

Significance
Understanding how new metabolic pathways emerge is one of the key issues in evolutionary and systems biology. The prevailing paradigm is that evolution capitalizes on the weak side activities of preexisting enzymes (i.e. underground reactions). However, the extent to which underground reactions provide novelties in the context of the entire cellular system has remained unexplored. In this study, we present a comprehensive computational model of the underground metabolism of Escherichia coli. Together with a high-throughput experimental survey across hundreds of nutrient environments we predicted and confirmed new functional states of metabolism in which underground reactions allow growth when their activity is increased. Our approach has important implications for biotechnological and medical applications, such as understanding gain-of-function mutations in tumor development.
Keywords: enzyme promiscuity, evolutionary innovation, molecular evolution, network evolution, phenotype microarray
Abstract
A central unresolved issue in evolutionary biology is how metabolic innovations emerge. Low-level enzymatic side activities are frequent and can potentially be recruited for new biochemical functions. However, the role of such underground reactions in adaptation toward novel environments has remained largely unknown and out of reach of computational predictions, not least because these issues demand analyses at the level of the entire metabolic network. Here, we provide a comprehensive computational model of the underground metabolism in Escherichia coli. Most underground reactions are not isolated and 45% of them can be fully wired into the existing network and form novel pathways that produce key precursors for cell growth. This observation allowed us to conduct an integrated genome-wide in silico and experimental survey to characterize the evolutionary potential of E. coli to adapt to hundreds of nutrient conditions. We revealed that underground reactions allow growth in new environments when their activity is increased. We estimate that at least ∼20% of the underground reactions that can be connected to the existing network confer a fitness advantage under specific environments. Moreover, our results demonstrate that the genetic basis of evolutionary adaptations via underground metabolism is computationally predictable. The approach used here has potential for various application areas from bioengineering to medical genetics.
How do new molecular pathways evolve? In the best-studied molecular networks, small-molecule metabolism, the prevailing paradigm is that new pathways are patched together from preexisting enzymes borrowed from different parts of the network (1–3). Central to this “patchwork” model of pathway evolution is the notion that many enzymes have limited substrate specificities and can catalyze, albeit at low rates, reactions other than those for which they have evolved (also referred to as enzyme promiscuity) (4). These so-called underground (5) or side activities are prevalent (6–8) and were shown to serve as starting points for the evolution of novel functions both in directed evolution experiments (9) and in the diversification of gene families in the wild (7). However, how the underground catalytic repertoire encoded in the genome can generate novelties within the context of the existing metabolic network remains unknown. Do underground reactions remain isolated, or can they potentially be wired into the native network and allow the organism to survive in novel environments? Furthermore, would it be possible to computationally predict the genetic basis of phenotypic evolution based on a detailed knowledge of the organism’s underground metabolism? Answering these questions requires both large-scale data on underground enzyme activities and systems-level approaches to analyze metabolic capabilities. Although systematic detection of underground activities by unbiased high-throughput approaches is not feasible at present, the accumulated knowledge of enzyme biochemistry in the well-studied prokaryote Escherichia coli provides a valuable resource of such nonnative enzyme activities (10). Thus, to explore the architecture of underground metabolism and its evolutionary potential, we compiled a comprehensive set of experimentally reported side activities of E. coli enzymes and integrated these reactions into a global metabolic network reconstruction of the same organism (11). Analysis of this extended network revealed that a substantial fraction of underground reactions can be fully integrated into the existing metabolism and participate in potential pathways that produce key precursors for cell growth. Using metabolic modeling, we then predicted specific environmental conditions under which such biologically relevant underground reactions confer a growth advantage, and hence deliver a phenotypic novelty. Our analyses revealed that the set of known underground reactions has a significant potential both to increase fitness in existing environments and to exploit new nutrient sources. A genome-wide gene overexpression screen across hundreds of carbon sources showed a good agreement with the model’s predictions, which illustrates that the genetic basis of phenotypic novelties can be predicted based on the knowledge of underground metabolism.
Results
Reconstructing the Underground Metabolic Network of E. coli.
To reconstruct a global metabolic network of E. coli that also incorporates underground reactions, we collected information for each E. coli enzyme on catalytic activities that have been detected in vitro and involve nonnative substrates (i.e., metabolites not considered as primary substrates of the enzyme) by database and literature mining (Materials and Methods). Altogether, we compiled 262 underground reactions and 277 novel compounds not present in the native network (Fig. 1 A and B and Dataset S1). Two analyses suggest that the assembled underground reactions occur at very low rates in E. coli, and hence they conceptually differ from enzyme multispecificity (12), where several reactions are catalyzed with similar efficiency (4). First, side reactions are catalyzed with significantly lower catalytic efficiency (kcat/Km) than native reactions of the same enzymes (∼220-fold pairwise difference, P < 0.001, Fig. 1C and SI Appendix, Table S1). Second, by compiling data from the E. coli Metabolome Database (13) (Materials and Methods), we found that metabolites introduced into the network via underground activities only are strongly underrepresented among empirically observed metabolites (22% versus 81% for native metabolites, P < 10−15, Fig. 1D; other metabolomics datasets yielded similar results, SI Appendix, Fig. S1). This suggests that these novel metabolites are either absent or present at very low abundances in the cell, hence remaining physiologically irrelevant. Because underground reactions occur at very low rates, they are unlikely to have essential roles in the wild-type background. Nevertheless, these side activities could potentially be enhanced by adaptive mutations (4) and thus provide raw material for network expansion.
Fig. 1.
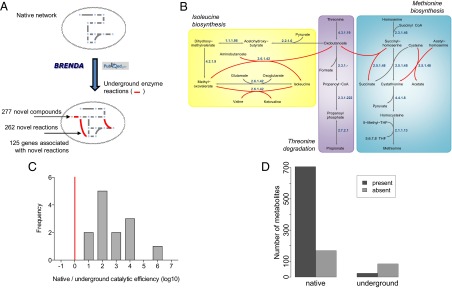
Reconstruction and validation of the E. coli underground metabolic network. (A) Schematic overview of the reconstruction process. Underground enzyme reactions were added to the native iAF1260 metabolic reconstruction of E. coli using the BRENDA (10) database and literature information. (B) Example of integrating underground reactions (red arrows) into the native network (gray arrows) as demonstrated in three interconnected amino acid metabolism pathways. Cofactors and small molecules (H2O, CO2, etc.) are not shown. Numbers next to the arrows denote the Enzyme Commission numbers associated with the corresponding reactions. (C) Distribution of the relative catalytic efficiency between native and underground substrates of the same enzyme (logarithm of the ratio between the kcat/Km values of the native and underground substrates). Red line represents equal catalytic efficiency. See also SI Appendix, Table S1. (D) Number of native and underground metabolites that are present/absent in the E. coli Metabolome Database (13). The difference is highly significant (P < 10−15, Fisher’s exact test). Underground metabolites are defined as those that are consumed and/or produced by underground reactions only.
Underground Reactions Can Often Be Wired into the Native Network.
In principle, novel biochemical reactions can introduce cross-wirings, create dead-ends, or remain isolated from the rest of the metabolic network (Fig. 2A). Our reconstruction suggests that enzyme side activities most often create cross-wirings: Forty-five percent of underground reactions can be fully connected to the network (see Fig. 1B for examples), and only 22% of them are completely isolated from native metabolism. What factors influence the network positions of cross-wiring underground reactions? Side activities are most frequently caused by substrate ambiguity (14), that is, when an enzyme catalyzes the same transformation on multiple structurally related substrates. This chemical constraint indeed yields nonrandom positioning at the network level: Native and underground activities of the same enzyme tend to participate in the same metabolic subsystem and are separated by fewer reaction steps than expected by chance (P < 10−4 and P = 0.0066, respectively, Fig. 2 B and C; also see SI Appendix, Fig. S2). Importantly, this result is based on a subset of the underground network that comes from a systematic substrate specificity screen (6) and hence is not distorted by potential investigation bias (Materials and Methods).
Fig. 2.

Network properties of underground reactions. (A) Connectedness of underground reactions in the native network. Reactions are either fully connected (cross-wiring) (i.e., both substrates and products are present in the native network), partially connected (i.e., either the substrates or the products are present), or isolated from the rest of the network (not connected). Connectedness was assessed for each underground reaction one by one. Nodes denote metabolites (blue, native; red, those associated with underground reactions only), and edges indicate reactions (gray, native; red, underground). (B and C) Distribution of the fraction of shared metabolic subsystem (B) and average network distance (C) in 10,000 samples of randomly assigned underground–native reaction pairs. Red lines indicate observed values for underground–native reaction pairs annotated to the same enzymes in our reconstruction. (D and E) Distribution of pairwise differences in yield (D) and length (E) of elementary pathways formed by native and underground reactions of the same enzyme. For each enzyme we calculated a score measuring the yield (and length) difference between two samples of pathways (one containing the underground and another one the native reaction of the enzyme). See Materials and Methods. Red line represents the null expectation of no difference. P = 0.21 and P = 0.45, respectively.
Underground Reactions Potentially Contribute to Biologically Relevant Pathways Akin to Native Ones.
The observation that many underground reactions can be wired into the native network raises the question of their potential biological relevance. Do they have pathway-level properties akin to native ones, and can they potentially contribute to the formation of key precursors needed for growth (biomass components)? We used elementary flux mode (EFM) analysis (15) to investigate these questions. EFM is a mathematical approach to decompose the network into biochemically relevant pathways that can operate in steady state from nutrient uptake to biomass component production. A sampling of such elementary paths showed that all underground reactions that can be fully wired into the native network and can carry flux in standard glucose medium also take part in at least one biomass-forming pathway. The chemical yields and lengths of pathways formed by native and underground reactions of the same enzyme are comparable to each other in standard glucose medium (Fig. 2 D and E). Taken together, a substantial fraction of the known underground catalytic repertoire of E. coli can be seamlessly integrated into the existing network and participate in biologically potentially relevant pathways.
However, the fact that underground reactions occur at very low rates suggests that they have not been exploited by evolution so far. Why should this be so? This issue is relevant because tradeoffs between novel and existing enzymatic functions are generally weak (4, 9) and can be readily resolved by gene duplication (16). We consider two alternative hypotheses to resolve this issue. First, underground reactions might interfere with existing processes (17) and are therefore disfavored by selection. Second, underground reactions might endow the cell with novel capabilities, but only under specific environmental conditions that the population has not regularly encountered during its evolutionary history.
No Evidence for the Detrimental Nature of Underground Reactions.
To test whether underground reactions tend to introduce harmful metabolites, we focused on metabolite toxicity, which has been implicated in the interference between a novel pathway and native metabolism (17). Toxicity of metabolites, as measured by IC50 values (half maximum inhibitory concentration), were predicted using a chemoinformatics tool trained on data measuring the susceptibility of E. coli against a diverse set of chemicals (18). Our analysis revealed no significant difference in toxicity between novel compounds introduced by underground reactions and native compounds associated with the same enzymes (P = 0.81, Fig. 3A). Thus, metabolite-induced damage is unlikely to pose a general limit on network expansion. A second possibility is that activation of underground reactions would incur a fitness cost by diverting metabolites from existing biomass-producing pathways. To address this scenario, we applied the EDGE algorithm (19), which identifies metabolic reactions that decrease growth when enforced to be active (i.e., higher flux). We found no support for this scenario: Underground reactions are not more likely to decrease growth when enforced to be active compared with native nonessential reactions of the same enzymes (P = 0.22; Materials and Methods).
Fig. 3.

Functional consequences of introducing underground reactions into the native network. (A) Metabolite toxicity. The plot shows the distribution of the pairwise differences in predicted IC50 between existing native and novel underground metabolites of the same enzymes. The vertical red line indicates the null expectation of no difference. P = 0.81, Wilcoxon rank-sum test (see Materials and Methods for details). (B) Heat map of in silico fitness advantages gained by adding single underground activities to the native network across different nutrient conditions. Only aerobic carbon sources are depicted here. Bright green squares indicate innovation, that is, utilization of a nutrient on which the native network showed no growth at all (i.e., fitness was zero). We note that u0192, although not being innovative under aerobic carbon sources, allows growth on l-arginine as a sole carbon source under anaerobic condition.
Predicting the Adaptive Potential of Underground Metabolism.
To test the impact of underground reactions on adaptation to specific environments we first systematically predicted growth phenotypes using flux balance analysis (FBA) (20) across a diverse set of environments. FBA is a modeling approach for analyzing metabolite flows from nutrient uptake toward production of metabolites in large-scale metabolic networks without the need for enzyme kinetic information. This modeling framework has been shown to be successful in predicting the growth capacity of wild-type E. coli across nutrient conditions and the viability of single-gene disruptions (11). The set of 2,754 environments defined here encompasses the complete range of carbon, nitrogen, sulfur, and phosphorus sources that can be imported into the network (Dataset S2) and thereby represents a comprehensive sample of the external nutrient space. As a baseline, we report that the native E. coli network shows in silico growth in 645 of these environments (Dataset S2). Next, we predicted the nutrient utilization profile for a metabolic network extended with all cross-wiring underground reactions. We note that because FBA seeks the most optimal flux distribution a larger network is expected to display slightly more optimal behavior under most conditions (i.e., pathways with somewhat higher biomass production can potentially be found in larger networks). To avoid such artifacts, we only considered fitness gains that are of at least 5% (Materials and Methods). The exhaustive list of investigated conditions enabled the identification of 19 otherwise nonutilizable nutrients on which the underground network allowed growth, hence introducing an innovation (21), and a further 31 environments where it provides a clear quantitative growth advantage (Dataset S2; see Fig. 3B for in silico fitness gains detected in aerobic carbon sources). Remarkably, incorporating the set of cross-wiring underground reactions into the native metabolic network increased its reaction content by 10.8% (i.e., from 1,257 to 1,393 intracellular reactions capable of carrying a flux) while concurrently expanding its scope of utilizable nutrients by 2.9% (from 645 to 664), underscoring the innovative potential of underground metabolism.
Next, we determined which underground reactions contribute to these novelties (Materials and Methods). We found that ∼15% of cross-wiring underground reactions confer an advantage when added individually to the network (Fig. 3B), and a further 5% do so in combination with other underground activities (Dataset S2). Furthermore, ∼11% contributed to metabolic innovations by increasing the scope of utilizable nutrients. In summary, these simulations suggest that an important fraction of the biochemically feasible evolutionary raw material in E. coli could potentially contribute to adaptation to novel nutrient environments with respect to growth.
Genome-Wide Experimental Screen Identifies Novel Phenotypes Conferred by Underground Activities.
To experimentally assess the role of underground reactions in adaptation to nutrient environments we performed an in vivo genome-wide screen to identify genes that enable growth on a new carbon source when strongly overexpressed. Our approach rests on the assumption that amplification of a single underground activity can confer a new phenotype under specific conditions. Indeed, strong gene overexpression through the viral promoter and high-copy plasmid that we apply here (typically three to four orders of magnitude increase in protein level; see SI Appendix) has been previously used to identify underground activities that provide metabolic suppression (22, 23). Following an established protocol (22, 24), a pooled collection of every E. coli ORF cloned into an expression vector (25) was tested for the provision of growth advantage in a large array of diverse carbon sources (∼4,300 ORFs in 194 conditions; see SI Appendix, Fig. S3 for a workflow). Following verification, we identified 17 ORFs whose overexpression improved growth in at least one of 17 specific carbon sources (9% of the investigated conditions; Table 1, Dataset S3, and SI Appendix, Fig. S4). Importantly, more than half of these novel growth phenotypes were conferred by enzyme-encoding genes with underground activities that are either biochemically already described for the corresponding E. coli enzyme or hypothesized based on reaction chemistry or evidence from orthologous enzymes (Fig. 4A; for more details see Dataset S4). Furthermore, six of these enzymes not only improved growth but also produced a metabolic innovation in one of the five carbon sources where wild-type E. coli was unable to grow (Fig. 4A). We therefore estimate that amplifying single underground enzyme activities expands the scope of utilizable carbon sources by ∼6% in this species (from 85 to 90 of the carbon sources experimentally tested here). This figure is expected to substantially underestimate the true evolutionary potential of underground metabolism in nutrient adaptations for at least four reasons. First, our screen captures only those innovations that can be accessed by increasing the activity of a single underground reaction. In principle, phenotypic novelties might also rely on multistep pathways and hence would go undetected in our screen. Indeed, our in silico screen identified three environments in which more than one underground reaction is jointly needed to confer a fitness benefit (Dataset S2). Second, underground activities with relatively modest beneficial effects might remain undetected in our assay. For example, there must be a lower threshold for fitness gains that is necessary for a clone within the pool of overexpression strains to overgrow the negative control. Third, overexpression is unlikely to cover the full dynamic range within which mutations can increase the catalytic efficiency of underground activities in nature. Finally, it is also possible that an underground activity with a potential fitness advantage remains silent because some of the required native reactions are unavailable for regulatory reasons.
Table 1.
Experimental results of the genome-wide overexpression screen
| Putative mode of action | No. of carbon sources |
| Native catalytic activity (pepQ, rihB) | 2 |
| Underground catalytic activity (bglB, dmlA, fumA, fumB, lacZ, leuB, mhpF, rbsK, ybfF, yihS) |
9 |
| Regulator of metabolic operon (bglJ) | 2 |
| Stress response (sspA, ycgZ) | 2 |
| Unknown mechanism (frdD, ygeN) | 2 |
Verified list of ORFs conferring a growth advantage in specific carbon sources when overexpressed. ORFs are grouped according to the putative mechanism of fitness gain. See Dataset S3 for more details.
Fig. 4.
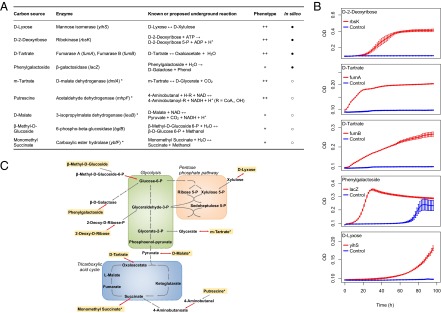
Underground enzyme activities conferring growth advantage in new carbon sources. (A) List of enzymes with catalytic side activities that enable (++) or improve (+) the utilization of specific carbon sources when amplified. Forty-four percent of the experimentally confirmed phenotypes were also predicted in silico (●), and the rest were identified experimentally (○). (B) Growth experiments validating the five computationally predicted carbon source–enzyme pairs presented in A. Red curves show the growth of cells overexpressing the predicted ORFs, and the blue one is the negative control (cells harboring the empty plasmid). Each curve represents the average of three biologically independent replicates and their SE. We note that growth dynamics is variable between carbon sources, akin to what is observed on native carbon sources (35). (C) Schematic view of central carbon metabolism (gray arrows) with underground reactions (red arrows) that confer a growth advantage on specific carbon sources (yellow highlight) when amplified. Asterisks in A and C denote cases where the carbon source is channeled into the native network via an underground reaction with indirect evidence. For details, see Dataset S4.
Experimentally Determined Metabolic Novelties Show Good Agreement with in Silico Predictions.
Computational predictions and results of the genome-wide survey showed a highly significant overlap (P < 10−13, SI Appendix, Table S2). In particular, modeling successfully predicted 44% of the carbon sources that can be used by the amplification of enzyme side activities (Fig. 4). For example, d-lyxose occurs rarely in nature (26) and wild-type E. coli is unable to degrade it. Our model predicts that establishing a pathway to use this nutrient only requires a single metabolic reaction, the side activity of mannose isomerase (YihS). Our experimental screen confirms that only the overexpression of yihS, but not of other genes, enables growth on d-lyxose. Novel metabolic phenotypes missed by our model are related to catalytic functions that have not been described in E. coli (Dataset S4), revealing that several underground reactions are yet to be discovered. Finally, we note that repeating the computational predictions using a reconstruction based on a more recent version of the E. coli native network (27) also yielded highly significant prediction accuracy (P < 10−7; see SI Appendix, Table S3 for details).
Discussion
The specificity of enzymes is inherently limited and the catalytic side activities stemming from this imperfection are thought to provide the raw material for the evolution of novel enzymatic functions (4). However, a hitherto uncharacterized portion of the catalytic raw material might be isolated from the rest of the metabolic network, produce harmful metabolites (17), or only contribute to the formation of pathways that are redundant with existing network parts (23). Because such underground activities are unlikely to contribute directly to adaptive novelties at the phenotypic level, they may never be realized by evolution. Our study attempts to systematically assess the biological relevance and evolutionary potential of underground reactions within the context of the entire metabolic network. We report that known underground reactions of E. coli can often be integrated into the native metabolic network and contribute to pathways producing precursors for cellular growth, with efficiencies comparable to native ones. Furthermore, as opposed to a previous case study (17), we found no evidence that underground reactions have a tendency to introduce harmful intermediates into the network or divert resources from growth-supporting pathways.
We found strong computational and experimental support for the notion that evolution can capitalize on underground reactions both to enhance growth in existing environments and to exploit completely novel nutrient sources. Furthermore, our in silico results suggest that the known underground repertoire of E. coli enzymes can substantially increase the range of utilizable carbon sources available to this organism (i.e., an ∼11% increase in network size expands the scope of available nutrients by ∼3%). We speculate that the contribution of underground metabolism to adaptation to new environments might be even more pronounced in eukaryotes, where metabolic network expansion by means of horizontal gene transfer has a less prevalent role compared with bacteria (28).
Underground metabolism could be exploited for additional functions beyond using novel nutrients for cellular growth. First, because many underground reactions generate a cross-wiring between existing network parts, these reactions might allow the network to react rapidly to perturbations in metabolite or enzyme concentrations (29). Second, evolution of new pathways via underground reactions may be important for the production of novel secondary metabolites (30).
A central challenge of evolutionary systems biology is to predict the phenotypic effect of mutations and potential routes of evolution (31, 32). Although important progress has been made in predicting the fitness effect and epistatic interactions of deleterious mutations in large-scale metabolic networks (31), the genetic basis of adaptive novelties has remained more elusive. Our work demonstrates that, based on the knowledge of underground metabolism, it is possible to predict both the range of novel metabolic phenotypes available to an organism in one mutational step and their genetic bases. Although our present information on underground metabolism is far from being complete, the overlap between in silico and in vivo identified genotype–phenotype pairs (Fig. 4A and SI Appendix, Table S2) suggests that our underground network already covers a significant part of the catalytic raw material available for short-term adaptation to novel environments.
Finally, the ability to computationally predict novel phenotypes based on knowledge of underground reactions also has important practical implications. For instance, systematic screens for enzyme side activities coupled with computational modeling could be used to reveal new pathways for industrially relevant compounds in new economically attractive growth environments. In addition, similar approaches might increase our understanding of the role of catalytic side activities and gain-of-function enzyme mutations in tumor evolution (33).
Materials and Methods
We reconstructed the underground metabolism of E. coli K-12 MG1655 (hereby termed iRN1260u) by extending the genome-scale metabolic network iAF1260 (11) with weak underground reactions from published experimental studies based on the BRENDA database (10) and literature (Datasets S1 and S5). The reconstruction is available as a computational SBML model (Dataset S6, also downloadable from http://group.szbk.u-szeged.hu/sysbiol/papp-balazs-lab-resources.html). Reactions were considered as underground reactions if they were listed in the BRENDA “substrate,” but not in the “natural substrates” section. Each reaction was examined as a whole for correct stoichiometry (i.e., mass and charge balance). To evaluate the correctness of the classification, we examined kinetic efficiency by kcat and Km values and metabolomics datasets for the occurrence of metabolites. Samples of elementary flux modes (i.e., minimal steady-state pathways) containing a reaction of interest were obtained using a modified algorithm of Kaleta et al. (34). We investigated the toxicity of both native and underground metabolites using a quantitative structure–activity relationship model developed to predict compound toxicity specifically in E. coli (EcoliTox web server) (18). The algorithm predicts IC50 based on molecule structure with high accuracy (R2 = 0.71). We applied FBA (20) to predict the contribution of underground metabolism to the adaptation to novel nutrient environments. Predictions were compared with results from a high-throughput gene-overexpression screen using the ASKA library (25) across ∼200 carbon sources following the protocol of Soo et al. (24) with modifications.
Detailed procedures of (i) the reconstruction and evaluation of the E. coli underground network, (ii) calculation of network distance and shared subsystems between native and underground reactions, (iii) identification of reactions capable of carrying a flux, (iv) elementary flux mode analysis, (v) metabolite toxicity analysis, (vi) EDGE analysis, and (vii) in silico and experimental surveys to identify novel phenotypes conferred by underground reactions are described in SI Appendix.
Supplementary Material
Acknowledgments
We thank István Nagy for DNA sequencing, Christoph Kaleta for useful suggestions on elementary flux mode sampling, and Martijn Huynen and Steve Oliver for insightful comments. This work was supported by a Netherlands Organisation for Scientific Research Veni grant (to R.A.N.), the Lendület Programme of the Hungarian Academy of Sciences, the Wellcome Trust (B.P. and C.P.), European Research Council (C.P.), the FP7 Initial Training Network METAFLUX (Metabolic Flux Analysis and Cancer) (F.P. and B.P.), the Hungarian Scientific Research Fund PD (B.K. and B.C.), Hungarian Academy of Sciences Postdoctoral Fellowship Programme SZ-039/2013 (B.B.), and Társadalmi Megújulás Operatív Program Grant 4.2.4. A/2-11-1-2012-0001 (to B.S.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. J.H.M. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1406102111/-/DCSupplemental.
References
- 1.Jensen RA. Enzyme recruitment in evolution of new function. Annu Rev Microbiol. 1976;30:409–425. doi: 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt S, Sunyaev S, Bork P, Dandekar T. Metabolites: A helping hand for pathway evolution? Trends Biochem Sci. 2003;28(6):336–341. doi: 10.1016/S0968-0004(03)00114-2. [DOI] [PubMed] [Google Scholar]
- 3.Rison SC, Teichmann SA, Thornton JM. Homology, pathway distance and chromosomal localization of the small molecule metabolism enzymes in Escherichia coli. J Mol Biol. 2002;318(3):911–932. doi: 10.1016/S0022-2836(02)00140-7. [DOI] [PubMed] [Google Scholar]
- 4.Khersonsky O, Tawfik DS. Enzyme promiscuity: A mechanistic and evolutionary perspective. Annu Rev Biochem. 2010;79:471–505. doi: 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- 5.D’Ari R, Casadesús J. Underground metabolism. BioEssays. 1998;20(2):181–186. doi: 10.1002/(SICI)1521-1878(199802)20:2<181::AID-BIES10>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 6.Kuznetsova E, et al. Genome-wide analysis of substrate specificities of the Escherichia coli haloacid dehalogenase-like phosphatase family. J Biol Chem. 2006;281(47):36149–36161. doi: 10.1074/jbc.M605449200. [DOI] [PubMed] [Google Scholar]
- 7.Huang R, et al. Enzyme functional evolution through improved catalysis of ancestrally nonpreferred substrates. Proc Natl Acad Sci USA. 2012;109(8):2966–2971. doi: 10.1073/pnas.1019605109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Macchiarulo A, Nobeli I, Thornton JM. Ligand selectivity and competition between enzymes in silico. Nat Biotechnol. 2004;22(8):1039–1045. doi: 10.1038/nbt999. [DOI] [PubMed] [Google Scholar]
- 9.Aharoni A, et al. The ‘evolvability’ of promiscuous protein functions. Nat Genet. 2005;37(1):73–76. doi: 10.1038/ng1482. [DOI] [PubMed] [Google Scholar]
- 10.Scheer M, et al. BRENDA, the enzyme information system in 2011. Nucleic Acids Res. 2011;39(Database issue):D670–D676. doi: 10.1093/nar/gkq1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feist AM, et al. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol. 2007;3:121. doi: 10.1038/msb4100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nam H, et al. Network context and selection in the evolution to enzyme specificity. Science. 2012;337(6098):1101–1104. doi: 10.1126/science.1216861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo AC, et al. ECMDB: The E. coli Metabolome Database. Nucleic Acids Res. 2013;41(Database issue):D625–D630. doi: 10.1093/nar/gks992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khersonsky O, Malitsky S, Rogachev I, Tawfik DS. Role of chemistry versus substrate binding in recruiting promiscuous enzyme functions. Biochemistry. 2011;50(13):2683–2690. doi: 10.1021/bi101763c. [DOI] [PubMed] [Google Scholar]
- 15.Schuster S, Fell DA, Dandekar T. A general definition of metabolic pathways useful for systematic organization and analysis of complex metabolic networks. Nat Biotechnol. 2000;18(3):326–332. doi: 10.1038/73786. [DOI] [PubMed] [Google Scholar]
- 16.Näsvall J, Sun L, Roth JR, Andersson DI. Real-time evolution of new genes by innovation, amplification, and divergence. Science. 2012;338(6105):384–387. doi: 10.1126/science.1226521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim J, Copley SD. Inhibitory cross-talk upon introduction of a new metabolic pathway into an existing metabolic network. Proc Natl Acad Sci USA. 2012;109(42):E2856–E2864. doi: 10.1073/pnas.1208509109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Planson AG, Carbonell P, Paillard E, Pollet N, Faulon JL. Compound toxicity screening and structure-activity relationship modeling in Escherichia coli. Biotechnol Bioeng. 2012;109(3):846–850. doi: 10.1002/bit.24356. [DOI] [PubMed] [Google Scholar]
- 19.Wagner A, et al. Computational evaluation of cellular metabolic costs successfully predicts genes whose expression is deleterious. Proc Natl Acad Sci USA. 2013;110(47):19166–19171. doi: 10.1073/pnas.1312361110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Price ND, Reed JL, Palsson BO. Genome-scale models of microbial cells: Evaluating the consequences of constraints. Nat Rev Microbiol. 2004;2(11):886–897. doi: 10.1038/nrmicro1023. [DOI] [PubMed] [Google Scholar]
- 21.Wagner A. The Origins of Evolutionary Innovations: A Theory of Transformative Change in Living Systems. Oxford: Oxford Univ Press; 2011. [Google Scholar]
- 22.Patrick WM, Quandt EM, Swartzlander DB, Matsumura I. Multicopy suppression underpins metabolic evolvability. Mol Biol Evol. 2007;24(12):2716–2722. doi: 10.1093/molbev/msm204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J, Kershner JP, Novikov Y, Shoemaker RK, Copley SD. Three serendipitous pathways in E. coli can bypass a block in pyridoxal-5′-phosphate synthesis. Mol Syst Biol. 2010;6:436. doi: 10.1038/msb.2010.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soo VW, Hanson-Manful P, Patrick WM. Artificial gene amplification reveals an abundance of promiscuous resistance determinants in Escherichia coli. Proc Natl Acad Sci USA. 2011;108(4):1484–1489. doi: 10.1073/pnas.1012108108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitagawa M, et al. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): Unique resources for biological research. DNA Res. 2005;12(5):291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- 26.Ahmed Z. Production of natural and rare pentoses using microorganisms and their enzymes. Electron J Biotechnol. 2001;4(2):1–16. [Google Scholar]
- 27.Orth JD, et al. A comprehensive genome-scale reconstruction of Escherichia coli metabolism—2011. Mol Syst Biol. 2011;7:535. doi: 10.1038/msb.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pál C, Papp B, Lercher MJ. Adaptive evolution of bacterial metabolic networks by horizontal gene transfer. Nat Genet. 2005;37(12):1372–1375. doi: 10.1038/ng1686. [DOI] [PubMed] [Google Scholar]
- 29.Wagner A, Fell DA. The small world inside large metabolic networks. Proc Biol Sci. 2001;268(1478):1803–1810. doi: 10.1098/rspb.2001.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weng JK, Philippe RN, Noel JP. The rise of chemodiversity in plants. Science. 2012;336(6089):1667–1670. doi: 10.1126/science.1217411. [DOI] [PubMed] [Google Scholar]
- 31.Papp B, Notebaart RA, Pál C. Systems-biology approaches for predicting genomic evolution. Nat Rev Genet. 2011;12(9):591–602. doi: 10.1038/nrg3033. [DOI] [PubMed] [Google Scholar]
- 32.Heckmann D, et al. Predicting C4 photosynthesis evolution: Modular, individually adaptive steps on a Mount Fuji fitness landscape. Cell. 2013;153(7):1579–1588. doi: 10.1016/j.cell.2013.04.058. [DOI] [PubMed] [Google Scholar]
- 33.Dang L, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kaleta C, de Figueiredo LF, Behre J, Schuster S, eds (2009) EFMEvolver: Computing Elementary Flux Modes in Genome-Scale Metabolic Networks (Gesellschaft für Informatik, Bonn), Vol 157, pp 179–189.
- 35.Vaas LA, Sikorski J, Michael V, Göker M, Klenk HP. Visualization and curve-parameter estimation strategies for efficient exploration of phenotype microarray kinetics. PLoS ONE. 2012;7(4):e34846. doi: 10.1371/journal.pone.0034846. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.