduction in endocytosis is reminiscent of a proposed vitamin D- and cyclic AMP-dependent autophagic mechanism that apparently inhibits HIV replication in response to a TLR8 agonist (30). Reduced endocytosis is also observed in macrophages from HIV-infected humanized mice (16). The finding that endocytosis in M2-polarized or TLR2 agonist-primed macrophages was similar to that in MDMs suggests that endocytosis most likely has no role in the anti-HIV effects in this setting.
We also investigated the role of anti-HIV-active IFN-α (34) and well-known HIV restriction factors (26). Neutralizing the IFN axis reversed the anti-HIV effects only modestly when triggering the TLR2 pathway; this occurred to a greater extent when triggering the TLR3 and -4 pathways but only minimally when triggering the TLR8 pathway. TLR3 and -4 triggering was associated with a fairly uniform upregulation of a number of HIV restriction factors and downregulation of cyclophilin A (Fig. 8). A lack of incorporation of cyclophilin A into viral particles in the presence of cyclosporine results in attenuated viral replication (35). All these restriction factors are encoded by IFN-stimulated genes (26) and may contribute to the TLR3- and TLR4-induced HIV-inhibitory effects. Surprisingly, IFN-α was not increased in response to the various TLR agonists; however, we observed a quite impressive increase of IFN-β in response to poly(I·C), as reported by Gendelman et al. (23). Irrespective of whether IFN levels were detectable or not, the partial reversion of the TLR3- and 4-mediated anti-HIV activity when neutralizing the IFN axis may be explained by prevention of the upregulation of IFN-dependent HIV restriction factors. The efficacy of blocking the IFN axis by this neutralizing antibody to IFN-αR is convincingly demonstrated by the complete prevention of poly(I·C)-dependent up- and downregulation of HIV restriction factors in MDMs which were pretreated with it (see Fig. S6B in the supplemental material).
As noted above, the anti-HIV effect of TLR8 triggering might occur via an autophagic process (30). Swaminathan et al. recently reported that TLR3 and -4 triggering upregulates microRNA miR-155, which targets several HIV-1-dependent factors involved in the early steps of the HIV replication cycle (29). Like us, they found that the suppression of infection, or a lack thereof, did not correlate with different effects on CD4 or CCR5 expression, type I interferon induction, or the production of proinflammatory cytokines or β-chemokines. For the sake of completeness, we note that Verani et al. demonstrated that the TLR4-dependent release of β-chemokines is a key factor for LPS-induced HIV inhibition (36). β-Chemokines, which are thought to act by interfering with either CCR5 binding or its downregulation, are unlikely to be the sole mechanism, since VSV envelope-pseudotyped particles were blocked, just as the HIV env-pseudotyped particles were. In that sense, Victoria et al. reported that the HIV-inhibitory effect of TLR2 activation was reversed only when the TLR2-triggered IL-10 and β-chemokines were neutralized at the same time (31). While there is a default anti-HIV program in polarized and TLR agonist-primed macrophages, their distinct biological properties make us believe that different anti-HIV mechanisms function in different settings.
Looking at the reduced permissiveness of polarized or primed macrophages, we speculate that the predominant presence of quiescent and thus permissive macrophages when HIV is transmitted contributes to the explosive HIV replication during the acute phase of HIV infection. Later, through an overall change in the milieu or TLR-dependent triggering, macrophages in HIV infection have an activated phenotype that supports productive HIV replication less well. This activated phenotype, however, may be passed on to neighboring cells, which may contribute in turn to the HIV-associated immune activation (37).
In summary, polarization renders macrophages susceptible to a second trigger that then renders them highly active. Here we demonstrate that the TLR2, -3, -4, -5, and -8 agonists reinforce the nonpermissive state of polarized macrophages to HIV. Although both polarization and activation of the TLR axis inhibit HIV replication before integration, one mechanism alone is unlikely to explain the anti-HIV effects. A thorough elucidation of the various mechanisms/differences is highly desirable for understanding their role in pathogenesis and for their potential application in immunomodulation, including as adjuvants in the future.
Supplementary Material
ACKNOWLEDGMENTS
The study was supported by the Theodor and Ida Herzog-Egli Stiftung, the Olga-Mayenfisch Stiftung, and the Swiss National Science Foundation (grant 31003A_135682/1).
We also express our special thanks to Huber Rehrauer (Functional Genomic Centers, Zurich, Switzerland) for creating the heat maps.
E.S. did all experiments, M.-A.R. and L.D. assisted E.S. in a large number of experiments, E.S. and R.F.S. outlined the concept of this work, and R.F.S. wrote the manuscript.
We declare no conflict of interest.
Footnotes
Published ahead of print 18 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01053-14.
REFERENCES
- 1.Pluddemann A, Mukhopadhyay S, Gordon S. 2011. Innate immunity to intracellular pathogens: macrophage receptors and responses to microbial entry. Immunol. Rev. 240:11–24. 10.1111/j.1600-065X.2010.00989.x [DOI] [PubMed] [Google Scholar]
- 2.Sica A, Mantovani A. 2012. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122:787–795. 10.1172/JCI59643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu J, Mohan C. 2010. Toll-like receptor signaling pathways—therapeutic opportunities. Mediators Inflamm. 2010:781235. 10.1155/2010/781235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sasai M, Yamamoto M. 2013. Pathogen recognition receptors: ligands and signaling pathways by Toll-like receptors. Int. Rev. Immunol. 32:116–133. 10.3109/08830185.2013.774391 [DOI] [PubMed] [Google Scholar]
- 5.McCoy CE, O'Neill LA. 2008. The role of Toll-like receptors in macrophages. Front. Biosci. 13:62–70. 10.2741/2660 [DOI] [PubMed] [Google Scholar]
- 6.Koppensteiner H, Brack-Werner R, Schindler M. 2012. Macrophages and their relevance in human immunodeficiency virus type I infection. Retrovirology 9:82. 10.1186/1742-4690-9-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mantovani A, Sica A, Locati M. 2005. Macrophage polarization comes of age. Immunity 23:344–346. 10.1016/j.immuni.2005.10.001 [DOI] [PubMed] [Google Scholar]
- 8.Martinez FO, Sica A, Mantovani A, Locati M. 2008. Macrophage activation and polarization. Front. Biosci. 13:453–461. 10.2741/2692 [DOI] [PubMed] [Google Scholar]
- 9.Cassol E, Cassetta L, Rizzi C, Alfano M, Poli G. 2009. M1 and M2a polarization of human monocyte-derived macrophages inhibits HIV-1 replication by distinct mechanisms. J. Immunol. 182:6237–6246. 10.4049/jimmunol.0803447 [DOI] [PubMed] [Google Scholar]
- 10.Herbein G, Varin A. 2010. The macrophage in HIV-1 infection: from activation to deactivation? Retrovirology 7:33. 10.1186/1742-4690-7-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graziosi C, Gantt KR, Vaccarezza M, Demarest JF, Daucher M, Saag MS, Shaw GM, Quinn TC, Cohen OJ, Welbon CC, Pantaleo G, Fauci AS. 1996. Kinetics of cytokine expression during primary human immunodeficiency virus type 1 infection. Proc. Natl. Acad. Sci. U. S. A. 93:4386–4391. 10.1073/pnas.93.9.4386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biglino A, Sinicco A, Forno B, Pollono AM, Sciandra M, Martini C, Pich P, Gioannini P. 1996. Serum cytokine profiles in acute primary HIV-1 infection and in infectious mononucleosis. Clin. Immunol. Immunopathol. 78:61–69. 10.1006/clin.1996.0009 [DOI] [PubMed] [Google Scholar]
- 13.Liovat AS, Rey-Cuille MA, Lecuroux C, Jacquelin B, Girault I, Petitjean G, Zitoun Y, Venet A, Barre-Sinoussi F, Lebon P, Meyer L, Sinet M, Muller-Trutwin M. 2012. Acute plasma biomarkers of T cell activation set-point levels and of disease progression in HIV-1 infection. PLoS One 7:e46143. 10.1371/journal.pone.0046143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC. 2006. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 12:1365–1371. 10.1038/nm1511 [DOI] [PubMed] [Google Scholar]
- 15.Hofer U, Speck RF. 2009. Disturbance of the gut-associated lymphoid tissue is associated with disease progression in chronic HIV infection. Semin. Immunopathol. 31:257–266. 10.1007/s00281-009-0158-3 [DOI] [PubMed] [Google Scholar]
- 16.Hofer U, Schlaepfer E, Baenziger S, Nischang M, Regenass S, Schwendener R, Kempf W, Nadal D, Speck RF. 2010. Inadequate clearance of translocated bacterial products in HIV-infected humanized mice. PLoS Pathog. 6:e1000867. 10.1371/journal.ppat.1000867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan SY, Speck RF, Ma MC, Goldsmith MA. 2000. Distinct mechanisms of entry by envelope glycoproteins of Marburg and Ebola (Zaire) viruses. J. Virol. 74:4933–4937. 10.1128/JVI.74.10.4933-4937.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schaefer BC, Mitchell TC, Kappler JW, Marrack P. 2001. A novel family of retroviral vectors for the rapid production of complex stable cell lines. Anal. Biochem. 297:86–93. 10.1006/abio.2001.5327 [DOI] [PubMed] [Google Scholar]
- 19.Moore JP, McKeating JA, Weiss RA, Sattentau QJ. 1990. Dissociation of gp120 from HIV-1 virions induced by soluble CD4. Science 250:1139–1142. 10.1126/science.2251501 [DOI] [PubMed] [Google Scholar]
- 20.Audige A, Schlaepfer E, Bonanomi A, Joller H, Knuchel MC, Weber M, Nadal D, Speck RF. 2004. HIV-1 does not provoke alteration of cytokine gene expression in lymphoid tissue after acute infection ex vivo. J. Immunol. 172:2687–2696. 10.4049/jimmunol.172.4.2687 [DOI] [PubMed] [Google Scholar]
- 21.Muller PY, Janovjak H, Miserez AR, Dobbie Z. 2002. Processing of gene expression data generated by quantitative real-time RT-PCR. Biotechniques 32:1372–1374, 1376, 1378–1379 [PubMed] [Google Scholar]
- 22.Vignali DA. 2000. Multiplexed particle-based flow cytometric assays. J. Immunol. Methods 243:243–255. 10.1016/S0022-1759(00)00238-6 [DOI] [PubMed] [Google Scholar]
- 23.Gendelman HE, Friedman RM, Joe S, Baca LM, Turpin JA, Dveksler G, Meltzer MS, Dieffenbach C. 1990. A selective defect of interferon alpha production in human immunodeficiency virus-infected monocytes. J. Exp. Med. 172:1433–1442. 10.1084/jem.172.5.1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gobeil LA, Lodge R, Tremblay MJ. 2013. Macropinocytosis-like HIV-1 internalization in macrophages is CCR5 dependent and leads to efficient but delayed degradation in endosomal compartments. J. Virol. 87:735–745. 10.1128/JVI.01802-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berthiaume EP, Medina C, Swanson JA. 1995. Molecular size-fractionation during endocytosis in macrophages. J. Cell Biol. 129:989–998. 10.1083/jcb.129.4.989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cobos Jimenez V, Booiman T, de Taeye SW, van Dort KA, Rits MA, Hamann J, Kootstra NA. 2012. Differential expression of HIV-1 interfering factors in monocyte-derived macrophages stimulated with polarizing cytokines or interferons. Sci. Rep. 2:763. 10.1038/srep00763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuitemaker H, Kootstra NA, Koppelman MH, Bruisten SM, Huisman HG, Tersmette M, Miedema F. 1992. Proliferation-dependent HIV-1 infection of monocytes occurs during differentiation into macrophages. J. Clin. Invest. 89:1154–1160. 10.1172/JCI115697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kornbluth RS, Oh PS, Munis JR, Cleveland PH, Richman DD. 1989. Interferons and bacterial lipopolysaccharide protect macrophages from productive infection by human immunodeficiency virus in vitro. J. Exp. Med. 169:1137–1151. 10.1084/jem.169.3.1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swaminathan G, Rossi F, Sierra LJ, Gupta A, Navas-Martin S, Martin-Garcia J. 2012. A role for microRNA-155 modulation in the anti-HIV-1 effects of Toll-like receptor 3 stimulation in macrophages. PLoS Pathog. 8:e1002937. 10.1371/journal.ppat.1002937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campbell GR, Spector SA. 2012. Toll-like receptor 8 ligands activate a vitamin D mediated autophagic response that inhibits human immunodeficiency virus type 1. PLoS Pathog. 8:e1003017. 10.1371/journal.ppat.1003017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Victoria S, Temerozo JR, Gobbo L, Pimenta-Inada HK, Bou-Habib DC. 2013. Activation of Toll-like receptor 2 increases macrophage resistance to HIV-1 infection. Immunobiology 218:1529–1536. 10.1016/j.imbio.2013.06.006 [DOI] [PubMed] [Google Scholar]
- 32.Martinez FO, Gordon S, Locati M, Mantovani A. 2006. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J. Immunol. 177:7303–7311. 10.4049/jimmunol.177.10.7303 [DOI] [PubMed] [Google Scholar]
- 33.Carter GC, Bernstone L, Baskaran D, James W. 2011. HIV-1 infects macrophages by exploiting an endocytic route dependent on dynamin, Rac1 and Pak1. Virology 409:234–250. 10.1016/j.virol.2010.10.018 [DOI] [PubMed] [Google Scholar]
- 34.Hughes R, Towers G, Noursadeghi M. 2012. Innate immune interferon responses to human immunodeficiency virus-1 infection. Rev. Med. Virol. 22:257–266. 10.1002/rmv.1708 [DOI] [PubMed] [Google Scholar]
- 35.Zhou D, Mei Q, Li J, He H. 2012. Cyclophilin A and viral infections. Biochem. Biophys. Res. Commun. 424:647–650. 10.1016/j.bbrc.2012.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verani A, Scarlatti G, Comar M, Tresoldi E, Polo S, Giacca M, Lusso P, Siccardi AG, Vercelli D. 1997. C-C chemokines released by lipopolysaccharide (LPS)-stimulated human macrophages suppress HIV-1 infection in both macrophages and T cells. J. Exp. Med. 185:805–816. 10.1084/jem.185.5.805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.d'Ettorre G, Paiardini M, Ceccarelli G, Silvestri G, Vullo V. 2011. HIV-associated immune activation: from bench to bedside. AIDS Res. Hum. Retroviruses 27:355–364. 10.1089/aid.2010.0342 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.