

Synopsis
Erythrocytes must regulate hemoglobin synthesis to limit the toxicities of unstable free globin chain subunits. This is particularly relevant in β-thalassemia, where β globin deficiency causes accumulation of free α globin, which forms intracellular precipitates that destroy erythroid precursors. Experimental evidence accumulated over more than 40 years indicates that erythroid cells can neutralize moderate amounts of free α globin through generalized protein quality control mechanisms including molecular chaperones, the ubiquitin proteasome system and autophagy. In many ways, β-thalassemia resembles protein aggregation disorders of nervous system, liver and other tissues, which occur when levels of unstable proteins exceed cellular compensatory mechanisms. It is likely that information gained through studies of non-erythroid protein aggregation disorders can be exploited to further understand and perhaps treat β-thalassemia.
Keywords: hemoglobin, thalassemia, protein quality control
Hemoglobin synthesis during erythroid maturation is tightly coordinated to maximize production of functional hemoglobin A (α2β2) and to balance the levels of individual globin subunits, which are unstable and cytotoxic in the absence of their partners. The importance of this homeostasis is illustrated in β-thalassemia, a common inherited anemia in which β globin gene mutations cause a relative excess of free α globin, which forms intracellular precipitates and reactive oxygen species.(1) Accumulation of free α chains destroys erythroid precursors in a process termed ineffective erythropoiesis and also shortens the lifespan of mature erythrocytes. The phenotype of β-thalassemia is largely determined by the degree of free α globin excess and ranges from minimal symptoms in β-thalassemia minor to severe β-thalassemia major. Interestingly, individuals who are heterozygous for a β globin null allele (β-thalassemia trait) are relatively unaffected, despite synthesis of two-fold excess α globin. This clinical observation indicates that erythroid precursors are able to neutralize significant amounts of free α globin. Defining the associated protective mechanisms will produce insights into basic erythroid biology and may also have implications for treating β-thalassemia.
Virtually all cell types detoxify and/or degrade potentially damaging unstable proteins through a process termed protein quality control. Numerous diseases ensue when the levels of unstable proteins exceed tissue capacities for quality control. Excessive protein misfolding, aggregation and consequent toxicities are linked to the pathogenesis of numerous diseases affecting nervous system, heart, pancreas, liver and other tissues (reviewed in (2-4)). In β-thalassemia, abundant excess α globin is destabilized by several mechanisms, including deficiency of its natural binding partner and autocatalytic oxidative stress. It is likely that this disease impacts erythroid protein quality control mechanisms and vice versa. Here we review experimental evidence that erythroid cells can mitigate the toxicity of excess α globin to a certain extent using generally conserved protein quality control pathways. We propose that β-thalassemia fits within a broader framework of protein aggregation disorders and that by applying the lessons learned from these non-erythroid diseases, we can extend our understanding of β-thalassemia pathogenesis and perhaps devise novel therapies for this common genetic disorder which causes significant morbidity in many areas of the world.(5) Conversely, closer examination of protein quality control using β thalassemia as a model system could provide new insights into relatively rare protein aggregation disorders such as amyotrophic lateral sclerosis and Huntington’s disease.
General principles of protein quality control: how cells handle unstable proteins
Basic principles of protein quality control are discussed briefly here and reviewed more extensively elsewhere (2-4, 6-8). Even under optimal circumstances, protein misfolding occurs frequently in all cells through “off-pathway” folding of otherwise normal polypeptides and random biosynthetic errors.(6, 8) High protein concentrations and rapid biosynthetic rates of proteins on polysomes increase non-productive associations that lead to misfolding and aggregation. These processes can be accelerated either during or after protein synthesis by various insults including destabilizing mutations, deficiency of endogenous binding partners and environmental stresses such as thermal and oxidant injury (Table 1). Note that several of these factors apply to β-thalassemia and other hemoglobinopathies.
Table 1. Causes of protein misfolding and aggregation.
Factors applicable to β-thalassemia are highlighted. For further information, see references (2-4, 6-8).
| Protein-intrinsic | |
|---|---|
| Mutated or incomplete proteins | Coding sequence mutations (missense or nonsense), synthetic errors, premature translation termination, proteolytic cleavage products |
| Lack of binding partner | Absence of obligate chaperone or multimeric complex partners, excess subunit synthesis |
| Postsynthetic damage | Oxidative or free radical damage, covalent modifications, denaturation, proteolytic cleavage |
| Misfolded proteins | Off-pathway folding - an inherent part of protein synthesis and existence |
| Intracellular conditions | |
| Thermal stress | Thermal denaturation |
| Reactive small molecules and free radicals | Oxidation, nitrosylation, glycation, deamidation, oxidative cross-linking |
| Extremes of ionic strength and pH | Favor multimer dissociation and protein denaturation |
| Macromolecular crowding | High protein synthetic rates |
| Presence of other misfolded or unfolded proteins | Nascent polypeptides (not yet fully folded), misfolded or mutated proteins, depletion of chaperones by other stresses |
Misfolded and aggregated proteins can be deleterious to the cell not only through loss of protein function, but also via toxic gain of function. They can interfere with other normal proteins through direct physical interactions or indirectly by sequestering and monopolizing protein quality control system components that regulate critical cellular functions (reviewed in (9)). Numerous interdependent elements contribute to protein quality control in virtually all cells. These include molecular chaperones, the ubiquitin-proteasome system (UPS) and lysosome/autophagy pathways.
Molecular Chaperones
Molecular chaperones represent a diverse and multi-functional class of proteins that bind “client” proteins to facilitate their folding and/or assembly into multi-subunit complexes (reviewed in (7, 10, 11)). Most chaperones, termed “public”, recognize a broad range of client proteins, frequently through interactions with hydrophobic surfaces. For example, ubiquitously expressed Hsp70 is estimated to bind up to 15-20% of newly synthesized proteins in mammalian cells.(12) Other “private” chaperones perform specialized functions by recognizing more limited repertoires of client proteins. Molecular chaperones participate in protein quality control through several mechanisms. They promote folding of many newly synthesized proteins and also help to repair proteins that become misfolded during cellular stress. Additionally, chaperones bind irreversibly damaged proteins to mitigate their potentially toxic effects, direct their subcellular localization to structures termed aggresomes and/or facilitate their degradation through lysosome-autophagy or ubiquitin-proteasome proteolytic systems.
The ubiquitin-proteasome system (UPS)
The UPS for protein degradation was initially recognized and characterized in reticulocytes and is now known to regulate numerous essential functions in all cells (reviewed in (8, 13)). For example, the UPS regulates cell growth, division and differentiation through precisely timed proteolysis of specific substrates. Oxygen sensitive UPS pathways regulate erythropoietin production to maintain circulating erythrocytes.(14) The UPS also participates in protein quality control by degrading misfolded and otherwise damaged proteins. Proteins are generally marked for degradation through covalent conjugation of polyubiquitin chains, which serve as a recognition signal for the 26S proteasome complex. Ubiquitin is joined to target proteins by the concerted action of E1 ubiquitin-activating enzymes, E2 ubiquitin-conjugating enzymes, and E3 ubiquitin-protein ligases, with the E3 ligase complex providing substrate specificity. E3 ubiquitin ligases can recognize substrates on their own or through interactions with chaperones and other adapter proteins, allowing for multiple layers of regulation and specificity for both normal and damaged proteins.
Autophagy
Autophagy refers to numerous processes in which cytoplasmic proteins or whole organelles are degraded by lysosomes (reviewed in (4, 15, 16)). Macroautophagy, the most commonly cited and best understood form, is a highly conserved pathway for bulk degradation of cytoplasmic contents, including organelles. During this process, the material to be degraded is enveloped in a double-membrane “autophagosome”, which then fuses with a lysosome to form an autolysosome. Chaperone-mediated autophagy represents direct translocation of chaperone-unfolded proteins into the lysosome for subsequent degradation. Microautophagy is the capture of small amounts of cytoplasmic material through direct invagination of lysosomal membranes. The most well defined function of autophagy is maintenance of the amino acid pool under starvation conditions through nonspecific bulk degradation. However recent work illustrates the importance of autophagy in a broad range of cellular functions. For example, during erythropoiesis, mitochondria are removed from reticulocytes through macroautophagy (discussed below). Autophagy mediated processes also participate in cellular protein quality control by removing misfolded and aggregated proteins, as illustrated in studies of neurodegenerative diseases.(4, 15)
Aggresomes
Until recently, protein aggregation was believed to be a passive process induced by association of sticky misfolded polypeptides. However, exciting new studies suggest that formation of aggregates may be a conserved, organized and regulated process whereby abnormal proteins are sequestered into organized structures termed aggresomes to inhibit harmful interactions with normal proteins and facilitate subsequent degradation (reviewed in (3, 17, 18)).
Aggresomes form when cellular capacity to remove or repair abnormal proteins is exceeded. These structures were initially described for abnormal membrane-bound proteins, including the cystic fibrosis transmembrane receptor,(19) and subsequently identified in a variety of diseases associated with abnormal cytoplasmic proteins.(20-28) In tissue culture models, aggresome formation is induced by expression of unstable proteins and/or inhibition of proteasomal degradation pathways.(19) Aggresomes surround centrioles (the microtubule-organizing center, MTOC) and may be enclosed in an intermediate filament cage. Aggresome formation is microtubule-dependent, and the current model holds that microtubule-associated motor proteins actively deliver dispersed protein precipitates to MTOC-associated aggresomes. Aggresomes contain ubiquitinated proteins, molecular chaperones and multiple components of the ubiquitin-proteasome pathway.
Current studies indicate a central role for aggresomes in protein quality control pathways. Proteasomes and chaperones such as Hsp70 and Hsp90 are present at the MTOC under basal conditions, perhaps to form the basis of an organized protein quality control center.(29, 30) Although aggresomes are relatively insoluble, their contents are dynamic. Photobleaching studies demonstrate the capacity for aggresome contents to exchange with soluble cytoplasmic compartments, perhaps through chaperone-mediated refolding.(31) The presence of proteasomes suggests that aggresomes may also serve as a proteolysis center. Aggresomes also appear to target abnormal proteins for degradation through autophagy. p62/SQSTM is a critical aggresome component that can bind polyubiquitinated proteins and polymerize around them, forming a shell to facilitate fusion with lysosomes.(32-35) In some cases, aggresomes can also sequester precipitated proteins that cannot be degraded, perhaps limiting their toxicity.(31) Overall, it appears that the functions of aggresomes and the ultimate fate of their contents depend on the cell type and the nature of the resident abnormal proteins.
Protein quality control pathways in disease
Disorders of protein aggregation have mainly been characterized in the central nervous system, but also involve numerous other tissues including liver, heart and pancreas (see Table 2 and (2, 4, 9, 10, 15, 18) for reviews). Despite divergent affected cell types and clinical features, several common themes emerge. First, abnormal disease-associated proteins are generally cleared through autophagy and/or the UPS. Clinical manifestations of cytotoxicity ensue when quality control systems become overloaded as cells age and accumulate excess aggregated protein over time. Second, abnormal proteins are frequently sequestered in aggresome-like structures, named according to the disease. For example, Mallory bodies in alcoholic liver disease and Lewy bodies in Parkinson’s disease resemble aggresomes morphologically and contain known aggresome components such as p62/SQSTM1.(24, 36) Third, diverse aggregation-prone proteins share similar mechanisms of cytotoxicity. One hypothesis is that these aggregates cause global inhibition of protein quality control systems through numerous potential mechanisms including sequestration of proteasomes and chaperones, physical clogging of proteasomes by aggregates, depletion of the free ubiquitin pool and increased oxidative stress.(9, 37-39) The result is misfolding and accumulation of “bystander” proteins and impaired degradation of critical physiological proteasome substrates such as cell cycle regulators, leading to cellular dysfunction and apoptosis.
Table 2.
Examples of protein Aggregation Diseases.
| Disease | Abnormal Protein | Cell type | Intracellular Inclusion |
|---|---|---|---|
| Alzheimer’s disease | Tau | Neurons | Neurofibrillar tangles |
| Amyotrophic lateral sclerosis (familial) | Superoxide dismutase 1 | Neurons | Bunina bodies |
| Parkinson’s disease | α-synuclein | Neurons | Lewy bodies |
| Huntington’s disease | Huntingtin (polyglutamine expansion) | Neurons | - |
| X-linked spinobulbar muscular atrophy (Kennedy’s disease) | Androgen receptor (polyglutamine expansion) | Neurons | - |
| Prion disease | Prion protein | Neurons | - |
| α1-antitrypsin deficiency | α1-antitrypsin | Hepatocytes | - |
| Alcoholic liver disease | Intermediate filaments | Hepatocytes | Mallory bodies |
| Wilson’s disease | ATP7B | Hepatocytes | - |
| β-thalassemia | α globin | Erythrocytes | - |
One interesting example of a protein aggregation disorder is familial amyotrophic lateral sclerosis (ALS) caused by mutations in superoxide dismutase 1 (SOD1).(40) These mutations cause toxicity not through loss of function, but rather, by generating a misfolded, aggregation-prone protein with enhanced ability to generate reactive oxygen species (reviewed in (41)), analogous to what occurs in β-thalassemia. Mutant SOD1 proteins are associated with aggresome-like structures (20) and cause global dysfunction of the ubiquitin proteasome system.(42, 43). Overexpression of the chaperone Hsp70 or Dorfin, a ubiquitin ligase that recognizes SOD1 mutants, can partially rescue the toxicity of SOD1 mutations by stabilizing the abnormal proteins or facilitating their degradation.(44, 45) In this way, efforts to define how protein quality control systems intersect with disease pathophysiology have elucidated new pathways for potential therapeutic manipulation.
Protein quality control systems in erythropoiesis
Erythroid maturation presents unique protein quality control challenges, including the high concentration of cytotoxic hemoglobin subunit proteins, oxidative stress associated with iron (both free and heme-bound), and the need to clear unnecessary non-globin proteins and organelles during terminal maturation. Accordingly, many of the principles and systems of protein quality control can be applied to normal erythroid development and associated diseases, particularly hemoglobinopathies such as β-thalassemia.
Molecular chaperones
The involvement of molecular chaperone systems in erythroid development is reviewed in (46). Reticulocytes can increase production of chaperones in response to stress through preferential mRNA translation, indicating the importance of protein quality control during late stage erythroid maturation.(47) “Public” chaperones such as Hsp70 facilitate erythroid maturation through regulation of key erythroid proteins.(48) Alpha hemoglobin stabilizing protein (AHSP) is an erythroid-specific “private” chaperone that specifically stabilizes free α globin subunits. Loss of AHSP in mice causes hemolytic anemia with globin precipitates (Heinz bodies) and exacerbates β-thalassemia.(49-51) It is likely that other important globin chaperones, both private and public, play important roles in erythroid cells.
Ubiquitin mediated proteolysis
The UPS was initially described in reticulocytes, a relatively simple anucleate cell that degrades multiple proteins as part of normal maturation.(52-54) More recent studies explore the role of proteasome-dependent protein degradation during erythropoiesis.(55, 56) Activated erythropoietin receptors may be cleared in part through proteasomal degradation.(57) Cytoskeletal proteins actin and tubulin are degraded by the UPS in reticulocytes,(55) and pharmacological proteasome inhibition interferes with numerous aspects of erythroid maturation in vitro, including enucleation.(56) Our general understanding of the UPS and its components is expanding rapidly and it likely that future studies will identify additional roles for targeted proteolysis in globin homeostasis and erythroid maturation.
Autophagy
Maturing erythroid cells eliminate not only soluble proteins, but also entire organelles. Reticulocytes degrade mitochondria and ribosomes by autophagy.(58-61) Targeted deletions of murine genes encoding macroautophagy pathway components, including Nix,(62, 63) Ulk1,(64) and Atg7,(65) interfere to varying degrees with physiological degradation of mitochondria during reticulocyte maturation. Morphological studies described later in this review illustrate lysosomal engulfment of α globin inclusions in β thalassemic erythroblasts, although the overall importance of autophagic pathways in degrading excess α globin is not known.
Proteolytic control of globin chain balance
The synthesis and subsequent fates of nascent globin chains have been analyzed extensively through pulse labeling studies in normal and thalassemic erythroid precursors.(66-71) In normal erythroid precursors, globin chain synthesis is relatively balanced, with some studies revealing slight excess of α chains in a soluble pool.(72, 73) The per-cell globin chain synthetic rates are highest in the basophilic erythroblast stage and decrease as the cells mature,(74-76) but globin synthesis and accumulation continues through the late stages, with reticulocytes accumulating as much as 7-20% of total hemoglobin content.(77) β-thalassemic erythroid precursors contain a larger pool of excess free α chains that are initially competent to form hemoglobin tetramers, but are unstable in the absence of their binding partner.(69, 78-80) These early studies provided the first evidence linking α globin excess to the pathophysiology of β-thalassemia.(81-83) Analysis of β-thalassemia trait and intermedia erythroid precursors at different stages of maturation revealed further insights into globin chain metabolism. In these studies, the α:β chain ratio increased during successive stages of maturation with the highest degrees of α globin excess found in circulating reticulocytes. Prolonged labeling experiments showed that in bone-marrow erythroblasts, β chains accumulate at a constant rate, but nascent α chain levels stabilize over time, so that the α:β imbalance decreased. This effect was highest in earlier maturation stages. These data indicate that β-thalassemic erythroid precursors, particularly at early developmental states, degrade excess α globin to prevent its accumulation. This protective mechanism becomes less effective during the course of erythroid maturation as globin chains accumulate and the capacity to remove excess α chains becomes overwhelmed.
Subsequent experiments characterized the proteolytic degradation of α globin. Hanash et al performed mixing studies of reticulocyte and normoblast lysates to show that earlier erythroid precursors have a greater capacity to degrade excess α chains.(84) This activity was specific for α globin and inhibited by proteolysis inhibitors.(85-87) Moreover, α globin proteolysis was higher in β-thalassemia erythroid precursors than in those of patients with other anemias or causes of reticulocytosis. Early experiments describing the ubiquitin-proteasome system in reticulocyte lysates used denatured globins or globin chains rendered unstable by incorporation of amino acid analogs.(52, 53, 88) These experiments all indicate that erythroid precursors have the capacity to degrade excess or structurally unstable globins.
Later studies by Schaeffer and colleagues, focused on the role of the UPS in α globin degradation. Pulse-chase studies of intact β thalassemia reticulocytes showed that they degrade endogenous α globin but not β globin, in an ATP-dependent fashion.(89, 90) Biochemical studies showed that β-thalassemia reticulocyte lysates can ubiquitinate added α globin at several sites to facilitate its degradation by proteasomes. (90-94) Moreover, mono-or poly ubiquitination of α globin was required for its proteasomal degradation.(94) Further work showed that free α, β, and γ globin chains can be ubiquitinated and degraded using in vitro reconstituted systems, and that this process can occur co-translationally or shortly after release of nascent chains from ribosomes.(95)
One implication of this research is that differences in proteolytic capacity for α globin may account for some of the phenotype heterogeneity in β-thalassemia and for interspecies differences in the manifestations of β-globin gene mutations. In humans, a 50% reduction in β globin synthesis usually results in asymptomatic β-thalassemia trait. In mice, a similar reduction in β globin gene dosage causes a more severe phenotype resembling β-thalassemia intermedia.(96, 97) Rouyer-Fessard et al have suggested that this interspecies difference may be due in part to reduced capacity for degrading unstable toxic α globin in mouse erythroblasts.(98, 99)
Of note, accumulation of free β globin chains in α thalassemia also poses protein quality control problems for erythroid precursors. For example, pulse-chase labeling of reticulocytes from a patient with α thalassemia showed normalization of globin chain accumulation through preferential degradation of β globin, similar to what is seen with α chains in β-thalassemia.(100) However, compared to α globin, free β–like globin chains are more stable and soluble, in part due their ability to form homotetramers. Compared to β thalassemia, α thalassemia is associated with fewer globin precipitates in erythroid precursors and less ineffective erythropoiesis. However, severe forms of α thalassemia are characterized by extensive HbH (β4) inclusions in circulating erythrocytes, causing shortened half-life and anemia. Further comparative studies are required to better define the similarities and differences in how protein quality control pathways handle excess free α and β globin chains.
Together, numerous studies over many years indicate that the UPS system can degrade unstable globins as a protective mechanism for hemoglobinopathies, particularly β-thalassemia. Of note, additional proteolytic systems may complement this UPS function. For example, oxidant-damaged hemoglobin can be degraded by intact reticulocytes and extracts in ATP- and ubiquitin-independent fashions, possibly through a cytosolic protease.(101, 102) Interestingly, this activity appears to be retained in mature erythrocytes.
Unstable globin aggregation
Inclusion bodies have long been considered a hallmark of the thalassemias. In β-thalassemia, small intracellular aggregates appear in early normoblasts and coalesce into large inclusions by late normoblast and reticulocyte stages.(103, 104) The size and frequency of inclusions correlate with disease severity.(103, 105) The inclusions, initially described as “stromal-bound” globins, were found to contain as much as 10% of all α globin synthesized in splenectomized β-thalassemia major patients.(106-108) Labeling studies showed that in β-thalassemia, aggregation-prone α globin enters the insoluble stromal fraction rapidly after synthesis (67, 78, 87, 109) and may be removed subsequently by ATP-dependent proteolytic pathways.
Klemes et al used unstable valine analogs to label rabbit reticulocytes and found that the resultant abnormal globins formed high molecular weight, non-membrane bound structures that were insoluble in mild detergents but soluble in harsher ionic detergents.(88) These aggregates increased in molecular weight and decreased in solubility with longer amino acid analog treatment and after ATP-depletion. In pulse-chase studies, the insoluble fraction was subject to ATP-dependent degradation. In β-thalassemic reticulocytes, similar ATP-dependent turnover of the stromal α globin fraction was seen in parallel with removal of soluble α globin.(89) It is not clear whether this turnover is mediated by cytosolic proteases or membrane/stromal associated systems, as other studies showed that a proteolytic activity for α globin also exists in purified stromal fractions.(87, 109) The most likely explanation is that soluble α chains are degraded by soluble proteasomal components, while the insoluble α chains are degraded by a combination of stromal-associated proteasomal and lysosomal pathways.
Wickramasinghe and others demonstrated by electron microscopy that β-thalassemic erythroblasts contained electrodense inclusions beginning at early polychromatic stages with increasing in size and frequency during subsequent maturation.(110, 111) These inclusions also occur to a lesser extent in β-thalassemia trait.(112) Remarkably, the α globin inclusions of β-thalassemia share numerous properties with classical aggresomes discovered years later in studies of other protein precipitation disorders (Figure 1). These include cytoplasmic, juxtanuclear localization in association with centrioles.(110, 112, 113) Immuno-electron microscopy confirmed that the β-thalassemic inclusions contain α globin (but not β globin) and also ubiquitin, consistent with the presence of ubiquitinated α globin chains targeted for degradation by the UPS.(114, 115) Ultrastructural studies also revealed β-thalassemic α globin inclusions in autophagic vacuoles, suggesting elimination by macroautophagy.(110, 112) Similar inclusions containing both α and β globin were found in patients with dominantly-inherited β-thalassemia caused by mutations that destabilize β globin chains.(116, 117) Ultrastructural studies of α-thalassemia 1 trait and HbH disease showed a similar pattern with excess β globin chains, with degree of precipitation proportional to globin chain imbalance.(118, 119) Similar appearing aggregates containing globin and non-globin proteins have also been reported in patients with congenital dyserythropoietic anemia.(115, 120, 121) Together, these studies indicate that pathologic erythroid precursors sequester unstable globin chains and possibly other abnormal proteins into aggresome-like structures resembling those observed in neurodegenerative and other protein aggregation disorders.
Figure 1. Electron microscopy reveals aggresome-like α globin inclusions in β-thalassemia.
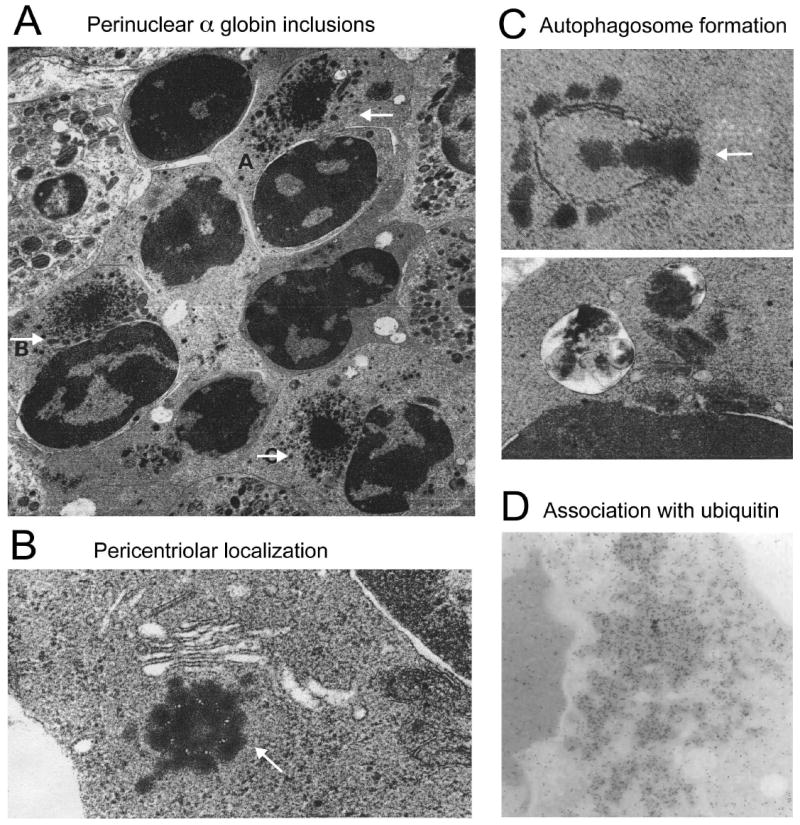
(A) Several late polychromatic erythroblasts, labeled A, B, and C, with intracytoplasmic perinuclear α-chain precipitates (adapted from Wickramasinghe SN, Bush V. Observations on the ultrastructure of erythropoietic cells and reticulum cells in the bone marrow of patients with homozygous beta-thalassaemia. Br J Haematol 1975;30(4):395-399, with permission); (B) precipitation of α-chains around the centriole of an erythroblast. The arrow indicates microtubule triplets in centriole cross-section (adapted from Wickramasinghe SN, Hughes M. Precipitation of alpha-chains on the centrioles of erythroblasts in beta-thalassaemia. Br J Haematol 1982;52(4):681-682, with permission); (C) Top: Autophagosome membrane formation partially enclosing precipitated α-chains; Bottom: Two autophagic vacuoles containing electrodense material, probably precipitated α chains (adapted from Wickramasinghe SN, Hughes M. Ultrastructural studies of erythropoiesis in beta-thalassaemia trait. Br J Haematol 1980;46(3):401-407, with permission); (D) Immunogold staining for ubiquitin within α globin precipitates of a β-thalassemic erythroblast (adapted from Wickramasinghe SN, Lee MJ. Evidence that the ubiquitin proteolytic pathway is involved in the degradation of precipitated globin chains in thalassaemia. Br J Haematol 1998;101(2):245-250, with permission).
Putting it all together
Many of the pioneering studies to understand globin chain maintenance in β-thalassemia and other hemoglobinopathies were performed when the field of protein quality control was in its infancy. For example, studies of Wickramasinghe and colleagues indicated that precipitated α globin accumulates in aggresomes years before this structure was appreciated or defined (Figure 1).(110, 112-114) Now, it appears that erythroid cells compartmentalize and metabolize unstable globins, at least in part, through generally conserved pathways, including the UPS and autophagy, which are studied most extensively in other protein aggregation disorders. How protein quality control likely modulates β thalassemia is illustrated in Figure 2. Early stage β-thalassemic erythroblasts neutralize excess α globin reasonably well via proteolytic systems and/or chaperones. As total globin levels increase during maturation, free α globin becomes compartmentalized into aggresome-like structures, which are modeled and eliminated via the UPS and autophagy. Ultimately, in severe forms of the disease, α globin precipitation and aggregation overwhelms compensatory detoxification mechanisms, resulting in cellular toxicities associated with ineffective erythropoiesis (Figure 3). Damage from unstable α globin may arise from its direct toxicities and also indirectly via its ability to inhibit protein quality control systems that are essential for various aspects of erythroid maturation. While erythroid protein quality control systems are not fully defined, they almost certainly contain both generally expressed components, such as ubiquitous proteases and public chaperones, as well as cell type-restricted molecules, such as AHSP and others.
Figure 2. Model for repair or elimination of free α globin by cellular protein quality control systems.
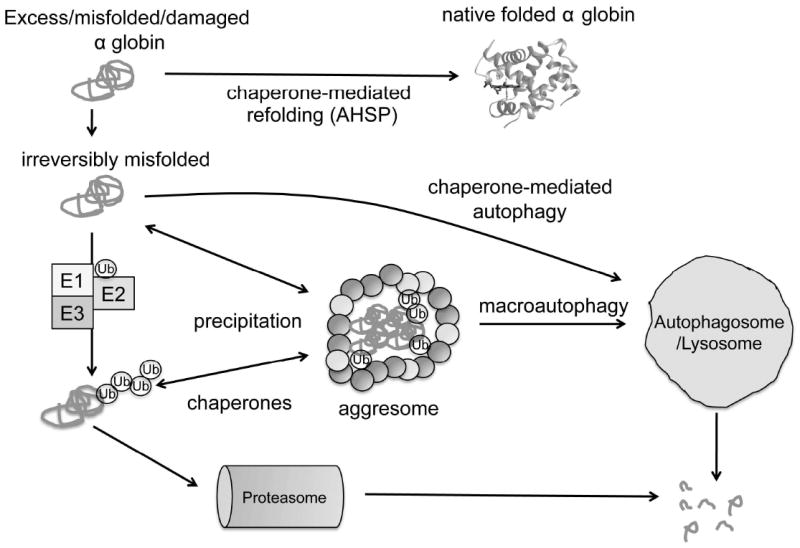
Misfolded free α globin may be stabilized by interactions with molecular chaperones, perhaps AHSP and/or generalized public chaperones. Irreversibly damaged protein may be removed either by chaperone-mediated autophagy or ubiquitin-proteasome (UPS) pathways. E1 ubiquitin-activating enzymes, E2 ubiquitin-conjugating enzymes, and E3 ubiquitin ligases mediate conjugation of polyubiquitin (Ub) chains to α globin, targeting it for proteasomal degradation via the UPS. In addition, insoluble α globin (+/- Ub) can be shuttled to aggresomes, which are subsequently cleared by macroautophagy.
Figure 3. Overloading of degradation systems leads to accumulation of α globin in β-thalassemia.
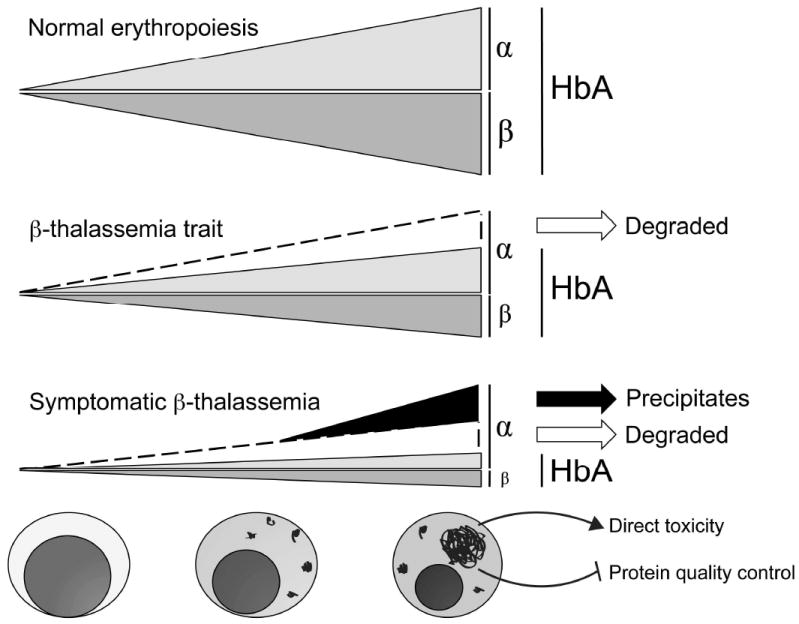
During normal erythropoiesis, α and β globin subunits are synthesized close to equally and join to form HbA (α2β2) tetramers. In β-thalassemia trait, excess α chains are synthesized, but removed by protein quality control systems and there is minimal pathology. In β-thalassemia, free α globin is degraded in early erythroid precursors in attempts to balance globin synthesis. As cellular maturation proceeds, free α chain levels overwhelm compensatory mechanisms, resulting in accumulation of toxic precipitates. Precipitated α globin can damage the cell either directly, or indirectly by interfering with the functions of protein quality control systems.
Future directions
We speculate that many general lessons learned from various protein misfolding and aggregation diseases can be applied to better understand β-thalassemia pathogenesis and discover new treatments. Recent technological advances such as development of animal disease models and high-throughput screening systems should allow closer examination of β-thalassemia, address questions arising from earlier work, and better define the mechanisms of α globin metabolism. Mouse models of β-thalassemia major and intermedia (96, 97) and methods for culture of human erythroid progenitors from β-thalassemia patients (122) may be used for genetic and pharmacologic dissection of pathways required for α globin detoxification in vivo. Mice with targeted deletions of key aggresome components (33, 123) and critical autophagy pathway genes (63-65) can be bred to β-thalassemia mouse models to determine the importance of these pathways in β-thalassemia. Additionally, shRNA approaches to disrupt these pathways can be used to study cultured human or mice erythroblasts. For example, it should be possible to configure high-throughput screens using small inhibitory RNAs to identify specific E3 ubiquitin ligases and other quality control components that regulate free α globin in erythroid cells. The UPS and aggresome formation pathways can also be manipulated in thalassemia using pharmacologic inhibitors or enhancers that are either in clinical use for other disorders (such as proteasome inhibitor bortezomib in multiple myeloma) or in development (18, 124-126).
Current high-resolution immunofluoresence microscopy can confirm whether α globin inclusions found in β-thalassemic erythroblasts are bona fide aggresomes. Insoluble protein aggregates from thalassemia patient samples can also be studied by mass spectrometry to identify constituent proteins with presumed roles in α globin quality control. This proteomic approach has yielded useful information in other unstable protein disorders.(127) For example, mass spectroscopy of Mallory bodies in alcoholic liver disease showed them to contain aggresomeassociated intermediate filaments and p62/SQSTM, a critical aggresome component.(35) In addition to identifying known aggresome components, mass spectroscopy of β-thalassemic aggregates might identify new α globin-specific chaperones or ubiquitin ligases.
As discussed earlier, excessive protein misfolding and aggregation can damage cells by inhibiting protein quality control systems. This pathophysiology may be particularly important in β-thalassemia, since erythroid precursors must degrade numerous proteins and organelles to undergo normal maturation. This hypothesis can be tested using newly developed in vivo fluorescent reporters that read out UPS activity.(128) Proteomic approaches can define whether UPS substrates accumulate in erythroid precursors. It may also be informative to determine whether some β-thalassemia phenotypes can be recapitulated by overexpression of non-globin aggregation-prone proteins in erythroid precursors.
In summary, protein quality control pathways are becoming increasingly appreciated as important modifiers of human disease and are likely to function during normal erythropoiesis and in β-thalassemia. In parallel, new technologies are rapidly enhancing the ability to study and manipulate how cells handle unstable proteins. With these new tools and perspectives in hand, it should be possible it should be possible to initiate new studies to better understand and eventually treat β-thalassemia.
Acknowledgments
We thank Stephen Liebhaber and Vijay Sankaran for helpful critiques of the manuscript. Work in our laboratory on globin chain metabolism and thalassemia is supported by National Institutes of Health Grants 5R01HL087427-04 (MJW), 5R01DK061692-08 (MJW) and 3T32GM007170-35S1 (EK), and by the Cooley’s Anemia Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weatherall DJ, Clegg JB. The thalassaemia syndromes. 4. Oxford ; Malden, MA: Blackwell Science; 2001. [Google Scholar]
- 2.Aigelsreiter A, Janig E, Stumptner C, et al. How a cell deals with abnormal proteins. Pathogenetic mechanisms in protein aggregation diseases. Pathobiology. 2007;74(3):145–158. doi: 10.1159/000103374. [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Mata R, Gao Y-S, Sztul E. Hassles with taking out the garbage: aggravating aggresomes. Traffic. 2002;3(6):388–396. doi: 10.1034/j.1600-0854.2002.30602.x. [DOI] [PubMed] [Google Scholar]
- 4.Ding W-X, Yin X-M. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. 2008;4(2):141–150. doi: 10.4161/auto.5190. [DOI] [PubMed] [Google Scholar]
- 5.Angastiniotis M, Modell B. Global epidemiology of hemoglobin disorders. Ann N Y Acad Sci. 1998;850:251–269. doi: 10.1111/j.1749-6632.1998.tb10482.x. [DOI] [PubMed] [Google Scholar]
- 6.Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16(6):574–581. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 7.McClellan AJ, Tam S, Kaganovich D, et al. Protein quality control: chaperones culling corrupt conformations. Nat Cell Biol. 2005;7(8):736–741. doi: 10.1038/ncb0805-736. [DOI] [PubMed] [Google Scholar]
- 8.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426(6968):895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 9.Dantuma N, Lindsten K. Stressing the ubiquitin/proteasome system. Cardiovascular Research. 2009 doi: 10.1093/cvr/cvp255. [DOI] [PubMed] [Google Scholar]
- 10.Barral JM, Broadley SA, Schaffar G, et al. Roles of molecular chaperones in protein misfolding diseases. Semin Cell Dev Biol. 2004;15(1):17–29. doi: 10.1016/j.semcdb.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 11.Broadley SA, Hartl FU. The role of molecular chaperones in human misfolding diseases. FEBS Letters. 2009;583(16):2647–2653. doi: 10.1016/j.febslet.2009.04.029. [DOI] [PubMed] [Google Scholar]
- 12.Thulasiraman V, Yang CF, Frydman J. In vivo newly translated polypeptides are sequestered in a protected folding environment. EMBO J. 1999;18(1):85–95. doi: 10.1093/emboj/18.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 14.Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 15.Jaeger PA, Wyss-Coray T. All-you-can-eat: autophagy in neurodegeneration and neuroprotection. Mol Neurodegener. 2009;4:16. doi: 10.1186/1750-1326-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10(12):524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 18.Olzmann JA, Li L, Chin LS. Aggresome formation and neurodegenerative diseases: therapeutic implications. Curr Med Chem. 2008;15(1):47–60. doi: 10.2174/092986708783330692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143(7):1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnston JA, Dalton MJ, Gurney ME, et al. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2000;97(23):12571–12576. doi: 10.1073/pnas.220417997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.García-Mata R, Bebök Z, Sorscher EJ, et al. Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J Cell Biol. 1999;146(6):1239–1254. doi: 10.1083/jcb.146.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Junn E, Lee SS, Suhr UT, et al. Parkin accumulation in aggresomes due to proteasome impairment. J Biol Chem. 2002;277(49):47870–47877. doi: 10.1074/jbc.M203159200. [DOI] [PubMed] [Google Scholar]
- 23.Ma J, Lindquist S. Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc Natl Acad Sci USA. 2001;98(26):14955–14960. doi: 10.1073/pnas.011578098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McNaught KSP, Shashidharan P, Perl DP, et al. Aggresome-related biogenesis of Lewy bodies. Eur J Neurosci. 2002;16(11):2136–2148. doi: 10.1046/j.1460-9568.2002.02301.x. [DOI] [PubMed] [Google Scholar]
- 25.Mukai H, Isagawa T, Goyama E, et al. Formation of morphologically similar globular aggregates from diverse aggregation-prone proteins in mammalian cells. Proc Natl Acad Sci USA. 2005;102(31):10887–10892. doi: 10.1073/pnas.0409283102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salomons FA, Menéndez-Benito V, Böttcher C, et al. Selective accumulation of aggregation-prone proteasome substrates in response to proteotoxic stress. Mol Cell Biol. 2009;29(7):1774–1785. doi: 10.1128/MCB.01485-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanaka M, Kim YM, Lee G, et al. Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J Biol Chem. 2004;279(6):4625–4631. doi: 10.1074/jbc.M310994200. [DOI] [PubMed] [Google Scholar]
- 28.Waelter S, Boeddrich A, Lurz R, et al. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12(5):1393–1407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wigley WC, Fabunmi RP, Lee MG, et al. Dynamic association of proteasomal machinery with the centrosome. J Cell Biol. 1999;145(3):481–490. doi: 10.1083/jcb.145.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wojcik C, Schroeter D, Wilk S, et al. Ubiquitin-mediated proteolysis centers in HeLa cells: indication from studies of an inhibitor of the chymotrypsin-like activity of the proteasome. Eur J Cell Biol. 1996;71(3):311–318. [PubMed] [Google Scholar]
- 31.Kaganovich D, Kopito R, Frydman J. Misfolded proteins partition between two distinct quality control compartments. Nature. 2008;454(7208):1088–1095. doi: 10.1038/nature07195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bjørkøy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171(4):603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 34.Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 35.Zatloukal K, Stumptner C, Fuchsbichler A, et al. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol. 2002;160(1):255–263. doi: 10.1016/S0002-9440(10)64369-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riley NE, Li J, Worrall S, et al. The Mallory body as an aggresome: in vitro studies. Exp Mol Pathol. 2002;72(1):17–23. doi: 10.1006/exmp.2001.2413. [DOI] [PubMed] [Google Scholar]
- 37.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292(5521):1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 38.Bennett EJ, Shaler TA, Woodman B, et al. Global changes to the ubiquitin system in Huntington’s disease. Nature. 2007;448(7154):704–708. doi: 10.1038/nature06022. [DOI] [PubMed] [Google Scholar]
- 39.Bennett EJ, Bence NF, Jayakumar R, et al. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Molecular Cell. 2005;17(3):351–365. doi: 10.1016/j.molcel.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 40.Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 41.Cleveland DW, Liu J. Oxidation versus aggregation - how do SOD1 mutants cause ALS? Nat Med. 2000;6(12):1320–1321. doi: 10.1038/82122. [DOI] [PubMed] [Google Scholar]
- 42.Cheroni C, Marino M, Tortarolo M, et al. Functional alterations of the ubiquitin-proteasome system in motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Hum Mol Genet. 2009;18(1):82–96. doi: 10.1093/hmg/ddn319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Urushitani M, Kurisu J, Tsukita K, et al. Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J Neurochem. 2002;83(5):1030–1042. doi: 10.1046/j.1471-4159.2002.01211.x. [DOI] [PubMed] [Google Scholar]
- 44.Niwa J-I, Ishigaki S, Hishikawa N, et al. Dorfin ubiquitylates mutant SOD1 and prevents mutant SOD1- mediated neurotoxicity. J Biol Chem. 2002;277(39):36793–36798. doi: 10.1074/jbc.M206559200. [DOI] [PubMed] [Google Scholar]
- 45.Bruening W, Roy J, Giasson B, et al. Up-regulation of protein chaperones preserves viability of cells expressing toxic Cu/Zn-superoxide dismutase mutants associated with amyotrophic lateral sclerosis. J Neurochem. 1999;72(2):693–699. doi: 10.1046/j.1471-4159.1999.0720693.x. [DOI] [PubMed] [Google Scholar]
- 46.Weiss MJ, Dos Santos CO. Chaperoning erythropoiesis. Blood. 2009;113(10):2136–2144. doi: 10.1182/blood-2008-09-115238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Banerji SS, Theodorakis NG, Morimoto RI. Heat shock-induced translational control of HSP70 and globin synthesis in chicken reticulocytes. Mol Cell Biol. 1984;4(11):2437–2448. doi: 10.1128/mcb.4.11.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ribeil JA, Zermati Y, Vandekerckhove J, et al. Hsp70 regulates erythropoiesis by preventing caspase-3- mediated cleavage of GATA-1. Nature. 2007;445(7123):102–105. doi: 10.1038/nature05378. [DOI] [PubMed] [Google Scholar]
- 49.Kihm AJ, Kong Y, Hong W, et al. An abundant erythroid protein that stabilizes free alpha-haemoglobin. Nature. 2002;417(6890):758–763. doi: 10.1038/nature00803. [DOI] [PubMed] [Google Scholar]
- 50.Yu X, Kong Y, Dore LC, et al. An erythroid chaperone that facilitates folding of alpha-globin subunits for hemoglobin synthesis. J Clin Invest. 2007;117(7):1856–1865. doi: 10.1172/JCI31664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kong Y, Zhou S, Kihm AJ, et al. Loss of alpha-hemoglobin-stabilizing protein impairs erythropoiesis and exacerbates beta-thalassemia. J Clin Invest. 2004;114(10):1457–1466. doi: 10.1172/JCI21982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ciehanover A, Hod Y, Hershko A. A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem Biophys Res Commun. 1978;81(4):1100–1105. doi: 10.1016/0006-291x(78)91249-4. [DOI] [PubMed] [Google Scholar]
- 53.Etlinger JD, Goldberg AL. A soluble ATP-dependent proteolytic system responsible for the degradation of abnormal proteins in reticulocytes. Proc Natl Acad Sci USA. 1977;74(1):54–58. doi: 10.1073/pnas.74.1.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hershko A, Heller H, Elias S, et al. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J Biol Chem. 1983;258(13):8206–8214. [PubMed] [Google Scholar]
- 55.Liu J, Guo X, Mohandas N, et al. Membrane remodeling during reticulocyte maturation. Blood. 2010;115(10):2021–2027. doi: 10.1182/blood-2009-08-241182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen CY, Pajak L, Tamburlin J, et al. The effect of proteasome inhibitors on mammalian erythroid terminal differentiation. Exp Hematol. 2002;30(7):634–639. doi: 10.1016/s0301-472x(02)00826-3. [DOI] [PubMed] [Google Scholar]
- 57.Walrafen P, Verdier F, Kadri Z, et al. Both proteasomes and lysosomes degrade the activated erythropoietin receptor. Blood. 2005;105(2):600–608. doi: 10.1182/blood-2004-03-1216. [DOI] [PubMed] [Google Scholar]
- 58.Gronowicz G, Swift H, Steck TL. Maturation of the reticulocyte in vitro. J Cell Sci. 1984;71:177–197. doi: 10.1242/jcs.71.1.177. [DOI] [PubMed] [Google Scholar]
- 59.Heynen MJ, Tricot G, Verwilghen RL. Autophagy of mitochondria in rat bone marrow erythroid cells. Relation to nuclear extrusion. Cell Tissue Res. 1985;239(1):235–239. doi: 10.1007/BF00214924. [DOI] [PubMed] [Google Scholar]
- 60.Kent G, Minick OT, Volini FI, et al. Autophagic vacuoles in human red cells. Am J Pathol. 1966;48(5):831–857. [PMC free article] [PubMed] [Google Scholar]
- 61.Takano-Ohmuro H, Mukaida M, Kominami E, et al. Autophagy in embryonic erythroid cells: its role in maturation. Eur J Cell Biol. 2000;79(10):759–764. doi: 10.1078/0171-9335-00096. [DOI] [PubMed] [Google Scholar]
- 62.Sandoval H, Thiagarajan P, Dasgupta SK, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454(7201):232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schweers RL, Zhang J, Randall MS, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA. 2007;104(49):19500–19505. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kundu M, Lindsten T, Yang C-Y, et al. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112(4):1493–1502. doi: 10.1182/blood-2008-02-137398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang J, Randall MS, Loyd MR, et al. Mitochondrial clearance is regulated by Atg7-dependent and - independent mechanisms during reticulocyte maturation. Blood. 2009;114(1):157–164. doi: 10.1182/blood-2008-04-151639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bank A, O’Donnell JV. Intracellular loss of free alpha chains in beta thalassemia. Nature. 1969;222(5190):295–296. doi: 10.1038/222295a0. [DOI] [PubMed] [Google Scholar]
- 67.Bargellesi A, Pontremoli S, Menini C, et al. Excess of alpha-globin synthesis in homozygous betathalassemia and its removal from the red blood cell cytoplasm. Eur J Biochem. 1968;3(3):364–368. doi: 10.1111/j.1432-1033.1968.tb19538.x. [DOI] [PubMed] [Google Scholar]
- 68.Chalevelakis G, Clegg JB, Weatherall DJ. Imbalanced globin chain synthesis in heterozygous betathalassemic bone marrow. Proc Natl Acad Sci USA. 1975;72(10):3853–3857. doi: 10.1073/pnas.72.10.3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Clegg JB, Weatherall DJ. Haemoglobin synthesis during erythroid maturation in -thalassaemia. Nature New Biol. 1972;240(101):190–192. doi: 10.1038/newbio240190a0. [DOI] [PubMed] [Google Scholar]
- 70.Kan YW, Nathan DG, Lodish HF. Equal synthesis of - and -globin chains in erythroid precursors in heterozygous -thalassemia. J Clin Invest. 1972;51(7):1906–1909. doi: 10.1172/JCI106993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wood WG, Stamatoyannopoulos G. Globin synthesis in fractionated Normoblasts of beta-thalassemia heterozygotes. J Clin Invest. 1975;55(3):567–578. doi: 10.1172/JCI107964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shaeffer JR. Evidence for soluble alpha-chains as intermediates in hemoglobin synthesis in the rabbit reticulocyte. Biochem Biophys Res Commun. 1967;28(4):647–652. doi: 10.1016/0006-291x(67)90363-4. [DOI] [PubMed] [Google Scholar]
- 73.Gill FM, Schwartz E. Free alpha-globin pool in human bone marrow. J Clin Invest. 1973;52(12):3057–3063. doi: 10.1172/JCI107504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Borsook H, Lingrel JB, Scaro JL, et al. Synthesis of haemoglobin in relation to the maturation of erythroid cells. Nature. 1962;196:347–350. doi: 10.1038/196347a0. [DOI] [PubMed] [Google Scholar]
- 75.Casale GP, Khairallah EA, Grasso JA. An analysis of hemoglobin synthesis in erythropoietic cells. Dev Biol. 1980;80(1):107–119. doi: 10.1016/0012-1606(80)90502-3. [DOI] [PubMed] [Google Scholar]
- 76.Nathan DG, Piomelli S, Gardner FH. The synthesis of heme and globin in the maturing human erythroid cell. J Clin Invest. 1961;40:940–946. doi: 10.1172/JCI104333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Skadberg O, Brun A, Sandberg S. Human reticulocytes isolated from peripheral blood: maturation time and hemoglobin synthesis. Lab Hematol. 2003;9(4):198–206. [PubMed] [Google Scholar]
- 78.Bank A. Hemoglobin synthesis in beta-thalassemia: the properties of the free alpha-chains. J Clin Invest. 1968;47(4):860–866. doi: 10.1172/JCI105779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bargellesi A, Pontremoli S, Menini C, et al. Kinetic evidence for the existence of alpha-globin pool in betathalassemic reticulocytes. Eur J Biochem. 1968;7(1):73–77. doi: 10.1111/j.1432-1033.1968.tb19576.x. [DOI] [PubMed] [Google Scholar]
- 80.Conconi F, Bargellesi A, Pontremoli S. Excess of alpha-globin synthesis in homozygous beta-thalassemia. Its cytoplasmic molecular forms. Eur J Biochem. 1968;5(3):409–414. doi: 10.1111/j.1432-1033.1968.tb00384.x. [DOI] [PubMed] [Google Scholar]
- 81.Nathan DG, Stossel TB, Gunn RB, et al. Influence of hemoglobin precipitation on erythrocyte metabolism in alpha and beta thalassemia. J Clin Invest. 1969;48(1):33–41. doi: 10.1172/JCI105972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rachmilewitz EA, Peisach J, Blumberg WE. Studies on the stability of oxyhemoglobin A and its constituent chains and their derivatives. J Biol Chem. 1971;246(10):3356–3366. [PubMed] [Google Scholar]
- 83.Bank A, Braverman AS, O’Donnell JV, et al. Absolute rates of globin chain synthesis in thalassemia. Blood. 1968;31(2):226–233. [PubMed] [Google Scholar]
- 84.Hanash SM, Rucknagel DL. Proteolytic activity in erythrocyte precursors. Proc Natl Acad Sci USA. 1978;75(7):3427–3431. doi: 10.1073/pnas.75.7.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Braverman AS, Lester D. Evidence for increased proteolysis in intact beta thalassemia erythroid cells. Hemoglobin. 1981;5(6):549–564. doi: 10.3109/03630268108991686. [DOI] [PubMed] [Google Scholar]
- 86.Loukopoulos D, Karoulias A, Fessas P. Proteolysis in thalassemia: studies with protease inhibitors. Annals of the New York Academy of Sciences. 1980;344:323–335. doi: 10.1111/j.1749-6632.1980.tb33672.x. [DOI] [PubMed] [Google Scholar]
- 87.Vettore L, De Matteis MC, Di Iorio EE, et al. Erythrocytic proteases: preferential degradation of alpha hemoglobin chains. Acta Haematol. 1983;70(1):35–42. doi: 10.1159/000206686. [DOI] [PubMed] [Google Scholar]
- 88.Klemes Y, Etlinger JD, Goldberg AL. Properties of abnormal proteins degraded rapidly in reticulocytes. Intracellular aggregation of the globin molecules prior to hydrolysis. J Biol Chem. 1981;256(16):8436–8444. [PubMed] [Google Scholar]
- 89.Shaeffer JR. Turnover of excess hemoglobin alpha chains in beta-thalassemic cells is ATP-dependent. J Biol Chem. 1983;258(21):13172–13177. [PubMed] [Google Scholar]
- 90.Shaeffer JR. ATP-dependent proteolysis of hemoglobin alpha chains in beta-thalassemic hemolysates is ubiquitin-dependent. J Biol Chem. 1988;263(27):13663–13669. [PubMed] [Google Scholar]
- 91.Shaeffer JR. Monoubiquitinated alpha globin is an intermediate in the ATP-dependent proteolysis of alpha globin. J Biol Chem. 1994;269(35):22205–22210. [PubMed] [Google Scholar]
- 92.Shaeffer JR. Heterogeneity in the structure of the ubiquitin conjugates of human alpha globin. J Biol Chem. 1994;269(47):29530–29536. [PubMed] [Google Scholar]
- 93.Shaeffer JR, Cohen RE. Ubiquitin aldehyde increases adenosine triphosphate-dependent proteolysis of hemoglobin alpha-subunits in beta-thalassemic hemolysates. Blood. 1997;90(3):1300–1308. [PubMed] [Google Scholar]
- 94.Shaeffer JR, Kania MA. Degradation of monoubiquitinated alpha-globin by 26S proteasomes. Biochemistry. 1995;34(12):4015–4021. doi: 10.1021/bi00012a020. [DOI] [PubMed] [Google Scholar]
- 95.Adachi K, Lakka V, Zhao Y, et al. Ubiquitylation of nascent globin chains in a cell-free system. J Biol Chem. 2004;279(40):41767–41774. doi: 10.1074/jbc.M405059200. [DOI] [PubMed] [Google Scholar]
- 96.Ciavatta DJ, Ryan TM, Farmer SC, et al. Mouse model of human beta zero thalassemia: targeted deletion of the mouse beta maj- and beta min-globin genes in embryonic stem cells. Proc Natl Acad Sci USA. 1995;92(20):9259–9263. doi: 10.1073/pnas.92.20.9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yang B, Kirby S, Lewis J, et al. A mouse model for beta 0-thalassemia. Proc Natl Acad Sci USA. 1995;92(25):11608–11612. doi: 10.1073/pnas.92.25.11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rouyer-Fessard P, Leroy-Viard K, Domenget C, et al. Mouse beta thalassemia, a model for the membrane defects of erythrocytes in the human disease. J Biol Chem. 1990;265(33):20247–20251. [PubMed] [Google Scholar]
- 99.Rouyer-Fessard P, Scott MD, Leroy-Viard K, et al. Fate of alpha-hemoglobin chains and erythrocyte defects in beta-thalassemia. Annals of the New York Academy of Sciences. 1990;612:106–117. doi: 10.1111/j.1749-6632.1990.tb24296.x. [DOI] [PubMed] [Google Scholar]
- 100.Sancar GB, Cedeno MM, Rieder RF. Rapid destruction of newly synthesized excess beta-globin chains in HbH disease. Blood. 1981;57(5):967–971. [PubMed] [Google Scholar]
- 101.Fagan JM, Waxman L. The ATP-independent pathway in red blood cells that degrades oxidant-damaged hemoglobin. J Biol Chem. 1992;267(32):23015–23022. [PubMed] [Google Scholar]
- 102.Fagan JM, Waxman L, Goldberg AL. Red blood cells contain a pathway for the degradation of oxidantdamaged hemoglobin that does not require ATP or ubiquitin. J Biol Chem. 1986;261(13):5705–5713. [PubMed] [Google Scholar]
- 103.Fessas P. Inclusions of hemoglobin erythroblasts and erythrocytes of thalassemia. Blood. 1963;21:21–32. [PubMed] [Google Scholar]
- 104.Yataganas X, Fessas P. The pattern of hemoglobin precipitation in thalassemia and its significance. Ann N Y Acad Sci. 1969;165(1):270–287. doi: 10.1111/j.1749-6632.1969.tb27797.x. [DOI] [PubMed] [Google Scholar]
- 105.Yataganas X, Fessas P. The pattern of hemoglobin precipitation in thalassemia and its significance. Annals of the New York Academy of Sciences. 1969;165(1):270–287. doi: 10.1111/j.1749-6632.1969.tb27797.x. [DOI] [PubMed] [Google Scholar]
- 106.Braverman AS, Schwartzberg L, Berkowitz R. Soluble and stroma-bound globin chains in mild and severe beta thalassemia. Hemoglobin. 1982;6(4):347–367. doi: 10.3109/03630268208996941. [DOI] [PubMed] [Google Scholar]
- 107.Fessas P, Loukopoulos D, Kaltsoya A. Peptide analysis of the inclusions of erythroid cells in betathalassemia. Biochim Biophys Acta. 1966;124(2):430–432. doi: 10.1016/0304-4165(66)90216-9. [DOI] [PubMed] [Google Scholar]
- 108.Fessas P, Loukopoulos D, Thorell B. Absorption spectra of inclusion bodies in beta-thalassemia. Blood. 1965;25:105–109. [PubMed] [Google Scholar]
- 109.Ballas SK, Burka ER, Gill FM. Abnormal red cell membrane proteolytic activity in severe heterozygous beta-thalassemia. J Lab Clin Med. 1982;99(2):263–274. [PubMed] [Google Scholar]
- 110.Wickramasinghe SN, Bush V. Observations on the ultrastructure of erythropoietic cells and reticulum cells in the bone marrow of patients with homozygous beta-thalassaemia. Br J Haematol. 1975;30(4):395–399. doi: 10.1111/j.1365-2141.1975.tb01853.x. [DOI] [PubMed] [Google Scholar]
- 111.Polliack A, Rachmilewitz EA. Ultrastructural studies in beta-thalassaemia major. Br J Haematol. 1973;24(3):319–326. doi: 10.1111/j.1365-2141.1973.tb01656.x. [DOI] [PubMed] [Google Scholar]
- 112.Wickramasinghe SN, Hughes M. Ultrastructural studies of erythropoiesis in beta-thalassaemia trait. Br J Haematol. 1980;46(3):401–407. doi: 10.1111/j.1365-2141.1980.tb05986.x. [DOI] [PubMed] [Google Scholar]
- 113.Wickramasinghe SN, Hughes M. Precipitation of alpha-chains on the centrioles of erythroblasts in betathalassaemia. Br J Haematol. 1982;52(4):681–682. doi: 10.1111/j.1365-2141.1982.tb03946.x. [DOI] [PubMed] [Google Scholar]
- 114.Wickramasinghe SN, Lee MJ. Evidence that the ubiquitin proteolytic pathway is involved in the degradation of precipitated globin chains in thalassaemia. Br J Haematol. 1998;101(2):245–250. doi: 10.1046/j.1365-2141.1998.00699.x. [DOI] [PubMed] [Google Scholar]
- 115.Wickramasinghe SN, Lee MJ, Furukawa T, et al. Composition of the intra-erythroblastic precipitates in thalassaemia and congenital dyserythropoietic anaemia (CDA): identification of a new type of CDA with intra-erythroblastic precipitates not reacting with monoclonal antibodies to alpha- and beta-globin chains. Br J Haematol. 1996;93(3):576–585. doi: 10.1046/j.1365-2141.1996.d01-1693.x. [DOI] [PubMed] [Google Scholar]
- 116.Ho PJ, Wickramasinghe SN, Rees DC, et al. Erythroblastic inclusions in dominantly inherited beta thalassemias. Blood. 1997;89(1):322–328. [PubMed] [Google Scholar]
- 117.Beris P, Miescher PA, Diaz-Chico JC, et al. Inclusion body beta-thalassemia trait in a Swiss family is caused by an abnormal hemoglobin (Geneva) with an altered and extended beta chain carboxy-terminus due to a modification in codon beta 114. Blood. 1988;72(2):801–805. [PubMed] [Google Scholar]
- 118.Wickramasinghe SN, Hughes M, Fucharoen S, et al. The fate of excess beta-globin chains within erythropoietic cells in alpha-thalassaemia 2 trait, alpha-thalassaemia 1 trait, haemoglobin H disease and haemoglobin Q-H disease: an electron microscope study. Br J Haematol. 1984;56(3):473–482. doi: 10.1111/j.1365-2141.1984.tb03977.x. [DOI] [PubMed] [Google Scholar]
- 119.Wickramasinghe SN, Hughes M, Hollan SR, et al. Electron microscope and high resolution autoradiographic studies of the erythroblasts in haemoglobin H disease. Br J Haematol. 1980;45(3):401–404. doi: 10.1111/j.1365-2141.1980.tb07160.x. [DOI] [PubMed] [Google Scholar]
- 120.Antonelou MH, Papassideri IS, Karababa FJ, et al. Ultrastructural characterization of the erythroid cells in a novel case of congenital anemia. Blood Cells Mol Dis. 2003;30(1):30–42. doi: 10.1016/s1079-9796(03)00006-8. [DOI] [PubMed] [Google Scholar]
- 121.Iolascon A, Martire B, Lee MJ, et al. Transfusion-dependent congenital dyserythropoietic anaemia with intraerythroblastic inclusions of a non-globin protein. Eur J Haematol. 2000;65(2):140–143. doi: 10.1034/j.1600-0609.2000.9c227.x. [DOI] [PubMed] [Google Scholar]
- 122.Salvatori F, Breveglieri G, Zuccato C, et al. Production of beta-globin and adult hemoglobin following G418 treatment of erythroid precursor cells from homozygous beta(0)39 thalassemia patients. Am J Hematol. 2009;84(11):720–728. doi: 10.1002/ajh.21539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang Y, Kwon S, Yamaguchi T, et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol Cell Biol. 2008;28(5):1688–1701. doi: 10.1128/MCB.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hideshima T, Bradner JE, Wong J, et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci USA. 2005;102(24):8567–8572. doi: 10.1073/pnas.0503221102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Colland F. The therapeutic potential of deubiquitinating enzyme inhibitors. Biochem Soc Trans. 38(Pt 1):137–143. doi: 10.1042/BST0380137. [DOI] [PubMed] [Google Scholar]
- 126.Testa U. Proteasome inhibitors in cancer therapy. Curr Drug Targets. 2009;10(10):968–981. doi: 10.2174/138945009789577909. [DOI] [PubMed] [Google Scholar]
- 127.Schessl J, Zou Y, McGrath MJ, et al. Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. J Clin Invest. 2008;118(3):904–912. doi: 10.1172/JCI34450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lindsten K, Menéndez-Benito V, Masucci MG, et al. A transgenic mouse model of the ubiquitin/proteasome system. Nat Biotechnol. 2003;21(8):897–902. doi: 10.1038/nbt851. [DOI] [PubMed] [Google Scholar]