

ABSTRACT
The ability of CD8+ T cells to effectively limit HIV-1 replication and block HIV-1 acquisition is determined by the capacity to rapidly respond to HIV-1 antigens. Understanding both the functional properties and regulation of an effective CD8+ response would enable better evaluation of T cell-directed vaccine strategies and may inform the design of new therapies. We assessed the antigen specificity, cytokine signature, and mechanisms that regulate antiviral gene expression in CD8+ T cells from a cohort of HIV-1-infected virus controllers (VCs) (<5,000 HIV-1 RNA copies/ml and CD4+ lymphocyte counts of >400 cells/μl) capable of soluble inhibition of HIV-1. Gag p24 and Nef CD8+ T cell-specific soluble virus inhibition was common among the VCs and correlated with substantial increases in the abundance of mRNAs encoding the antiviral cytokines macrophage inflammatory proteins MIP-1α, MIP-1αP (CCL3L1), and MIP-1β; granulocyte-macrophage colony-stimulating factor (GM-CSF); lymphotactin (XCL1); tumor necrosis factor receptor superfamily member 9 (TNFRSF9); and gamma interferon (IFN-γ). The induction of several of these mRNAs was driven through a coordinated response of both increased transcription and stabilization of mRNA, which together accounted for the observed increase in mRNA abundance. This coordinated response allows rapid and robust induction of mRNA messages that can enhance the CD8+ T cells' ability to inhibit virus upon antigen encounter.
IMPORTANCE We show that mRNA stability, in addition to transcription, is key in regulating the direct anti-HIV-1 function of antigen-specific memory CD8+ T cells. Regulation at the level of RNA helps enable rapid recall of memory CD8+ T cell effector functions for HIV-1 inhibition. By uncovering and understanding the mechanisms employed by CD8+ T cell subsets with antigen-specific anti-HIV-1 activity, we can identify new strategies for comprehensive identification of other important antiviral genes. This will, in turn, enhance our ability to inhibit virus replication by informing both cure strategies and HIV-1 vaccine designs that aim to reduce transmission and can aid in blocking HIV-1 acquisition.
INTRODUCTION
In acute HIV-1 infection, CD8+ T cells are associated with controlling initial HIV-1 viremia (1, 2), exerting selective pressure on virus replication (3–5), mediating antigen-specific virus inhibition (6), and predicting CD4+ T cell decline (7). Moreover, the magnitude of the acute CD8+ T cell response correlates with the subsequent disease course (8–10). CD8+ T cells are also associated with long-term control of virus replication at low or undetectable levels in a population of HIV+ individuals known as virus controllers (VCs) (6, 11–16). Studying the regulation of CD8+ T cell responses in these VCs provides the opportunity to discover mechanisms of durable control of HIV-1. Previous research has shown that the CD8+ T cell population in VCs is heterogeneous in its ability to inhibit virus replication and that distinct T cells are responsible for virus inhibition (17–20). Further defining specific features of the select CD8+ T cells responsible for the potent control of viremia in VCs will impact the design of efficacious HIV-1 vaccines and therapies (4, 21, 22).
Cells respond to changes in their environment through dynamic regulation of gene expression. Two regulatory processes drive changes in gene expression at the level of mRNA abundance: transcription of new mRNAs and decay of new and existing RNA (23). Control of gene expression is important for the immune system, as rapid initiation of responses is crucial for timely control of infection and prolonged responses can prove detrimental (24). The coordinated regulation of transcription and RNA decay is better able to provide balanced cellular responses than either one independently. A number of studies have demonstrated the importance of regulating both transcription and RNA decay in the immune response (25–30).
In this study, we evaluated the antigen specificity, antiviral activity, and regulation of gene expression of the soluble CD8+ T cell responses. We consequently have begun to define the roles of transcriptional and posttranscriptional gene regulation in genes that correspond to virus inhibition within a cohort of virus controller patients. Gag p24 and Nef-specific CD8+ T cell-mediated virus inhibition was associated with increased abundance of mRNAs encoding macrophage inflammatory proteins (MIP-1α, MIP-1αP, and MIP-1β), gamma interferon (IFN-γ), lymphotactin (XCL1), tumor necrosis factor receptor superfamily member 9 (TNFRSF9), and granulocyte-macrophage colony-stimulating factor (GM-CSF). The abundance of the mRNAs of these cytokines was dependent on changes in both transcription and mRNA decay, with evidence for potential differences in the regulation of mRNA between Nef- and Gag-specific CD8+ T cells.
MATERIALS AND METHODS
Patient cohorts.
Eleven antiretroviral therapy (ART)-naive HIV-1-infected virus controllers (Table 1) (maintaining plasma HIV-1 loads of <5,000 RNA copies/ml and CD4+ lymphocyte counts of >400 cells/μl) and one ART-experienced individual (VC15) enrolled through the Infectious Diseases Clinic at the Duke University Medical Center and with CD8+ T cell-mediated virus inhibition were studied here. VC15 was previously on ART but naturally controlled his/her viremia (maintaining a VL of <5,000 copies/ml and a CD4 count of >800 cells/μl) for two years posttherapy before being enrolled in our study. While in the study, VC15 had VLs of 1,590 to 2,950 copies/ml and CD4 counts of 721 to 801 cells/μl. VC27 maintained VLs of <100 to 2,690 copies/ml with the exception of two draw dates on which his/her VLs were 5,190 and 5,360 copies/ml. Seven VCs were HLA typed, and only two of these patients had alleles known to be associated with CD8+ T cell control. Three patients (VC28, VC29, and VC30) maintained viral loads below 48 copies/ml (infected for >6 years, with viral loads measured for >8 months). Healthy uninfected donors were recruited through the Duke Virologic Basis for Specific Immune Defects in AIDS program (kindly provided by Kent Weinhold, Department of Surgery, Duke University). The studies were reviewed and approved by the Duke University Medical Center Institutional Review Board, and all participants provided written informed consent.
TABLE 1.
HIV-1 VCs
| Patient ID (HIV-1+)a | Geographic location (presumed clade B) | Viral load (HIV-1 RNA copies/ml)b | CD4 count (cells/μl)b | Yr of diagnosis |
|---|---|---|---|---|
| VC9 | USA | 1,020 | 719 | 2001 |
| VC11 | USA | 783 | 750 | 2006 |
| VC15 | USA | 2,950 | 721 | |
| VC16 | USA | 878 | 671 | 2006 |
| VC20 | USA | 1,040 | 704 | 2007 |
| VC23 | USA | <48–444c | 462–467c | |
| VC24 | USA | 2,810c | 686–749 | 2004 |
| VC26 | USA | 131 | 569 | 2009 |
| VC27 | USA | 5,360 | 963 | 2002 |
| VC28 | USA | <48 | 966–1347c | 1999 |
| VC29 | USA | <48 | 1,420–1,690c | 2007 |
| VC30 | USA | <48 | 1,345–1,573c | 2004 |
Virus controllers were recruited from the Adult Infectious Diseases Clinic, Duke University Medical Center, and had presumed clade B infections (due to the geographical location).
Viral loads and CD4 counts are measured values for samples used in experiments.
A range of samples from multiple draw dates were used. One of the VL values could not be determined for one of the draw dates used for VC24. During 5.8 years of enrollment, VC24 had virus loads of 661 to 2,810 copies/ml.
Transmitted/founder virus.
Replication-competent virus stocks from a full-length infectious molecular clone (IMC) expressing a transmitted/founder virus (CH040.c) were generated as described previously (6, 17, 31, 32).
Antigen-specific sVIA.
HIV-1-specific soluble virus inhibition assays (sVIAs) with primary CD8+ T cells were performed as previously described (6) with slight modifications. Twelve pools of HIV-1 peptides (potential T cell epitopes [PTE]; NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH [HIV-1 PTE peptides]) (33) represent the following HIV-1 regions (based on HXB2 numbering): Env1, amino acids 4 to 296 (gp120); Env2, amino acids 297 to 488 (gp120); Env3, amino acids 489 to 602 (gp41); Env4, amino acids 603 to 840 (gp41); Gag1, amino acids 1 to 128 (p17); Gag2, amino acids 131 to 361 (p24); Gag3, amino acids 362 to 486 (p17); Pol1, amino acids 1 to 152 (protease); Pol2, amino acids 156 to 447 (p51); Pol3, amino acids 452 to 709 (p51 plus p15); Pol4, amino acids 711 to 988 (p31); and Nef, amino acids 1 to 193. These pools were used (0.2 μg/ml) to stimulate isolated activated CD8+ T cells from HIV-1+ virus controllers and seronegative patients for 5.5 h. Poststimulation, CD8+ cells were placed in the upper chambers of a 96-well transwell plate, with the bottom chamber containing TZM-bl cells, DEAE-dextran, and CH040.c virus, which was added at the same time as the CD8+ T cells. After 48 h, the TZM-bl cells were lysed and the firefly luciferase content of the lysate was measured. Virus inhibition was calculated as the log reduction in relative light units (RLU) (luciferase) of wells with CD8+ T cells compared to control wells without CD8+ T cell effectors. The cutoff for significant virus inhibition (>0.39-log-unit reduction) was determined using seronegative control subjects.
Multiparameter intracellular-cytokine-staining assay.
Flow cytometric analyses of HIV-1-specific CD8+ T cells were performed as previously described (5, 6, 17). Briefly, peripheral blood mononuclear cells (PBMCs) were stimulated with the PTE peptide pools as described above for 5.5 h. Stimulation with 0.2 μg/ml staphylococcal enterotoxin B, also for 5.5 h, was used as a positive control. The titer of each antibody was determined to obtain the saturating concentration used for the final staining. Stimulations were conducted in the presence of 0.5 μg/ml anti-CD107a phycoerythrin (PE)-Cy5 (clone H4A3; eBioscience), 5 μg/ml brefeldin A (Sigma), and 4 μl/6 ml Golgi Stop (BD Pharmingen) for 5.5 h at 37°C in 5% CO2. After washing, the cells were stained with a viability indicator (LIVE/DEAD Fixable Aqua Dead Cell Stain Kit; Molecular Probes) in phosphate-buffered saline for 20 min at room temperature. The cells were then washed and stained for 20 min at 4°C with a surface stain cocktail containing anti-CD3–APC-H7 (clone SK7; BD Biosciences), anti-CD4-BV605 (clone RPA-T4; BD Horizon), anti-CD8-PacBlue (clone RPA-T8; BD Pharmingen), anti-CD27-Cy7-PE (clone M-T271; BD Pharmingen), anti-CD45RO-ECD (clone UCHL1; Beckman Coulter), and anti-CCR7-Alexa Fluor 700 (clone 150503; BD Pharmingen). The PBMCs were subsequently washed twice and then fixed and permeabilized with fixation/permeabilization solution (BD Cytofix/Cytoperm) for 20 min at 4°C. After incubation, the cells were washed twice in Perm/Wash buffer (BD Perm/Wash) diluted 1:10 with distilled water. The cells were then stained with anti-IFN-γ–BV650 (4S.B3; Biolegend), anti-interleukin 2 (IL-2)–allophycocyanin (APC) (clone MQ1-17H12; BD Pharmingen), anti-MIP-1α-fluorescein (clone 93342; R&D Systems), anti-MIP-1β-PE (clone D21-1351; BD Pharmingen), and anti-TNF-α–peridinin chlorophyll protein (PerCP)–cyanine 5.5 (clone MAb11; eBioscience) for 45 min at 4°C. After washing and fixation, all the samples were acquired on a custom-made LSRII (BD Bioscience, San Jose, CA) within the next 24 h. Gates were set to include singlet events, lymphocytes, live CD3+ cells, and CD4+ and CD8+ subsets. From the total CD4+ and CD8+ populations, the naive subset was identified as CD45RO- CD27+. This subset was excluded from the subsequent analysis, including only the memory population. Antigen-specific populations were identified within the memory population as single-function cells shown in the sequential single cytokine/chemokine/degranulation gates. Responses were considered positive if the percentage of antigen-specific cells was 3-fold above the background and greater than 0.05% after background subtraction. Data analysis was performed using FlowJo 9.6.4 software (TreeStar Inc.).
4sU incorporation and PCR analysis.
To assess transcription and mRNA decay independently, 4-thiouridine (4sU) (200 μM) was added 1 h prior to harvest of cells (4.5 h after the start of peptide stimulation). Cells were harvested in 1 ml of TRIzol, and RNA was extracted following the manufacturer's protocols (Life Technologies). Separation of 4sU-labeled RNA from unlabeled RNA was performed using a highly efficient (>90%) biotinylation method as described previously (34) with minor modifications. Notably, the RNA was DNase (Ambion) treated after initial RNA extraction from cells, and streptavidin MyOne C1 Dynabeads (Invitrogen) were used to extract biotinylated 4sU RNA. Subsequently, three populations of RNAs for each sample were reverse transcribed into cDNA using an iScript kit (Bio-Rad): total RNA (T), labeled RNA (L), and unlabeled RNA (U). Real-time PCR was performed on each population of RNA and then normalized for the relative amount of input RNA. The abundance of mRNA was represented by the measurement of the total RNA sample (T). Net transcription was represented by the labeled fraction of mRNA (L). The decay rate (DR) was calculated from measurements of labeled (L) and unlabeled (U) mRNA, as a function of L/U: ln(1 − L/U). An apparent RNA half-life was calculated using the decay rate, −t × [ln(2)/DR], where t is the time of 4sU incorporation (1 h for the purposes of these experiments). Two assumptions of this method are that transcription and stability are constant over the period of measurement. These assumptions result in more conservative conclusions regarding changes in stability between samples, especially among short-lived mRNAs. The mRNA stabilities calculated using 4sU-based measurements correlate very well with those calculated using other established methods, such as actinomycin D (34). To compensate for a bias against labeling of short or U-depleted mRNAs, we calculated the stochastic likelihood of labeling based on conservative incorporation rates of 1 4sU for every 100 uridines, taken from previous publications (34–36) and our own measurements (data not shown), using an equation described previously (37). All half-lives were normalized to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), where the GAPDH half-life was defined as 8 h based on previous publications and other global analyses (38, 39). This method led to calculated half-lives that were consistent across the patients tested. The average apparent half-life (in hours) of RANTES mRNA was 5.56 ± 0.73, the half-life of the IFN-γ mRNA was 0.63 ± 0.039, that of MIP-1α mRNA was 0.82 ± 0.10, that of MIP-1αP mRNA was 1.05 ± 0.19, that of MIP-1β mRNA was 2.30 ± 0.37 h, that of GM-CSF mRNA was 0.96 ± 0.12, and that of TNFRSF9 mRNA was 1.57 ± 0.20 (see Fig. 5A). As expected, the half-life of XCL mRNA was more varied among different patients due to the longer XCL half-life (average, 8.39 h) and the fact that RNA stability calculations are prone to greater error with longer intrinsic mRNA survival.
FIG 5.

Increases in RNA stability of IFN-γ, MIP1α, MIP1α-AP, MIP1β, and GM-CSF contribute to the observed increase in RNA expression. (A) Calculated apparent half-lives are consistent across unstimulated cells from patient cohorts. Apparent half-lives were calculated for the unstimulated CD8+ T cells from 12 VCs and 3 seronegative samples. The lines represent the mean values, while the error bars represent standard errors of the mean. (B) Raw GAPDH-normalized apparent half-life values for unstimulated and 5.5-h p24-stimulated CD8+ T cells from VC30. The results are from three independent experiments. The lines represent the median values. (C) Values for the fold change in apparent half-life of p24-stimulated CD8+ T cells compared to unstimulated cells from patients with 5.5-h p24-specific inhibition (triangles, n = 6: VC11, -23, -26, -27, -28, and -29) and those without (circles, n = 5: 3 HIV-1-seronegative patients and VC16 and -24). The lines indicate the medians. (D) Values for the fold change in apparent half-life of 5.5-h Nef-stimulated CD8+ T cells compared to unstimulated cells from patients with Nef-specific inhibition (diamonds, n = 4: VC26, -27, -29, and -16) and those without (squares, n = 7: 3 HIV-1-seronegative patients and VC11, -23, -28, and -24). The lines indicate the medians. The P values are indicative of significant difference in fold changes in apparent half-life in patients with antigen-specific inhibition compared to those without. The P values were calculated using the Wilcoxon exact test, controlling for false discovery using the Benjamini and Hochberg method. (E and F) The sum of changes in transcription and stability fully accounts for observed changes in mRNA abundance in Gag p24 and Nef. Boxes indicate the median as well as the upper and lower quartiles, with whiskers being minimum and maximum values. Lines at y = 1 indicate no change in value (compared to that for unstimulated). The observed total mRNA abundance across VCs, the predicted mRNA abundances based on the observed transcription rates only, the predicted mRNA abundances based on the measured apparent half-lives (stability) only, and the predicted mRNA abundances based on calculations that included observed net transcription and apparent half-life values are shown. All values were normalized to the observed total mRNA abundance of GAPDH of each message.
Time course prediction modeling.
4sU labeling was performed at 1-h intervals for 6.5 h after stimulation (see Fig. 7A), resulting in continuous measurement of transcription. The amounts of RNA in each population were measured as described above. Predictions of RNA abundance that assumed a constant decay rate were determined based on a previously developed model (40) that uses the unstimulated RNA abundance and then iteratively adds amounts of transcribed RNA in each hour and subtracts a decay rate dependent on the amount of decayed RNA in that hour: for time i, Ti = T0 + [Ni − Ti − 1 × DR0]i, where T0 is the initial measured total, Ni is the net transcription at time i, and DR0 is the initial calculated decay rate. Because several of the mRNAs are short (∼1,000 bp) and contain relatively few uridines, we corrected for the stochastic likelihood of failing to include a 4sU in place of a uridine for an entire mRNA, as outlined previously (37). Subsequently, modeling the optimized decay rate solved for the decay rate in the above equation, since the totals are observed: DRoptimized = (Ti−1 + Ni − Ti)/−Ti−1.
FIG 7.

Temporal expression of antiviral cytokines is quicker and more robust in CD8+ T cells with higher RNA stability. (A) Diagram of time course experiments. Cells were labeled with 4sU for 1-h segments after stimulation for 6.5 h with p24 peptide, and then the RNA was separated to measure the net transcription. The levels of total mRNA were predicted using a prestimulation total, net transcription over each hour-long time period, and the prestimulation decay rate. These predictions were then compared to observed total mRNA abundances. (B and C) For unchanged (RANTES) (B) or transcriptionally induced (TNFRSF9) (C) genes, the model (red lines) accurately predicted the observed levels of mRNA abundance (blue lines). (D to F) For IFN-γ (D), MIP-1α (E), and MIP-1β (F), however, the levels of total mRNA observed were much greater than a constant decay rate predicted. (G to K) We calculated optimized mRNA stabilities based on the observed mRNA totals using the following formula: DRoptimized = (Ti − 1 + Ni − Ti)/−Ti − 1, where Ti is the measured total mRNA at time i and Ni is the measured net transcription at time i. The optimized stability of mRNA increased in coordination with increases in net transcription, so that mRNAs underwent a “peaked” stability response that correlated with the transcriptional induction. Error bars indicate standard error.
Data from two biological replicates were independently treated through the modeling and then averaged using fold change measurements in predicted and observed abundances.
Statistical analyses.
Statistical analyses were performed using GraphPad Prism (GraphPad Software) and SAS v9.3 (SAS Institute). Correlations between the cytokine expression level and virus inhibition were calculated using the Spearman's rank correlation coefficient (GraphPad Software). Appropriate SAS PROC tests were used to calculate raw P values using Wilcoxon exact tests and for controlling the false-discovery rate (FDR) using the Benjamini and Hochberg method (41).
RESULTS
Soluble HIV-1 inhibition from p24- and Nef-specific CD8+ T cells.
We first evaluated the antigen specificity of CD8+ T cells that inhibit HIV-1 via soluble mechanisms by using an HIV-1-specific transwell sVIA (6). Primary CD8+ T cells from VCs were stimulated with Env, Nef, Pol, or Gag HIV-1 PTE peptide (33) pools for 5.5 h and then tested for the ability to inhibit an R5-tropic clade B founder virus, CH040.c (31, 32). In 11 of 12 VC patients (Table 1), antigen-specific stimulation of CD8+ T cells mediated soluble inhibition of HIV-1 replication (Table 2). The most common epitope specificities of cells that mediated soluble virus inhibition were Gag p24 (Gag2) (9 out of 12 VC patients) and Nef (7 out of 12 VC patients). The second most common CD8+ T cell specificities that inhibited virus replication were Gag p17 (Gag1) and protease (Pol1) (4 out of 12 VC patients each).
TABLE 2.
Antigen-specific CD8+ T cell inhibition of HIV-1 replication
| Patient ID | Reduction value (log10)a |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unstimulated | Env |
Gag |
Pol |
Nef | |||||||||
| E1 | E2 | E3 | E4 | G1 | G2 | G3 | P1 | P2 | P3 | P4 | |||
| VC9 | <0.39 | <0.39 | <0.39 | 0.44 | 0.53 | 0.45 | 0.64 | <0.39 | 0.56 | 0.63 | 0.52 | 0.61 | 0.74 |
| VC11 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | 0.40 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 |
| VC15 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | 0.42 | <0.39 | <0.39 | 0.45 | 0.50 | <0.39 | 0.47 |
| VC16 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | 0.44 | <0.39 | <0.39 | <0.39 | 0.42 |
| VC20 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | 0.48 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | 0.42 |
| VC23 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | 0.40 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 |
| VC24 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 |
| VC26 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | 0.43 | 0.40 | <0.39 | <0.39 | <0.39 | 0.45 |
| VC27 | <0.39 | <0.39 | <0.39 | <0.39 | 0.54 | 0.59 | 0.63 | <0.39 | 0.46 | <0.39 | 0.42 | 0.41 | 0.57 |
| VC28 | <0.39 | <0.39 | 0.47 | 0.41 | 0.42 | 0.43 | 0.42 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 |
| VC29 | <0.39 | 0.51 | 0.41 | <0.39 | <0.39 | 0.41 | 0.47 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | 0.49 |
| VC30 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | 0.45 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 | <0.39 |
| Totalb | 0 | 1 | 2 | 2 | 3 | 4 | 9 | 1 | 4 | 2 | 3 | 2 | 7 |
The log10 reduction values for HIV-1 peptide-specific soluble inhibition by CD8+ T cells from HIV-1 VCs are shown. An sVIA was used to measure the inhibitory capacities of antigen-stimulated primary CD8+ T cells from 12 HIV-1+ virus controllers. Virus inhibition was measured as log reduction in CH040.c (transmitted/founder virus) replication without CD8+ T cell effectors. Values of >0.39-log-unit reduction in virus replication compared to a no-CD8+ T cell control indicate significant inhibition and are reported (boldface). A value of <0.39 (lightface) indicates a nonsignificant log10 reduction value. The peptide pools were divided as follows to represent HIV-1 regions based on the HXB2 amino acid number: Env1 (E1), amino acids 4 to 296 (gp120); Env2 (E2), amino acids 297 to 488 (gp120); Env3 (E3), amino acids 489 to 602 (gp41); Env4 (E4), amino acids 603 to 840 (gp41); Gag1 (G1), amino acids 1 to 128 (p17); Gag2 (G2), amino acids 131 to 361 (p24); Gag3 (G3), amino acids 362 to 486 (p17); Pol1 (P1), amino acids 1 to 152 (protease); Pol2 (P2), amino acids 156 to 447 (p51); Pol3 (P3), amino acids 452 to 709 (p51 plus p15); Pol4 (P4), amino acids 711 to 988 (p31); Nef, amino acids 1 to 193.
Total number of values of >0.39-log-unit reduction in virus replication compared to a no-CD8+ T cell control.
HIV-1 antigen-specific CD8+ T cells have increased expression of IFN-γ, MIP-1α, MIP-1αP, MIP-1β, GM-CSF, XCL1, and TNFRSF9 that correlate with virus inhibition.
The effector function of CD8+ T cells includes the antiviral activity of cytokines (42–44). In order to identify correlates of antiviral control, we assessed the expression of a panel of cytokines, including MIP-1α, MIP-1αP, MIP-1β, IFN-γ, XCL1, GM-CSF, RANTES, and TNFRSF9. We and others have shown that IFN-γ and the β-chemokines MIP-1α and MIP-1β are associated with CD8+ T cell inhibition of HIV-1 (17, 42, 44–48). MIP-1α, MIP-1αP, and MIP-1β bind CCR5 and block the entry of R5-tropic viruses (including transmitted/founder viruses) in CD4+ T cells (44, 49, 50). The gene copy number of MIP-1αP varies greatly among the population and has been linked to HIV-1 infection and disease progression (51). XCL1 is a recently characterized CD8+ T cell-derived anti-HIV chemokine that works to block HIV-1 attachment via direct interaction with gp120 (52). The interaction between TNFRSF9 and its ligand, TNFSF9, is known to enhance the proliferation and activity of CD8+ T cells (53), while GM-CSF can also influence the T cell response (54). To determine whether these cytokines are upregulated in CD8+ T cells from VCs with antigen-specific antiviral activity, we first measured mRNA cytokine levels, in three independent experiments, in primary CD8+ T cells from a virus controller patient, VC30, after a 5.5-h Gag p24-peptide stimulation and compared the cytokine expression to that of unstimulated autologous cells. Compared to unstimulated cells, p24-stimulated CD8+ T cells had increases in mRNAs for IFN-γ, MIP-1α, MIP-1αP, MIP-1β, TNFRSF9, XCL1, and GM-CSF (Fig. 1A). In contrast, no change was seen in the mRNA expression of RANTES, another β-chemokine with HIV-1-suppressive activity (44).
FIG 1.
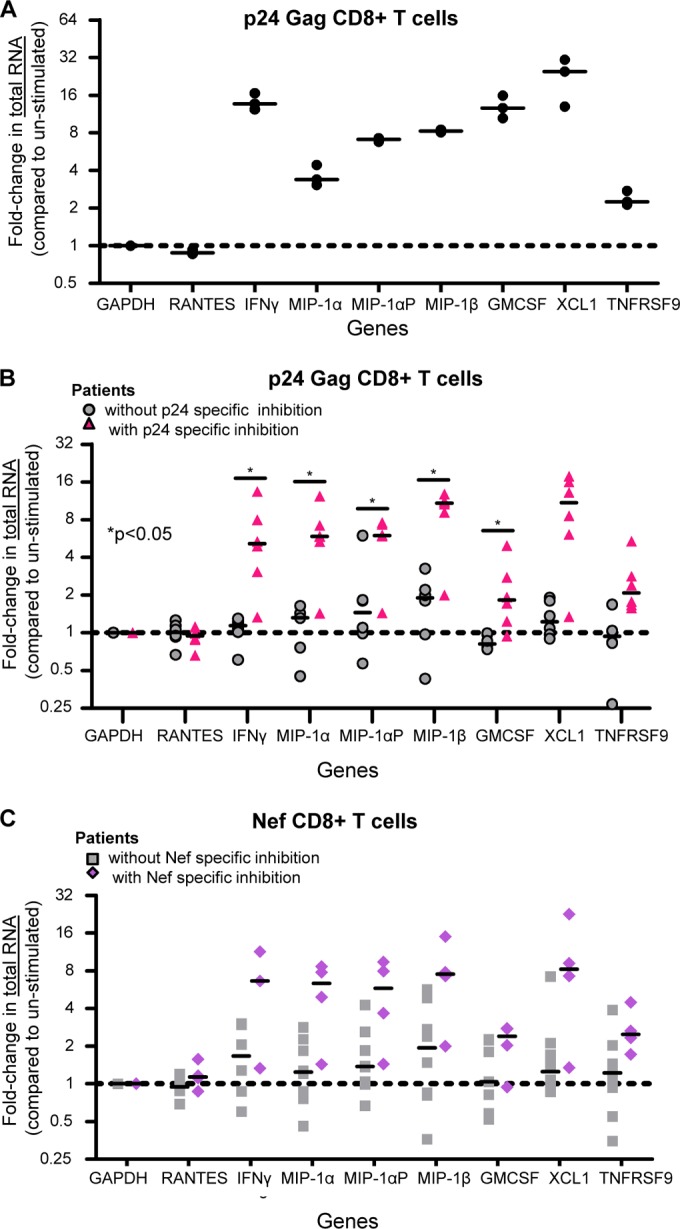
CD8+ T cells from VCs with antigen-specific inhibition have increased expression of IFN-γ, MIP1α, MIP1α-AP, MIP1β, GM-CSF, TNFRSF9, and XCL1 mRNAs. (A) Values for fold changes (5.5 h p24 stimulation/unstimulated) in total mRNA abundance (real-time [RT]-PCR) in CD8+ T cells from an HIV-1+ virus controller (VC30). The results are from three independent experiments. The lines represent the median values. (B) Fold changes in total mRNA levels (5.5-h p24-stimulated CD8+ T cells compared to unstimulated cells) from patients with p24-specific inhibition (triangles, n = 6: VC11, -23, -26, -27, -28, and -29) and those without (circles, n = 5: 3 HIV-1-seronegative patients and VC16 and -24). The lines indicate the medians. *, P < 0.05. (C) Values for the fold change in mRNA levels (5.5-h Nef-stimulated CD8+ T cells compared to unstimulated cells) from patients with Nef-specific inhibition (diamonds, n = 4: VC26, -27, -29, and -16) and those without (squares, n = 7: 3 HIV-1-seronegative patients and VC11, -23, -28, and -24). The lines indicate the medians. mRNA abundance was determined via primer-specific PCR. The P values are indicative of significant differences in fold changes in abundance in patients with antigen-specific inhibition compared to those without. The P values were calculated using the Wilcoxon exact test, controlling for false discovery using the Benjamini and Hochberg method.
We next measured the levels of mRNA expression of the aforementioned cytokines and the CXCR4-interacting cytokines macrophage-derived chemokine (MDC) (55) and thymus- and activation-regulated chemokine (TARC) (56) in unstimulated and 5.5-h-stimulated (Gag p24, Nef, or Gag p17) CD8+ T cells from 10 additional VC patients. For the Gag p24 experiments, we divided the patients into two groups: those with p24-specific soluble virus inhibition (VC11, -23, -26, -27, -28, and -29; n = 6) and those without (3 HIV-1-seronegative patients, VC16, and VC24; n = 5) (Table 2). The amounts of mRNA in p24-stimulated CD8+ T cells from VCs without p24-specific inhibition and seronegative patients were largely unchanged compared to unstimulated cells (Fig. 1B). However, the levels of the mRNAs that encode IFN-γ, MIP-1α, MIP-1αP, MIP-1β, GM-CSF, TNFRSF9, and XCL1 increased in p24-stimulated CD8+ T cells from VCs with p24-specific inhibition compared to unstimulated autologous cells (Fig. 1B). Comparing the fold increases in cells from patients with p24-specific inhibition to those from patients without p24-specific inhibition, the increases in IFN-γ, MIP-1α, MIP-1αP, MIP-1β, and GM-CSF are statistically significantly (P < 0.05; Wilcoxon exact test controlled for the false-discovery rate using the Benjamini and Hochberg method [41]) higher in those with antigen-specific inhibition. CD8+ T cells from VCs with p24-specific inhibition had increased mRNA levels for at least three of the cytokines, indicating some degree of polyfunctionality in their responses. We next compared Nef-stimulated CD8+ T cells from patients with Nef-specific inhibition (VC26, -27, -29, and -16; n = 3) and those without (3 HIV-1-seronegative patients and VC11, -23, -28, and -24; n = 7). Nef-stimulated cells had an upregulation in mRNA expression for the queried genes, similar to p24-stimulated cells, although these values did not reach statistical significance when corrected for multiple comparisons; however, some of the unadjusted P values did reach significance (Fig. 1C and Table 3). These changes were specific, as no change was observed in mRNA levels of RANTES in any of the subject cohorts or under any stimulation conditions, consistent with our previous report that RANTES did not correlate with virus inhibition (6). The levels of MDC and TARC were also unchanged (data not shown) and serve as additional negative controls. Because Gag p17 (Gag3)-specific inhibition was not common among the VCs (Table 2), we compared the abundances of cytokine mRNAs in p24- and Nef-stimulated CD8+ T cells to those in autologous Gag p17-stimulated cells to determine if the observed changes in gene expression were global responses to antigen stimulation or were antigen specific. The fold changes in mRNA abundance observed when p24- and Nef-stimulated cells were compared to p17-stimulated cells (data not shown) mirrored those in comparison to unstimulated cells, indicating that the upregulation of gene expression is antigen specific. The increased expression of IFN-γ, MIP-1α, MIP-1αP, MIP-1β, and GM-CSF was statistically significant in p24- compared to p17-stimulated CD8+ T cells (P < 0.05; Wilcoxon exact test, controlled for the false-discovery rate using the Benjamini and Hochberg method [41]); however, the observed increased abundance in Nef-stimulated samples did not reach statistical significance.
TABLE 3.
P values for fold changes in total RNA, transcription rate, and RNA stability
| Variable | Gene | Fold change P valuea |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Gag p24-stimulated cells |
Nef-stimulated cells |
||||||||
| Compared to unstimulated |
Compared to Gag p17 |
Compared to unstimulated |
Compared to Gag p17 |
||||||
| Raw | FDR corrected | Raw | FDR corrected | Raw | FDR corrected | Raw | FDR corrected | ||
| Total mRNA | IFN-γ | 0.004 | 0.030 | 0.008 | 0.030 | 0.042 | 0.091 | 0.117 | 0.185 |
| MIP-1α | 0.004 | 0.030 | 0.008 | 0.030 | 0.042 | 0.091 | 0.183 | 0.257 | |
| MIP-1αP | 0.004 | 0.030 | 0.016 | 0.054 | 0.042 | 0.091 | 0.262 | 0.349 | |
| MIP-1β | 0.004 | 0.030 | 0.008 | 0.030 | 0.042 | 0.091 | 0.067 | 0.127 | |
| GM-CSF | 0.004 | 0.030 | 0.008 | 0.030 | 0.164 | 0.245 | 0.833 | 0.875 | |
| TNFRSF9 | 0.017 | 0.054 | 0.032 | 0.080 | 0.024 | 0.068 | 0.833 | 0.875 | |
| XCL1 | 0.017 | 0.054 | 0.008 | 0.030 | 0.073 | 0.133 | 0.383 | 0.454 | |
| Net transcription | IFN-γ | 0.004 | 0.030 | 0.008 | 0.030 | 0.315 | 0.389 | 0.383 | 0.454 |
| MIP-1α | 0.052 | 0.104 | 0.008 | 0.030 | 0.109 | 0.180 | 0.667 | 0.727 | |
| MIP-1αP | 0.004 | 0.030 | 0.008 | 0.030 | 0.230 | 0.312 | 0.383 | 0.454 | |
| MIP-1β | 0.004 | 0.030 | 0.008 | 0.030 | 0.073 | 0.133 | 0.117 | 0.185 | |
| GM-CSF | 0.052 | 0.104 | 0.008 | 0.030 | 0.788 | 0.848 | 0.517 | 0.595 | |
| TNFRSF9 | 0.017 | 0.054 | 0.008 | 0.030 | 0.315 | 0.389 | 1.000 | 1.000 | |
| XCL1 | 0.004 | 0.030 | 0.008 | 0.030 | 0.315 | 0.389 | 0.183 | 0.257 | |
| Apparent half-life | IFN-γ | 0.017 | 0.054 | 0.095 | 0.163 | 0.024 | 0.068 | 0.033 | 0.080 |
| MIP-1α | 0.082 | 0.147 | 0.151 | 0.235 | 0.109 | 0.180 | 0.033 | 0.080 | |
| MIP-1αP | 0.004 | 0.030 | 0.032 | 0.080 | 0.315 | 0.389 | 0.033 | 0.080 | |
| MIP-1β | 0.052 | 0.104 | 0.222 | 0.306 | 0.024 | 0.068 | 0.067 | 0.127 | |
| GM-CSF | 0.537 | 0.609 | 0.095 | 0.163 | 0.927 | 0.950 | 0.183 | 0.257 | |
| TNFRSF9 | 0.177 | 0.257 | 1.000 | 1.000 | 0.927 | 0.950 | 0.667 | 0.727 | |
| XCL1 | 0.429 | 0.500 | 0.310 | 0.389 | 0.164 | 0.245 | 0.667 | 0.727 | |
P values were calculated using the Wilcoxon exact test to compare fold changes for Gag p24- and Nef-stimulated cells for patients with and without antigen-specific inhibition. Two P values are reported: the raw P value, which is not corrected for multiple comparisons, and the FDR-corrected P value, which controls for the false-discovery rate. The FDR-corrected P values were calculated using the Benjamini and Hochberg method (41). Boldface indicates significance (P < 0.05), and lightface indicates nonsignificance (P > 0.05).
We have previously shown that antiviral activity is strongly associated with increased numbers of memory CD8+ T cells expressing CD107a or MIP-1β (17) and that soluble-protein expression of MIP-1α, MIP-1β, and IFN-γ strongly correlates with contact-mediated CD8+ T cell virus inhibition (48), as well as soluble CD8+ T cell-mediated virus inhibition (6). Here, we also determined whether cell surface staining of MIP-1β and CD107 was associated with CD8+ T cells that mediate virus inhibition. We measured the expression of these cytokines in a memory CD8+ T cell subset of virus controllers with and without p24-specific inhibition by multiparameter flow cytometry. The percentage of CD8+ T cells from patient VC23 (Gag p24-specific inhibition) expressing MIP-1β compared to unstimulated cells increased from 2.7% to 4.7% of memory cells (Fig. 2), while the expression of CD107 increased from 0.5% to 2.8%. These changes were not seen in CD8+ T cells without soluble virus inhibition (Fig. 2, VC24). Thus, MIP-1β and CD107 are associated with memory CD8+ T cell antigen-specific soluble inhibition of HIV-1, consistent with cytokine mRNA levels translating to changes in protein expression.
FIG 2.

Expression of MIP-1β and CD107 proteins in p24-stimulated and unstimulated (Un-stim) memory CD8+ T cells is also increased. (A) CD8+ T cells from VC23 (p24-specific inhibition) and VC24 (no p24-specific inhibition) were analyzed by multiparameter flow cytometry after a 5.5-h p24 stimulation. Live CD3+ cells were gated for a CD8 single-positive population, to which an exclusion gate was applied for naive cells (CD27+ CD45RO−). The expression of MIP-1β or CD107, markers previously shown to be independently associated with virus inhibition, was measured in the nonexcluded (memory) cells. (B) Summary of percent increases in cytokine expression in p24-stimulated cells compared to unstimulated cells. Expression of MIP-1β and CD107 protein increased in memory cells of VCs with p24-specific inhibition. This increase in MIP-1β mirrors the increases seen in MIP-1β RNA.
CD8+ T cells with antigen-specific inhibition exhibit an increased net transcription for IFN-γ, MIP-1α, MIP-1αP, MIP-1β, GM-CSF, XCL1, and TNFRSF9.
To investigate the mechanisms behind the upregulation of IFN-γ, MIP-1α, MIP-1αP, MIP-1β, GM-CSF, TNFRSF9, and XCL1 mRNAs in CD8+ T cells with p24- and Nef-specific inhibition of HIV-1, we performed 4sU analysis after antigen stimulation (34). 4sU analysis uses incorporation of 4sU into nascent RNAs to physically isolate mRNAs synthesized before 4sU addition from those synthesized after 4sU addition. For a fixed period of 4sU incorporation, comparing 4sU-labeled mRNA across samples allows the calculation of net transcription (total transcription minus the amount of 4sU-labeled RNA that decays prior to cell harvest) while using the ratio of labeled to unlabeled mRNA to infer a decay rate (34). The method has been employed extensively to simultaneously measure mRNA transcription and decay across a variety of systems (34, 40, 57, 58) with greater reproducibility than using actinomycin D (34). We added 4sU to cells 4.5 h after the start of antigen stimulation and then allowed 4sU incorporation for 1 h before lysing the cells and harvesting RNA. In triplicate experiments in CD8+ T cells from patient VC30, we observed highly reproducible increases in the net transcription of several cytokines in p24-stimulated compared to autologous unstimulated cells (Fig. 3A). Notably, however, the change in net transcription was consistently less than the change we observed in total mRNA abundance. This trend was apparent in the other VCs with p24-specific inhibition: in a majority of the cases in which there was a significant increase in gene expression (compared to patients without antigen-specific inhibition), we also observed a less dramatic, albeit significant (P < 0.05; Wilcoxon exact test, controlling for the FDR [41]), increase in the rate of net transcription (Fig. 3B and Table 3). The exceptions to this observation were XCL1 and TNFRSF9, for which the measured changes in net transcription were greater than the changes in abundance. The same trend was also seen in patients with Nef-specific inhibition, although the changes in net transcription were smaller and did not reach statistical significance (Fig. 3C). In patients without antigen-specific virus inhibition, we observed no changes in net transcription. Comparison to Gag p17-stimulated cells (data not shown) mirrored comparison to unstimulated autologous cells. The notable difference between changes in total mRNA and net mRNA transcription (Fig. 4A and B) indicated the potential for the contribution of additional regulatory mechanisms to the control of gene expression. Though there are several possibilities for the other mechanism(s) involved, including the temporal dynamics of transcription, one logical explanation is stabilization of the mRNA itself.
FIG 3.
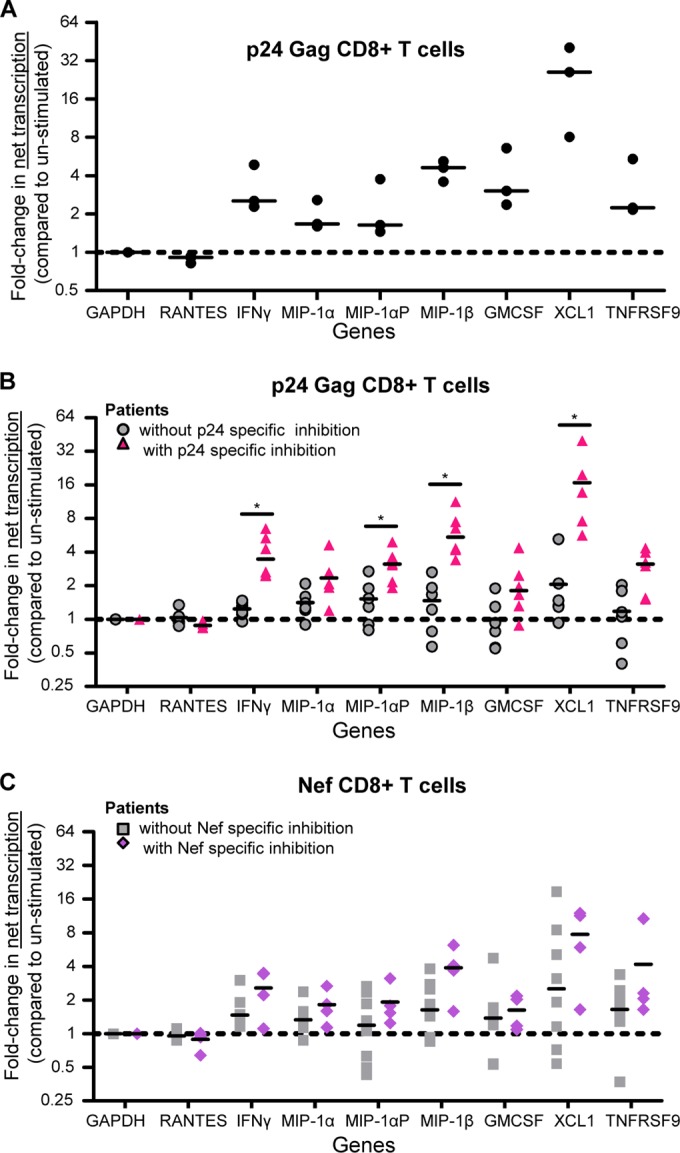
CD8+ T cells from VCs with antigen-specific inhibition exhibit increased net transcription for IFN-γ, MIP1α, MIP1α-AP, MIP1β, GM-CSF, TNFRSF9, and XCL1. (A) Fold change (5.5-h p24-stimulated/unstimulated) in net transcription in CD8+ T cells from an HIV-1+ virus controller (VC30). The results are from three independent experiments. The lines represent the median values. (B) Values for fold change (5.5-h p24-stimulated CD8+ T cells compared to unstimulated cells) in net transcription from patients with p24-specific inhibition (triangles, n = 6: VC11, -23, -26, -27, -28, and -29) and those without (circles, n = 5: 3 HIV-1-seronegative patients and VC16 and -24). The lines indicate the medians. *, P < 0.05. (C) Values for the fold change (5.5-h Nef-stimulated CD8+ T cells compared to unstimulated cells) in net transcription from patients with Nef-specific inhibition (diamonds, n = 4: VC26, -27, -29, and -16) and those without (squares, n = 7: 3 HIV-1-seronegative patients and VC11, -23, -28, and -24). The lines indicate the medians. Net transcription was determined via primer-specific PCR of 4sU-containing RNA. The P values are indicative of a significant difference in fold change in net transcription in patients with antigen-specific inhibition compared to those without. The P values were calculated using a Wilcoxon exact test, controlling for false discovery using the Benjamini and Hochberg method.
FIG 4.
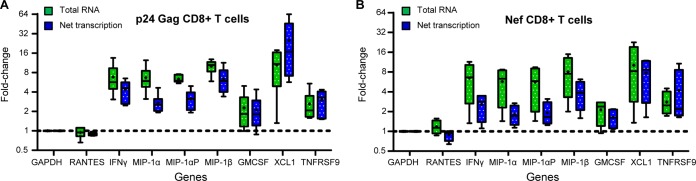
Measured changes in the transcription rate do not match observed changes in mRNA abundance. Boxes indicate the median as well as the upper and lower quartiles, with whiskers being minimum and maximum values. Lines at y = 1 indicate no change in value (compared to that for unstimulated). The change in total mRNA abundance (green) from Gag p24-stimulated (A) and Nef-stimulated (B) CD8+ T cells from VCs with antiviral activity does not match the change in net transcription (blue) for MIP-1α, MIP-1αP, MIP-1β, IFN-γ, and GM-CSF. For each of these messages, increases in total mRNA for these markers were 1.5- to 3-fold more than the net transcription. In comparison, total mRNA levels from an mRNA that is known to be transcriptionally induced, TNFRSF9, increased similarly to net transcription. The same is seen for XCL-1. This indicates that other mechanisms of regulation may play a role in induction of MIP-1α, MIP-1αP, MIP-1β, IFN-γ, and GM-CSF.
mRNAs encoding IFN-γ, MIP-1α, MIP-1αP, and MIP-1β are stabilized after stimulation, contributing to the observed increase of RNA expression.
The observed expression of mRNA is the balance between synthesis of new mRNA (transcription) and degradation (decay) of mRNA over time (59). To investigate the role of decay, we used the ratio of 4sU-labeled mRNA to unlabeled mRNA to calculate mRNA decay rates for the selected panel of antiviral cytokines in CD8+ T cells. Using these decay rates, we calculated an apparent half-life, the instantaneous measure of the predicted half-life given the cellular conditions, of each message with and without antigen stimulation. For messages from unstimulated cells from VCs and seronegative patients, these apparent half-lives were consistent across the patients tested (Fig. 5A). For example, in the 12 VCs and 2 HIV-1-seronegative subjects examined, the average apparent half-life of the IFN-γ mRNA was 0.63 ± 0.039 h (Fig. 5A), similar to previously published values for the stability of IFN-γ mRNA (60–62). We next calculated the decay rates from VC30 CD8+ T cells without stimulation and from autologous cells 4.5 to 5.5 h after stimulation with Gag p24 (Fig. 5B). We observed a consistent increase in the apparent half-life after stimulation with Gag p24 for IFN-γ, MIP-1α, MIP-1αP, MIP-1β, and GM-CSF. We extended this analysis to additional virus controller patients and HIV-1-seronegative individuals. After stimulation, cells from VCs without an antigen-specific antiviral CD8+ T cell response had mRNA half-lives that remained highly consistent with those in unstimulated cells. Cells with a p24-specific response showed increases in stability, with MIP-1αP showing the only statistically significant increase (Fig. 5C) (P < 0.05; Wilcoxon exact test, controlling for the FDR [41]). While changes in the apparent half-life in Nef-stimulated (compared to unstimulated) cells did not reach significance when comparing patients with Nef-specific inhibition to those without and correcting for multiple comparisons, the values for IFN-γ and MIP-1β did reach significance when they were not adjusted for comparisons (Fig. 5D and Table 3). In addition, compared to Gag p17-stimulated cells, the increases in stability observed in Nef-stimulated CD8+ T cells were significant for IFN-γ, MIP-1α, and MIP-1αP (Table 3). Taken together, the sum of the change in net transcription and the change in mRNA stability corresponded to the differences in the mRNA abundances after antigen stimulation (Fig. 5E and F).
Virus inhibition correlates with changes in total RNA, transcription, and half-life.
Since the changes in RNA abundance, net transcription, and apparent mRNA half-life were distinct in patients with and without antigen-specific inhibition, we next determined if these measurements correlated with antigen-specific virus inhibition. We examined whether changes in total RNA abundance, transcription, and half-life correlated with the ability of CD8+ T cells to inhibit HIV-1 replication through soluble mechanisms (Spearman's rank correlation coefficients per gene) (Fig. 6). Among the seven cytokines tested, the fold changes in the total mRNA abundances of MIP-1α (r = 0.6636; P = 0.026), MIP-1αP (r = 0.7198; P = 0.0125), MIP-1β (r = 0.6273; P = 0.0388), and XCL1 (r = 0.6636; P = 0.26) strongly correlated with soluble inhibition of p24-stimulated cells that have HIV-1-inhibitory activity (Fig. 6A, top row). Change in the net transcription of the mRNAs encoding MIP-1αP (r = 0.6727; P = 0.0233), MIP-1β (r = 0.700; P = 0.0165), XCL1 (r = 0.6273;P = 0.0388), and TNFRSF9 (r = 0.6091;P = 0.0467) also significantly correlated with soluble inhibition by p24-stimulated CD8+ T cells (Fig. 6A, bottom row). In Nef-stimulated CD8+ T cells, the abundance of TNFRSF9 (r = 0.6364; P = 0.0353), net transcription of IFN-γ (r = 0.6182; P = 0.0426), and mRNA half-life of MIP-1α (r = 0.7364; P = 0.0098) significantly correlated with CD8+ virus inhibition (Fig. 6B). RANTES was included as a negative control, and as expected, no correlations were observed (Fig. 6A and B).
FIG 6.

Virus inhibition correlates with changes in total RNA, net transcription, and apparent half-life. (A) Fold changes in p24-stimulated cells that significantly correlated with virus inhibition. (Top row) In p24-stimulated cells, the fold changes (compared to unstimulated cells) in mRNA abundances of MIP1α, MIP1α-AP, MIP1β, and XCL1 correlated with soluble virus inhibition (measured as log10 reduction in virus replication), while RANTES did not. mRNA abundance was determined via primer-specific PCR. (Bottom row) Fold changes (p24 stimulated/unstimulated) in net transcription of MIP1α-AP, MIP1β, XCL1, and TNFRSF9 correlate with soluble inhibition, while RANTES again does not. The rate of transcription was determined by primer-specific PCR of 4sU-labeled RNA (described in Materials and Methods). (B) Fold changes in Nef-stimulated cells that correlate with virus inhibition. In Nef-stimulated cells, the fold change (compared to unstimulated cells) in the mRNA abundance of TNFRSF9, net transcription of IFN-γ, and apparent half-life of MIP1α correlates with soluble virus inhibition (measured as log10 reduction in virus replication), while RANTES does not. The log10 reduction in virus replication was determined via sVIA, as described in Materials and Methods. The r values were calculated using Spearman correlation. Trendlines are shown.
Temporal dynamics of CD8+ T cell responses.
Measuring mRNA stability at fixed times after stimulation allowed us to observe differences in the apparent half-life in the immediately preceding hour. However, the possibility remained that transcription rate dynamics occurring before the addition of 4sU at 4.5 h could explain the observed differences in the apparent half-life. To independently observe the impacts of transcription and stability on total mRNA abundance, we performed 4sU analysis on p24-stimulated CD8+ T cells from VC20 every hour for a full 6.5-hour time course to capture all net transcription (Fig. 7A). From this, we quantified the total mRNA abundance and net transcription at each hour (starting at 0.5 h) after stimulation. Using the decay rate of unstimulated cells (Fig. 5A) and the initial abundance and net transcription for every hour, we predicted the mRNA abundance at each time point, assuming no change in mRNA stability and compared to the measured total (Fig. 7). In cases where mRNA abundances are constant or derive primarily from transcriptional variation, this model is highly accurate (40). Two controls confirmed the accuracy of the model for both stable (RANTES) (Fig. 7B) and a known transcriptionally driven (TNFRSF9) message (Fig. 7C). Substantial deviations from the predictions given by the model suggest that RNA stability is the likely variable. If the observed totals are greater than predicted, it suggests the RNA is stabilized, while if they are less than predicted, it suggests the RNA is destabilized. As shown in Fig. 7D to F, the predicted levels of RNA for MIP-1α, MIP-1β, and IFN-γ were substantially less than the RNA abundances that were observed, suggesting that the RNA is stabilized. For example, the predicted levels of IFN-γ and MIP-1β were approximately half the observed values (Fig. 7D and F), while the observed abundance of MIP-1α was ∼25% higher than the model predicted (Fig. 7E). These results suggest that substantial stabilization of the target mRNAs took place in these cells. We further incorporated the variance in stability we observed across VCs, predicting abundances using apparent half-lives that represented the extremes of the observed unstimulated stabilities in the VCs (Fig. 5A). These extreme values still could not mimic the observed results.
We subsequently allowed stability to vary and solved the equation for the optimized mRNA stability for the observed total mRNA abundances. While RANTES and TNFRSF9 minimally change in mRNA stability (Fig. 7G and H), IFN-γ, MIP-1α, and MIP-1β robustly increased in stability, in coordination with the induction of transcription (Fig. 7I to K). Most messages followed a peaked response, where stability was highest during the time of greatest transcription, resulting in maximal increases in mRNA abundance (Fig. 7G to K).
DISCUSSION
We report here that a cohort of virus controllers have substantial antigen-specific Gag p24 and Nef CD8+ T cell-mediated antiviral responses that, through soluble mechanisms, inhibit viral replication. Strongly associated with this antigen-specific antiviral activity are increases in mRNA abundances of IFN-γ, MIP-1α, MIP-1αP, MIP-1β, GM-CSF, TNFRSF9, and XCL1. Several of these cytokines, such as the β-chemokines (MIP-1α, MIP-1αP, and MIP-1β) and XCL1 could play pivotal roles in the ability of CD8+ T cells to inhibit virus at entry. Through 4sU RNA analysis, we report the novel observation that the expression of these cytokines is controlled at the level of RNA abundance through coordinated regulation of both transcription and mRNA stability, enabling a rapid and robust antiviral response.
Our findings that Gag p24- and Nef-specific CD8+ T cells are most associated with HIV-1 inhibition agree with previous research (63–67). We previously demonstrated that Gag- and Nef-dominant soluble activity mediated by CD8+ T cells during acute HIV-1 infection corresponded to the breadth of virus inhibition, as well as immune pressure against transmitted founder viruses, but that this activity was diminished by 6 months postinfection in the patients examined (6). Others have also reported early Gag and Nef CD8+ T cell antiviral activity in acute infection (68, 69). In addition, MIP-1β, which correlated with inhibition in this study, correlated with initial CD8+ T cell antiviral responses in acute HIV-1 infection (6) and initial viremic control (5). The observance of the similarity of soluble responses in acute patients and VCs suggests that the soluble antiviral activity of CD8+ T cells that broadly develop during acute infection may be maintained in virus controller patients. This evidence complements an earlier report that long-term nonprogressors maintain functional cytotoxic CD8+ T cells that are lost in progressors (70) and indicates that further longitudinal studies to investigate soluble CD8+ T cell response retention are warranted. Notably, we also found that Pol-specific CD8+ T cells from some virus controllers mediated antigen-specific virus inhibition. Borthwick et al. recently reported that an HIV conserved immunogen vaccine (prime-boost) strategy induced CD8+ T cell virus inhibition that correlated with both Gag and Pol CD8+ T cells (71).
The expression of cytokines has long been associated with soluble antiviral functions. The regulation of these cytokines, however, is poorly understood. We assessed transcription and decay rates for mRNAs in unstimulated and stimulated (p24, p17, and Nef) CD8+ T cells and found that both mechanisms drive induction of key cytokines, with maximum increases in stability and net transcription occurring at the same time. This induction of gene expression via the cooperation of signaling pathways to bring about increased transcription and enhanced stability to induce multiple cytokines with antiviral activity is an interesting contrast to the usual gene expression buffering seen in eukaryotes (72).
There are several mechanisms known to enhance transcription (73, 74). One plausible explanation for the observed increase in transcription could be the presence of epigenetic marks at the loci of these genes in memory cells that contribute to a primed state, where these cells are poised to display effector functions upon antigen reencounter (75). We have previously shown that treatment of CD8+ T cells with valproic acid, a chemical that alters histone acetylation, impacts the ability of CD8+ T cells to inhibit HIV-1 (16). Alternatively, there could be an increase in a transcription factor(s) that regulates the expression of genes relevant to CD8+ T cell effector function. Further experiments will determine if epigenetic marks near the loci of antiviral genes or the expression of relevant transcription factors are altered in these cells.
These data suggest that a concomitant signaling pathway may result from regulation of the stability of mRNAs encoding HIV-1 antiviral cytokines by predictable transacting factors. RNA stability is primarily controlled by RNA binding proteins (RBPs) and microRNAs (miRNAs). IFN-γ, MIP-1α, MIP-1αP, and MIP-1β have long 3′ untranslated regions (UTRs) and contain AU-rich elements (AREs) (76), well-known regulatory sites for RBPs, including HuR (77) and TTP (78), and miRNAs (79). For example, TTP is known to bind an ARE in the 3′ UTR of IFN-γ and to destabilize it in T cells stimulated with CD3 (60). The RBP Roquin is known to regulate IFN-γ in CD8+ T cells (80), recognizing a specific structural element to drive constitutive decay of target messages (81). While the RNA abundances of these RBPs did not change in these studies (data not shown), it is known that posttranslational modifications can affect their mRNA targeting (82, 83). Changes in miRNA regulation may also underlie changes in mRNA stability. In previous reports, elimination of Dicer, a protein required for miRNA processing, resulted in CD8+ T cells that responded faster after anti-CD3 and anti-CD28 stimulation but that were not able to resolve activating responses (84). After stimulation, miR-130/301 (84) and miR-155 (85) are strongly upregulated in CD8+ T cells. Additionally, large-scale differences in miRNA profiles were observed in naive, effector, and memory CD8+ T cells (86). Identifying the important regulatory RBPs and miRNAs driving the posttranscriptional responses in HIV-1 antigen-stimulated CD8+ T cells would be a key step in fully understanding the regulation of antiviral cytokine responses.
These results bring up an interesting concept of possible differences among epitope-driven responses by different CD8+ T cell subpopulations. While changes in total RNA did occur in CD8+ T cells with p24- and Nef-stimulated inhibition, statistical analyses revealed a possible difference in the regulation of RNA abundances in p24- and Nef-specific cells. We observed that there was a marked statistically significant increase in the net transcription for several mRNAs encoding antiviral cytokines in cells with p24-specific inhibition of virus while changes in Nef-stimulated cells were less dramatic and not significant. Interestingly, comparing p24- and Nef-stimulated cells to autologous p17-stimulated cells, we observed significant changes in the stability of IFN-γ, MIP-1α, and MIP-1αP in Nef-stimulated cells while we observed significance in the fold change in stability of MIP-1αP only in p24-stimulated cells (Table 3). These observations fit with previous reports that Nef responses are the first to arise in acute infections (87), which may be a reflection of the rapid expression that can be garnered through regulation of RNA stability, allowing a more immediate display of effector function. However, further studies are needed to examine the potential differences in the p24- and Nef-specific CD8+ T cell responses to determine if regulation by transcription and RNA stability is dependent on antigen specificity and, if so, whether the stage of memory cell differentiation (88) of these antigen-specific CD8+ T cells is a factor.
Induction of CD8+ T cells capable of inhibiting HIV-1 replication is important for both development of cure therapies (89) and HIV-1 vaccine strategies. Here, we report on the antigen specificity, cytokine signature, and regulation of CD8+ T cells that inhibit virus replication in virus controllers. The finding that RNA stability is involved in the CD8+ T cell response allows possible future identification of other markers of CD8+ T cell effector function using techniques aimed at globally identifying mRNAs that exhibit increased stabilization and other posttranscriptional changes in CD8+ T cells with anti-HIV-1 effector function. The results of this approach can be applied to the design of vaccine immunogens in order to target well-defined HIV-1-specific CD8+ T cells that mediate the stabilization and rapid release of β-chemokines and other antiviral cytokines upon antigen encounter to block HIV-1 acquisition.
ACKNOWLEDGMENTS
This work was supported by R01/R56 NIH grant AI-52779 (G.D.T.), an NIH F31 fellowship (1F31AI106519-01) (T.L.P.), and Center for AIDS Research (P30 AI 64518) and Duke Interdisciplinary Research Training Program in AIDS (NIH IRTPA T32) grant 5T32AI007392 (J.B.). Work by J.B. was also supported by grants from the National Cancer Institute (grant number R01 CA157268 to J.D.K.) and the National Science Foundation (0842621 to J.D.K.).
We thank Kent Weinhold, Director of the Duke CFAR, and Coleen Cunningham and John Bartlett, Directors of the Duke CFAR Clinical Core, and the patients, physicians, and staff of the Duke Adult Infectious Diseases Clinic (Sunita Patil, Gary Cox, Nathan Thielman, Cameron Wolfe, Elizabeth Livingston, Brianna Norton, Kristen Dicks, Mehri McKellar, Vivian Chu, Jason Stout, and Ann Mosher) for virus controller patient recruitment. We thank John Kappes, Christina Ochsenbauer, and the UAB CFAR Virology Core for HIV-1 infectious molecule clones and Cavin Ward-Caviness for helpful discussions.
Footnotes
Published ahead of print 4 June 2014
REFERENCES
- 1.Borrow P, Lewicki H, Hahn BH, Shaw GM, Oldstone MB. 1994. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J. Virol. 68:6103–6110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koup RA, Safrit JT, Cao Y, Andrews CA, McLeod G, Borkowsky W, Farthing C, Ho DD. 1994. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 68:4650–4655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goonetilleke N, Liu MK, Salazar-Gonzalez JF, Ferrari G, Giorgi E, Ganusov VV, Keele BF, Learn GH, Turnbull EL, Salazar MG, Weinhold KJ, Moore S, Letvin N, Haynes BF, Cohen MS, Hraber P, Bhattacharya T, Borrow P, Perelson AS, Hahn BH, Shaw GM, Korber BT, McMichael AJ. 2009. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J. Exp. Med. 206:1253–1272. 10.1084/jem.20090365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McMichael AJ, Borrow P, Tomaras GD, Goonetilleke N, Haynes BF. 2010. The immune response during acute HIV-1 infection: clues for vaccine development. Nat. Rev. Immunol. 10:11–23. 10.1038/nri2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferrari G, Korber B, Goonetilleke N, Liu MK, Turnbull EL, Salazar-Gonzalez JF, Hawkins N, Self S, Watson S, Betts MR, Gay C, McGhee K, Pellegrino P, Williams I, Tomaras GD, Haynes BF, Gray CM, Borrow P, Roederer M, McMichael AJ, Weinhold KJ. 2011. Relationship between functional profile of HIV-1 specific CD8 T cells and epitope variability with the selection of escape mutants in acute HIV-1 infection. PLoS Pathog. 7:e1001273. 10.1371/journal.ppat.1001273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freel SA, Picking RA, Ferrari G, Ding H, Ochsenbauer C, Kappes JC, Soderberg K, Weinhold KJ, Cunningham CK, Denny TN, Crump JA, Cohen MS, McMichael AJ, Haynes BF, Tomaras GD. 2012. Initial HIV-1 antigen-specific CD8+ T cells in acute HIV-1 inhibit transmitted founder virus replication. J. Virol. 86:6835–6846. 10.1128/JVI.00437-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang H, Wu H, Hancock G, Clutton G, Sande N, Xu X, Yan H, Huang X, Angus B, Kuldanek K, Fidler S, Denny TN, Birks J, McMichael A, Dorrell L. 2012. Antiviral inhibitory capacity of CD8+ T cells predicts the rate of CD4+ T-cell decline in HIV-1 infection. J. Infect. Dis. 206:552–561. 10.1093/infdis/jis379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Landay AL, Mackewicz CE, Levy JA. 1993. An activated CD8+ T cell phenotype correlates with anti-HIV activity and asymptomatic clinical status. Clin. Immunol. Immunopathol. 69:106–116. 10.1006/clin.1993.1157 [DOI] [PubMed] [Google Scholar]
- 9.Pollack H, Zhan MX, Safrit JT, Chen SH, Rochford G, Tao PZ, Koup R, Krasinski K, Borkowsky W. 1997. CD8+ T-cell-mediated suppression of HIV replication in the first year of life: association with lower viral load and favorable early survival. AIDS 11:F9–F13. 10.1097/00002030-199701000-00002 [DOI] [PubMed] [Google Scholar]
- 10.Ndhlovu ZM, Chibnik LB, Proudfoot J, Vine S, McMullen A, Cesa K, Porichis F, Jones RB, Alvino DM, Hart MG, Stampouloglou E, Piechocka-Trocha A, Kadie C, Pereyra F, Heckerman D, De Jager PL, Walker BD, Kaufmann DE. 2013. High-dimensional immunomonitoring models of HIV-1-specific CD8 T-cell responses accurately identify subjects achieving spontaneous viral control. Blood 121:801–811. 10.1182/blood-2012-06-436295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deeks SG, Walker BD. 2007. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity 27:406–416. 10.1016/j.immuni.2007.08.010 [DOI] [PubMed] [Google Scholar]
- 12.Lambotte O, Boufassa F, Madec Y, Nguyen A, Goujard C, Meyer L, Rouzioux C, Venet A, Delfraissy JF. 2005. HIV controllers: a homogeneous group of HIV-1-infected patients with spontaneous control of viral replication. Clin. Infect. Dis. 41:1053–1056. 10.1086/433188 [DOI] [PubMed] [Google Scholar]
- 13.Barker E, Mackewicz CE, Reyes-Teran G, Sato A, Stranford SA, Fujimura SH, Christopherson C, Chang SY, Levy JA. 1998. Virological and immunological features of long-term human immunodeficiency virus-infected individuals who have remained asymptomatic compared with those who have progressed to acquired immunodeficiency syndrome. Blood 92:3105–3114 [PubMed] [Google Scholar]
- 14.Pereyra F, Addo MM, Kaufmann DE, Liu Y, Miura T, Rathod A, Baker B, Trocha A, Rosenberg R, Mackey E, Ueda P, Lu Z, Cohen D, Wrin T, Petropoulos CJ, Rosenberg ES, Walker BD. 2008. Genetic and immunologic heterogeneity among persons who control HIV infection in the absence of therapy. J. Infect. Dis. 197:563–571. 10.1086/526786 [DOI] [PubMed] [Google Scholar]
- 15.Tomaras GD, Lacey SF, McDanal CB, Ferrari G, Weinhold KJ, Greenberg ML. 2000. CD8+ T cell-mediated suppressive activity inhibits HIV-1 after virus entry with kinetics indicating effects on virus gene expression. Proc. Natl. Acad. Sci. U. S. A. 97:3503–3508. 10.1073/pnas.97.7.3503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saunders KO, Freel SA, Overman RG, Cunningham CK, Tomaras GD. 2010. Epigenetic regulation of CD8(+) T-lymphocyte mediated suppression of HIV-1 replication. Virology 405:234–242. 10.1016/j.virol.2010.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freel SA, Lamoreaux L, Chattopadhyay PK, Saunders K, Zarkowsky D, Overman RG, Ochsenbauer C, Edmonds TG, Kappes JC, Cunningham CK, Denny TN, Weinhold KJ, Ferrari G, Haynes BF, Koup RA, Graham BS, Roederer M, Tomaras GD. 2010. Phenotypic and functional profile of HIV-inhibitory CD8 T cells elicited by natural infection and heterologous prime/boost vaccination. J. Virol. 84:4998–5006. 10.1128/JVI.00138-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Killian MS, Johnson C, Teque F, Fujimura S, Levy JA. 2011. Natural suppression of human immunodeficiency virus type 1 replication is mediated by transitional memory CD8+ T cells. J. Virol. 85:1696–1705. 10.1128/JVI.01120-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buckheit RW, III, Salgado M, Silciano RF, Blankson JN. 2012. Inhibitory potential of subpopulations of CD8+ T cells in HIV-1-infected elite suppressors. J. Virol. 86:13679–13688. 10.1128/JVI.02439-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barker TD, Weissman D, Daucher JA, Roche KM, Fauci AS. 1996. Identification of multiple and distinct CD8+ T cell suppressor activities: dichotomy between infected and uninfected individuals, evolution with progression of disease, and sensitivity to gamma irradiation. J. Immunol. 156:4476–4483 [PubMed] [Google Scholar]
- 21.Saunders KO, Rudicell RS, Nabel GJ. 2012. The design and evaluation of HIV-1 vaccines. AIDS 26:1293–1302. 10.1097/QAD.0b013e32835474d2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Girard MP, Osmanov S, Assossou OM, Kieny MP. 2011. Human immunodeficiency virus (HIV) immunopathogenesis and vaccine development: a review. Vaccine 29:6191–6218. 10.1016/j.vaccine.2011.06.085 [DOI] [PubMed] [Google Scholar]
- 23.Keene JD. 2001. Ribonucleoprotein infrastructure regulating the flow of genetic information between the genome and the proteome. Proc. Natl. Acad. Sci. U. S. A. 98:7018–7024. 10.1073/pnas.111145598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hao S, Baltimore D. 2009. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol. 10:281–288. 10.1038/ni.1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson P. 2010. Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nat. Rev. Immunol. 10:24–35. 10.1038/nri2685 [DOI] [PubMed] [Google Scholar]
- 26.Carrick D, Lai W, Blackshear P. 2004. The tandem CCCH zinc finger protein tristetraprolin and its relevance to cytokine mRNA turnover and arthritis. Arthritis Res. Ther. 6:248. 10.1186/ar1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mukherjee N, Lager PJ, Friedersdorf MB, Thompson MA, Keene JD. 2009. Coordinated posttranscriptional mRNA population dynamics during T-cell activation. Mol. Syst. Biol. 5:288. 10.1038/msb.2009.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papadaki O, Milatos S, Grammenoudi S, Mukherjee N, Keene JD, Kontoyiannis DL. 2009. Control of thymic T cell maturation, deletion and egress by the RNA-binding protein HuR. J. Immunol. 182:6779–6788. 10.4049/jimmunol.0900377 [DOI] [PubMed] [Google Scholar]
- 29.Phillips K, Kedersha N, Shen L, Blackshear PJ, Anderson P. 2004. Arthritis suppressor genes TIA-1 and TTP dampen the expression of tumor necrosis factor, cyclooxygenase 2, and inflammatory arthritis. Proc. Natl. Acad. Sci. U. S. A. 101:2011–2016. 10.1073/pnas.0400148101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE, Haynes BF, Blackshear PJ. 1996. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity 4:445–454. 10.1016/S1074-7613(00)80411-2 [DOI] [PubMed] [Google Scholar]
- 31.Edmonds TG, Ding H, Yuan X, Wei Q, Smith KS, Conway JA, Wieczorek L, Brown B, Polonis V, West JT, Montefiori DC, Kappes JC, Ochsenbauer C. 2010. Replication competent molecular clones of HIV-1 expressing Renilla luciferase facilitate the analysis of antibody inhibition in PBMC. Virology 408:1–13. 10.1016/j.virol.2010.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ochsenbauer C, Edmonds TG, Ding H, Keele BF, Decker J, Salazar MG, Salazar-Gonzalez JF, Shattock R, Haynes BF, Shaw GM, Hahn BH, Kappes JC. 2012. Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J. Virol. 86:2715–2728. 10.1128/JVI.06157-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li F, Malhotra U, Gilbert PB, Hawkins NR, Duerr AC, McElrath JM, Corey L, Self SG. 2006. Peptide selection for human immunodeficiency virus type 1 CTL-based vaccine evaluation. Vaccine 24:6893–6904. 10.1016/j.vaccine.2006.06.009 [DOI] [PubMed] [Google Scholar]
- 34.Dölken L, Ruzsics Z, Rädle B, Friedel CC, Zimmer R, Mages J, Hoffmann R, Dickinson P, Forster T, Ghazal P, Koszinowski UH. 2008. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14:1959–1972. 10.1261/rna.1136108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jr, Jungkamp AC, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141:129–141. 10.1016/j.cell.2010.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ascano M, Hafner M, Cekan P, Gerstberger S, Tuschl T. 2012. Identification of RNA–protein interaction networks using PAR-CLIP. Wiley Interdiscip. Rev. RNA 3:159–177. 10.1002/wrna.1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller C, Schwalb B, Maier K, Schulz D, Dumcke S, Zacher B, Mayer A, Sydow J, Marcinowski L, Dolken L, Martin DE, Tresch A, Cramer P. 2011. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol. 7:458. 10.1038/msb.2010.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dani C, Blanchard JM, Piechaczyk M, El Sabouty S, Marty L, Jeanteur P. 1984. Extreme instability of myc mRNA in normal and transformed human cells. Proc. Natl. Acad. Sci. U. S. A. 81:7046–7050. 10.1073/pnas.81.22.7046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharova LV, Sharov AA, Nedorezov T, Piao Y, Shaik N, Ko MS. 2009. Database for mRNA half-life of 19,977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells. DNA Res. 16:45–58. 10.1093/dnares/dsn030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rabani M, Levin JZ, Fan L, Adiconis X, Raychowdhury R, Garber M, Gnirke A, Nusbaum C, Hacohen N, Friedman N, Amit I, Regev A. 2011. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. 29:436–442. 10.1038/nbt.1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57:289–300 [Google Scholar]
- 42.Kinter AL, Ostrowski M, Goletti D, Oliva A, Weissman D, Gantt K, Hardy E, Jackson R, Ehler L, Fauci AS. 1996. HIV replication in CD4+ T cells of HIV-infected individuals is regulated by a balance between the viral suppressive effects of endogenous beta-chemokines and the viral inductive effects of other endogenous cytokines. Proc. Natl. Acad. Sci. U. S. A. 93:14076–14081. 10.1073/pnas.93.24.14076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rubbert A, Weissman D, Combadiere C, Pettrone KA, Daucher JA, Murphy PM, Fauci AS. 1997. Multifactorial nature of noncytolytic CD8+ T cell-mediated suppression of HIV replication: beta-chemokine-dependent and -independent effects. AIDS Res. Hum. Retroviruses 13:63–69. 10.1089/aid.1997.13.63 [DOI] [PubMed] [Google Scholar]
- 44.Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. 1995. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science 270:1811–1815. 10.1126/science.270.5243.1811 [DOI] [PubMed] [Google Scholar]
- 45.Diaz LS, Stone MR, Mackewicz CE, Levy JA. 2003. Differential gene expression in CD8+ cells exhibiting noncytotoxic anti-HIV activity. Virology 311:400–409. 10.1016/S0042-6822(03)00177-6 [DOI] [PubMed] [Google Scholar]
- 46.Katz BZ, Salimi B, Gadd SL, Huang CC, Kabat WJ, Kersey D, McCabe C, Heald-Sargent T, Katz ED, Yogev R. 2011. Differential gene expression of soluble CD8+ T-cell mediated suppression of HIV replication in three older children. J. Med. Virol. 83:24–32. 10.1002/jmv.21933 [DOI] [PubMed] [Google Scholar]
- 47.Martinez-Marino B, Foster H, Hao Y, Levy JA. 2007. Differential gene expression in CD8(+) cells from HIV-1-infected subjects showing suppression of HIV replication. Virology 362:217–225. 10.1016/j.virol.2006.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saunders KO, Ward-Caviness C, Schutte RJ, Freel SA, Overman RG, Thielman NM, Cunningham CK, Kepler TB, Tomaras GD. 2011. Secretion of MIP-1beta and MIP-1alpha by CD8(+) T-lymphocytes correlates with HIV-1 inhibition independent of coreceptor usage. Cell. Immunol. 266:154–164. 10.1016/j.cellimm.2010.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. 1996. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272:1955–1958. 10.1126/science.272.5270.1955 [DOI] [PubMed] [Google Scholar]
- 50.Nibbs RJ, Yang J, Landau NR, Mao JH, Graham GJ. 1999. LD78beta, a non-allelic variant of human MIP-1alpha (LD78alpha), has enhanced receptor interactions and potent HIV suppressive activity. J. Biol. Chem. 274:17478–17483. 10.1074/jbc.274.25.17478 [DOI] [PubMed] [Google Scholar]
- 51.Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, Nibbs RJ, Freedman BI, Quinones MP, Bamshad MJ, Murthy KK, Rovin BH, Bradley W, Clark RA, Anderson SA, O'Connell RJ, Agan BK, Ahuja SS, Bologna R, Sen L, Dolan MJ, Ahuja SK. 2005. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science 307:1434–1440. 10.1126/science.1101160 [DOI] [PubMed] [Google Scholar]
- 52.Guzzo C, Fox J, Lin Y, Miao H, Cimbro R, Volkman BF, Fauci AS, Lusso P. 2013. The CD8-derived chemokine XCL1/lymphotactin is a conformation-dependent, broad-spectrum inhibitor of HIV-1. PLoS Pathog. 9:e1003852. 10.1371/journal.ppat.1003852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laderach D, Movassagh M, Johnson A, Mittler RS, Galy A. 2002. 4-1BB co-stimulation enhances human CD8(+) T cell priming by augmenting the proliferation and survival of effector CD8(+) T cells. Int. Immunol. 14:1155–1167. 10.1093/intimm/dxf080 [DOI] [PubMed] [Google Scholar]
- 54.Wanjalla CN, Goldstein EF, Wirblich C, Schnell MJ. 2012. A role for granulocyte-macrophage colony-stimulating factor in the regulation of CD8(+) T cell responses to rabies virus. Virology 426:120–133. 10.1016/j.virol.2012.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pal R, Garzino-Demo A, Markham PD, Burns J, Brown M, Gallo RC, DeVico AL. 1997. Inhibition of HIV-1 infection by the beta-chemokine MDC. Science 278:695–698. 10.1126/science.278.5338.695 [DOI] [PubMed] [Google Scholar]
- 56.Imai T, Baba M, Nishimura M, Kakizaki M, Takagi S, Yoshie O. 1997. The T cell-directed CC chemokine TARC is a highly specific biological ligand for CC chemokine receptor 4. J. Biol. Chem. 272:15036–15042. 10.1074/jbc.272.23.15036 [DOI] [PubMed] [Google Scholar]
- 57.Veloso A, Biewen B, Paulsen MT, Berg N, Carmo de Andrade Lima L, Prasad J, Bedi K, Magnuson B, Wilson TE, Ljungman M. 2013. Genome-wide transcriptional effects of the anti-cancer agent camptothecin. PLoS One 8:e78190. 10.1371/journal.pone.0078190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. 2011. Global quantification of mammalian gene expression control. Nature 473:337–342. 10.1038/nature10098 [DOI] [PubMed] [Google Scholar]
- 59.Ross J. 1995. mRNA stability in mammalian cells. Microbiol. Rev. 59:423–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ogilvie RL, SternJohn JR, Rattenbacher B, Vlasova IA, Williams DA, Hau HH, Blackshear PJ, Bohjanen PR. 2009. Tristetraprolin mediates interferon-gamma mRNA decay. J. Biol. Chem. 284:11216–11223. 10.1074/jbc.M901229200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mavropoulos A, Sully G, Cope AP, Clark AR. 2005. Stabilization of IFN-gamma mRNA by MAPK p38 in IL-12- and IL-18-stimulated human NK cells. Blood 105:282–288. 10.1182/blood-2004-07-2782 [DOI] [PubMed] [Google Scholar]
- 62.Lee SK, Silva DG, Martin JL, Pratama A, Hu X, Chang PP, Walters G, Vinuesa CG. 2012. Interferon-gamma excess leads to pathogenic accumulation of follicular helper T cells and germinal centers. Immunity 37:880–892. 10.1016/j.immuni.2012.10.010 [DOI] [PubMed] [Google Scholar]
- 63.Saez-Cirion A, Sinet M, Shin SY, Urrutia A, Versmisse P, Lacabaratz C, Boufassa F, Avettand-Fenoel V, Rouzioux C, Delfraissy JF, Barre-Sinoussi F, Lambotte O, Venet A, Pancino G. 2009. Heterogeneity in HIV suppression by CD8 T cells from HIV controllers: association with Gag-specific CD8 T cell responses. J. Immunol. 182:7828–7837. 10.4049/jimmunol.0803928 [DOI] [PubMed] [Google Scholar]
- 64.Julg B, Williams KL, Reddy S, Bishop K, Qi Y, Carrington M, Goulder PJ, Ndung'u T, Walker BD. 2010. Enhanced anti-HIV functional activity associated with Gag-specific CD8 T-cell responses. J. Virol. 84:5540–5549. 10.1128/JVI.02031-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Janes H, Friedrich DP, Krambrink A, Smith RJ, Kallas EG, Horton H, Casimiro DR, Carrington M, Geraghty DE, Gilbert PB, McElrath MJ, Frahm N. 2013. Vaccine-induced gag-specific T cells are associated with reduced viremia after HIV-1 infection. J. Infect. Dis. 208:1231–1239. 10.1093/infdis/jit322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adland E, Carlson JM, Paioni P, Kloverpris H, Shapiro R, Ogwu A, Riddell L, Luzzi G, Chen F, Balachandran T, Heckerman D, Stryhn A, Edwards A, Ndung'u T, Walker BD, Buus S, Goulder P, Matthews PC. 2013. Nef-specific CD8+ T cell responses contribute to HIV-1 immune control. PLoS One 8:e73117. 10.1371/journal.pone.0073117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Malhotra U, Li F, Nolin J, Allison M, Zhao H, Mullins JI, Self S, McElrath MJ. 2007. Enhanced detection of human immunodeficiency virus type 1 (HIV-1) Nef-specific T cells recognizing multiple variants in early HIV-1 infection. J. Virol. 81:5225–5237. 10.1128/JVI.02564-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Turk G, Ghiglione Y, Falivene J, Socias ME, Laufer N, Coloccini RS, Rodriguez AM, Ruiz MJ, Pando MA, Giavedoni LD, Cahn P, Sued O, Salomon H, Gherardi MM. 2013. Early Gag immunodominance of the HIV-specific T-cell response during acute/early infection is associated with higher CD8+ T-cell antiviral activity and correlates with preservation of the CD4+ T-cell compartment. J. Virol. 87:7445–7462. 10.1128/JVI.00865-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lichterfeld M, Yu XG, Cohen D, Addo MM, Malenfant J, Perkins B, Pae E, Johnston MN, Strick D, Allen TM, Rosenberg ES, Korber B, Walker BD, Altfeld M. 2004. HIV-1 Nef is preferentially recognized by CD8 T cells in primary HIV-1 infection despite a relatively high degree of genetic diversity. AIDS 18:1383–1392. 10.1097/01.aids.0000131329.51633.a3 [DOI] [PubMed] [Google Scholar]
- 70.Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, Lederman MM, Benito JM, Goepfert PA, Connors M, Roederer M, Koup RA. 2006. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 107:4781–4789. 10.1182/blood-2005-12-4818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Borthwick N, Ahmed T, Ondondo B, Hayes P, Rose A, Ebrahimsa U, Hayton EJ, Black A, Bridgeman A, Rosario M, Hill AV, Berrie E, Moyle S, Frahm N, Cox J, Colloca S, Nicosia A, Gilmour J, McMichael AJ, Dorrell L, Hanke T. 2014. Vaccine-elicited human T cells recognizing conserved protein regions inhibit HIV-1. Mol. Ther. 22:464–475. 10.1038/mt.2013.248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sun M, Schwalb B, Pirkl N, Maier KC, Schenk A, Failmezger H, Tresch A, Cramer P. 2013. Global analysis of eukaryotic mRNA degradation reveals Xrn1-dependent buffering of transcript levels. Mol. Cell 52:52–62. 10.1016/j.molcel.2013.09.010 [DOI] [PubMed] [Google Scholar]
- 73.Chen K, Rajewsky N. 2007. The evolution of gene regulation by transcription factors and microRNAs. Nat. Rev. Genet. 8:93–103. 10.1038/nrg1990 [DOI] [PubMed] [Google Scholar]
- 74.Berger SL. 2007. The complex language of chromatin regulation during transcription. Nature 447:407–412. 10.1038/nature05915 [DOI] [PubMed] [Google Scholar]
- 75.Sallusto F, Geginat J, Lanzavecchia A. 2004. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu. Rev. Immunol. 22:745–763. 10.1146/annurev.immunol.22.012703.104702 [DOI] [PubMed] [Google Scholar]
- 76.Shaw G, Kamen R. 1986. A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell 46:659–667. 10.1016/0092-8674(86)90341-7 [DOI] [PubMed] [Google Scholar]
- 77.Tenenbaum SA, Carson CC, Lager PJ, Keene JD. 2000. Identifying mRNA subsets in messenger ribonucleoprotein complexes by using cDNA arrays. Proc. Natl. Acad. Sci. U. S. A. 97:14085–14090. 10.1073/pnas.97.26.14085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stoecklin G, Tenenbaum SA, Mayo T, Chittur SV, George AD, Baroni TE, Blackshear PJ, Anderson P. 2008. Genome-wide analysis identifies interleukin-10 mRNA as target of tristetraprolin. J. Biol. Chem. 283:11689–11699. 10.1074/jbc.M709657200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297. 10.1016/S0092-8674(04)00045-5 [DOI] [PubMed] [Google Scholar]
- 80.Chang P-P, Lee SK, Hu X, Davey G, Duan G, Cho J-H, Karupiah G, Sprent J, Heath WR, Bertram EM, Vinuesa CG. 2012. Breakdown in repression of IFN-γ mRNA leads to accumulation of self-reactive effector CD8+ T cells. J. Immunol. 189:701–710. 10.4049/jimmunol.1102432 [DOI] [PubMed] [Google Scholar]
- 81.Leppek K, Schott J, Reitter S, Poetz F, Hammond MC, Stoecklin G. 2013. Roquin promotes constitutive mRNA decay via a conserved class of stem-loop recognition motifs. Cell 153:869–881. 10.1016/j.cell.2013.04.016 [DOI] [PubMed] [Google Scholar]
- 82.Masuda K, Abdelmohsen K, Kim MM, Srikantan S, Lee EK, Tominaga K, Selimyan R, Martindale JL, Yang X, Lehrmann E, Zhang Y, Becker KG, Wang J-Y, Kim HH, Gorospe M. 2011. Global dissociation of HuR-mRNA complexes promotes cell survival after ionizing radiation. EMBO J. 30:1040–1053. 10.1038/emboj.2011.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Abdelmohsen K, Pullmann R, Lal A, Kim HH, Galban S, Yang X, Blethrow JD, Walker M, Shubert J, Gillespie DA, Furneaux H, Gorospe M. 2007. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol. Cell 25:543–557. 10.1016/j.molcel.2007.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang N, Bevan MJ. 2010. Dicer controls CD8+ T-cell activation, migration, and survival. Proc. Natl. Acad. Sci. U. S. A. 107:21629–21634. 10.1073/pnas.1016299107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gracias DT, Stelekati E, Hope JL, Boesteanu AC, Doering TA, Norton J, Mueller YM, Fraietta JA, Wherry EJ, Turner M, Katsikis PD. 2013. The microRNA miR-155 controls CD8+ T cell responses by regulating interferon signaling. Nat. Immunol. 14:593–602. 10.1038/ni.2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wu H, Neilson JR, Kumar P, Manocha M, Shankar P, Sharp PA, Manjunath N. 2007. miRNA profiling of naive, effector and memory CD8 T cells. PLoS One 2:e1020. 10.1371/journal.pone.0001020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mlotshwa M, Riou C, Chopera D, de Assis Rosa D, Ntale R, Treunicht F, Woodman Z, Werner L, van Loggerenberg F, Mlisana K, Abdool Karim S, Williamson C, Gray CM. 2010. Fluidity of HIV-1-specific T-cell responses during acute and early subtype C HIV-1 infection and associations with early disease progression. J. Virol. 84:12018–12029. 10.1128/JVI.01472-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xie J, Lu W, Samri A, Costagliola D, Schnuriger A, da Silva BC, Blanc C, Larsen M, Theodorou I, Rouzioux C, Autran B. 2010. Distinct differentiation profiles of HIV-Gag and Nef-specific central memory CD8+ T cells associated with HLA-B57/5801 and virus control. AIDS 24:2323–2329. 10.1097/QAD.0b013e32833e5009 [DOI] [PubMed] [Google Scholar]
- 89.Purcell DF, Elliott JH, Ross AL, Frater J. 2013. Towards an HIV cure: science and debate from the International AIDS Society 2013 symposium. Retrovirology 10:134. 10.1186/1742-4690-10-134 [DOI] [PMC free article] [PubMed] [Google Scholar]