

Abstract
Ribonucleoside analogues bearing terminal alkynes, including 7-ethynyl-8-aza-7-deazaadenosine (7-EAA), are useful for RNA modification applications. However, although alkyne- and triazole-bearing ribonucleosides are in widespread use, very little information is available on the impact of these modifications on RNA structure. By solving crystal structures for RNA duplexes containing these analogues, we show that, like adenosine, 7-EAA and a triazole derived from 7-EAA base pair with uridine and are well-accommodated within an A-form helix. We show that copper-catalyzed azide/alkyne cycloaddition (CuAAC) reactions with 7-EAA are sensitive to the RNA secondary structure context, with single-stranded sites reacting faster than duplex sites. 7-EAA and its triazole products are recognized in RNA template strands as adenosine by avian myoblastosis virus reverse transcriptase. In addition, 7-EAA in RNA is a substrate for an active site mutant of the RNA editing adenosine deaminase, ADAR2. These studies extend our understanding of the impact of these novel nucleobase analogues and set the stage for their use in probing RNA structure and metabolism.
Ribonucleoside analogues bearing terminal alkynes are useful for RNA modification applications.1,2 The alkyne serves as a substrate for copper-catalyzed azide/alkyne cycloaddition (CuAAC or “click”) reactions, allowing for further manipulation of the RNA structure and its properties.3−14 Terminal alkynes can be incorporated into RNA enzymatically or using modified phosphoramidites.4,13,15 Using the latter approach, our lab was able to tune the properties of an siRNA with 7-ethynyl-8-aza-7-deazaadenosine (7-EAA) as well as 1,2,3-triazoles from CuAAC reactions of a 7-EAA-containing guide strand (Figure 1).4 Although alkyne- and triazole-bearing ribonucleosides are in widespread use, very little information is available on the impact of these modifications on RNA structure. Here, we address this issue for 7-EAA and 7-EAA-derived triazoles by solving crystal structures of RNA duplexes containing these analogues. In addition, we show that 7-EAA in RNA is an excellent substrate for CuAAC reactions and is more reactive than 5-ethynyluridine (5-EU), a nucleoside analogue commonly used to modify RNA with alkynes.5 However, we note that the rate of triazole formation with this analogue is lower in duplex RNA than in single strands, suggesting that the CuAAC reaction could be used to probe the secondary structure context of specific adenosines. Finally, we evaluated the efficacy of the reaction of RNA containing 7-EAA and 7-EAA-derived triazoles with two different RNA processing enzymes, a reverse transcriptase and an adenosine deaminase, acting on RNA. These experiments show that, as expected given the site of modification on the Hoogsteen face of the nucleobase, avian myoblastosis virus reverse transcriptase (AMV-RT) reads 7-EAA and a 7-EAA-derived triazole as adenosine in a template strand. However, at low dNTP concentrations, AMV-RT pauses at the 7-EAA site, and inhibition of primer extension is more pronounced for a 7-EAA-derived triazole. We found that the R455A mutant of human adenosine deaminase that acts on RNA 2 (ADAR2) efficiently deaminates 7-EAA in RNA but does not react with a 7-EAA-derived triazole. These studies extend our understanding of the impact of these nucleobase analogues on RNA structure and reactivity.
Figure 1.

7-Ethynyl-8-aza-7-deazaadenosine (7-EAA) is readily converted to 7-triazolyl-8-aza-7-deazaadenosines in RNA via CuAAC reaction.4
Results and Discussion
X-ray Crystallography of Duplex RNAs Bearing 7-EAA and 7-EAA Triazole
The positioning of the 7-alkyne and 7-triazole appendages on the Hoogsteen face of 8-aza-7-deazaadenosine suggested that these modifications would lie in the major groove in duplex RNA, although this had yet to be established by crystallographic analysis of a modified RNA duplex. Initial characterization of 7-EAA- and 7-EAA triazole-modified RNAs indicated that these analogues had minimal effect on the thermal stability of RNA duplexes (decrease in TM of 1–3 °C for 12 bp duplexes compared to that of adenosine) while maintaining adenosine-like pairing specificity.4 However, because each modification adds a new π system to the base that could influence stacking by, for instance, inducing sliding between adjacent base pairs, it was important to evaluate their impact on RNA structure.
Using the 7-EAA phosphoramidite, we replaced the adenosine at the fifth nucleotide position from the 5′ end of a self-complementary 16 nt RNA strand similar in sequence to one previously crystallized (Figure 2A).4,16 A fraction of the 7-EAA-containing RNA was then subjected to a CuAAC reaction with N-ethylpiperidine azide to give a 16 nt RNA bearing the N-ethylpiperidine 7-EAA triazole (Figure 3A). After purification by polyacrylamide gel electrophoresis, 16 nt RNAs bearing 7-EAA, 7-EAA triazole, and adenosine (for comparison) were crystallized. Crystallization was carried out using the sitting-drop method with the native and 7-EAA-modified RNAs crystallizing under the conditions previously reported (10% 2-methyl-2,4-pentanediol (MPD), 40 mM Na-cacodylate, pH 7, 12 mM spermine, 80 mM SrCl2, and 20 mM MgCl2 equilibrated against a reservoir containing 35% MPD).16 To obtain high-quality crystals of the 7-EAA triazole-modified duplex, a change in the crystallization conditions was necessary by replacing 80 mM SrCl2 with 80 mM KCl and reducing the reservoir concentration of MPD from 35 to 20%.
Figure 2.

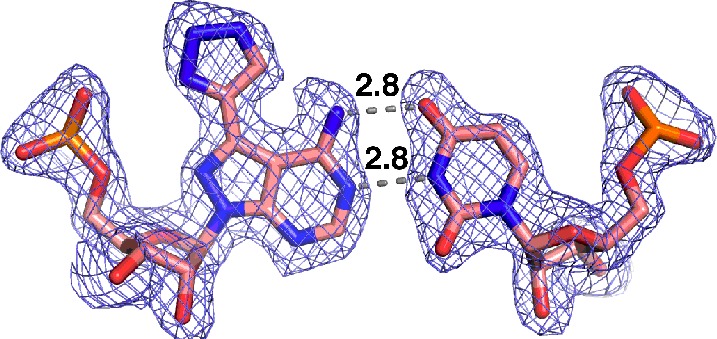
Crystal structure of 7-EAA-modified RNA. (A) Sequence of RNA used for crystallization, where N corresponds to the site of 7-EAA incorporation. (B) Quality of the electron density map at 1.85 Å resolution (contoured at 1σ) for the 7-EAA-containing duplex (7-EAA nucleoside has salmon-colored carbons). (C) 7-EAA:U pair with distances in angstroms between H-bonding sites. The final model is displayed with the 2Fo – Fc electron density map contoured at 1σ. (D) Side view of the 7-EAA:U pair showing flanking base pairs and stacking distances in angstroms. (E) Top view of nucleotides shown in panel D illustrating the stacking environment of 7-EAA. Carbon atoms are colored salmon.
Figure 3.

Crystal structure of 7-EAA-triazole modified RNA. (A) N-Ethylpiperidine 7-EAA triazole modification incorporated. (B) Electron density at 1.70 Å resolution (2Fo – Fc, contoured at 1σ) observed for the 7-EAA triazole:U pair with distances in angstroms between H-bonding sites. (C) Shown in purple is residual Fo – Fc electron density contoured at 3σ observed near the 7-EAA triazole:U pair. (D) Side view of the 7-EAA triazole:U pair showing flanking base pairs and stacking distances in angstroms. (E) Top view of nucleotides shown in panel D illustrating the stacking environment of 7-EAA triazole. Carbon atoms are colored salmon.
All three 16 nt RNAs crystallized in space group C2 with similar unit cell parameters (Supporting Information Table 1). The crystals contain three 16 mers in the asymmetric unit (A–C), with strands B and C forming one duplex, while strand A is situated at the crystallographic 2-fold axis to generate an A–A duplex. The original native unmodified structure was solved by molecular replacement using a previously published RNA structure (PDB ID: 1YZD).16 Subsequent 7-EAA and 7-EAA triazole-modified structures were solved using the native structure as a phasing model. The structures reveal that all three RNAs assemble into a classical A-type helix, with all nucleotides participating in base pair interactions. Representative electron density of the 7-EAA B–C duplex is shown in Figure 2B and clearly reveals the ethynyl group off carbon 7 of the 8-aza-7-deazaadenine ring at nucleotide position 5 (Figure 2C). 7-EAA in the 16 bp duplex adopts an “anti” conformation about the glycosidic bond and has a 3′ endo ribose pucker (Figure 2C). Normal adenosine-like pairing with uridine is observed with an N6–O4 distance of 3.1 Å and an N1–N3 distance of 2.8 Å (Figure 2C). This pairing geometry results in the expected major groove localization of the 7-ethyne substituent, with this group stacked on the guanosine in the same strand immediately 5′ to the position of the analogue (Figure 2D,E).
As seen in the 7-EAA structure, the 7-EAA triazole analogue is found with an anti glycosidic bond conformation, a 3′-endo sugar pucker, and normal base pairing with uridine (Figure 3B). Although the 7-triazole is clearly defined by the experimental electron density, we did not observe complete density for the N-ethylpiperidine-containing appendage, suggesting that this group is mobile within the major groove. However, residual elongated electron density is observed near the triazole projecting along the strand in the 5′ direction. Although this density is too large to accommodate water molecules, it is too small and ambiguous to confidently model in the N-ethylpiperidine moiety extending from the triazole (Figure 3C). This electron density, which is found in all three strands of the 7-EAA triazole in the asymmetric unit, is not found in the electron density maps for the other structures (with 7-EAA or A at the variable nucleotide position). Thus, we tentatively favor a conformation for the appendage that places the flexible N-ethylpiperidine within the major groove overlapping this residual density. Importantly, the 7-triazole appears to partially stack on the 5′ guanosine and has not otherwise altered the stacking of the base within the helix (compared to that of A or 7-EAA; see below) (Figure 3D,E).
Superposition of the 16 nt RNA strands from the three different crystal structures underscores their similarity and how well the 7-EAA and 7-EAA triazole analogues are accommodated within an A-form helical geometry (Figure 4). The 7-EAA 16 mer overlays onto the native strand with a root-mean-squared deviation (RMSD) of 0.274 Å for 335 equivalent atoms from the 16 nt strand, whereas the 7-EAA triazole-modified 16 mer superimposes with an RMSD of 0.483 Å. The largest deviations are found at the extreme ends of the duplex (Figure 4).
Figure 4.
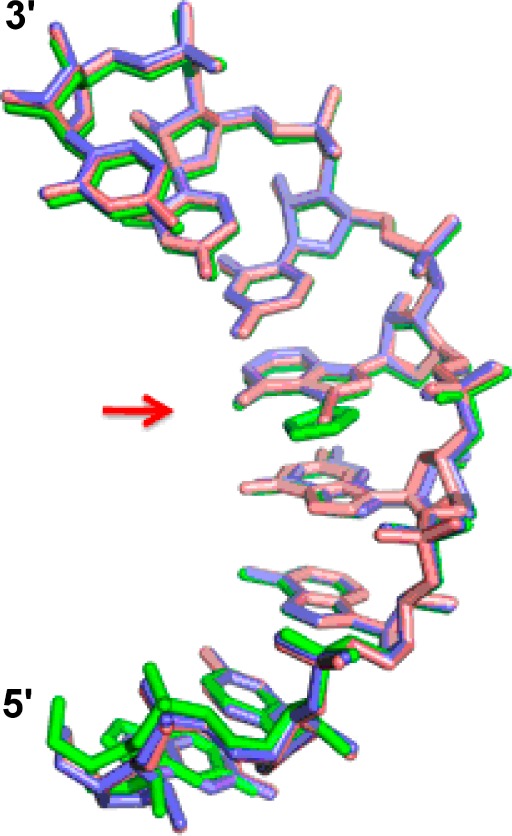
Overlay of strands from three unique RNA structures solved here containing adenosine (blue), 7-EAA (pink), or 7-EAA triazole (green) at the variable position indicated by the arrow. For clarity, only residues 1–8 of one strand of the palindromic sequence are shown.
CuAAC Reactions with 7-EAA-Containing RNA
While preparing samples for crystallization, we noted an unusually slow CuAAC reaction at high concentrations of the 7-EAA-modified self-complementary 16 nt strand (∼1 mM), with an increase in reaction rate at lower strand concentrations. In addition, it appeared from the structure of the 7-EAA-containing duplex that access to the ethyne is partially occluded (Figure 2B). These observations suggested that CuAAC reactions with this analogue might be less efficient in duplexes than in single strands. This was tested by comparing the reaction of a 7-EAA-containing 21 nt single-stranded RNA (not self-complementary) to that of the same strand in a duplex with a complementary 21 nt DNA (Figure 5A). We found that duplex formation slows the CuAAC reaction (7-EAA-containing RNA kobs = 0.14 ± 0.01 min–1; 7-EAA-containing RNA + DNA complement kobs = 0.06 ± 0.01 min–1) (Figure 5B). The effect is not observed with a control DNA that is not complementary to the 7-EAA-containing RNA (Supporting Information Figure 1). However, the phenomenon is not strictly a feature of the major groove location of the ethyne, as has been suggested for slow reactions for 5-ethynyl-2′-deoxyuridine (5-EdU) in duplex DNA.17 Our previously reported N2-propargyl-2-aminopurine derivative,15 which places an alkyne in the minor groove of a duplex, also shows a faster reaction in single-stranded RNA than in a duplex (Supporting Information Figure 2). Thus, the slower CuAAC reaction observed for the duplex compared to that for single-stranded RNA is not simply due to limited access to a major groove-localized alkyne, but it is more likely due to reduced flexibility in the duplex, as has been suggested for CuAAC reactions at the RNA 2′ position.7
Figure 5.
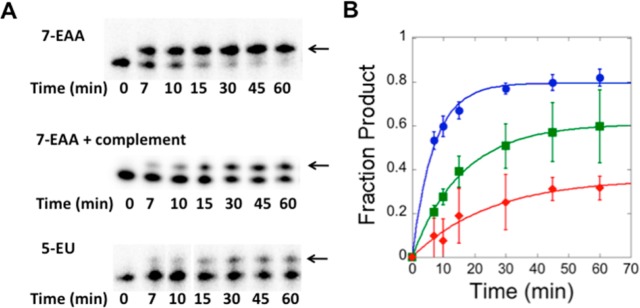
CuAAC reactions with 7-EAA-modified RNA. (A) Progress of CuAAC reactions with 32P-labeled 21 nt RNA and N-ethylpiperidine azide assessed by gel electrophoresis. The arrow indicates the location of the triazole product in the gel. (B) Comparison of CuAAC reaction kinetics for 7-EAA-containing RNA (blue), 7-EAA-containing RNA plus complementary DNA (green), and 5-EU-containing RNA (red). See the text for fitted rate constants.
On the other hand, we noted that CuAAC reactions with 7-EAA in single-stranded RNA proceeded rapidly and to a high yield. Indeed, 7-EAA in single-stranded RNA reacts faster and to a higher yield than does 5-EU in a similar CuAAC reaction (5-EU-containing RNA kobs = 0.04 ± 0.03 min–1) (Figure 5A,B). The slow 5-EU reaction was not surprising, as others have observed incomplete conversion to product for 5-EdU-containing DNA.18 We also observed a faster CuAAC reaction with 7-EAA in a different RNA sequence, indicating that this effect is general and not an artifact of the original sequence tested (Supporting Information Figure 3). The superior efficiency of the 7-EAA reaction compared to that of 5-EU is not observed only with N-ethylpiperidine azide, as other azides also react faster with the 7-EAA RNA (Supporting Information Figure 4).
Enzymatic Reactions of 7-EAA and 7-EAA Triazole-Containing RNAs
Our crystal structures indicated that 7-EAA and a 7-EAA triazole are accommodated well within an A-form helix. This suggested that these analogues might be processed by nucleic acid-modifying enzymes that recognize adenosine within the context of A-form helices. We tested this notion using AMV-RT and human ADAR2, an RNA editing adenosine deaminase. If the 7-EAA-modified nucleosides serve as substrates for these enzymes, then one could envision future studies using these analogues in a variety of ways to probe RNA structure and metabolism (see below).
Reverse transcriptases make multiple contacts to their template strands.19 However, these contacts are primarily in the minor groove, where 7-EAA and 7-EAA triazole are identical to adenosine. Also, DNA polymerases are known to faithfully copy 7-substituted 7-deazapurines.20,21 Because 7-EAA and 7-EAA triazole base pair like adenosine, one would expect a reverse transcriptase to incorporate thymidine opposite them. Indeed, AMV-RT faithfully incorporates T opposite both 7-EAA and 7-EAA triazole and extends to the end of a modified template strand (Figure 6).
Figure 6.
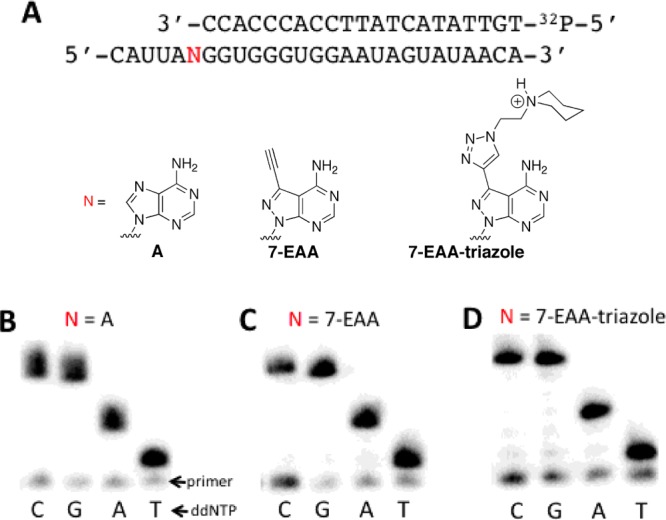
Incorporation of T opposite 7-EAA and 7-EAA triazole by AMV-RT reaction. (A) Sequences of strands employed in primer extension assay, with N indicating the site of the variable nucleotide (A, 7-EAA, or 7-EAA triazole). (B–D) Primer extension results for reactions containing different ddNTPs and different template strands, with N = A, 7-EAA, or 7-EAA-triazole.
It occurred to us that if it were possible to use a CuAAC reaction to convert 7-EAA into a nucleoside analogue that caused a reverse transcription stop, then one could use this as a means of identifying accessible/flexible sites in RNA where the CuAAC reaction appears to be most efficient. This would be particularly useful in testing ideas about ADAR selectivity (see below). To determine if a 7-EAA CuAAC reaction product could pause AMV-RT, we chose to test a biotinylated triazole (Figure 7A). This triazole is more sterically demanding than the N-ethylpiperdine triazole described above. Furthermore, addition of a biotin-binding protein could enhance the effect of the modified nucleotide. We found that under standard primer extension reaction conditions (10 μM dNTPs, 42 °C, 45 min) extension to full-length product was observed with minimal inhibition at the modified position in the template RNA, including for the biotinylated triazole in the presence of monomeric streptavidin (Figure 7B). These reactions were carried out using a primer whose 3′ end is four nucleotides from the modified position in the template. However, under reaction conditions known to be more sensitive to nucleotide modification in the template strand22 (1 μM dNTPs, 37 °C, 5 min), 7-EAA caused a small inhibition of primer extension (23 ± 4% of total extension stopped at site of modification), whereas the biotinylated triazole induced a 44 ± 2% stop, and the biotinylated triazole + streptavidin inhibited primer extension by 65 ± 8%. Therefore, it is possible to increase AMV-RT pausing at a 7-EAA site using a CuAAC reaction.
Figure 7.

Controlling the extent of pausing by AMV-RT at a 7-EAA site. (A) Sequences of strands employed in primer extension assay, with N indicating the site of the variable nucleotide. (B) Primer extension results for reactions containing 10 μM dNTPs at 42 °C for 45 min. Lanes are labeled as follows for N: −, labeled primer only, no extension; A, adenosine; 7, 7-EAA; B, biotin triazole; and B + S, biotin triazole + monomeric streptavidin. (C) Primer extension results for reactions containing 1 μM dNTPs at 37 °C for 5 min; lanes are labeled the same as in panel B, and the arrow indicates a pause site. (D) Quantification of inhibition of primer extension under the conditions used in panel C; the average for at least three independent primer extension reactions ± standard deviation is plotted.
ADARs are RNA editing adenosine deaminases that bind and process duplex substrates using a base-flipping mechanism.23 Deamination of adenosine generates inosine that is decoded as guanosine during translation. Thus, this reaction can lead to codon changes (recoding) in mRNA and the introduction of amino acids into a gene product not encoded in the gene. The basis for ADAR selectivity for reaction at specific adenosines in RNA substrates is not fully understood and may be controlled by the intrinsic conformational flexibility of the reactive nucleotides.24 A nucleoside analogue that could serve as an ADAR substrate and an independent probe of conformational flexibility would be valuable to test ideas about ADAR selectivity. The R455A mutant of human ADAR2 has been shown to accept adenosine analogues with bulky 7-position modifications.25 To determine if 7-EAA or the 7-EAA triazole could be deaminated by this enzyme, we incorporated each at a known editing site in a model substrate derived from the human glutamate receptor B subunit (GluR B) pre-mRNA. A primer extension RNA editing assay was then used to evaluate the reaction outcome. Interestingly, hADAR2 R455A deaminates the 7-EAA substrate at a rate similar to that for the adenosine-containing substrate (7-EAA: kobs = 1.1 ± 0.2 min–1; A: kobs = 0.6 ± 0.1 min–1) (Figure 8). However, no product was observed for the reaction with the 7-EAA triazole-containing RNA, likely indicating the capacity of the ADAR2 active site had been exceeded with this analogue (Figure 8). Importantly, our observation that 7-EAA in RNA can be both an ADAR substrate and a substrate for a CuAAC reaction suggests that this compound will be useful to probe the role of accessibility/flexibility in determining ADAR substrate specificity. Studies along these lines are currently underway in our laboratory.
Figure 8.

Effect of 7-EAA and 7-EAA triazole on the ADAR2-catalyzed adenosine deamination reaction. (A) ADAR reaction. (B) Sequence of ADAR substrate used, with the variable nucleotide position indicated by N. (C) Primer extension assay results with R455A hADAR2 reaction products for N = A, 7-EAA, or 7-EAA triazole. (D) Plot of product formation vs time for different R455A hADAR2 reactions (blue squares, A; green diamonds, 7-EAA; red circles, 7-EAA triazole).
Conclusions
In summary, by solving crystal structures for RNA duplexes containing 7-EAA and a triazole derived from it, we show that these analogues have adenosine-like base pairing with uridine and are well-accommodated within an A-form helix. CuAAC reactions with 7-EAA in RNA are generally faster than similar reactions with 5-EU-containing RNA and are sensitive to the secondary structure context of the alkyne. AMV-RT recognizes 7-EAA and its triazole products in RNA template strands as adenosine. At low dNTP concentration, pausing by AMV-RT is observed at a 7-EAA site, with increased pausing at a 7-EAA-derived triazole. In addition, 7-EAA in RNA is a substrate for the R455A mutant of the RNA editing adenosine deaminase, ADAR2. These studies extend our understanding of the impact of these novel nucleobase analogues and set the stage for their future use in probing RNA structure and metabolism.
Methods
General
Unless otherwise stated, reagents were purchased from Fisher Scientific, Sigma-Aldrich, or Life Technologies. Brown Centrex columns, Plate Crystalbridge well inserts, and ComboPlate 24-well polystyrene protein crystallization plates with lids for sitting-drop vapor diffusion were purchased from VWR. 5-EU phosphoramidite was purchased from Berry & Associates. T4 polynucleotide kinase and RNase inhibitor were purchased from New England Biolabs. γ-[32P]ATP was purchased from PerkinElmer Life Sciences. AMV-RT and dNTP mix were purchased from Promega. Oligodeoxyribonucleotides (21 nt complementary DNA, 21 nt control DNA, and GluR B pre-mRNA primers) were purchased from Integrated DNA Technologies. Engineered monomeric streptavidin 2 (mSA2) protein was purchased from KeraFAST. Nucleic Acid Mini Screen (NAM), MPD, and 3 in. wide crystal clear sealing tape were purchased from Hampton Research. Storage phosphor imaging plates were imaged using a Molecular Dynamics 9400 Typhoon phosphorimager from Molecular Dynamics, a Typhoon 8600 scanner from Molecular Dynamics, or a Typhoon FLA 9000 from GE Healthcare. Data were analyzed using Molecular Dynamics ImageQuant 5.2 software. MALDI mass spectrometry of oligonucleotide samples was carried out at the Campus Mass Spectrometry Facilities, UC Davis. ESI mass spectrometry of oligonucleotide samples was carried out at either the Campus Mass Spectrometry Facilities, UC Davis, or at Novatia, LLC. Oligonucleotide masses were determined using Mongo Oligo Mass Calculator v2.06. The human ADAR2 R455A protein was purified as previously described.25 All gels displayed in this article are denaturing gels.
CuAAC Reactions
For the CuAAC reaction to prepare the 7-EAA-triazole-modified 16 nt RNA and 7-EAA triazole-containing GluR B pre-mRNA, crude 7-EAA-containing 16 nt RNA or purified 7-EAA-containing GluR B pre-mRNA was mixed with copper sulfate, sodium ascorbate, tris(3-hydroxypropyltriazolylmethyl)amine, and N-ethylpiperidine azide so that the final concentrations were as follows: copper sulfate at 0.4 mM, sodium ascorbate at 4 mM, tris(3-hydroxypropyltriazolylmethyl)amine at 4 mM, N-ethylpiperidine azide at 0.2 mM, 2 mM Tris-HCl (pH 8), and 50 μM RNA. For the 7-EAA triazole-containing GluR B pre-mRNA, the reaction was allowed to proceed for 6 h, and then an equal volume of stop solution containing 80% (v/v) formamide and 10 mM EDTA was added. The product was purified as previously described.26 For the 7-EAA-triazole modified 16 nt RNA, the reaction was allowed to proceed for 7 h, the sample was concentrated 5-fold, and an equal volume of stop solution containing 80% (v/v) formamide and 10 mM EDTA was added. Samples were gel-purified as described above. For the 7-EAA biotin triazole-containing GluR B pre-mRNA, crude 7-EAA-containing GluR B pre-mRNA was mixed with copper sulfate, sodium ascorbate, tris(3-hydroxypropyltriazolylmethyl)amine, and biotin azide so that the final concentrations were as follows: copper sulfate at 20 mM, sodium ascorbate at 200 mM, tris(3-hydroxypropyltriazolylmethyl)amine at 200 mM, biotin azide at 10 mM, and 20% (v/v) DMSO. The reaction was allowed to proceed for 4 h, and then an equal volume of stop solution containing 80% (v/v) formamide and 10 mM EDTA was added. The product was purified as previously described except that it was ethanol precipitated instead of Sep-pak desalted.26 MALDI-MS [M + H]+ calcd for 16 nt RNA bearing the 7-EAA triazole, 5258.8; found, 5258.2. ESI-MS calcd for 7-EAA triazole-containing GluR B pre-mRNA, 8924.6; found, 8923.7 and calcd for 7-EAA biotin triazole-containing GluR B pre-mRNA, 9169.1; found, 9169.6.
Crystallization of RNA Duplexes
RNAs for crystallography were self-hybridized in DEPC-treated water by heating at 95 °C for 5 min and allowed to slowly cool to RT. Crystallization experiments were carried out using the sitting-drop vapor diffusion method at ambient temperature using the same procedure as previously described.16 For the native 16 nt RNA and 7-EAA-containing 16 nt RNA, equal volumes of Hampton Nucleic Acid Mini Screen (NAM) reagent 24 were mixed with 300 μM duplex RNA. Drops were equilibrated against 35% (v/v) MPD in the reservoir. Crystals were observed overnight for the native 16 nt RNA and after 4 days for the 7-EAA-containing 16 nt RNA. For the 7-EAA-triazole-modified 16 nt RNA, an equal volume of Hampton Nucleic Acid Mini Screen (NAM) reagent 14 was mixed with 300 μM duplex RNA, and the drop was equilibrated against 20% (v/v) MPD in the reservoir. Crystals were observed 5 days following setup of the tray.
Data Collection and Structure Determination
X-ray diffraction data from all three crystals were collected on beamline 7-1 at the Stanford Synchrotron Radiation Lightsource (SSRL) at 100 K. Diffraction intensities were processed and scaled with the software packages XDS and XSCALE, respectively.27 Originally the data were indexed, processed, and scaled in space group R32, with unit cell parameters a = b = 43.2 Å and c = 125.8 Å (hexagonal setting) and with one RNA 16 mer in the crystallographic asymmetric unit (the crystal 2-fold generated the palindromic duplex). Although the data scaled well as R32 (Rmerge < 5%), subsequent refinement after structure solution resulted in the Rfree value incapable of decreasing less than ∼30%, possibly indicating the wrong space group. Therefore, data was reanalyzed and reprocessed in the lower symmetry space group C2, with unit cell parameters a = 75.0 Å, b = 43.1 Å, c = 48.8 Å, and β = 120.98°, with three RNA 16 mers (1.5 duplexes) in the crystallographic unit (VM = 2.0 Å3/Da, solvent content ∼60%). Strands B and C generated one duplex, and strand A was situated at the crystal 2-fold to generate an A–A duplex. Subsequent structure refinement resulted in Rfree values ∼24%, suggesting that the correct space group is C2 with pseudo R32 symmetry.28 Data collection and processing statistics are listed in Table 1 in the Supporting Information. The original native structure was solved by molecular replacement using the program PHASER.29 The search model consisted of a single-stranded 8 mer RNA structure (PDB ID: 1YZD)16 that had the same palindromic sequence as the unknown but was modified by 2′-amino ribose on position 6. The native structure was used as a phasing model to solve the two base-modified structures: 7-EAA modified and 7-EAA-triazole modified 16 nt RNAs. The atomic models were built with the molecular graphics program COOT30 and refined using REFMAC.31 Geometric restraint libraries, used in refinement for modified bases, were generated using CCP4 programs Library Sketcher and libchek to create dictionary files. Refinement was carried out with local noncrystallographic symmetry (NCS) restraints. Removing the NCS restraints resulted in increasing the Rfree value and did not improve the electron density. Final refinement statistics are listed in Table 1 in the Supporting Information. The 7-EAA-modified base showed clear electron density to incorporate the two extra carbon atoms in the adenine-modified base at position 5. The 7-EAA-triazole-modified structure showed clear electron density for the triazole group off carbon-7. However, weak broken electron density in the major groove near the triazole group prevented us from confidently building the piperidine moiety and suggests that the piperidine group is flexible or adopts multiple conformations. This weak piperidine electron density is observed in all three RNA strands in the asymmetric unit for the C2 crystal and was completely absent in the R32 space group refinement.
32P Labeling of Oligonucleotides
The 5-EU-containing 21 nt RNA, 7-EAA-containing 21 nt RNA, and GluR B pre-mRNA primer (60 pmoles) were 5′-end-labeled by incubating the oligonucleotides with T4 polynucleotide kinase and [γ-32P]ATP (6000 Ci mmol–1) for 1 h at 37 °C. Samples were then filtered through a G25 column and purified on a 19% (w/v) denaturing polyacrylamide gel. Bands were visualized by storage phosphor autoradiography, excised, crushed, and soaked overnight at 4 °C using a solution containing 500 mM NH4OAc, 0.1% (w/v) SDS, and 0.1 mM EDTA. Samples were then phenol–chloroform extracted, ethanol precipitated, washed with 70% (v/v) ethanol, lyophilized to dryness, and redissolved in DEPC-treated water.
CuAAC Reaction Kinetics
For both 5-EU- and 7-EAA-containing 21 nt RNA, the labeled and unlabeled RNA were mixed together to a concentration of 17 μM and allowed to equilibrate to 4 °C. For both 5-EU- and 7-EAA-containing 21 nt RNA with complementary DNA, labeled and unlabeled RNA were mixed together with 21 nt complementary DNA to a concentration of 17 μM RNA and 30 μM DNA, heated at 95 °C, and then allowed to cool slowly to 4 °C. Next, a solution of copper sulfate, sodium ascorbate, tris(3-hydroxypropyltriazolylmethyl)amine, and N-ethylpiperidine azide that was pre-equilibrated to 4 °C was mixed with the RNA so that the final concentrations were as follows: copper sulfate at 0.4 mM, sodium ascorbate at 4 mM, tris(3-hydroxypropyltriazolylmethyl)amine at 4 mM, N-ethylpiperidine azide at 0.2 mM, Tris-HCl (pH 8) at 2 mM, and RNA at 8.5 μM. For both 5-EU- and 7-EAA-containing 21 nt RNA with complementary DNA, the final concentration of the DNA was 15 μM. Reactions were allowed to proceed for specified times (7, 10, 15, 30, 45, and 60 min), and then an equal volume of stop solution was added containing 80% (v/v) formamide and 10 mM EDTA followed by freezing in liquid nitrogen. Samples were resolved on a 19% (w/v) denaturing polyacrylamide gel. Gels were dried, and bands were imaged using storage phosphor imaging plates. Gel bands were quantified using ImageQuant. Data were fitted to the equation [P]t = α[1 – exp(−kobs·t)], where [P]t is the fraction reacted at time t, α is the fitted reaction end point, and kobs is the fitted rate constant using KaleidaGraph. Each experiment was carried out in triplicate, and the rate constants reported in the text are average values ± standard deviations. The mass of triazole products for these reactions have been confirmed by MALDI-MS [M + Na]+ calcd for the 5-EU/N-ethylpiperidine triazole-containing 21 nt RNA, 6946.0; found, 6945.2; for 7-EAA triazole, see Ibarra-Soza et al.4
ADAR-Catalyzed Deamination Kinetics with Modified RNA
The GluR B pre-mRNA, 7-EAA-containing GluR B pre-mRNA, and 7-EAA triazole-containing GluR B pre-mRNA were separately hybridized to the GluR B pre-mRNA complement by dissolving the RNAs in buffer containing 10 mM Tris-HCl (pH 7.5), 0.1 mM EDTA, and 200 mM NaCl at a concentration of 180 nM for each of the RNAs. The samples were then heated at 95 °C for 5 min and allowed to slowly cool to RT. Deamination reactions were carried out using the following concentrations: 15 mM Tris-HCl, pH 7.1, 3% (v/v) glycerol, 0.5 mM DTT, 60 mM KCl, 1.5 mM EDTA, 0.003% (w/v) NP-40, 160 units/mL RNase inhibitor, 1.0 μg mL–1 tRNA, 10 nM RNA, and 260 nM hADAR2 R455A.25 RNA was incubated at 30 °C for 30 min prior to the addition of hADAR2 R455A. hADAR2 R455A was added, and the reaction was allowed to proceed for various times (0.5, 1, 3, 5, 10, 20, and 30 min) at 30 °C. Reactions were stopped by the addition of an equal volume of 1% (w/v) SDS at 95 °C, and then the solution was incubated at 95 °C for 2 min. Samples were phenol–chloroform extracted and ethanol precipitated. A 70% (v/v) ethanol wash was carried out, and the samples were lyophilized to dryness. Each RNA (≤0.1 pmol) was resuspended in 1× Promega AMV-RT buffer, and ≤0.8 pmol of GluR B pre-mRNA 21 nt primer was added. The resulting solution was incubated at 62 °C for 15 min. Extent of editing was evaluated using AMV-RT-catalyzed ddNTP incorporation (see below).32
AMV-RT-Catalyzed ddNTP Incorporation
For each reaction, dNTPs, ddNTPs, 5× Promega AMV-RT buffer, and AMV-RT were added so that the concentrations in the reaction were as follows: 20 nM RNA, ∼80 nM 32P-labeled 21 nt GluR B pre-mRNA primer, 10 mM of the 3 dNTPs, 10 mM of ddNTP for the fourth base, 1× Promega AMV-RT buffer, and 5 units of AMV-RT. These reactions were incubated at 42 °C for 45 min. Formamide loading buffer was added, and the samples were heated at 95 °C for 5 min. Samples were resolved on a 12% (w/v) polyacrylamide gel. Gels were dried, and bands were imaged using storage phosphor imaging plates. Each experiment was carried out in duplicate. For hADAR2 R455A deamination reactions, the same protocol as above was used with the following modifications. RNA from deamination reactions (≤0.1 pmol) was resuspended to a final concentration ≤10 nM. The three dNTPs used were dCTP, dATP, and dGTP, and the ddNTP used was ddTTP. Bands were quantified using ImageQuant. Data were fitted to the equation [P]t = α[1 – exp(−kobs·t)], where [P]t is the fraction edited at time t, α is the fitted reaction end point, and kobs is the fitted rate constant using KaleidaGraph. Each experiment was carried out in triplicate, and the rate constants reported in the text are average values ± standard deviations.
AMV-RT-Catalyzed Primer Extension Reactions
RNA template (40 nM) was incubated with ∼160 nM 32P-labeled GluR B pre-mRNA primer and 1× Promega AMV buffer for 15 min at 62 °C. Samples were then cooled on ice for 5 min. For template RNA bearing the biotin triazole in the presence of engineered monomeric streptavidin 2 (mSA2) protein, 0.3 μg of protein was added to the sample and allowed to incubate for 5 min. Samples were then mixed with AMV-RT and dNTP mix so that the final concentrations were as follows: 20 nM RNA, ∼80 nM 32P-labeled GluR B pre-mRNA 18 nt primer, 10 μM dNTPs, 1× Promega AMV-RT buffer, and 5 units of AMV-RT for the standard extension conditions protocol and 20 nM RNA, ∼80 nM 32P-labeled GluR B pre-mRNA 18 nt primer, 1 μM dNTPs, 1× Promega AMV-RT buffer, and 5 units of AMV-RT for the low [dNTP] conditions protocol. These reactions were incubated at 42 °C for 45 min (standard) or 37 °C for 5 min ([low dNTP]). Formamide loading buffer was added, and the samples were heated at 95 °C for 5 min. Samples were resolved on a 12% (w/v) polyacrylamide gel. Gels were dried, and bands were imaged using storage phosphor imaging plates. Each experiment was carried out in duplicate.
Acknowledgments
P.A.B. acknowledges the National Institutes of Health for financial support in the form of grant R01-GM061115. We acknowledge A. Ball-Jones and A. Jean-Gilles for technical assistance. We would also like to thank J. Wedekind for helpful insight and discussions. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Supporting Information Available
Results of CuAAC reactions with a minor groove-localized alkyne ± a DNA complement; reaction results with various azides and RNA containing 7-EAA or 5-EU; detailed method for the purification of RNA; and X-ray crystallography data collection, phasing, and refinement statistics. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Coordinates have been deposited in the PDB under the following accession codes: 4NFO, 4NFP, and 4NFQ for the native, 7-EAA modified, and 7-EAA-triazole modified RNAs, respectively.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Phelps K.; Morris A.; Beal P. A. (2012) Novel modifications in RNA. ACS Chem. Biol. 7, 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammel M.; Hang H. C. (2013) Chemical reporters for biological discovery. Nat. Chem. Biol. 9, 475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammel M.; Hang H.; Conrad N. K. (2012) Chemical reporters for monitoring RNA synthesis and poly(A) tail dynamics. ChemBioChem. 13, 112–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra-Soza J. M.; Morris A. A.; Jayalath P.; Peacock H.; Conrad W. E.; Donald M. B.; Kurth M. J.; Beal P. A. (2012) 7-Substituted 8-aza-7-deazaadenosines for modification of the siRNA major groove. Org. Biomol. Chem. 10, 6491–6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao C. Y.; Salic A. (2008) Exploring RNA transcription and turnover in vivo by using click chemistry. Proc. Natl. Acad. Sci. U.S.A. 105, 15779–15784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu D.; Zhou L.; Wang W.; Wang Z.; Wang G.; Chi W.; Zhang B. (2013) 5-Ethynylcytidine as a new agent for detecting RNA synthesis in live cells by “click” chemistry. Anal. Biochem. 434, 128–135. [DOI] [PubMed] [Google Scholar]
- Winz M.-L.; Samanta A.; Benzinger D.; Jaschke A. (2012) Site-specific terminal and internal labeling of RNA by poly(A) polymerase tailing and copper-catalyzed or copper-free strain-promoted click chemistry. Nucleic Acids Res. 40, e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sagheer A. H.; Brown T. (2010) New strategy for the synthesis of chemically modified RNA constructs exemplified by hairpin and hammerhead ribozymes. Proc. Natl. Acad. Sci. U.S.A. 107, 15329–15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motorin Y.; Burhenne J.; Teimer R.; Koynov K.; Willnow S.; Weinhold E.; Helm M. (2011) Expanding the chemical scope of RNA:methyltransferases to site-specific alkynylation of RNA for click labeling. Nucleic Acids Res. 39, 1943–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutisya D.; Selvam C.; Kennedy S. D.; Rozners E. (2011) Synthesis and properties of triazole-linked RNA. Bioorg. Med. Chem. Lett. 21, 3420–3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onizuka K.; Shibata A.; Taniguchi Y.; Sasaki S. (2011) Pin-point chemical modification of RNA with diverse molecules through the functionality transfer reaction and the copper-catalyzed azide-alkyne cycloaddition reaction. Chem. Commun. 47, 5004–5006. [DOI] [PubMed] [Google Scholar]
- Paredes E.; Das S. R. (2011) Click chemistry for rapid labeling and ligation of RNA. ChemBioChem. 12, 125–131. [DOI] [PubMed] [Google Scholar]
- Yamada T.; Peng C. G.; Matsuda S.; Addepalli H.; Jayaprakash K. N.; Alam M. R.; Mills K.; Maier M. A.; Charisse K.; Sekine M.; Manoharan M.; Rajeev K. G. (2011) Versatile site-specific conjugation of small molecules to siRNA using click chemistry. J. Org. Chem. 76, 1198–1211. [DOI] [PubMed] [Google Scholar]
- Curanovic D.; Cohen M.; Singh I.; Slagle C. E.; Leslie C. S.; Jaffrey S. R. (2013) Global profiling of stimulus-induced polyadenylation in cells using a poly(A) trap. Nat. Chem. Biol. 9, 671–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock H.; Maydanovych O.; Beal P. A. (2010) N2-Modified 2-aminopurine ribonucleosides as groove-modulating adenosine replacements in duplex RNA. Org. Lett. 12, 1044–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherghe C. M.; Krahn J. M.; Weeks K. M. (2005) Crystal structures, reactivity and inferred acylation transition states for 2′-amine substituted RNA. J. Am. Chem. Soc. 127, 13622–13628. [DOI] [PubMed] [Google Scholar]
- Wirges C. T.; Gramlich P. M. E.; Gusmiedl K.; Gierlich J.; Burley G. A.; Carell T. (2007) Pronounced effect of DNA hybridization on click reaction efficiency. QSAR Comb. Sci. 26, 1159–1164. [Google Scholar]
- Gierlich J.; Burley G. A.; Gramlich P. M. E.; Hammond D. M.; Carell T. (2006) Click chemistry as a reliable method for the high-density postsynthetic functionalization of alkyne-modified DNA. Org. Lett. 8, 3639–3642. [DOI] [PubMed] [Google Scholar]
- Nowak E.; Potrzebowski W.; Konarev P. V.; Rausch J. W.; Bona M. K.; Svergun D. I.; Bujnicki J. M.; Le Grice S. F. J.; Nowotny M. (2013) Structural analyisis of monomeric retroviral reverse transcriptase in complex with an RNA/DNA hybrid. Nucleic Acids Res. 41, 3874–3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thum O.; Jaeger S.; Famulok M. (2001) Functionalized DNA: a new replicable biopolymer. Angew. Chem., Int. Ed. 40, 3990–3993. [DOI] [PubMed] [Google Scholar]
- Bergen K.; Steck A. L.; Strutt S.; Baccaro A.; Welte W.; Diederichs K.; Marx A. (2012) Structures of KlenTaq DNA polymerase caught while incorporating C5-modified pyrimidine and C7-modified 7-deazapurine nucleoside triphosphates. J. Am. Chem. Soc. 134, 11840–11843. [DOI] [PubMed] [Google Scholar]
- Maden B. E. H.; Corbett M. E.; Heeney P. A.; Pugh K.; Ajuh P. M. (1995) Classical and novel approaches to the detection and localization of the numberous modified nucleotides in eukaryotic ribosomal RNA. Biochimie 77, 22–29. [DOI] [PubMed] [Google Scholar]
- Goodman R. A.; Macbeth M. R.; Beal P. A. (2012) ADAR proteins: structure and catalytic mechanism. Curr. Top. Microbiol. Immunol. 353, 1–33. [DOI] [PubMed] [Google Scholar]
- Kuttan A.; Bass B. L. (2012) Mechanistic insights into editing-site specificity of ADARs. Proc. Natl. Acad. Sci. U.S.A. 109, E3295–E3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokharel S.; Jayalath P.; Maydanovych O.; Goodman R. A.; Wang S. C.; Tantillo D. J.; Beal P. A. (2009) Matching active site structure to substrate analog for an RNA editing reaction. J. Am. Chem. Soc. 131, 11882–11891. [DOI] [PubMed] [Google Scholar]
- Mizrahi R. A.; Phelps K. J.; Ching A. Y.; Beal P. A. (2012) Nucleoside analog studies indicate mechanistic differences between RNA-editing adenosine deaminases. Nucleic Acids Res. 40, 9825–9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. (2010) XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart P. H.; Grosse-Kunstleve R. W.; Lebedev A. A.; Murshudov G. N.; Adams P. D. (2008) Surprises and pitfalls arising from (pseudo)symmetry. Acta Crystallogr., Sect. D: Biol. Crystallogr. 64, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr., Sect. D: Biol. Crystallogr. 63, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubak P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maden B. E. (2001) Mapping 2′-O-methyl groups in ribosomal RNA. Methods 25, 374–382. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.