

Abstract
The conversion of alkynes to their corresponding vinyl triflates in the presence of stoichiometric TMS-triflate was greatly facilitated by the triflate salt of several transition metal catalysts most especially Zn(OTf)2. Products are formed in high regioselectivity under mild conditions. Internal alkynes bearing an aryl substituent afford vinyl triflates with a modest preference for the Z-isomer especially with larger substituents. A mechanism is put forward to explain the unique role of silicon in this system.
Vinyl triflates continue to play an important role in organic synthesis most especially in C–C bond formation using palladium-catalyzed coupling reactions.1 These intermediates are usually produced by trapping ketone enolates with various trifluoromethanesulfonylating agents.2 Fewer methods exist for their formation from alkynes. Early efforts involved the addition of trifluoromethanesulfonic acid (triflic acid) to an alkyne.3 Besides the highly acidic conditions, the method suffers from several drawbacks including the requirement of very low temperatures (−100 °C) and exacting workup conditions to prevent isomerization. In some cases, the use of excess alkyne is required.4 Additions of sulfonic acids to alkynes in the presence of Au5 and Rh6 have also been reported to give the E-isomer as a major product. Very recently, the formation of vinyl triflates using diaryl iodonium triflate reagents under mild conditions was reported independently by Gaunt7 and Liu.8 In these methods, the iodonium reagent donates its aryl ligand to the substrate providing an excellent route to tri- and tetra-substituted alkenes. In seeking a strategy to form vinyl triflates from alkynes without ligand transfer, we were inspired by recent reports that vinyl triflates were formed as intermediates in iridium and iron catalyzed alkyne hydrations.9 There is also evidence that vinyl triflates can be obtained from aryl alkynes and trimethylsilyl triflate (TMSOTf) albeit under fairly harsh conditions and lengthy reaction times.10
Finally, a report by Rahaim and Shaw demonstrated that TMSOTf reacts with terminal alkynes to give alkynyl-TMS products in the presence of an amine base.11 In examining this and previous reports, we suspected that Shaw’s method involved the intermediacy of vinyl triflate B (Scheme 1). We have previously observed the propensity of vinyl triflates to produce alkynes in the presence of mild amine bases.12 Thus, we reasoned that the absence of base should make possible the formation of vinyl triflates (Scheme 1). Indeed, as described here, we found that alkynes react with TMSOTf to rapidly afford vinyl triflates under exceptionally mild conditions in the presence of a variety of transition metal salts.
Scheme 1. Strategy for Metal Catalyzed Alkyne Conversion to Vinyl Triflate.

We initially studied the conversion of phenyl acetylene (1) to vinyl triflate 1a in the presence of TMSOTf (1 equiv).10 This transformation required 48 h at room temperature in acetonitrile to achieve 80%–90% conversion. This same reaction in the presence of 10% Cu(OTf)2 afforded 1a in 75% conversion in 1 h (Table 1, entry 1).
Table 1. Screening of Metal Catalysts and Silyl Reagents.
| entry | catalyst | silyl | silyl-OTf (equiv) | react time (min) | conva (%) |
|---|---|---|---|---|---|
| 1 | Cu(OTf)2 | Me3Si | 1.0 | 60 | 75 |
| 2 | Cu(OTf)2 | Me3Si | 1.5 | 50 | 90 |
| 3 | CuI | Me3Si | 1.0 | 60 | 53 |
| 4 | CuI | Me3Si | 1.5 | 50 | 67 |
| 5 | CuOTf | Me3Si | 1.0 | 60 | 74 |
| 6 | CuOTf | Me3Si | 1.5 | 50 | 76 |
| 7 | Sc(OTf)3 | Me3Si | 1.5 | 50 | 43 |
| 8 | Rh[COD]2Cl2 | Me3Si | 1.5 | 20 | 68 |
| 9 | Zn(OTf)2 | Me3Si | 1.5 | 20 | 91 |
| 10 | Zn(OTf)2 | Et3Si | 1.5 | 210 | 76 |
| 11 | Zn(OTf)2 | iPr3Si | 1.5 | 600 | 64 |
| 12 | Zn(OTf)2 | tBu2PhSi | 1.5 | 1440 | 53b |
Determined by 1H NMR.
Reaction incomplete after 24 h.
Reactions with excess TMSOTf (1.5 equiv) led to an improved conversion (90%) in 50 min. We then examined other catalysts in this reaction, and to our surprise we found that copper(I) (entries 3–6) and a variety of other metals also expedite this transformation. For example, scandium(III) triflate and Rh[Cod]2Cl2 with excess TMSOTf (1.5 equiv) afforded reasonable conversions to vinyl triflate 1a in less than 1 h (entries 7 and 8). However, optimal results were obtained with Zn(OTf)2 leading to a 91% conversion in 20 min (entry 9).
Bulkier silyl triflate reagents appear to slow the reaction considerably (entries 10–12). Thus, while TMSOTf leads to complete conversion in 20 min, reactions of 1 with triethylsilyl triflate require 210 min to give a similar conversion to vinyl triflate 1a. With still bulkier silyl triflates, high conversions to 1a were only achieved after extended reaction times (24–48 h). Importantly, reactions of 1 performed with catalytic Zn(OTf)2 using KOTf (2.5 equiv) gave no reaction. Similarly, reactions of 1 with stoichiometric amounts of Zn(OTf)2 left only starting material. Finally, reactions of 1 with 10% Zn(OTf)2 in the presence of TMSCl (1.5 equiv) afforded only 15–20% conversion to vinyl triflate 1a even after extended times. Based on these control studies, it seems that the silyl reagent serves as more than a soluble source of triflate in the present reaction.
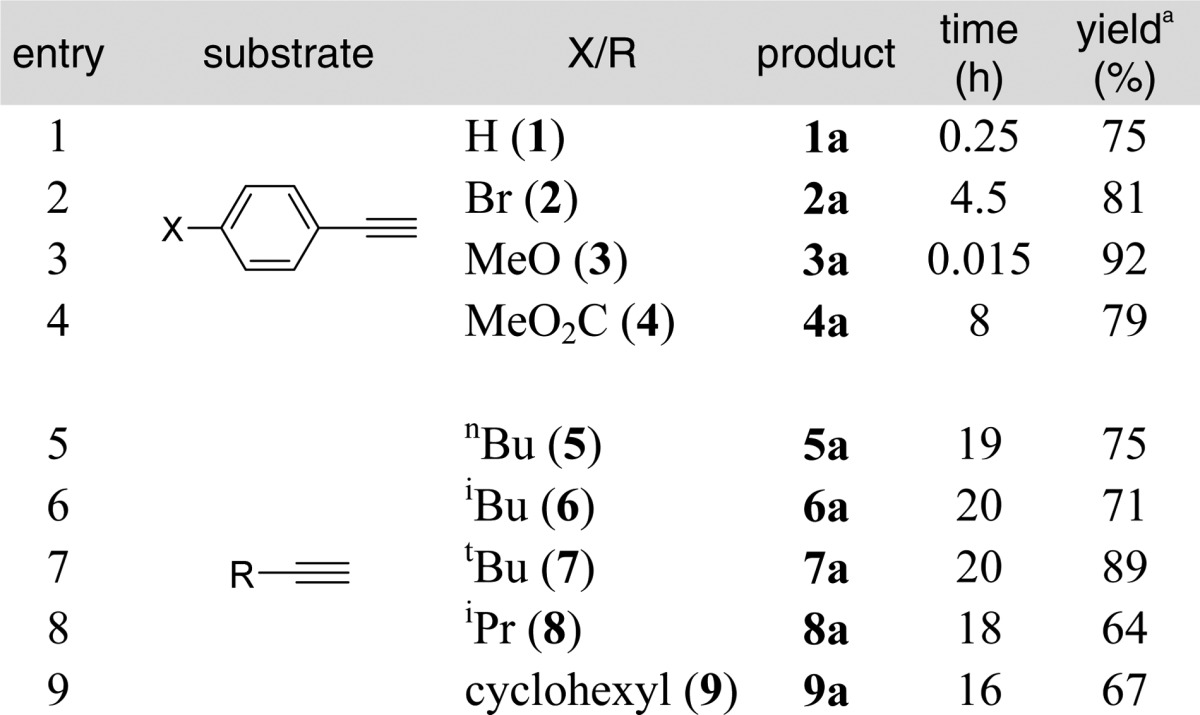
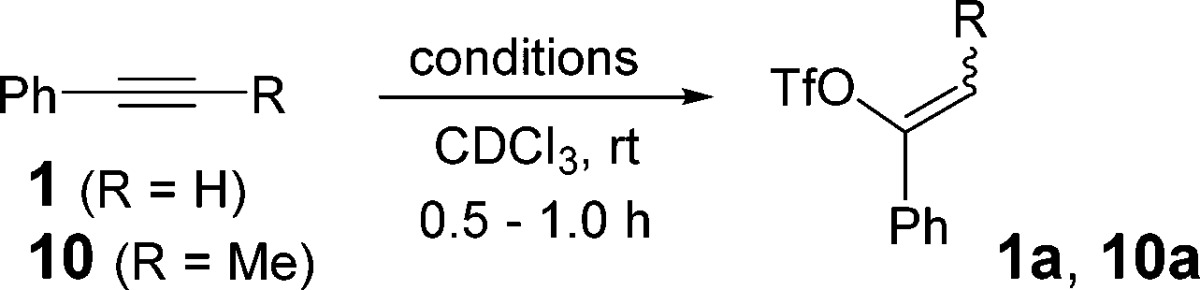
We next examined the generality of this method with various aryl and alkyl alkynes (Tables 2 and 3). Terminal aryl alkynes were rapidly converted to vinyl triflate products especially with an electron-donating group on the aromatic ring (Table 2, entry 3). Terminal alkynes bearing alkyl groups (entries 5–9) required substantially longer times (16–20 h) to produce the corresponding triflates in good yields.13 Internal alkynes also reacted slowly and appeared to favor products of Z-geometry especially with increasing bulkiness of alkyl substituents. We observed an increase in Z-selectivity from smaller groups such as methyl (Table 3, entry 1) to isopropyl alkyne 12 which gave nearly a 5:1 ratio in favor of Z-vinyl triflate 12a (entry 3). An erosion of the Z-selectivity with extended reaction times (>20 h) was also noted.
Table 2. Reactions with Terminal Alkynes.
Isolated yields.
Table 3. Reactions with Internal Alkynes.

| entry | R | product | silyl | Z/E | time (h) | conva (%) |
|---|---|---|---|---|---|---|
| 1 | Me (10) | 10a | Me3Si | 2.1 | 15 | 75 |
| 2 | Et (11) | 11a | Me3Si | 3.0 | 14 | 81 |
| 3 | iPr (12) | 12a | Me3Si | 4.9 | 5 | 92 |
| 4 | tBu (13) | 13a | Me3Si | – | – | 0b |
| 5 | Me (10) | 10a | Et3Si | 2.1 | 6 | 75 |
| 6 | Me (10) | 10a | iPr3Si | 2.3 | 6 | 23 |
Isolated yields.
Only decomposition observed.
In our studies of this reaction, we noticed that the product yield diminished substantially as greater efforts were made to rigorously exclude water. When the zinc catalyst was subjected to high vacuum (0.01 Torr) overnight to remove trace moisture, the conversion of 1 to 1a decreased to 39% (Table 4, entry 1). The addition of 0.25 equiv of water to this reaction led to an 81% conversion. Similar conversions were observed in the presence of 0.50 and 0.75 equiv of water (entries 5 and 7). Internal alkyne 10 exhibited similar rate enhancements in the presence of water (Table 4, R = Me). However, lower diastereoselectivity was observed as water content was increased. It appears that the optimal yield and diastereoselectivity are achieved with the unmodified commercially available catalyst.
Table 4. Role of Water in Vinyl Triflate Formation.
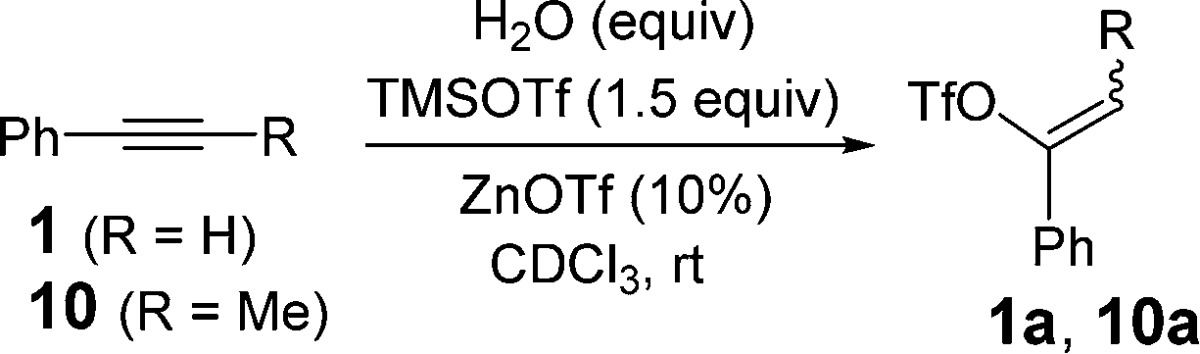
| entry | H2O (equiv) | R | Z/Ea | time (h) | conva (%) |
|---|---|---|---|---|---|
| 1 | – | H | – | 0.25 | 39 |
| 2 | – | Me | 3.0 | 3.0 | 36 |
| 3 | 0.25 | H | – | 0.25 | 81 |
| 4 | 0.25 | Me | 1.6 | 3.0 | 91 |
| 5 | 0.50 | H | – | 0.25 | 79 |
| 6 | 0.50 | Me | 1.2 | 3.0 | 90 |
| 7 | 0.75 | H | – | 0.25 | 75 |
| 8 | 0.75 | Me | 1.1 | 3.0 | 91 |
| 9 | 1.0 | H | – | 0.25 | 83 |
| 10 | 1.0 | Me | 1.3 | 3.0 | 83 |
| 11 | 1.5 | H | – | 0.25 | 87 |
| 12 | 1.5 | Me | 1.0 | 3.0 | 73 |
Determined by 1H NMR.
One interpretation for the role of water in this system is that it hydrolyzes TMSOTf to give triflic acid as the key reactant produced in a controlled manner throughout the reaction. To assess this mechanistic possibility, we attempted to mimic the hypothesized formation of minute quantities of triflic acid produced under the reaction conditions. This was accomplished by the slow addition of a dilute solution of triflic acid (0.0010 M in chloroform) to alkyne 10 (Table 5, entry 1). Under these conditions, vinyl triflate 10a was formed but predominantly as the E-isomer (Z/E = 0.25). This is a complete reversal of selectivity relative to our Zn(OTf)2 catalyzed reaction (Table 3, entry 1). To further evaluate the role of the metal catalyst in this system, substrate 1 was reacted under identical conditions to those optimized for terminal alkynes (Table 2) except that TMSOTf was replaced with triflic acid. This reaction resulted in the formation of vinyl triflate product 1a in 90% conversion (Table 5, entry 2) which is comparable to the reaction with TMSOTf (Table 1, entry 9). However, this same reaction in the absence of Zn(OTf)2 resulted in the formation of product 1a in a much lower conversion (50%) along with several side products (Table 5, entry 4).14 When the same reactions were performed using internal alkyne 10, product 10a was rapidly formed but with no selectivity (entries 3 and 5). These experiments suggest that both Zn(OTf)2 and an electrophilic silylating agent are necessary for the (Z)-selectivity observed in the present reaction.
Table 5. Experiments with TfOH in Chloroform.
| entry | conditions | R | Z/Ea | conva (%) |
|---|---|---|---|---|
| 1 | TfOH (1 equiv, 0.0010 M) | Me | 0.25 | 100 |
| 2 | TfOH (1 equiv), Zn(OTf)2b | H | – | 90 |
| 3 | TfOH (1 equiv), Zn(OTf)2b | Me | 1 | 100 |
| 4 | TfOH (1 equiv) | H | – | 50c |
| 5 | TfOH (1 equiv) | Me | 1 | 100 |
Determined by 1H NMR.
0.20 equiv.
Starting material completely consumed; several side products are observed (see ref (14)).
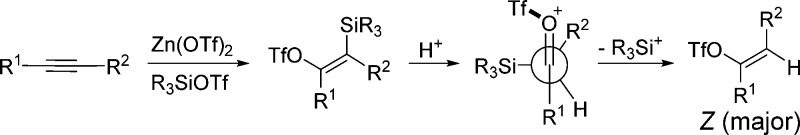
We suggest an alternative role for water in the present reaction. Instead of producing triflic acid, we expect that water will react rapidly with TMSOTf to yield a protonated silanol (Me3SiOH2+). Studies suggest that silanols are relatively basic and that the protonated form possesses a silicon geometry resembling a silicon cation.15 Judging from this previous literature report, we argue that silicon (and not proton) serves as the primary electrophile for reactions with metal vinyl intermediates A (Scheme 1) to produce vinyl silane B.16 This step should be slower with hindered silyl triflate agents which is consistent with our results (Table 1, entries 9–12). Intermediate B is then protonated to give oxonium ion C (Scheme 2).17 This species then undergoes desilylation most optimally when the silyl group in C is orthogonal to the carbonyl unit. This can occur in two conformations; however, the conformer which minimizes eclipsing interaction between the R1 and R2 groups is likely preferred (Scheme 2). Elimination of the silyl in this more favored conformer then affords the vinyl triflate product with the Z-isomer as the kinetic product.
Scheme 2. Proposed Mechanism for Vinyl Triflate Formation.

In summary, we described a mild reaction for the preparation of vinyl triflates from alkynes catalyzed by several metal triflates. The method is general to both aliphatic and aromatic terminal alkynes as well as internal alkynes bearing an aryl substituent. For Z-diastereoselectivity, the reaction requires silyl triflates pointing to a unique role for silicon in the mechanism. Further studies suggest that silicon initially serves as a target for the nucleophilic vinyl metal intermediate allowing the catalyst to be regenerated. Subsequent removal of silicon from the substrate in the presence of limited amounts of water leads to the observed triflate product. Due to the continuing importance of vinyl triflates, we believe the present method will find valuable applications in organic synthesis.
Acknowledgments
We thank the National Institutes of Health (GM110651) and the ACS-PRF (51785) for financial support.
Supporting Information Available
Experimental procedures and characterization data of all new compounds; copies of 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Ritter K. Synthesis 1993, 735. [Google Scholar]; b Dounay A. B.; Overman L. E. Chem. Rev. 2003, 103, 2945. [DOI] [PubMed] [Google Scholar]; c Nicolaou K. C.; Frederick M. O.; Burtoloso A. C. B.; Denton R. M.; Rivas F.; Cole K. P.; Aversa R. J.; Gibe R.; Umezawa T.; Susuki T. J. Am. Chem. Soc. 2008, 130, 7466. [DOI] [PubMed] [Google Scholar]
- a Wright M. E.; Pulley S. R. J. Org. Chem. 1989, 54, 2886. [Google Scholar]; b Comins D. L.; Dehghani A. Tetrahedron Lett. 1992, 33, 6299. [Google Scholar]; c Specklin S.; Bertus P.; Weibel J.-M.; Pale P. J. Org. Chem. 2008, 73, 7845. [DOI] [PubMed] [Google Scholar]; d Foti C. J.; Comins D. L. J. Org. Chem. 1995, 60, 2656. [Google Scholar]
- a Summerville R. H.; Schleyer P. V. J. Am. Chem. Soc. 1974, 96, 1110. [Google Scholar]; b Crisp G. T.; Meyer A. G. Synthesis 1994, 667. [Google Scholar]; c Vasilyev A. V.; Walspurger S.; Chassaing S.; Pale P.; Sommer J. Eur. J. Org. Chem. 2007, 5740. [Google Scholar]
- Stang P. J.; Summerville R. J. Am. Chem. Soc. 1969, 91, 4600. [Google Scholar]
- Cui D.-M.; Meng Q.; Zheng J.-Z.; Zhand C. Chem. Commun. 2009, 1577. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Moschetta E. G.; Rioux R. R. ChemCatChem 2013, 5, 3005. [Google Scholar]
- Suero G. M.; Bayle D. E.; Collins S. L. B.; Gaunt J. M. J. Am. Chem. Soc. 2013, 135, 5332. [DOI] [PubMed] [Google Scholar]
- Xu Z.-F.; Cai C.-X.; Liu J.-T. Org. Lett. 2013, 15, 2096. [DOI] [PubMed] [Google Scholar]
- a Gao Q.; Li S.; Pan Y.; Xu Y.; Wang H. Tetrahedron 2013, 69, 3375. [Google Scholar]; b Park J.; Yeon J.; Lee P. H.; Lee K. Tetrahedron 2013, 54, 4414. [Google Scholar]
- This transformation has been reported to take place in the absence of a catalyst though long reaction times (48 h) are required.Rivers J. Australian National University Undergraduate Research Journal 2011, 3, 47. [Google Scholar]
- Rahaim R. J.; Shaw J. T. J. Org. Chem. 2008, 73, 2912. [DOI] [PubMed] [Google Scholar]
- Maity P.; Lepore S. D. J. Org. Chem. 2009, 74, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- We note that vinyl triflate products were isolated in nearly pure form by a simple filtration of the reaction mixture. This was accomplished by the addition of a small amount of silica gel to the reaction upon its completion to scavenge the unreacted silyl triflate. The resulting slurry was then filtered through a cotton plug.
- The side products are likely due to the formation of vinyl cation intermediates that subsequently react with alkyne starting material. See refs (3) and (4).
- Xie Z.; Bau R.; Reed C. A. J. Chem. Soc., Chem. Commun. 1994, 2519. [Google Scholar]
- Cyclic intermediates similar to B have been reported:; a Yoshida H.; Tanino K.; Ohshita J.; Kunai A. Angew. Chem., Int. Ed. 2004, 43, 5052. [DOI] [PubMed] [Google Scholar]; b Almeida G.; Sletten E. M.; Nakamura H.; Palaniappan K. L.; Bertozzi C. R. Angew. Chem., Int. Ed. 2012, 51, 2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilyev A. V.; Walspurger S.; Chassaing S.; Pale P.; Sommer J. Eur. J. Org. Chem. 2007, 5740. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.