

Abstract

A systematic process is introduced to compare 13C NMR spectra of two (or more) candidate samples of known structure to a natural product sample of unknown structure. The process is designed for the case where the spectra involved can reasonably be expected to be very similar, perhaps even identical. It is first validated by using published 13C NMR data sets for the natural product 4,6,8,10,16,18-hexamethyldocosane. Then the stereoselective total syntheses of two candidate isomers of the related 4,6,8,10,16-pentamethyldocosane natural product are described, and the process is applied to confidently assign the configuration of the natural product as (4S,6R,8R,10S,16S). This is accomplished even though the chemical shift differences between this isomer and its (16R)-epimer are only ±5–10 ppb (±0.005–0.01 ppm).
Introduction
Several years ago, Kitching and co-workers isolated two polymethylated docosanes from the cuticular extract of the cane beetle Antitrogus parvulus.1 The compounds were identified as a 4,6,8,10,16,18-hexamethyldocosane and a 4,6,8,10,16-pentamethyldocosane (Figure 1), and synthesis established the anti,anti,anti-orientation of the methyl groups at the 4,6,8,10-positions for both compounds.
Figure 1.

Potential structures of cane beetle hydrocarbon natural products: 4,6,8,10,16,18-hexamethyldocosane is 1 or 3; 4,6,8,10,16-pentamethyldocosane is 2 or 4.
For the hexamethyl compound, the 16- and 18-methyl groups were shown to be syn, but their configuration relative to the other four methyl groups was not established. Similarly, the configuration of the 16-methyl group of the pentamethyldocosane relative to the other methyl groups was not elucidated. Thus, the open questions were whether the hexamethyl compound was 1 or 3 and whether the pentamethyl compound was 2 or 4.1 (Due to CIP priority changes, 16R-1 corresponds to 16S-2, while 16S-3 and 16R-4 correspond.)
Synthetic chemists soon rallied to this challenge,2 and Herber and Breit established by head-to-head comparison of synthetic and natural samples that the natural hexamethyldocosane was the (4S,6R,8R,10S,16R,18S)-stereoisomer 1, rather than epimer 3.2 Burgess and Negishi also described stereoselective syntheses of 1.
Meanwhile, in complementary work, one of our groups tackled the stereoselective syntheses of 2 and 4 to test methodology and to establish whether that natural product was the (16S)- or the (16R)-isomer.3
Once the syntheses of 2 and 4 were completed, we faced a problem because no natural sample was available for head-to-head comparison. Accordingly, the only option was to compare tabular lists of 13C NMR chemical shift data for the two synthetic samples with the natural sample. Not surprisingly, these data lists are very similar.3 Indeed, so similar that we had to consider the possibility that the spectra of synthetic 2 and 4 were substantially identical and that any tiny observed differences were due to error. If that were true, then the structure of the natural product could not be assigned. Even if the spectra of 2 and 4 could be proved to be different, how do we know which is the natural product if we have no natural sample to compare head-to-head under identical conditions?
This type of assignment problem is common for natural products that have remote stereocenters or local symmetry that is broken only at a distant point.4 The usual solution is to treat the distant parts of the molecule independently and to assign absolute configurations of each part separately.5 However, this requires derivatization (to make Mosher esters, for example) or at least mixing with shift reagents. Subsequent recovery of precious natural or synthetic samples in pure form can be an issue. At the most basic level, it requires functional groups for derivatization or complexation, and these are absent in 1–4.
Here we provide a solution to the problem of identifying a natural product or other unknown compound by matching its 1D 13C NMR spectrum with two spectra from a pair of known compounds. The solution is generally applicable but is specifically designed for the case where the spectra of the two known samples are extremely similar, potentially even identical. Two common complications are addressed: systematic differences in calibration (chemical shift referencing) between spectra and, importantly, the differential temperature dependence of 13C chemical shifts.
First, we lay out a stepwise process for comparing pairs of 13C NMR data sets that allows one to decide whether the spectra are the same or different. If they are different, then the differences are articulated and applied for structure assignment. We claim that the method requires only standard tables of 13C NMR chemical shifts. Second, to validate this claim, we show that published data sets from Kitching and Breit can be used to assign the structure of 1 without any head-to-head sample comparison. Third, we provide full details of the synthesis of 2 and 4, and finally, fourth, we apply the new method to rigorously show without the aid of a natural sample that the natural product is (4S,6R,8R,10S,16S)-pentamethyldocosane 2. Remarkably, the method succeeds even though the largest difference in chemical shift in the 13C NMR spectra of isomers 2 and 4 is only about 10 ppb! When real differences in chemical shifts are this small, it is critical to account for possible differences in sample temperature.
Results and Discussion
The current standard for comparison of 13C NMR spectra of two similar candidate samples with the spectrum of a natural sample is simply raw subtraction. The set of chemical shifts of the natural product spectrum is subtracted from each set of chemical shifts of the candidate spectra, resulting in two sets of differences that are typically shown as scatter plots. The two plots are then compared by standard deviation or other means, and then the natural product is assigned the structure of the candidate with the smaller differences.
For pairs of spectra from samples like 1/3 or 2/4, this process is inadequate for at least three reasons. First, it assumes that there are real and reliable differences in the spectra of the two candidate isomers, but there may not be. Second, it assumes without justification that all errors are random and therefore will tend to cancel in the final comparison. Third, it treats all differences in chemical shifts as equal in importance, regardless of the locations of the carbon atoms involved. However, it is clear, for example, that a difference in chemical shift in 1/3 or 2/4 at Me10 or Me16 should get more weight than a difference at the chain terminus methyl groups (C1 or C22). In other words, some chemical shift differences are more valuable than others. But which ones? And how do you know?
Systematic Comparison of Very Similar 13C NMR Spectral Data Sets
Figure 2 illustrates a three-step process for rigorous comparison of data sets of 13C NMR spectra of two samples of known structure with each other and then with a third sample of unknown structure. The goals are to both assign the structure of the unknown compound and to confidently articulate that the assignment is correct. The initial discussion here presents the generalized process, so it is necessarily abstract. However, each of the steps of the processes will be illustrated concretely in the following section on validation.
Figure 2.

Steps and possible outcomes of a process for comparing different samples whose 13C NMR spectra are very similar and possibly identical. Samples S and R are two different candidates of known structure that are compared to a natural product NP of unknown structure.
In Figure 2, the two candidate samples of known structure (typically isomers made by total synthesis) are generically called S and R, and the unknown sample (typically a natural product) is called NP. To the extent possible, the resonances of the spectra are assigned to their associated carbon atoms by standard means. The spectra of the synthetic samples must be recorded under the same conditions and at the same nominal probe temperature (within about 1 K). The temperature of the NP spectrum is critical but does not need to be known in advance because it emerges from the analysis.6
Step 1 is to compare the spectra of the candidate samples R and S not to the natural product but to each other. The goals are to determine whether the candidate spectra are the same or different and, if different, to articulate the differences. Start by subtracting the chemical shifts of R from S (or the reverse) and gauge the experimental uncertainty in the chemical shift values. Record duplicate spectra if needed to estimate this. If the subtractions are all zero within the expected experimental error, then the spectra are substantially identical. This means that the two candidate spectra cannot be used to assign the configuration of NP. Further subtraction of S and R from NP is pointless, and another means of assignment is needed.
If some subtractions are not zero, then group all resonances into three categories based on their differences in their chemical shifts. The chemical shifts of each pair of resonances in S and R are either (1) the same, (2) uncertain, or (3) reliably different. Establish chemical shift limits for the three categories based on the estimated error and common sense analysis of the data. Going forward, the “same” resonances are used only as controls; you know that the natural product is one of these two samples, so it has to have all of the “same” resonances too. The “reliably different” resonances are used only for assignment; the natural product has either one set or the other. These values will ultimately be obtained again when the natural product chemical shifts are subtracted from those of the wrong candidate isomer, so they are called “mismatch” values. The “uncertain” resonances are not used at all; this category is simply a place to cull differences that are too close to call.
Before advancing to step 2, check the carbon atom assignments of the various resonances, which should pass the sniff test of chemical common sense. For example, if the structures are very similar (like 1/3 and 2/4, for example), then it cannot be that all of the pairs of chemical shifts are reliably different. Likewise, the “reliably different” and “same” resonances should not be randomly distributed about the structures. The carbons of the “reliably different” resonances should cluster around the parts of the molecules that are different, while the carbons of the “same” resonances should cluster in regions that are remote from the differences.
In steps 2 and 3, we compare the spectra of the candidate samples to that of the NP. This is where standard comparisons usually start; however, we are ahead of the game because we already know that our comparisons are meaningful (that is, S and R do not have identical spectra) and we know what to compare (the reliably different resonances).
In step 2, we control for temperature and calibration differences of the candidate and natural samples. These controls are performed by subtracting members of the “same” group of chemical shifts in S (or R, they are the same after all) from the corresponding chemical shifts of the natural product NP. In principle, the subtractions should all be zero. In practice, there are three possible outcomes: First, if all of the values are zero (or close to zero), then both the temperature and calibration errors are small. In that case, proceed directly to step 3 (final comparison). Second, if the values are small and constant (either positive or negative), then there is a calibration difference between the synthetic and natural samples. Standardize the chemical shifts of NP if the calibration error is significant compared to the values of the “reliably different” chemical shifts. Third, if the subtraction values of the “same” groups give variable values, then there is probably a difference in sample temperature between the different experiments. This has to be accounted for when the values of the “same” subtractions approach or exceed the above “reliably different” values. Temperature effects on 13C NMR spectra of alkanes are variable, ranging from 0 to about 20 ppb per degree K in either direction (that is, upfield or downfield with increasing T), reflecting the change in conformation populations with temperature.7 So in two different samples with very similar 13C NMR spectra, the chemical shift differences caused by small changes in sample temperature could easily exceed the real differences. Clearly the spectra of the candidate samples have to be collected at the same sample temperature as the spectrum of the natural product.
What to do if you identify a temperature difference between your synthetic samples (sample temperature known) and the natural product sample (sample temperature unknown)? Fortunately, the list of the “same” chemical shifts of the candidate samples allows the sample temperature of the NP sample to be estimated retrospectively. This process will be illustrated below.
In the final step 3, simply subtract the “reliably different” chemical shifts of R and of S from those of the NP. If all of the subtraction values equal zero within the experimental uncertainty, then this is the natural product match. If all subtraction values equal the reliably different values identified in step 1, then this is the mismatch. No other result is possible; there cannot be subtractions that neither match (zero) nor mismatch (reliably different).8
Validation of the Method with Published Breit/Kitching Data Sets
A corollary of the assignment method in Figure 2 is that neither new samples nor new experiments are needed if complete data sets are already published (provided that the sample temperatures are the same in the data sets to be compared). Thus, prior to using the method to assign 2/4, we validated it with the complete data sets published for the related compounds 1/3 from Breit and Kitching.
Three lists of 13C NMR peaks were used in the validation, and these are shown in Figure 3a. First, Kitching provided the spectrum of the isolated natural product and assigned the various resonances.1 Second, Kitching also provided a spectrum of a synthetic mixture of four diastereomers: 1, 3, and their epimers at C10.1 Third, Breit and Herbert stereoselectively made the two individual candidate isomers 1 and 3 and recorded their 13C NMR spectra (data in Table 1).2 Kitching observed in his mixed sample that some of the carbons appeared as single peaks even though four isomers were present. This proves a key assumption of our method, that at least some of the carbons in the isomers have identical chemical shifts. Kitching also saw various doubled resonances, eight of which he assigned to the epimers at C16/18. Because Kitching’s sample was made as a mixture, he could not know which resonances belonged to which isomers, so he could not assign the natural product structure at that point.
Figure 3.

(a) Published 13C NMR data sets for hexamethyldocosane and (b) Breit’s head-to-head comparison of the natural sample with the synthetic candidates.
Table 1. Step 1 of the Comparison Process: Subtraction of 13C NMR Data Set 1 from Set 3 and Classification of the Chemical Shifts by Magnitude of the Differences.
| C | anti | syn | δ3 – δ1 (ppb) | categorya |
|---|---|---|---|---|
| C22 | 14.181 | 14.181 | 0 | same |
| C1 | 14.389 | 14.389 | 0 | same |
| Meb | 19.553 | 19.553 | 0 | same |
| Meb | 19.570 | 19.570 | 0 | same |
| Meb | 19.589 | 19.589 | 0 | same |
| Meb | 19.647 | 19.649 | 2 | UC |
| C2 | 20.080 | 20.080 | 0 | same |
| Mec | 20.302 | 20.302 | 0 | same |
| C21 | 23.072 | 23.072 | 0 | same |
| C12 | 26.928 | 26.940 | 12 | RD |
| C14 | 27.063 | 27.070 | 7 | RD |
| C8 | 27.294 | 27.294 | 0 | same |
| C6 | 27.294 | 27.294 | 0 | same |
| C20 | 29.176 | 29.176 | 0 | same |
| C4 | 29.715 | 29.715 | 0 | same |
| C18 | 29.982 | 29.982 | 0 | same |
| C16 | 29.982 | 29.982 | 0 | same |
| C10 | 29.995 | 30.002 | 7 | RD |
| C13 | 30.346 | 30.365 | 19 | RD |
| C19 | 36.573 | 36.570 | –3 | UC |
| C15 | 36.876 | 36.878 | 2 | UC |
| C11 | 37.876 | 37.885 | 9 | RD |
| C3 | 40.222 | 40.222 | 0 | same |
| C17 | 45.225 | 45.222 | –3 | UC |
| C5 | 45.552 | 45.550 | –2 | UC |
| C9 | 45.579 | 45.562 | –17 | RD |
| C7 | 46.535 | 46.537 | 2 | UC |
UC is uncertain; RD is reliably different.
Me groups 4,6,8,10.
Me groups 16,18.
Breit concluded that assigning the configuration of 3 by the usual raw subtraction of chemical shifts of synthetic and natural samples was not possible because the spectra of 1 and 3 were too similar. Instead, Breit ingeniously inserted a narrow-bore capillary tube containing the natural product (provided by Kitching) inside standard-bore NMR tubes containing either of the two synthetic isomers, then recorded a single spectrum of the two sample components (Figure 3b). The (16R,18S) configuration 1 was assigned to the natural product because no peaks were doubled in the spectrum of the sample of 1 with the natural product capillary inserted, whereas a lone peak was doubled in the corresponding spectrum of 3 with the capillary. The tube-in-tube experiments suggest that the stereoisomers 1 and 3 have 27 chemical shifts that are the same and only one that is significantly different; however, we show presently that more differences can be identified.
Together, the data sets provide an extremely rare (perhaps the only) case study where spectra of pairs of diastereomers with similar spectra have been collected in three different ways: as individual, pure samples (Breit), truly mixed (Kitching, but admixed with two other isomers), and artificially mixed (Breit, tube-in-tube). Our goal was to do what was not possible by raw subtraction: to confidently assign the structure of the hexamethyldocosane natural product from the data sets of the individual samples alone.
Exemplifying the process in Figure 2, step 1 is to compare the spectra of Breit’s two individual synthetic samples of 1 and 3 to each other. Table 1 shows his complete data lists2 along with the subtraction results. Breit is correct—the spectra are strikingly similar. At the ±1 ppb level, about half of the subtraction results are exactly zero, while the largest differences are less than 20 ppb (+19 ppb for C13 and −17 ppb for C9). The many zeros show that Breit’s two spectra were recorded with the same sample temperature and consistently calibrated.
Prior to comparison with the natural product spectrum, the subtraction values are placed in the three groups: (1) the same group for <2 ppb difference (15 resonances), (2) the uncertain group (UC) for 2–6 ppb difference (6 resonances), and (3) the reliably different group (RD) for >7 ppb difference (6 resonances). The subtraction results from the reliably different group (RD, in red) and uncertain group (UC, in green) are placed with their assigned carbon atoms on the hexamethyldocosane structure in Figure 4. Unlabeled carbons are all in the “same” group.
Figure 4.
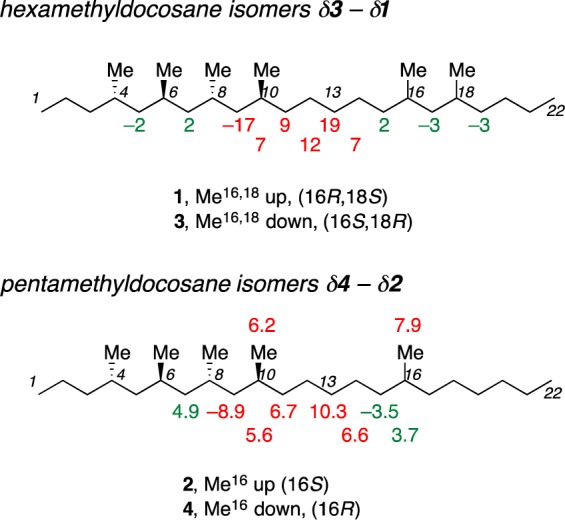
Chemical shift differences (in ppb) and assigned carbon atoms on the structures of the hexamethyldocosane (top) and pentamethyldocosane (bottom) candidate isomers. The uncertain resonances are labeled in green, and reliably different resonances are in red. The unmarked resonances are in the “same” group.
Notice how the reliably different resonances are not randomly distributed or clustered at the ends. Instead, they cluster in the region C9–C14. This makes sense given the structural difference between the two isomers. Further, even the “uncertain” resonances cluster immediately on either side of the reliably different resonances, implying that these tiny differences (2–6 ppb) may also be real. Still, the six reliably different resonances suffice for the analysis.
Strikingly, most of the uncertain and reliably different resonances in the hexamethyldocosane isomers 1 and 3 belong to methylene carbons. Every methylene group along the chain from C5 to C20 lights up as UC or RD. In contrast, only one of the six methines (C10 RD class) and one of the six methyl groups (not shown because it was not assigned, UC class) lights up in this analysis.
In step 2, calibration, we subtract the “same” chemical shifts identified in Table 1 on the synthetic samples from the corresponding chemical shifts of the natural product. These and the remaining subtraction tables are shown in the Supporting Information. The result is a small, constant difference of about 3 ppb. This means that Breit’s and Kitching’s spectra were recorded at the same sample temperature and that 3 ppb is the calibration correction. We applied this correction going forward; however, the same final result is obtained without it because the calibration error (3 ppb) is smaller than the differences in values for the reliably different resonances (±7–19 ppb).
Finally, to complete the retrospective assignment in step 3, we subtract the reliably different chemical shifts of Breit’s pure samples from Kitching’s natural product, with the results shown in columns four (NP – 1) and five (NP – 3) of Table 2. For evaluation purposes, we include both the UC and RD resonances; however, for assignment we use only the RD set. From Figure 2, we expect that one of the two sets of values will be the match and one will be the mismatch. In principle, the match subtraction results are all zero, while the mismatch results are the values δ3 – δ1 (from Table 1 or Figure 4). The actual values match the expected results in both columns within about ±2 ppb; NP – 1 is the match and NP – 3 is the mismatch. Without question, the hexamethyldocosane is 1.
Table 2. Retrospective Assignment of Hexamethyldocosane by the Comparison Process.
| C | mismatcha | category | NP – 1 | NP – 3 | Kitching mixture |
|---|---|---|---|---|---|
| Mea | 2 | UC | 2 | 2 | 3 |
| C12 | 12 | RD | 0 | 12 | 14 |
| C14 | 7 | RD | –2 | 5 | 6 |
| C10 | 7 | RD | 0 | 7 | b |
| C13 | 19 | RD | 0 | 19 | 21 |
| C19 | –3 | UC | –1 | –4 | b |
| C15 | 2 | UC | 2 | 4 | 13 |
| C11 | 9 | RD | 0 | 9 | 10 |
| C17 | –3 | UC | –1 | –4 | b |
| C5 | –2 | UC | –3 | –5 | b |
| C9 | –17 | RD | 0 | –17 | –20 |
| C7 | 2 | UC | 4 | 6 | 3 |
From the comparisons in steps 1 and 2, taken from Table 1.
Not doubled in Kitching’s mixture.
Now let us compare our method of identifying different peaks to Breit’s and Kitching’s mixture experiments. Specifically, are the reliably different resonances identified by this method real? Recall that Breit’s artificial mixture method (tube-in-tube) identified only one different resonance, which is that of C13. Indeed, at +19 ppb this has the largest magnitude of any difference. Separate resonances were not observed in Breit’s tube-in-tube experiments for C9 or C12 even though these differ by −17 and +12 ppb, respectively, reflecting the price paid in spectral resolution for the use of concentric samples.
The last column of Table 2 lists the ppb differences in the doubled resonances in Kitching mixture sample. These should equal the mismatch values in column 2 (Breit’s two synthetic isomers) and column 5 (NP – 3, the difference between the natural product and the wrong isomer). Of the eight doubled resonances assigned to C16,18 epimers in Kitching’s mixture, five are identified as reliably different and three as uncertain. The magnitudes of the differences match within about 2 ppb. This cannot be coincidence. Clearly Kitching assigned these resonances properly, and we identified them faithfully from Breit’s data set.
The Breit/Kitching data sets do not match perfectly; one reliably different resonance from Breit is not listed as doubled in Kitching’s mixture (C10, which is very close to C16 and C18 making firm assignment difficult in the mixture spectrum), and one pair of resonances in Kitching is classed as uncertain rather than reliably different (C15, +13 ppb difference in Kitching, +2 ppb difference in Breit). This simply shows that having more reliably different resonances increases the confidence of the method. It is not important to find all of the real differences, only that the differences found are reliable.
In summary, the standard raw subtraction method for assignment could not be used with Breit’s samples 1 and 3 to assign the configuration of the natural product. However, the method outlined in Figure 2 succeeded. This success was enabled in no small part by the high quality of the published data sets from both Breit and Kitching. Breit has already assigned 1 securely by the tube-in-tube method, so here it is the validation of the method that counts.
Strikingly, the new method of controlled subtraction of the data was comparable in effectiveness to recording spectra of true mixtures in identifying different (doubled) resonances and was considerably better than the tube-in-tube method. Indeed, the tube-in-tube method identified only one of the six resonances with differences >6 ppb. We speculate that this may be due to the poorer field inhomogeneity inevitable with two tubes. Regardless, the results show in hindsight that no mixing of the candidate and natural samples (either real or artificial) is needed. By careful, controlled comparison of published data sets, we could identify differences in chemical shift at the low ppb level, close to the experimental level of resolution. The upshot is a secure structure confirmation without access to a natural sample and without new experiments.
Total Synthesis of Pentamethyldocosane Candidate Isomers
The Manchester group has recently communicated the synthesis of the candidate isomers 2 and 4 of the pentamethyldocosane natural product.3 The synthesis is summarized here with additional details. Complete experimental information and compound characterization data are found in the Supporting Information.
The two epimers 2 and 4 were made convergently with a late stage formation of the C(11)–C(12) bond. The synthesis of the requisite C(1)–C(11) fragment is based on chemistry developed for the stereoselective synthesis of hydrocarbons with methyl substituents disposed at 1,3,5-positions down an aliphatic chain.9 The key step in this synthesis is the reaction of (E)-5-benzyloxy-2,4-dimethylpent-2-enyl bromide with aldehyde 9. Such reactions are selective for the formation of (E)-1,5-anti-products when mediated by the low valence bismuth species formed by reduction of bismuth(III) iodide with zinc powder.9,10
The synthesis of the left-half C1–C11 is summarized in Scheme 1. Ozonolysis of the tert-butyldimethylsilyl ether 6(11) of (S)-citronellol with a reductive workup gave (S)-6-tert-butyldimethylsilyloxy-4-methylhexanol 7.12 Elimination of hydrogen iodide from the corresponding iodide followed by desilylation and hydrogenation gave (S)-3-methylhexanol 8.13 Oxidation then gave the aldehyde 9(13) that was used immediately. This synthesis of 9 is straightforward and scalable.
Scheme 1. Synthesis of the 1-Iodo-2,4,6,8-tetramethyl-undecane 22.

Reagents and conditions: (i) TBSCl, imid., THF, rt, 16 h (98%); (ii) O3, DCM, MeOH, −78 °C, 2 h, NaBH4, −78 °C to rt, 3 h (98%); (iii) (a) I2, Ph3P, imid., DCM, rt, 2 h; (b) KOtBu, THF, rt, 16 h; (c) 4 M aq HCl, THF, rt, 3 h; (d) H2, Pd/C, THF, rt, 16 h (60% from 7); (iv) Dess–Martin periodinane, DCM, NaHCO3, rt, 30 min; (v) BiI3, Zn powder, THF, rt, 2 h, add 9 and 10, THF, reflux, 2 h (60%); (vi) 4-O2NC6H4CO2H, Ph3P, DIAD, THF, rt, 16 h (70%); (vii) 2 N aq NaOH, THF, 50 °C, 1 h (70%); (viii) naphth., Li, THF, −25 °C, add 11, 2 h (78%); (ix) TIPSCl, imid., THF, rt, 16 h (94%); (x) [Rh(NBD)Diphos-4]BF4 (cat.), DCM, H2, 950 psi, rt, 5 h (16, 70%; 18, 16%); (xi) TsCl, DMAP, THF, rt, 16 h (93%); (xii) CuI, MeLi·LiI, 0 °C, 17, 0 °C–rt, 16 h (21%); (xiii) 4 M aq HCl, dioxane, THF, rt, 16 h (91%); (xiv) TsCl, DMAP, DCM, rt, 16 h (85%); (xv) NaI, acetone, reflux, 16 h (90%).
The bismuth-mediated reaction of aldehyde 9 with the pent-2-enyl bromide 10(9) gave a major product identified on the basis of precedent,10 as the (3E,6S)-2,6-anti-2,4,8-trimethylundec-3-en-6-ol 11. Minor products were detected at the 5% level but could not easily be separated. The (3E,6S)-undecenol 11 was converted into its (3E,6R)-epimer 15 by treatment with 4-nitrobenzoic acid under Mitsunobu conditions followed by hydrolysis of the intermediate 4-nitrobenzoate 14. The (3E,6R)-epimer 15 could be distinguished from the (3E,6S)-undecenol 11 by 1H NMR and was shown to be about 5% of the product mixture from the reaction between the aldehyde 9 and the bromide 10.
Reductive cleavage of the benzyl ether 11 followed by selective silylation of the primary alcohol 12 gave the triisopropylsilyl ether 13. Following precedents established by Evans14 and used in preliminary studies,9 hydrogenation of the alkenol 13 using [Rh(NBD)diphos-4]BF4 as the catalyst gave a mixture of the 4-epimers of the 2,4,8-trimethylundecan-6-ols 16 and 18, in isolated yields of 75% and 16%, respectively. Structures were assigned to these products by analogy with our earlier work9 and were confirmed by comparison of the 13C NMR spectra of later intermediates, in particular of the 2,4,6,8-tetramethylundecanol 20, with published data.2
The remaining methyl group was introduced by treatment of the toluene p-sulfonate 17, prepared from the alcohol 16, with a higher order methyl cuprate.15 This reaction proceeds with inversion to provide the anti,anti,anti-2,4,6,8-tetramethylundecanyl silyl ether 19. The yield of the cuprate reaction was low (21%), perhaps because of steric hindrance due to the flanking 4- and 8-methyl substituents, but sufficient product was obtained to complete the syntheses. Desilylation gave the alcohol 20 that was converted into the iodide 22 via the toluene p-sulfonate 21.
The syntheses of the enantiomeric C(12)–C(22) fragments and completion of the syntheses of the 16-epimeric 4,6,8,10,16-pentamethyldocosanes 2 and 4 are outlined in Scheme 2. Alkylation of butyl phenyl sulfone using the (R)- and (S)-iodides (R)-23 and (S)-2316 that are available from the corresponding enantiomers of citronellol gave the dodecenyl sulfones 24 and 31 as mixtures of epimers at C(4).
Scheme 2. Completion of Syntheses of the 4,6,8,10-Pentamethyldocosanes 2 and 4.

Reagents and conditions: (i) nBuS(O)2Ph, THF, DMPU, nBuLi, −40 °C, 30 min, 23, −40 °C to rt, 16 h (24, 95%; 31, 90%); (ii) O3, DCM, MeOH, −78 °C, then NaBH4, −78 °C to rt, 16 h (25, 85%; 32, 87%); (iii) Na/Hg, MeOH, rt, 16 h [(S)-26, 73%; (R)-26, 70%]; (iv) TsCl, DMAP, DCM, rt, 16 h [(S)-27, 99%; (R)-27, 98%]; (v) NaI, acetone, reflux, 2 h [(S)-28, 90%; (R)-28, 92%]; (vi) MeS(O)2Ph, THF, nBuLi, −40 °C, 30 min, add 28, rt, 16 h [(S)-29, 73%; (R)-29, 75%]; (vii) nBuLi, hexane, THF, DMPU, −40 °C, 30 min, add 22, −40 °C to rt, 16 h (30, 45%; 33, 34%); (viii) Na/Hg, MeOH, rt, 16 h (2, 83%; 4, 85%).
The alkenes in 24 and 31 were ozonized with a reductive workup to give the alcohols 25 and 32, and then reduction by sodium amalgam gave the enantiomeric 4-methyldecanols (S)- and (R)-26.17 These were converted into the sulfones (S)- and (R)-29 via the corresponding toluene p-sulfonates and iodides,18 and alkylation of these sulfones using the 1-iodo-2,4,6,8-tetramethylundecane 22 gave the 12-phenylsulfonylpentamethyldocosanes 30 and 33. Reductive removal of the phenylsulfonyl group gave the 4,6,8,10,16-pentamethyldocosanes 2 and 4.3 These final samples have 1H NMR spectra at 700 MHz that are indistinguishable, but the subsequent 13C NMR analysis showed that their isomeric purity is at least 95%.
Assignment of the Structure of the Natural Pentamethyldocosane
The data sets used to assign the pentamethyldocosane structure are summarized in Figure 5. Again there are data sets of the natural product and the four-isomer mixture from Kitching.1 We also had the originally published data sets for 2 and 4 at the time of communication.3 To complete the assignment, we recorded eight new data sets on the same two samples used for the original data sets.
Figure 5.

Four 13C NMR data sets used to assign the pentamethyldocosane.
To control for both temperature effects and inherent reproducibility, we recorded four new 13C NMR spectra on synthetic samples 2 and 4 (two each, 1 week apart) at 175 MHz in CDCl3 with a nominal probe temperature of 298 K. These data are fully tabulated in the Supporting Information alongside those of the published spectra of isomers 2 and the natural product. Comparing the spectra of the same samples recorded 1 week apart gave an idea of the random error of the experiments, which was strikingly low. We could reproduce the list of chemical shifts of each isomer to the level of about ±1 ppb. To control for temperature, we also recorded duplicate pairs of data sets at 293 K; again these were reproducible to the level of about ±1 ppb.
We work first with the new data sets at 298 K. The step 1 subtraction revealed that the differences between the candidate isomers of 2 and 4 are considerably smaller than those of 1 and 3. Seven of the 27 resonances differed by 5–10 ppb, and these “reliably different” resonances are shown in red adjacent to their assigned carbon atoms in Figure 4 (bottom) and also listed in Table 3. At the next level down, three additional resonances in the “uncertain” group shown in green differed by 3–4 ppb. The remaining 17 resonances in the “same” set differed by <2.5 ppb (most by <1.5 ppb).
Table 3. Matching and Mismatching by Subtraction of the Reliably Different Chemical Shifts of Spectra of the Pentamethyldocosane Natural Product (NP) with the Synthetic Samples 2 (16S) and 4 (16R) (data in ppb)a.
| resonance | 2 – 4 | NP – 2 | NP – 4 |
|---|---|---|---|
| Me-10 | 6 | 0 | 6 |
| Me-16 | 7 | –1 | 7 |
| C14 | 6 | –1 | 5 |
| C10 | 5 | –1 | 4 |
| C13 | 10 | –2 | 8 |
| C11 | 6 | –1 | 5 |
| C9 | –9 | –1 | –10 |
Standardization corrections before rounding in ppb are 2.3 and 1.6 (see Supporting Information). Prior to subtraction, values of synthetic sample peaks were rounded to 1 ppb to match the natural product (NP) spectrum.
As before, all of the resonances with the differences exceeding 2.5 ppb belong to carbons within or between the groups of stereocenters of 2, and none belongs to carbons on either end of the long chain (Figure 4). This is sensible. The largest difference of all, 10.3 ppb, belongs to C13, which is halfway between the stereocenters at C10 and C16. This is the same carbon that gave the largest difference in 1/3, but in that pair of compounds the difference was about twice as large (+19 ppb). Also as before, the uncertain resonances are just to the left and right of the reliably different resonances along the chain. This time, only half (5) of the reliably different and uncertain resonances belong to methylene groups. The other half are methyl groups (3) and methines (2).
Validation that these tiny subtraction differences are real and not error again comes from the relevant mixture sample of Kitching. The pentamethyldocosane mixture is again four isomers, epimers at C16 (the relevant center) and C10. Kitching assigned seven doubled peaks to C16 isomers, and these are the same seven resonances that differ by ≥5 ppb in our subtraction. Thus, Kitching again correctly identified the peaks that differ in the natural product and its C16 epimer.
At 187 MHz, Kitching did not observe doubling of any of the three peaks that differ by 3–5 ppb in our spectra. We suspect that these differences are meaningful because they show up reliably when subtracting the duplicate spectra recorded at 175 MHz. However, the final assignment ultimately comes back to comparison with Kitching’s published spectra of the natural product. He observed doubled peaks in 2 wherever chemical shift differences were >5 ppb, so we used that level as the cutoff for a reliable difference in further comparisons. This error level is crucial; if it is raised to just 10 ppb (0.01 ppm), then the spectra of 2 and 4 are identical.
In short, by comparing in step 1 the various new spectra of samples 2 and 4, we proved that the samples had very similar but not identical spectra and we deduced that differences >5 ppb were meaningful for comparisons with published data lists. Indeed, the spectra are so similar that even small differences in sample temperatures (as low as 2–3 K) can complicate comparisons because the magnitudes of temperature effects on chemical shifts can exceed the actual differences (see below).
Moving to step 2, we compared the new 298 K spectra of 2 and 4 to Kitching’s spectrum for the natural product.5b First, we ascertained that Kitching’s spectrum and ours were recorded at the same temperature by comparing the group of 20 resonances with the “same” chemical shifts. These resonances had the same chemical shifts (±2 ppb) in all three samples, so the natural product spectrum was recorded at the same temperature as the spectra of the two synthetic samples. These data were corrected for small calibration errors (see Supporting Information), though again the correction is not crucial to the outcome.9
Then in step 3 we compared the subtraction data for the seven reliably different resonances, as shown in Table 3. The results of subtractions NP – 2 and NP – 4 are shown in columns 3 and 4. In NP – 2 all of the values are zero or nearly zero; this is the match. In NP – 4, the values are very close to the reliably different values established in step 1 (column 2); this is the mismatch. Clearly the natural product is 2, not 4. Accordingly, the hexa- and pentamethyldocosane natural products 1 and 2 have the same configuration at C16.
Easy Assignment of the Docosanes by the Difference-of-Differences Method
The usual way to compare spectra of two very similar samples to an unknown that is presumed to match one and not the other is to subtract pairs of the same chemical shifts in different samples, as we did in Table 2. We have previously suggested that subtraction of pairs of different chemical shifts in the same sample from each other is also a valuable tool.4d This tool is simple because the whole assignment is reduced to comparison of one number from each sample. Because two chemical shifts are subtracted, it gives the largest possible ppb difference in the spectrum, and most importantly, any calibration error cancels in the subtraction. So this method works without standardization of the spectra being compared.
In the 2/4 pair, the largest difference is obtained by subtracting the resonance of C9 (the only resonance with a negative value) from C13 (the resonance with the largest positive value). This difference is 15.222 ppm for 2 and 15.240 for 4. The natural product difference is 15.222 ppm, matching 2.
Such subtractions of the absolute values give two large numbers that when compared have a small difference. Because only the difference is meaningful, it may be more convenient to look at the difference of differences in values of the chemical shifts. In other words, subtract the differences of C13–C9 in 2 and 4 from each other.19 At four decimal places: 15.2239 – 15.2215 = 0.0194 ppm, or 19.4 ppb. This is the mismatch result, rounded to 19 ppb for comparison with the natural product. As usual, the match result is zero. Now without any standardization, subtracting the NP value from 2 gives −1 ppb (match result) and subtracting the NP value from 4 gives 18 ppb (mismatch result).
Likewise, for hexamethyldocosane, the same two carbons give an almost double difference of differences of 36 ppb. Subtracting the natural product value from 1 gives 0 ppb (match result) and subtracting the NP value from 3 gives 36 ppb (mismatch result).
Temperature Effects
When comparing pairs of spectra with such tiny chemical shift differences, it is crucial to account for possible differences in sample temperature. We show this by comparing the newly recorded sets of spectra of 2 and 4 to the original published spectra.3 As is common, these original spectra were recorded at ambient temperature, without variable temperature control.
The originally published spectrum of 2 matched the new duplicate spectra at 298 K (±2 ppb) almost as well as the duplicate spectra matched each other (±1 ppb). However, the published spectrum of 4 did not match the new duplicate spectra of 4 nearly as well as the duplicates matched each other. Disconcertingly, according to the raw subtraction analysis (subtract all chemical shifts and calculate a standard deviation), the published spectrum of 4 is actually closer to the published spectrum of the natural product than to the new spectra of the same sample. This is disconcerting because 4 is not the natural product.
We deduced that the original sample temperature of isomer 2 matched the sample temperature of the new spectra of 2, while the sample temperature of isomer 4 did not. This is evident by comparing the “same” set of resonances in the original and new spectra of 2 and 4; many of them are not the same.
To retrospectively estimate the sample temperature of the published spectrum of 4, we recorded pairs of 13C NMR spectra of both candidate isomers at 293 K (5 K lower sample temperature), 1 week apart. Again, the spectra of the same samples recorded 1 week apart were identical down to the 1–2 ppb level. The subtraction values for 4 are shown in Figure 6; the values for 2 are very similar (see Supporting Information).
Figure 6.

Differences in chemical shifts for spectra of 4 recorded with sample temperatures 5 K apart. The values are listed as δ 298 K – δ 293 K in ppb.
By subtracting the chemical shifts obtained from the pairs of new spectra at 293 and 298 K from each other, we learned that 21 of the 27 resonances of 4 shifted by ≥5 ppb (≥1 ppb K–1). At 56 and 57 ppb (>10 ppb K–1), the resonances for C6 and C8 exhibit the largest temperature effects. Notice that these difference are more than 5 times larger than the largest real difference (10 ppb) in the spectra of the two isomers shown in Figure 4 and Table 2. Most of the other shifts were positive (that is, downfield shift with increasing temperature), but the two end methyl groups (C1 and C22) and one of the methylene groups (C21) experienced small negative shifts. These kinds of shifts are typical for other alkanes.4
In the published spectrum of 4, every one of the seven resonances that did not vary with temperature matched the corresponding resonances of the new spectra of 4 (within 2 ppb) at both 293 and 298 K, and the 20 temperature-dependent resonances in the published spectrum fell roughly 40% of the way in between the corresponding resonances of the new spectra at 293 and 298 K. This means that the published spectrum of 4 must have been recorded at about 295 K. This exercise shows how to estimate an unknown sample temperature retrospectively given available candidate sample spectra.
To show the importance of temperature control in candidate versus natural samples, consider the two deliberately mistaken subtraction analyses in Figure 7 taken from the new data sets and the natural product set. Here we show the scatter plots from raw subtraction (that is, the current standard method of data analysis). The results when the new spectrum of 2 recorded at 293 K is subtracted from the NP spectrum at 298 K are shown with the blue diamonds. This is the case where the spectra of the correct candidate isomer and the natural product are compared, but the spectra are recorded at different temperatures (right structure, wrong temperature).
Figure 7.
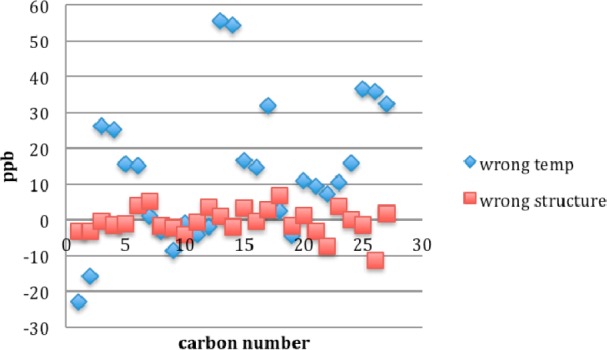
Overwhelming effect of sample temperature. The scatter plot for the wrong structure at the right temperature (red squares) looks much better than the plot for the right structure at the wrong temperature (blue diamonds). Wrong temp is NP (298 K) – 2 (293 K); wrong structure is NP (298 K) – 4 (298 K).
Now compare this to the subtraction of the wrong candidate isomer 4 from the natural product with spectra recorded at identical temperatures, shown with the red boxes. This is the case where the temperature matches, but the structures do not (right temperature, wrong structure). By any measure (peak-by-peak comparison of differences, average difference, standard deviation), the second pair of spectra (red boxes) “match” better than the first pair (blue diamonds). In other words, the usual raw subtraction process assigns the wrong structure to the natural product because it fails to account for the temperature effects. Chemical shift differences as a function of temperature are not random errors and therefore cannot be expected to cancel in any raw subtraction analysis.
Conclusions
In summary, we have postulated, validated, and then applied a systematic process for comparing 13C NMR spectra of two (or more) candidate samples of known structure with the spectrum of another compound of unknown structure (for example, a natural product). The process is designed for cases where the spectra of the candidates are very similar, perhaps even identical. In such cases, raw subtraction with scatter plot visualization is the current standard for assignment. This method always come down on the side of one candidate or the other. Indeed, even if the candidate samples have identical spectra, an assignment will still result simply because of error. Accordingly, when spectra are similar, there is a low level of confidence that an assignment by the raw subtraction method is correct.
In the new method, the spectra of the candidates are first compared to each other to firmly establish that they are different and then to articulate the differences. These differences should be sensible based on the structures being compared. Next, a subset of the resonances (the “same” group) is used to standardize the natural product spectrum to the candidate spectra and to determine whether sample temperatures of the spectra are the same. Finally, another subset of resonances (the “reliably different” group) is used to assign the structure of the natural product.
The assignment does not rely on a statistical analysis such as determination of a standard deviation. Instead, all of the key resonances of the natural product are expected to match one candidate isomer and not to match the other. The result is both an assignment of the natural product structure and a high level of confidence that the assignment is correct. The method reveals differences that are comparable to those revealed by recording spectra of mixed samples, a gold-standard process that is rarely practical either because no natural sample is available or because the synthetic and natural samples are simply too precious to mix.
The spectra compared in this work have real differences of only ±5–20 ppb. Such small differences are not usually considered meaningful and are typically ascribed to random error. However, provided that sample temperature differences are corrected for, these small differences are demonstrably real and therefore reliable. NMR probes around the world probably have ambient temperatures in the range of at least 293–303 K, a 10 K range. Over this range, the temperature dependence of 13C resonances can change chemical shifts by over ±100 ppb (±0.1 ppm). When spectra recorded at different sample temperatures are subtracted, real temperature effects (which are not random) are easily mistaken for random error. Conservatively, then, whenever chemical shift differences between candidate and natural product spectra are less than about ±500–1,000 ppb (±0.5–1 ppm), the sample temperature needs to be accounted for in comparisons.
Given the advantages over the standard raw subtraction analysis, we suggest that this systematic process should become standard practice for assignment in cases where two (or more) candidate structures for a natural product can reasonably be expected to have very similar or identical spectra.
Finally, we caution that both (or all) of the candidate samples have to be made for the secure assignment. One cannot simply make and then match one candidate to the natural product structure. This is an uncontrolled process. How do we know which resonances are the same or reliably different if we do not know whether the candidate and the natural product are the same or different? Only by circular reasoning. The spectra of 2 and 4 in this work, for example, easily “match” the relevant natural product spectrum when compared to it in isolation (see the “wrong” structure in Figure 7). Only when the other candidates 1 and 3 are introduced does it become clear that 2 and 4 are the mismatches and 1 and 3 are the matches.
As we have stated previously,4d assigning structures by proof has the same pitfalls as proving mechanisms. In formal logic, a structure (or mechanism) can only be disproved, never proved. The final proof comes from disproof of all sensible structure candidates (or mechanism candidates) but one.
Experimental Section
(3S)-1-tert-Butyldimethylsilyloxy-3,7-dimethyloct-6-ene 6(11)
Imidazole (479 mg, 7.0 mmol) was added to (S)-citronellol 5 (1.0 g, 6.4 mmol) in THF (25 mL), and the solution stirred at ambient temperature for 10 min before tert-butyldimethylsilyl chloride (1.06 g, 7.0 mmol) was added. The reaction mixture was stirred at room temperature for 16 h, diluted with ether (25 mL) and partitioned between ether (20 mL) and saturated aqueous sodium hydrogen carbonate. The organic layer was washed with brine (20 mL) and dried (Na2SO4). After concentration under reduced pressure, chromatography of the residue eluting with light petroleum (100%) gave the title compound 6(11) (1.7 g, 98%) as a colorless oil, Rf 0.3 (100% light petroleum), [α]20D −1.7 (c 0.2 in CHCl3); lit.11 value: [α]20D −1.42 (c 1.01 in CHCl3) (found: M+ – C4H9, 213.1675. C12H25OSi requires M, 231.1669); νmax 1462, 1377, 1361, 1254, 1094, 987, 897, 833, and 773 cm–1; δH (500 MHz, CDCl3) 0.00 (6 H, s, 2 × SiCH3), 0.82 (3 H, d, J 7, 3-CH3), 0.85 [9 H, s, SiC(CH3)3], 1.11 (1 H, m, 3-H), 1.24–1.32 and 1.51 (each 2 H, m, 4-H2 or 5-H2), 1.55 and 1.63 (each 3 H, s, 8-H3 or 7-CH3), 1.85–1.99 (2 H, m, 2-H2), 3.60 (2 H, m, 1-H2) and 5.04 (1 H, t, J 6.9, 6-H); δC (125 MHz, CDCl3) −5.3, −5.2, 17.7, 18.4, 19.6, 25.5, 25.8, 26.0, 29.1, 37.2, 39.9, 61.5, 124.9 and 131.1; m/z (EI) 213 (M+ – 57, 100%).
(4S)-6-tert-Butyldimethylsilyloxy-4-methylhexan-1-ol 7(12)
Ozone from a generator was bubbled through a solution of the alkene 6 (1.3 g, 4.8 mmol) in dichloromethane and methanol (1:1; 30 mL) at −78 °C for approximately 2 h until the solution turned blue. Oxygen was then bubbled through the solution at −78 °C until the blue color disappeared. Solid sodium borohydride (910 mg, 24 mmol) was added at −78 °C, and the reaction mixture was allowed to warm to room temperature and stirred for 3 h. After concentration under reduced pressure, the residue was partitioned between saturated aqueous sodium hydrogen carbonate (20 mL) and ether (30 mL). The organic layer was washed with brine (20 mL), dried (Na2SO4) and concentrated under reduced pressure. Chromatography of the residue eluting with light petroleum/ether (4:1) gave the title compound 7(12) (1.7 g, 98%) as a colorless oil, Rf 0.5 (1:1 light petroleum/ether), [α]20D +1.2 (c 0.2 in CHCl3) (found: M+, 247.2090. C13H31O2Si requires M, 247.2088); νmax 3325, 1462, 1387, 1361, 1253, 1093, 1006, 938, 896, 833, and 773 cm–1; δH (500 MHz, CDCl3) 0.00 (6 H, s, 2 × SiCH3), 0.84 [9 H, s, SiC(CH3)3], 0.86 (3 H, d, J 7.0, 4-CH3), 1.14 (1 H, m, 4-H), 1.30 (2 H, m, 3-H2), 1.53 (4 H, m, 2-H2 and 5-H2) and 3.59 (4 H, m, 1-H2, 6-H2); δC (100 MHz, CDCl3) −5.2, 18.4, 19.7, 26.0, 29.3, 30.3, 33.0, 39.9, 61.4 and 63.4; m/z (ES+) 269 (M+ + 23, 100%) and 247 (M+ + 1, 60).
(3S)-3-Methylhexan-1-ol 8(13)
Iodine (4.21 g, 16.6 mmol) was added portionwise to triphenylphosphine (4.33 g, 16.6 mmol), imidazole (1.73 g, 25.4 mmol) and alcohol 7 (3.14 g, 12.7 mmol) in dichloromethane (70 mL), and the mixture was stirred at room temperature for 2 h. Saturated aqueous sodium sulfite (30 mL) and ether (50 mL) were added, and the mixture was partitioned between water and ether. The organic layer was washed with brine (50 mL), dried (Na2SO4) and concentrated under reduced pressure. Chromatography eluting with light petroleum gave (4S)-6-tert-butyldimethylsilyloxy-4-methyl-1-iodohexane (3.7 g, 82%) as a colorless oil, Rf 0.2 (light petroleum), [α]20D +3.6 (c 0.2 in CHCl3) (found: M+ – C4H9, 299.0308. C9H20IOSi requires M, 299.0323); νmax 1462, 1388, 1361, 1254, 1214, 1174, 1094, 1006, 939, 896, 832, and 772 cm–1; δH (500 MHz, CDCl3) 0.00 (6 H, s, 2 × SiCH3), 0.86 [9 H, s, SiC(CH3)3], 0.86 (3 H, d, J 7.0, 4-CH3), 1.20 (1 H, m, 4-H), 1.31 (2 H, m, 3-H2), 1.52 (2 H, m, 2-H2), 1.79 (2 H, m, 5-H2), 3.12 (2 H, m, 1-H2) and 3.58 (2 H, m, 6-H2); δC (125 MHz, CDCl3) −5.3(2), 7.5, 18.3, 19.6, 26.0, 28.7, 31.2, 37.9, 39.7 and 61.4; m/z (EI) 299 (M+ – 57, 5%). A portion of this iodide (3.0 g, 8.4 mmol) was dissolved in THF (40 mL), potassium tert-butoxide (2.9 g, 25 mmol) was added, and the reaction mixture was stirred at room temperature for 16 h. The reaction mixture was partitioned between water (70 mL) and ether. The organic layer was washed with brine (50 mL), dried (Na2SO4) and concentrated under reduced pressure to yield a colorless oil identified as (4S)-6-tert-butyldimethylsilyloxy-4-methylhex-1-ene. Without purification, this alkene was dissolved in THF (30 mL), and aqueous hydrogen chloride (4 M; 2.2 mL, 9.0 mmol) was added. The reaction mixture was stirred at room temperature for 3 h, neutralized using aqueous sodium hydrogen carbonate and partitioned between saturated sodium hydrogen carbonate (50 mL) and ether. The organic layer was washed with brine (40 mL), dried (Na2SO4) and concentrated under reduced pressure. Chromatography of the residue eluting with petrol/ether (1:1) gave (3S)-3-methylhex-5-en-1-ol as a colorless oil. This alkene was dissolved in THF (20 mL) and Pd/C (10% Pd by weight; 150 mg) was added. The reaction mixture was stirred under an atmosphere of hydrogen at room temperature for 16 h. The suspension was then filtered, and the filtrate was concentrated under reduced pressure to give the title compound 8(13) (1.88 g, 60% overall yield) as a colorless oil, Rf 0.5 (1:1 light petroleum/ether), [α]20D −1.1 (c 0.2 in CHCl3) lit.13 value: [α]20D −0.94 (c 0.91, CHCl3) (found: M+ – H2O, 98.1087. C7H14 requires M, 98.1090); νmax 3323, 1458, 1378, 1120, 1054, 1008, 960, 836, and 738 cm–1; δH (500 MHz, CDCl3) 0.78 (3 H, t, J 7.0, 6-H3), 0.80 (3 H, d, J 6.8, 3-CH3), 1.03 (1 H, m, 3-H), 1.16–1.31 (4 H, m, 4-H2 and 5-H2), 1.50 (2 H, m, 2-H2) and 3.59 (2 H, m, 1-H2); δC (125 MHz, CDCl3) 14.4, 19.6, 20.0, 29.2, 39.4, 40.0 and 61.3; m/z (EI) 98 (M+ – 18, 10%).
(3E,2R,6S,8S)-1-Benzyloxy-2,4,8-trimethylundec-3-en-6-ol 11
Sodium hydrogen carbonate (212 mg, 2.5 mmol) and Dess–Martin periodinane (261 mg, 0.62 mmol) were added to the alcohol 8 (63 mg, 0.54 mmol) in dichloromethane (5 mL), and the reaction mixture was stirred at room temperature for 30 min. Saturated aqueous sodium hydrogen carbonate (4 mL) and sodium bisulfite (4 mL) were added, and the mixture was extracted with dichloromethane (2 × 10 mL). The organic extract was washed with brine (15 mL), dried (Na2SO4) and concentrated under reduced pressure to yield (3S)-3-methylhexanal 9. Zinc powder (80 mg, 1.22 mmol) was suspended in a solution of bismuth(III) iodide (637 mg, 1.08 mmol) in THF (4 mL), and the mixture was stirred vigorously at room temperature for 1 h during which time the orange/gray suspension turned black. The 5-benzyloxy-2,4-dimethylpent-2-enyl bromide 10(9a) (102 mg, 0.36 mmol) and the 3-methylhexanal 9 in THF (2 mL) were added, and the reaction mixture was stirred under reflux for 2 h. After cooling to room temperature, the mixture was concentrated under reduced pressure to give a black slurry. Chromatography of this residue eluting with light petroleum/ether (7:3) gave the title compound 11 (67 mg, 60%) as a colorless oil, containing ca. 5% of its (6R)-epimer 15 (1H, 13C NMR) and other minor components (up to 5%), Rf 0.5 (7:3 light petroleum/ether), [α]20D −6.7 (c 0.2 in CHCl3) (found: M+ + Na, 341.2453. C21H34O2Na requires M, 341.2452); νmax 3436, 1454, 1378, 1273, 1205, 1089, 1028, 901, 832, and 735 cm–1; δH (400 MHz, CDCl3) major epimer 11 0.82 (6 H, m, 8-CH3 and 11-H3), 0.88 (3 H, d, J 7.0, 2-CH3), 1.09 (1 H, m, 7-H), 1.19–1.29 (5 H, m, 8-H, 9-H2 and 10-H2), 1.41 (1 H, m, 7-H′), 1.60 (3 H, s, 4-CH3), 1.75 (1 H, br. s, OH), 1.86 (1 H, dd, J 12.8 and 10.0, 5-H), 2.11 (1 H, dd, J 12.8 and 7.2, 5-H′), 2.69 (1 H, m, 2-H), 3.15 (1 H, dd, J 9.1 and 7.8, 1-H), 3.23 (1 H, dd, J 9.1 and 7.3, 1-H′), 3.63 (1 H, m, 6-H), 4.40 and 4.43 (each 1 H, d, J 12, OHCHPh), 4.97 (1 H, d, J 8.9, 3-H) and 7.25 (5 H, m, ArH); minor epimer 15 0.92 (3 H, d, J 6.5, 2-CH3), 1.94 (1 H, dd, J 13.6 and 9.1, 5-H) and 4.44 (2 H, s, OCH2Ph); δC (100 MHz, CDCl3) major epimer 11 14.5, 16.5, 17.4, 20.0, 20.3, 29.5, 33.2, 39.1, 44.6, 48.5, 66.0, 73.0, 75.3, 127.5, 127.6, 128.4, 132.1, 132.6 and 138.5; minor epimer 15 29.1, 40.2, 48.8 and 65.6; m/z (ES+) 341 (M+ + 23, 100%).
(3E,2R,6R,8S)-1-Benzyloxy-2,4,8-trimethylundec-3-en-6-ol 15
Di-isopropyl diazodicarboxylate (51 mg, 0.25 mmol) was added to a suspension of alcohol 11 (40 mg, 0.13 mmol), 4-nitrobenzoic acid (32 mg, 0.19 mmol) and Ph3P (66 mg, 0.25 mmol) in THF at room temperature, and the reaction mixture was stirred at room temperature for 16 h. After concentration under reduced pressure, chromatography of the residue eluting with light petroleum/ether (90:10) gave the 4-nitrobenzoate 14 (41 mg, 70%) as a colorless oil, Rf 0.8 (4:1 light petroleum/ether); δH (400 MHz, CDCl3) 0.60 (3 H, d, J 6.8, 8-CH3), 0.79 (3 H, t, J 7.3, 11-H3), 0.84 (3 H, d, J 6.6, 2-CH3), 1.00–1.33 (6 H, m, 7-H2, 9-H2 and 10-H2), 1.64 (3 H, s, 4-CH3), 1.70 (1 H, m, 8-H), 2.00–2.40 (2 H, m, 5-H2), 2.56 (1 H, m, 2-H), 3.14 (2 H, m, 1-H2), 4.39 (2 H, s, OCH2Ph), 4.91 (1 H, d, J 9.3, 3-H), 5.34 (1 H, m, 6-H), 7.23 (5 H, m, ArH), 8.10 (2 H, d, J 8.6, ArH) and 8.19 (2 H, d, J 9.1, ArH).
The ester 14 (41 mg, 0.09 mmol) was dissolved in THF (1.5 mL), and aqueous sodium hydroxide (2 N; 1 mL) was added. The reaction mixture was stirred at 50 °C for 1 h then concentrated under reduced pressure. Chromatography of the residue eluting with light petroleum/ether (80:20) gave the title compound 15 (41 mg, 70%) as a colorless oil containing ca. 5% of the (6S)-epimer 11 (1H and 13C NMR), Rf 0.5 (7:3 light petroleum/ether); δH (400 MHz, CDCl3) major epimer 15 0.80–0.84 (6 H, m, 8-CH3 and 11-H3), 0.92 (3 H, d, J 6.6, 2-CH3), 1.03–1.30 (6 H, m, 7-H2, 9-H2 and 10-H2), 1.39 (1 H, m, 8-H), 1.55 (1 H, br. s, OH), 1.61 (3 H, s, 4-CH3), 1.94 (1 H, dd, J 9.1 and 13.6, 5-H), 2.08 (1 H, dd, J 4.3 and 13.4, 5-H′), 2.68 (1 H, m, 2-H), 3.23 (2 H, m, 1-H2), 3.70 (1 H, m, 6-H), 4.44 (2 H, s, OCH2Ph), 5.02 (1 H, d, J 9.1, 3-H) and 7.26 (5 H, m, ArH); minor epimer 11 0.88 (3 H, d, J 7.0, 2-CH3) and 1.86 (1 H, dd, J 10.0 and 12.8, 5-H); δC (125 MHz, CDCl3) major epimer 15 14.4, 16.6, 18.0, 19.3, 20.1, 29.1, 33.2, 40.2, 44.6, 48.8, 66.1, 73.0, 75.2, 127.5, 128.4, 131.8, 132.7 and 138.6; minor epimer 11 29.5, 39.0, 48.3 and 66.5.
(3E,2R,6S,8S)-2,4,8-Trimethylundec-3-ene-1,6-diol 12
Lithium metal (12 mg, 1.7 mmol) was added in small pieces to naphthalene (290 mg, 2.3 mmol) in THF (3 mL), and the reaction mixture was stirred at room temperature until the lithium was completely dissolved. The resulting dark green solution of lithium naphthalenide was cooled to −25 °C, and the benzyl ether 11 (90 mg, 0.28 mmol) in THF (2 mL) was added. The reaction mixture was stirred at −25 °C for 2 h, and saturated aqueous ammonium chloride (5 mL) and water (5 mL) were added. The mixture was extracted with ether, and the organic extract was washed with brine (10 mL), dried (Na2SO4) and concentrated under reduced pressure. Chromatography of the residue eluting with light petroleum/ether (20:80) gave the title compound 12 (50 mg, 78%) as a colorless gum, Rf 0.3 (3:7 light petroleum/ether), [α]20D +26.7 (c 0.4 in CHCl3) (found: M+ + Na, 251.1983. C14H28O2Na requires M, 251.1982); νmax 3308, 1455, 1378, 1072, 1031, 831, and 739 cm–1; δH (400 MHz, CDCl3) 0.81–0.88 (9 H, m, 2-CH3, 8-CH3 and 11-H3), 0.95–1.30 (6 H, m, 7-H2, 9-H2 and 10-H2), 1.55 (1 H, m, 8-H), 1.57 (3 H, s, 4-CH3), 1.90 (3 H, m, 5-H and 2 × OH), 2.09 (1 H, dd, J 12.7 and 3.3, 5-H′), 2.60 (1 H, m, 2-H), 3.23 (1 H, dd, J 9.8 and 8.6, 1-H), 3.47 (1 H, dd, J 9.8 and 6.2, 1-H′), 3.72 (1 H, m, 6-H) and 4.91 (1 H, d, J 9.0, 3-H); δC (100 MHz, CDCl3) 14.4, 16.6, 16.7, 20.0, 20.3, 29.5, 35.4, 39.1, 45.0, 48.5, 66.4, 67.8, 131.2 and 134.2; m/z (ES+) 251 (M+ + 23, 100%).
(3E,2R,6S,8S)-2,4,8-Trimethyl-1-tri-isopropylsilyloxyundec-3-en-6-ol 13
Imidazole (75 mg, 1.1 mmol) was added to the diol 12 (50 mg, 0.22 mmol) in THF (4 mL), and after 10 min, tri-isopropylsilyl chloride (51 mg, 0.24 mmol) was added at 0 °C. The reaction mixture was stirred at room temperature for 16 h then concentrated under reduced pressure. Chromatography of the residue eluting with light petroleum/ether (4:1) gave the title compound 13 (80 mg, 94%) as a colorless oil, Rf 0.7 (4:1 light petroleum/ether), [α]20D +9.6 (c 0.2 in CHCl3) (found: M+ + H, 385.3503. C23H49O2Si requires M, 385.3496); νmax 1461, 1381, 1089, 1065, 1013, 995, 881, 785, 680, and 658 cm–1; δH (400 MHz, CDCl3) 0.83 (6 H, m, 8-CH3 and 11-H3), 0.87 (3 H, d, J 7, 2-CH3), 0.98 [21 H, m, 3 × OSiCH(CH3)2], 1.10–1.35 (6 H, m, 7-H2, 9-H2 and 10-H2), 1.54 (1 H, m, 8-H), 1.61 (3 H, s, 4-CH3), 1.86 (1 H, dd, J 12.8 and 9.8, 5-H), 1.88 (1 H, s, OH), 2.10 (1 H, dd, J 12.8 and 3.2, 5-H′), 2.54 (1 H, m, 2-H), 3.37 and 3.42 (each 1 H, dd, J 6.9 and 9.2, 1-H), 3.64 (1 H, m, 6-H) and 4.96 (1 H, d, J 9.1, 3-H); δC (100 MHz, CDCl3) 12.0, 14.4, 16.6, 17.0, 18.0, 20.0, 20.2, 29.5, 35.8, 39.1, 44.4, 48.5, 66.0, 68.4, 132.1 and 132.4; m/z (ES+) 407 (M+ + 23, 100%).
(2R,4R,6S,8S)- and (2R,4S,6S,8S)-2,4,8-Trimethyl-1-tri-isopropylsilyloxyundecan-6-ol 16 and 18
The alkene 13 (80 mg, 0.21 mmol) followed by [Rh(NBD)Diphos-4]BF4 (7.5 mg, 0.01 mmol) and DCM (3 mL) were added to a boiling tube with stirrer bar, and the tube was placed inside a steel screw-cap high pressure bomb. The pressure gauge block was attached, and the bomb was flushed three times with hydrogen and then filled with hydrogen to 950 psi. The reaction mixture was stirred at room temperature for 5 h then concentrated under reduced pressure. Chromatography of the residue eluting with light petroleum/ether (4:1) gave the title compound 18 (15 mg, 16%) as a colorless oil, Rf 0.5 (4:1 light petroleum/ether), [α]20D +4.2 (c 0.2 in CHCl3) νmax 3351, 1462, 1380, 1100, 1067, 1013, 996, 882, 787, 680, and 658 cm–1; δH (500 MHz, CDCl3) 0.81–0.86 (12 H, m, 2-CH3, 4-CH3, 8-CH3 and 11-H3), 0.99 [21 H, m, 3 × OSiCH(CH3)2], 1.04–1.33 (10 H, m, 3-H2, 5-H2, 7-H2, 9-H2, and 10-H2), 1.50 (1 H, m, 8-H), 1.62–1.72 (2 H, m, 2-H and 4-H), 3.36 (1 H, dd, J 6.6 and 9.5, 1-H), 3.47 (1 H, dd, J 5.7 and 9.5, 1-H′) and 3.73 (1 H, m, 6-H); δC (100 MHz, CDCl3) 12.0, 14.4, 17.5, 18.1, 20.0, 20.2, 26.6, 29.4, 33.4, 39.1, 42.0, 44.9, 46.2, 67.6 and 68.7; m/z (ES+) 409 (M+ + 23, 100%). The second fraction was the title compound 16 (61 mg, 70%) as a colorless oil, Rf 0.45 (4:1 light petroleum/ether), [α]20D +7.8 (c 0.2 in CHCl3) (found: M+ + Na, 409.3486. C23H50O2SiNa requires M, 409.3473); νmax 3325, 1461, 1379, 1245, 1100, 1067, 1012, 995, 918, 881, 784, 679, and 658 cm–1; δH (400 MHz, CDCl3) 0.81–0.84 (12 H, m, 2-CH3, 4-CH3, 8-CH3 and 11-H3), 0.99 [21 H, m, 3 × OSiCH(CH3)2], 1.05–1.35 (10 H, m, 3-H2, 5-H2, 7-H2, 9-H2 and 10-H2), 1.40 (1 H, br. s, OH), 1.53 (1 H, m, 8-H), 1.61–1.70 (2 H, m, 2-H and 4-H), 3.38 (1 H, dd, J 9.8 and 6.1, 1-H), 3.43 (1 H, dd, J 9.8 and 6.1, 1-H′) and 3.73 (1 H, m, 6-H); δC (100 MHz, CDCl3) 12.0, 14.4, 16.4, 18.1, 19.2, 20.0, 20.4, 26.8, 29.3, 33.5, 38.9, 40.4, 45.8, 46.6, 67.9 and 69.4; m/z (ES+) 409 (M+ + 23, 100%).
(2R,4R,6S,8S)-2,4,8-Trimethyl-1-tri-isopropylsilyloxyundecan-6-yl Toluene p-Sulfonate 17
Toluene p-sulfonyl chloride (205 mg, 1.1 mmol) and 4-dimethylaminopyridine (202 mg, 1.65 mmol) were added to the alcohol 16 (143 mg, 0.37 mmol) in dichloromethane (4 mL) at room temperature, and the reaction was stirred at room temperature for 16 h. After concentration under reduced pressure, chromatography of the residue eluting with light petroleum/ether (4:1) gave the title compound 17 (185 mg, 93%) as a colorless oil, Rf 0.8 (4:1 light petroleum/ether), [α]20D +3.2 (c 0.2 in CHCl3) (found: M+ + H, 541.3738. C30H57O4SSi requires M, 541.3741); νmax 1598, 1462, 1362, 1186, 1175, 1096, 1067, 920, 880, 813, 760, and 679 cm–1; δH (400 MHz, CDCl3) 0.72–0.77 (12 H, m, 2-CH3, 4-CH3, 8-CH3 and 11-H3), 0.98 [21 H, m, 3 × OSiCH(CH3)2], 1.04–1.21 (5 H, m, 8-H, 9-H2 and 10-H2), 1.27–1.49 (7 H, m, 3-H2, 4-H, 5-H2 and 7-H2), 1.58 (1 H, m, 2-H), 2.37 (3 H, s, ArCH3), 3.37 (2 H, d, J 6.0, 1-H2), 4.64 (1 H, m, 6-H), 7.25 (2 H, d, J 7.9, ArH) and 7.72 (2 H, d, J 7.8, ArH); δC (100 MHz, CDCl3) 11.0, 13.2, 15.2, 17.0, 18.5, 18.8, 20.6, 25.6, 27.9, 32.3, 37.7, 39.5, 41.2, 42.3, 68.2, 80.7, 126.7, 128.6, 133.9 and 143.3; m/z (ES+) 563 (M+ + 23, 100%).
(2R,4S,6R,8S)-2,4,6,8-Tetramethyl-1-tri-isopropylsilyloxyundecane 19
Copper(I) iodide (224 mg, 1.18 mmol) was placed in a round-bottom flask. The flask was evacuated and purged with nitrogen three times. Tetrahydrofuran (2 mL) was added followed by methyl lithium·lithium iodide complex (1.6 M, 1.3 mL, 2.13 mmol) at 0 °C to produce a clear solution. The toluene p-sulfonate 17 (64 mg, 0.12 mmol) in THF (1 mL) was added. The reaction mixture was stirred at 0 °C for 1 h, allowed to warm to room temperature and then stirred for 16 h. Saturated aqueous ammonium chloride (10 mL) was added, and the mixture filtered through a pad of Celite then partitioned between water and ether. The organic layer was washed with brine (10 mL), dried (Na2SO4) and concentrated under reduced pressure. Chromatography of the residue eluting with light petroleum (100%) gave the title compound 19 (10 mg, 21%) as a colorless oil, Rf 0.7 (100% petrol), [α]20D +14.7 (c 0.2 in CHCl3) (found: M+ – C3H7, 341.3229. C21H45OSi requires M, 341.3234); νmax 1461, 1378, 1098, 1067, 1012, 994, 918, 881, and 783 cm–1; δH (500 MHz, CDCl3) 0.74 (9 H, m, 3 × CH3), 0.82 (6 H, m, 2 × CH3), 0.99 [21 H, m, 3 × OSiCH(CH3)2], 0.95–1.28 (8 H, m, 6-H, 7-H2, 8-H, 9-H2 and 10-H2), 1.42 (1 H, m, 4-H), 1.48–1.54 (4 H, m, 3-H2 and 5-H2), 1.64 (1 H, m, 2-H), 3.35 (1 H, dd, J 6.2 and 9.3, 1-H) and 3.45 (1H, dd, J 6.2 and 9.3, 1-H′); δC (100 MHz, CDCl3) 11.0, 13.4, 15.8, 17.1, 18.5(2), 18.6, 19.1, 26.2, 26.3, 28.7, 32.5, 39.2, 40.4, 44.5, 45.6 and 68.2; m/z (EI) 341 (M+ – 43, 100%).
(2R,4S,6R,8S)-2,4,6,8-Tetramethylundecan-1-ol 20(2b)
The silyl ether 19 (75 mg, 0.19 mmol) was dissolved in THF (2 mL), aqueous hydrogen chloride (4 M, 0.24 mL, 0.96 mmol) in dioxane was added, and the mixture was stirred at room temperature for 16 h. The mixture was concentrated under reduced pressure, and chromatography of the residue eluting with light petroleum/ether (7:3) gave the title compound 20(2b) (40 mg, 91%) as a colorless oil, Rf 0.5 (60:40 light petroleum/ether), [α]20D +30 (c 0.2 in CHCl3), lit.2b value: [α]20D +22.7 (c 1.01 in CHCl3) (found: M+ – H2O, 210.2342. C15H30 requires M, 210.2342); νmax 3335, 1463, 1379, 1037, 812, and 735 cm–1; δH (400 MHz, CDCl3) 0.72 (3 H, d, J 7.0, CH3), 0.75 (6 H, d, J 7.0, 2 × CH3), 0.80 (3 H, t, J 6.5, 11-H3), 0.85 (3 H, d, J 6.5, 2-CH3), 0.89–1.28 (10 H, m, 5-H2, 6-H, 7-H2, 8-H, 9-H2 and 10-H2), 1.41 (1 H, m, 4-H), 1.48–1.56 (2 H, m, 3-H2), 1.66 (1 H, m, 2-H), 3.35 (1 H, m, 1-H) and 3.40 (1 H, m, 1-H′); δC (100 MHz, CDCl3) 14.4, 16.8, 19.5(2), 19.6, 20.1, 27.3(2), 29.7, 33.5, 40.3, 41.5, 45.6, 46.6 and 69.2.
(2R,4S,6R,8S)-2,4,6,8-Tetramethylundecyl Toluene p-Sulfonate 21
Toluene p-sulfonyl chloride 20 (25 mg, 0.13 mmol) and DMAP (19 mg, 0.15 mmol) were added to the alcohol 20 (20 mg, 0.088 mmol) in dichloromethane (2 mL), and the mixture was stirred at room temperature for 16 h. The reaction mixture was concentrated under reduced pressure, and chromatography of the residue eluting with light petroleum/ether (4:1) gave the title compound 21 (28 mg, 85%) as a colorless oil, Rf 0.7 (80:20 light petroleum/ether), [α]20D +9.5 (c 0.2 in CHCl3) (found: M+ + Na, 405.2433. C22H38O3NaS requires M, 405.2434); νmax 1596, 1458, 1360, 1188, 1174, 1098, 962, 812, 665, and 654 cm–1; δH (400 MHz, CDCl3) 0.65, 0.68, 0.74, and 0.77 (each 3 H, d, J 7.0, 2-CH3, 4-CH3, 6-CH3 or 8-CH3), 0.80 (3 H, t, J 7.3, 11-H3), 0.86–1.45 (13 H, m, 3-H2, 4-H, 5-H2, 6-H, 7-H2, 8-H, 9-H2 and 10-H2), 1.78 (1 H, m, 2-H), 2.38 (3 H, s, ArCH3), 3.72 (1 H, dd, J 6.8 and 9.3, 1-H), 3.77 (1 H, dd, J 5.8 and 9.3 1-H′), 7.25 (2 H, d, J 7.8, ArH) and 7.72 (2 H, d, J 8.3, ArH); δC (100 MHz, CDCl3) 14.4, 16.4, 19.2, 19.4, 19.6, 20.1, 21.7, 27.0, 27.2, 29.7, 30.3, 40.2, 40.8, 45.5, 46.1, 75.8, 127.9, 129.8, 133.2 and 144.6; m/z (ES+) 405 (M+ + 23, 100%).
(2R,4S,6R,8S)-1-Iodo-2,4,6,8-tetramethylundecane 22
Sodium iodide (22 mg, 0.15 mmol) was added to the toluene p-sulfonate 21 (28 mg, 0.07 mmol) in acetone (2 mL), and the mixture was stirred under reflux for 16 h. The mixture was then partitioned between hexane and water, and the organic layer was washed with brine (10 mL), dried (Na2SO4) and concentrated under reduced pressure. Chromatography of the residue eluting with light petroleum (100%) gave the title compound 22 (21 mg, 90%) as a colorless oil, Rf 0.8 (100% light petroleum), [α]20D +12 (c 0.2 in CHCl3) (found: M+, 338.1462. C15H31I requires M, 338.1465); νmax 1457, 1378, 1193, 962, 816, and 739 cm–1; δH (500 MHz, CDCl3) 0.73 (9 H, m, 3 × CH3), 0.82 (3 H, t, J 7.0, 11-H3), 0.90 (3 H, d, J 6.5, 2-CH3), 0.92–1.28 (11 H, m, 4-H, 5-H2, 6-H, 7-H2, 8-H, 9-H2 and 10-H2), 1.41 (1 H, m, 2-H), 1.49 (2 H, m, 3-H2), 3.07 (1 H, dd, J 3.2 and 9.5, 1-H) and 3.15 (1 H, dd, J 4.7 and 9.5, 1-H′); δC (125 MHz, CDCl3) 14.4, 18.8, 19.5(2), 19.6, 20.1, 20.4, 27.2, 27.5, 29.7, 32.3, 40.2, 44.8, 45.5 and 46.0; m/z (EI) 338 (M+, 5%).
(R)- and (S)-8-Iodo-2,6-dimethyloct-2-ene (R)-23 and (S)-2316
Sodium iodide (1.83 g, 12.2 mmol) was added to the toluene p-sulfonate ester of (R)-citronellol20 (1.90 g, 6.1 mmol) in acetone (15 mL). The reaction mixture was stirred under reflux for 16 h then concentrated under reduced pressure and partitioned between hexane (30 mL) and aqueous Na2SO3 (15 mL). The organic layer was washed with brine (20 mL), dried (Na2SO4) and concentrated under reduced pressure. Chromatography of the residue eluting with light petroleum gave the title compound (R)-2316a (1.44 g, 90%), as a colorless oil, Rf 0.8 (light petroleum), [α]20D −5.2 (c 1.0 in CHCl3) lit.16a value: [α]20D −14.34 (neat) (found: M+, 266.0533. C10H19I requires M, 266.0526); νmax 1449, 1377, 1178, 984, 826, and 733 cm–1; δH (400 MHz, CDCl3) 0.82 (3 H, d, J 6.6, 6-CH3), 1.11 (1 H, m, 5-H), 1.27 (1 H, m, 5-H′), 1.49 (1 H, m, 4-H), 1.54 (3 H, s, 2-CH3 or 1-H3), 1.59 (1 H, m, 4-H′), 1.62 (3 H, s, 2-CH3 or 1-H3), 1.83 (1 H, m, 6-H), 1.93 (2 H, m, 7-H2), 3.11 (1 H, m, 8-H), 3.19 (1 H, m, 8-H′) and 5.02 (1 H, m, 3-H); δC (100 MHz, CDCl3) 5.3, 17.7, 18.7, 25.3, 25.7, 33.6, 36.3, 40.9, 124.5 and 131.5.
The same procedure using sodium iodide (2.7 g, 18.3 mmol) and the toluene p-sulfonate ester of (S)-citronellol21 (2.9 g, 9.2 mmol) gave the title compound (S)-2316b (2.21 g, 92%), Rf 0.8 (light petroleum), [α]20D +7.6 (c 0.6 in CHCl3) (found: M+, 266.0534. C10H19I requires M, 266.0526), with spectroscopic data identical to those of the (R)-enantiomer.
(7R,4RS)-7,11-Dimethyl-4-phenylsulfonyldodec-10-ene 24
n-Butyllithium (2.94 mL, 1.6 M in hexane, 4.70 mmol) was added to n-butyl phenyl sulfone (776 mg, 3.91 mmol) in dry THF (14 mL) and DMPU (2 mL) at −40 °C, and the mixture was stirred for 30 min. The iodide (R)-23 (1.25 g, 4.7 mmol) in THF (4 mL) was added, and the reaction mixture was allowed to warm to room temperature and was stirred for 16 h. Saturated aqueous ammonium chloride (10 mL) was added, and the mixture was partitioned between water (10 mL) and ether (20 mL). The organic layer was washed with brine (20 mL), dried (Na2SO4) and concentrated under reduced pressure. Chromatography of the residue eluting with light petroleum/ether (4:1) gave the title compound 24 (1.23 g, 95%) as a colorless oil, as a 50:50 mixture of 4-epimers (1H and 13C NMR), Rf 0.4 (4:1 light petroleum/ether), [α]20D −1.7 (c 1.6 in CHCl3) (found: M+ + H, 337.2194. C20H33O2S requires M, 337.2196); νmax 1446, 1377, 1302, 1143, 1083, 1024, 998, 756, 725, and 690 cm–1; δH (500 MHz, CDCl3) 0.73 (1.5 H, d, J 6.3, 7-CH3), 0.74 (1.5 H, d, J 6.6, 7-CH3), 0.81 (3 H, t, J 7.3, 1-H3), 0.98–1.54 (9 H, m, 2-H2, 6-H2, 7-H, 8-H2 and 9-H2), 1.51 and 1.61 (each 3 H, s, 11-CH3 or 12-H3), 1.70–1.93 (4 H, m, 3-H2 and 5-H2), 2.80 (1 H, m, 4-H), 4.98 (1 H, m, 10-H), 7.49 (2 H, t, J 7.8, ArH), 7.58 (1 H, t, J 7.3, ArH) and 7.82 (2 H, d, J 7.5, ArH); δC (100 MHz, CDCl3) 14.0(2), 17.7, 19.2, 19.4, 20.1, 25.4(2), 25.7, 29.9, 30.0, 32.5(2), 33.7, 33.9, 36.5, 36.9, 64.6, 64.7, 124.6(2), 128.8, 129.1, 131.3(2), 133.5 and 138.3; m/z (ES+) 359 (M+ + 23, 100%).
(4R,7RS)-4-Methyl-7-phenylsulfonyldecan-1-ol 25
The alkene 24 (460 mg, 1.37 mmol) was dissolved in dichloromethane and methanol (1:1, 20 mL), and the solution was cooled to −78 °C. Ozone was bubbled through the solution until it turned blue. Oxygen was then bubbled through the solution at −78 °C until the reaction became colorless. Sodium borohydride (250 mg, 6.61 mmol) was added, and the mixture allowed to warm to room temperature and stirred for 16 h. The mixture was partitioned between ether (20 mL) and brine (20 mL), and the organic layer washed with brine, dried (Na2SO4) and concentrated under reduced pressure. Chromatography of the residue eluting with light petroleum/ether (1:4) gave the title compound 25 (364 mg, 85%) as a colorless oil, as a 50:50 mixture of 7-epimers (1H and 13C NMR), Rf 0.4 (4:1 light petroleum/ether), [α]20D −2.9 (c 0.2 in CHCl3) (found: M+ + H, 313.1833. C17H29O3S requires M, 313.1832); νmax 3394, 1447, 1380, 1286, 1141, 1083, 758, 726, and 691 cm–1; δH (500 MHz, CDCl3) 0.75 (1.5 H, d, J 6.9, 4-CH3), 0.78 (1.5 H, d, J 5.0, 4-CH3), 0.80 (3 H, t, J 7.6, 10-H3), 1.02–1.58 (11 H, m, 3-H2, 4-H, 5-H2, 6-H2, 8-H2 and 9-H2), 1.71–1.83 (2 H, m, 2-H2), 2.81 (1 H, m, 7-H), 3.54 (2 H, t, J 6.6, 1-H2), 7.50 (2 H, t, J 7.5, ArH), 7.59 (1 H, t, J 7.5, ArH) and 7.82 (2 H, d, J 7.5, ArH); δC (100 MHz, CDCl3) 14.0(2), 15.3, 19.3, 19.4, 20.1, 25.4, 29.9, 30.1(2), 32.5, 32.7, 33.8, 33.9, 63.2, 64.6(2), 65.9, 128.8, 129.1, 133.5 and 138.2; m/z (ES+) 335 (M+ + 23, 100%).
(S)- and (R)-4-Methyldecan-1-ol (S)-26 and (R)-26.17
Sodium amalgam (10%; 10.0 g, 34.6 mmol) was added to the sulfone 25 (360 mg, 1.15 mmol) in anhydrous methanol (30 mL), and the mixture was stirred at room temperature for 16 h. The solution was transferred to another flask and concentrated under reduced pressure. The residue was partitioned between saturated aqueous ammonium chloride (40 mL) and ether (40 mL). The organic layer was washed with brine (20 mL), dried (Na2SO4) and concentrated under reduced pressure. Chromatography of the residue eluting with light petroleum/ether (4:1) gave the title compound (S)-26 (143 mg, 73%) as a colorless oil, Rf 0.6 (1:1 light petroleum/ether), [α]20D −3.9 (c 0.3 in CHCl3) lit.17 value: [α]20D −1.1 (c 5.33 in CHCl3) (found: M+ – H2O, 154.1723. C11H22 requires M, 154.1716); νmax 3314, 1459, 1378, 1057, 898, and 723 cm–1; δH (400 MHz, CDCl3) 0.80 (3 H, d, J 6.6, 4-CH3), 0.81 (3 H, t, J 6.6, 10-H3), 1.01–1.60 (15 H, m, 2-H2, 3-H2, 4-H, 5-H2, 6-H2, 7-H2, 8-H2 and 9-H2) and 3.59 (2 H, t, J 6.8, 1-H2); δC (100 MHz, CDCl3) 14.1, 19.7, 22.7, 27.0, 29.7, 30.4, 32.0, 32.6, 33.0, 37.0 and 63.5; m/z (EI) 154 (M+ – 18, 5%), 126 (20) and 91 (100).
The same procedure using the sulfone 32 (540 mg, 1.73 mmol) gave the title compound (R)-26 (206 mg, 70%), Rf 0.6 (1:1 light petroleum/ether), [α]20D +4.7 (c 0.3 in CHCl3) (found: M+ – H2O, 154.1714. C11H22 requires M, 154.1716), with spectroscopic data identical to those of the (S)-enantiomer.
(S)- and (R)-4-Methyldec-1-yl Toluene p-Sulfonate (S)-27 and (R)-27
Toluene p-sulfonyl chloride (166 mg, 0.87 mmol) and DMAP (127 mg, 1.04 mmol) were added to the alcohol (S)-26 (100 mg, 0.58 mmol) in dichloromethane (8 mL) at room temperature, and the mixture was stirred at room temperature for 16 h. The mixture was concentrated under reduced pressure, and chromatography of the residue eluting with light petroleum/ether (4:1) gave the title compound (S)-27 (187 mg, 99%) as a colorless oil, Rf 0.7 (7:3 light petroleum/ether), [α]20D +3.1 (c 0.4 in CHCl3) (found: M+ + Na, 349.1812. C18H30O3NaS requires M, 349.1808); νmax 1598, 1465, 1358, 1187, 1174, 1096, 961, 914, and 812 cm–1; δH (400 MHz, CDCl3) 0.73 (3 H, d, J 6.6, 4-CH3), 0.81 (3 H, t, J 7.0, 10-H3), 0.97–1.27 (13 H, m, 3-H2, 4-H, 5-H2, 6-H2, 7-H2, 8-H2 and 9-H2), 1.51–1.65 (2 H, m, 2-H2), 2.38 (3 H, s, ArCH3), 3.94 (2 H, t, J 6.6, 1-H2), 7.28 (2 H, m, ArH) and 7.72 (2 H, m, ArH); δC (100 MHz, CDCl3) 14.1, 19.4, 21.7, 22.7, 26.5, 26.9, 29.6, 31.9, 32.3, 32.5, 36.8, 71.1, 127.9, 129.8, 133.2 and 144.6; m/z (ES+) 349 (M+ + 23, 70%).
The (R)-alcohol (R)-26 (150 mg, 0.87 mmol), toluene p-sulfonyl chloride (248 mg, 1.3 mmol) and DMAP (191 mg, 1.51 mmol) similarly gave the (R)-enantiomer (R)-27 (277 mg, 98%), Rf 0.7 (7:3 light petroleum/ether), [α]20D −3.8 (c 0.4 in CHCl3) (found: M+ + Na, 349.1810. C18H30O3NaS requires M, 349.1808), with spectroscopic data identical to those of the (S)-enantiomer.
(S)- and (R)-1-Iodo-4-methyldecane (S)-28 and (R)-28.18
Sodium iodide (1.06 g, 7.04 mmol) was added to the toluene p-sulfonate (S)-27 (1.15 g, 3.52 mmol) in acetone (15 mL), and the mixture was stirred under reflux for 2 h. The mixture was then concentrated and partitioned between hexane (30 mL) and saturated aqueous sodium sulphite (15 mL). The organic layer was washed with brine (10 mL), dried (Na2SO4) and concentrated under reduced pressure to give the title compound (S)-28 (891 mg, 90%) as a colorless oil, Rf 0.8 (light petroleum), [α]20D +2.6 (c 0.2 in CHCl3) (found: M+, 282.0837. C11H23I requires M, 282.0839); νmax 1460, 1378, 1234, 1173, 926, and 724 cm–1; δH (400 MHz, CDCl3) 0.79 (3 H, d, J 6.6, 4-CH3), 0.81 (3 H, t, J 6.6, 10-H3), 1.01–1.36 (13 H, m, 3-H2, 4-H, 5-H2, 6-H2, 7-H2, 8-H2 and 9-H2), 1.65–1.85 (2 H, m, 2-H2) and 3.10 (2 H, dt, J 2.5 and 7.1, 1-H2); δC (100 MHz, CDCl3) 7.7, 14.1, 19.7, 22.7, 27.0, 29.7, 31.3, 32.0, 32.1, 36.9 and 37.9; m/z (EI) 282 (M+, 5%).
The toluene p-sulfonate (R)-27 (1.73 g, 5.3 mmol) and sodium iodide (1.6 g, 10.6 mmol) similarly gave the (R)-enantiomer (R)-28 (1.37 g, 92%), Rf 0.8 (light petroleum), [α]20D −1.7 (c 0.2 in CHCl3) (found: M+, 282.0835. C11H23I requires M, 282.0839), with spectroscopic data identical to those of the (S)-enantiomer.
(S)- and (R)-1-Phenylsulfonyl-4-methylundecane (S)-29 and (R)-29
n-Butyllithium (240 mL, 1.6 M in hexane, 0.38 mmol) was added to methyl phenyl sulfone (50 mg, 0.32 mmol) in THF (3 mL) at −40 °C. The mixture was stirred for 30 min, and the iodide (S)-28 (108 mg, 0.38 mmol) in THF (1 mL) was added. The reaction mixture was allowed to warm to room temperature and was stirred for 16 h. Saturated aqueous ammonium chloride (5 mL) was added, and the mixture was partitioned between water (2 mL) and ether (10 mL). The organic layer was washed with brine (10 mL), dried (Na2SO4) and concentrated under reduced pressure. Chromatography of the residue eluting with light petroleum/ether (4:1) gave the title compound (S)-29 (72 mg, 73%) as a colorless oil, Rf 0.5 (60:40 light petroleum/ether), [α]20D −6.4 (c 0.2 in CHCl3) (found: M+ + H, 311.2032. C18H31O2S requires M, 311.2040); νmax 1463, 1446, 1377, 1305, 1144, 1086, 1024, 998, 794, 745, 727, and 688 cm–1; δH (400 MHz, CDCl3) 0.73 (3 H, d, J 6.6, 5-CH3), 0.81 (3 H, t, J 7.0, 11-H3), 0.94–1.32 (15 H, m, 3-H2, 4-H2, 5-H, 6-H2, 7-H2, 8-H2, 9-H2 and 10-H2), 1.55–1.65 (2 H, m, 2-H2), 3.02 (2 H, t, J 8.1, 1-H2), 7.51 (2 H, m, ArH), 7.59 (1 H, m, ArH) and 7.84 (2 H, m, ArH); δC (100 MHz, CDCl3) 14.1, 19.5, 22.7, 23.0, 25.8, 27.0, 29.6, 31.9, 32.5, 36.4, 36.9, 56.4, 128.1, 129.3, 133.6 and 139.3; m/z (ES+) 333 (M+ + 23, 100%).
Methyl phenyl sulfone (75 mg, 0.48 mmol) and the (R)-iodide (R)-28 (162 mg, 0.57 mmol) similarly gave the (R)-enantiomer (R)-29 (110 mg, 75%), Rf 0.5 (60:40 light petroleum/ether), [α]20D +5.2 (c 0.2 in CHCl3) (found: M+ + H, 311.2034. C18H31O2S requires M, 311.2040), with spectroscopic data identical to those of the (S)-enantiomer.
(7S,11RS,13R,15S,17R,19S)-7,13,15,17,19-Pentamethyl-11-phenylsulfonydocosane 30
n-Butyllithium (33 mL, 1.6 M in hexane, 0.053 mmol) was added to the (S)-sulfone (S)-29 (13 mg, 0.044 mmol) in dry THF (0.5 mL) and DMPU (0.5 mL) at −40 °C. The mixture was stirred for 30 min, and then the iodide 22 (18 mg, 0.053 mmol) in THF (0.5 mL) was added. The mixture was allowed to warm to room temperature and was then stirred for 16 h. Saturated aqueous ammonium chloride (3 mL) was added, and the mixture was partitioned between water (2 mL) and ether (5 mL). The organic layer was washed with brine (10 mL), dried (Na2SO4) and concentrated under reduced pressure. Chromatography of the residue eluting with light petroleum/ether (4:1) gave the title compound 30 (10 mg, 45%) as a colorless oil, as a mixture of 11-epimers, Rf 0.6 (4:1 light petroleum/ether), [α]20D −3.7 (c 0.2 in CHCl3) (found: M+ + H, 521.4380. C33H61O2S requires M, 521.4387); νmax 1462, 1379, 1304, 1145, 1086, 764, 727, and 691 cm–1; δH (500 MHz, CDCl3) 0.64–0.84 (21 H, m, 1-H3, 7-CH3, 13-CH3, 15-CH3, 17-CH3, 19-CH3 and 22-H3), 0.86–1.80 (33 H, br. m, 2-H2, 3-H2, 4-H2, 5-H2, 6-H2, 7-H, 8-H2, 9-H2, 10-H2, 12-H2, 13-H, 14-H2, 15-H, 16-H2, 17-H, 18-H2, 19-H, 20-H2 and 21-H2), 2.91 (1 H, m, 11-H), 7.49 (2 H, t, J 7.5, ArH), 7.57 (1 H, t, J 6.9, ArH) and 7.81 (2 H, d, J 7.9, ArH); δC (100 MHz, CDCl3) 14.1, 14.4, 19.0, 19.4, 19.5, 19.6 (2), 20.1, 22.7, 24.2, 27.0, 27.2, 27.3, 27.9, 28.1, 29.0, 29.7(2), 32.0, 32.5, 36.2, 36.9, 37.0, 40.2, 40.3, 45.8, 46.3, 62.7, 128.9(2), 129.1, 133.5 and 138.3; m/z (ES+) 543 (M+ + 23, 100%).
(4S,6R,8R,10S,16S)-4,6,8,10,16-Pentamethyldocosane 2
Sodium amalgam (20%; 85 mg, 0.518 mmol) was added to the sulfone 30 (9 mg, 0.017 mmol) in MeOH (2 mL) at room temperature. The reaction mixture was stirred at room temperature for 6 h and then concentrated under reduced pressure. Saturated aqueous ammonium chloride (4 mL) was added, and mixture was partitioned between saturated aqueous ammonium chloride (6 mL) and hexane (6 mL). The organic layer was washed with brine (10 mL), dried (Na2SO4) and concentrated under reduced pressure. Chromatography of the residue eluting with hexane gave the title compound 2 (5 mg, 83%) as a colorless oil, Rf 1.0 (100% petrol), [α]20D +23.3 (c 1.2 in CHCl3); νmax 1463, 1378, 1260, 1094, 1018, and 799 cm–1; δH (500 MHz, CDCl3) 0.72–0.82 (21 H, m, 7 × CH3), 0.89–1.07 (10 H, m), 1.13–1.30 (20 H, m), 1.32–1.42 (2 H, m) and 1.42–1.55 (3 H, m); δC (100 MHz, CDCl3) 14.1212, 14.3854, 19.5518, 19.5682, 19.5882, 19.6502, 19.7286, 20.0803, 22.6990, 27.0508, 27.0672, 27.1128, 27.2933, 29.3003, 29.6987, 29.7169, 30.0012, 30.3457, 31.9639, 32.7567, 37.0948, 37.1003, 37.8848, 40.2229, 45.5496, 45.5660 and 46.5428; m/z (AP-) 367 (M+ – 13, 100%).
(7S,4RS)-7,11-Dimethyl-4-phenylsulfonyldodec-10-ene 31
Following the procedure outlined for the synthesis of sulfone 24, the iodide (S)-23 (1.9 g, 7.1 mmol) and butyl phenyl sulfone (1.2 g, 5.9 mmol) gave the title compound 31 (1.7 g, 90%) as a 50:50 mixture of 4-epimers (1H NMR), Rf 0.4 (4:1 light petroleum/ether), [α]20D +3.0 (c 1.6 in CHCl3) (found: M+ + Na, 359.2025. C20H32O2NaS requires M, 359.2016); νmax 3063, 1446, 1378, 1303, 1144, 1084, 757, 726, and 691 cm–1; δH (500 MHz, CDCl3) 0.65 and 0.67 (each 1.5 H, d, J 6.7, 7-CH3), 0.75 (3 H, t, J 7.3, 1-H3), 0.90–1.55 (9 H, m, 2-H2, 6-H2, 7-H, 8-H2 and 9-H2), 1.52 and 1.64 (each 3 H, s, 11-CH3 or 12-H3), 1.72–1.95 (4 H, m, 3-H2 and 5-H2), 2.81 (1 H, m, 4-H), 5.01 (1 H, m, 10-H), 7.51 (2 H, t, J 7.8, ArH), 7.60 (1 H, t, J 7.3, ArH) and 7.85 (2 H, d, J 7.5, ArH); δC (100 MHz, CDCl3) 14.0, 17.7, 19.2, 19.4, 20.1, 25.4(2), 25.7, 25.9, 30.0, 32.3, 32.5, 33.8, 33.9, 36.5, 36.9, 64.6, 64.7, 124.6, 128.8, 129.1, 131.3(2), 133.5 and 138.3; m/z (ES+) 359 (M+ + 23, 100%).
(4S,7RS)-4-Methyl-7-phenylsulfonyldecan-1-ol 32
Following the procedure outlined for the synthesis of alcohol 25, the alkene 31 (690 mg, 2.1 mmol) gave the title compound 32 (559 mg, 87%), a colorlesss oil, as a mixture of 7-epimers, Rf 0.4 (4:1 light petroleum/ether), [α]20D +4.7 (c 0.2 in CHCl3) (found: M+ + H, 313.1831. C17H29O3S requires M, 313.1832); νmax 3490, 1725, 1586, 1447, 1380, 1287, 1140, 1084, 1024, 999, 758, 726, and 690 cm–1; δH (500 MHz, CDCl3) 0.76 and 0.78 (each 1.5 H, d, J 6.8, 4-CH3), 0.81 (3 H, t, J 7.6, 10-H3), 1.04–1.61 (11 H, m, 3-H2, 4-H, 5-H2, 6-H2, 8-H2 and 9-H2), 1.72–1.84 (2 H, m, 2-H2), 2.82 (1 H, m, 7-H), 3.56 (2 H, t, J 6.6, 1-H2), 7.49 (2 H, m, ArH), 7.61 (1 H, m, ArH) and 7.82 (2 H, m, ArH); δC (100 MHz, CDCl3) 14.0(2), 19.2, 19.4, 20.1, 25.4, 29.9, 30.1(2), 30.2, 32.4, 32.7(2), 33.8, 33.9, 63.2, 64.6, 128.8, 129.1, 133.5 and 138.3; m/z (ES+) 335 (M+ + 23, 100%).
(7R,11RS,13R,15S,17R,19S)-7,13,15,17,19-Pentamethyl-11-phenylsulfonyldocosane 33
n-Butyllithium (50 mL, 1.6 M in hexane, 0.08 mmol) was added to the (R)-sulfone (R)-29 (20 mg, 0.07 mmol) in THF (0.8 mL) and DMPU (0.8 mL) at −40 °C, and the reaction mixture was stirred for 30 min. The iodide 22 (27 mg, 0.8 mmol) in THF (0.8 mL) was added, and the reaction mixture allowed to warm to room temperature and stirred for 16 h. Saturated aqueous ammonium chloride (5 mL) was added, and the mixture was partitioned between water (3 mL) and ether (8 mL). The organic layer was washed with brine (15 mL), dried (Na2SO4) and concentrated under reduced pressure. Chromatography of the residue eluting with light petroleum/ether (4:1) gave the title compound 33 (11 mg, 34%) as a colorless oil, as a mixture of 11-epimers, Rf 0.6 (4:1 light petroleum/ether) (found: M+ + H, 521.4371. C33H61O2S requires M, 521.4387); νmax 1463, 1378, 1304, 1145, 1086, 727, and 691 cm–1; δH (500 MHz, CDCl3) 0.63–0.83 (21 H, m, 1-H3, 7-CH3, 13-CH3, 15-CH3, 17-CH3, 19-CH3 and 22-H3), 0.87–1.80 (33 H, m, 2-H2, 3-H2, 4-H2, 5-H2, 6-H2, 7-H, 8-H2, 9-H2, 10-H2, 12-H2, 13-H, 14-H2, 15-H, 16-H2, 17-H, 18-H2, 19-H, 20-H2 and 21-H2), 2.91 (1 H, br m, 11-H), 7.49 (2 H, t, J 7.5, ArH), 7.58 (1 H, m, ArH) and 7.82 (2 H, d, J 7.5, ArH); δC (100 MHz, CDCl3) 14.1, 14.4, 19.0, 19.2, 19.6(3), 20.1, 22.7, 24.2, 27.0, 27.2, 27.9, 28.2, 29.7(2), 31.9, 32.5, 36.9(2), 37.0, 40.2, 40.3, 45.6, 45.9, 46.3, 46.7, 62.7, 128.9(2), 129.1, 133.4 and 138.3; m/z (ES+) 543 (M+ + 23, 100%).
(4S,6R,8R,10S,16R)-4,6,8,10,16-Pentamethyldocosane 4
Sodium amalgam (20%; 104 mg, 0.641 mmol) was added to the sulfone 33 (11 mg, 0.021 mmol) in MeOH (2 mL) at room temperature, and the reaction mixture was stirred at room temperature for 6 h. After concentration under reduced pressure, saturated aqueous ammonium chloride (4 mL) was added, and mixture was partitioned between saturated aqueous ammonium chloride (6 mL) and hexane (6 mL). The organic layer was washed with brine (10 mL), dried (Na2SO4) and concentrated under reduced pressure. Chromatography of the residue eluting with hexane (100%) gave the title compound 4 (6 mg, 85%) as a colorless oil, Rf 1.0 (light petroleum), [α]20D −4.8 (c 1.1 in CHCl3); νmax 1464, 1378, 1260, 1094, 1018, 799, and 725 cm–1; δH (500 MHz, CDCl3) 0.72–0.83 (21 H, m, 7 × CH3), 0.89–1.06 (10 H, m), 1.11–1.30 (20 H, m), 1.41 (2 H, m) and 1.49 (3 H, m); δC (100 MHz, CDCl3) 14.1266, 14.3890, 19.5354, 19.5518, 19.5773, 19.6338, 19.7176, 20.0766, 22.6990, 27.0472, 27.0599, 27.1037, 27.2629, 27.2689, 29.6951(2), 29.9757, 30.3311, 31.9603, 32.7439, 37.0884, 37.0966, 37.8702, 40.2101, 45.5296, 45.5533 and 46.5191; m/z (AP-) 367 (M+ – 13, 100%).
Acknowledgments
We thank Profs. Kitching and Breit and their co-workers for their careful accumulation and compilation of 13C NMR data. They could never have anticipated that their data would be subjected to (and stand up to) scrutiny at the low ppb level. The Manchester work was supported by the Engineering and Physical Sciences Research Council (grant number EP/I007989/1), and the Pittsburgh group thanks the NIH NIGMS.
Supporting Information Available
Full details of experiments and compound characterization, copies of spectra on intermediates and final products, and complete lists of published and new data sets with relevant subtractions. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Chow S.; Fletcher M. T.; Lambert L. K.; Gallagher O. P.; Moore C. J.; Cribb B. W.; Allsopp P. G.; Kitching W. J. Org. Chem. 2005, 70, 1808–1827. [DOI] [PubMed] [Google Scholar]; b Fletcher M. T.; Chow S.; Lambert L. K.; Gallagher O. P.; Cribb B. W.; Allsopp P. G.; Moore C. J.; Kitching W. Org. Lett. 2003, 5, 5083–5086. [DOI] [PubMed] [Google Scholar]
- a Herber C.; Breit B. Eur. J. Org. Chem. 2007, 3512–3519. [Google Scholar]; b Herber C.; Breit B. Angew. Chem., Int. Ed. 2005, 44, 5267–5269. [DOI] [PubMed] [Google Scholar]; c Zhou J.; Zhu Y.; Burgess K. Org. Lett. 2007, 9, 1391–1393. [DOI] [PubMed] [Google Scholar]; d Zhu G.; Liang B.; Negishi E.-i. Org. Lett. 2008, 10, 1099–1101. [DOI] [PubMed] [Google Scholar]
- Basar N. B.; Liu H.; Negi D.; Sirat H. M.; Morris G. A.; Thomas E. J. Org. Biomol. Chem. 2012, 10, 1743–1745. [DOI] [PubMed] [Google Scholar]
- Selected examples:; a Fletcher C. J.; Wheelhouse K. M. P.; Aggarwal V. K. Angew. Chem., Int. Ed. 2013, 52, 2503–2506. [DOI] [PubMed] [Google Scholar]; b Li G.; Hsung R. P. Org. Lett. 2009, 11, 4616–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Curran D. P.; Sui B. J. Am. Chem. Soc. 2009, 131, 5411–5413. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Curran D. P.; Zhang Q.; Lu H.; Gudipati V. J. Am. Chem. Soc. 2006, 128, 9943–9956. [DOI] [PubMed] [Google Scholar]; e Mulzer J.; Kaselow U.; Graske K.-D.; Kühne H.; Sieg A.; Martin H. J. Tetrahedron 2004, 60, 9599–9614. [Google Scholar]
- Kobayashi Y.; Czechtizky W.; Kishi Y. Org. Lett. 2003, 5, 93–96. [DOI] [PubMed] [Google Scholar]
- We did not control for concentration of the NMR samples in this work because the samples are dilute hydrocarbons. However, if needed, it is possible to correct for differences in concentration between the natural sample and the synthetic samples just as is described for temperature in the Supporting Information.
- a de Haan J. W.; van de Ven L. J. M.; Wilson A. R. N.; van der Hout-Lodder A. E.; Altona C.; Faber D. H. Org. Magn. Res. 1976, 8, 477–482. [Google Scholar]; b Schneider H. J.; Freitag W. J. Am. Chem. Soc. 1976, 98, 478–481. [Google Scholar]
- Looking at step 3 backwards, the natural product is being used as a standard to compare the spectra of A and B. If the NP is really one or the other or A and B, then the only result can be the values already measured in the direct A/B comparison.
- a Basar N.; Donnelly S.; Sirat H. M.; Thomas E. J. Org. Biomol. Chem. 2013, 11, 8476–8505. [DOI] [PubMed] [Google Scholar]; b Basar N.; Donnelly S.; Liu H.; Thomas E. J. Synlett 2010, 575–578. [Google Scholar]
- a Donnelly S.; Thomas E. J.; Fielding M. Tetrahedron Lett. 2004, 45, 6779–6782. [Google Scholar]; b Wada M.; Ohki H.; Akiba K.-y. Bull. Chem. Soc. Jpn. 1990, 63, 1738–1747. [Google Scholar]; c Miyoshi N.; Nishio M.; Murakami S.; Fukuma T.; Wada M. Bull. Chem. Soc. Jpn. 2000, 73, 689–692. [Google Scholar]; d Wada M.; Honna M.; Kuramoto Y.; Miyoshi N. Bull. Chem. Soc. Jpn. 1997, 70, 2265–2267. [Google Scholar]; e Bhuyan P. J.; Prajapati D.; Sandhu J. S. Tetrahedron Lett. 1993, 34, 7975–7976. [Google Scholar]; f Wada M.; Ohki H.; Akiba K.-y. Tetrahedron Lett. 1986, 27, 4771–4774. [Google Scholar]; g Wada M.; Akiba K.-y. Tetrahedron Lett. 1985, 26, 4211–4212. [Google Scholar]
- Matsushima Y.; Itoh H.; Nakayama T.; Horiuchi S.; Eguchi T.; Kakinuma K. J. Chem. Soc., Perkin Trans. I 2002, 949–958. [Google Scholar]
- Kambe T.; Maruyama T.; Naganawa A.; Asada M.; Seki A.; Maruyama T.; Nakai H.; Toda M. Chem. Pharm. Bull. 2011, 59, 1494–1508. [DOI] [PubMed] [Google Scholar]
- Williams D. R.; Nold A. L.; Mullins R. J. J. Org. Chem. 2004, 69, 5374–5382. [DOI] [PubMed] [Google Scholar]
- a Evans D. A.; Morrissey M. M.; Dow R. L. Tetrahedron Lett. 1985, 26, 6005–6008. [Google Scholar]; b Evans D. A.; Morrissey M. M. J. Am. Chem. Soc. 1984, 106, 3866–3868. [Google Scholar]; c Brown J. M. Angew. Chem., Int. Ed. 1987, 26, 190–203. [Google Scholar]
- a Lipshutz B. H.; Kozlowski J. A.; Breneman C. M. J. Am. Chem. Soc. 1985, 107, 3197–3204. [Google Scholar]; b Lipshutz B. H.; Wilhelm R. S.; Kozlowski J. A. Tetrahedron 1984, 40, 5005–5038. [Google Scholar]
- a Mori K.; Masuda S.; Suguro T. Tetrahedron 1981, 37, 1329–1340. [Google Scholar]; b Chow S. Y.; Williams H. J.; Huang Q.; Nanda S.; Scott I. A. J. Org. Chem. 2005, 70, 9997–1003. [DOI] [PubMed] [Google Scholar]
- Raederstorff D.; Shu A. Y. L.; Thompson J. E.; Djerassi C. J. Org. Chem. 1987, 52, 2337–2346. [Google Scholar]
- For the racemic iodide 28 see:Hedenström E.; Edlund H.; Lund S.; Abersten M.; Persson D. J. Chem. Soc., Perkin Trans. 1 2002, 1810–1817. [Google Scholar]
- Adding the absolute values of the two “reliably different” numbers gives the same number.
- Denmark S. E.; Regens C. S.; Kobayashi T. J. Am. Chem. Soc. 2007, 129, 2774–2776. [DOI] [PubMed] [Google Scholar]
- Santangelo E. M.; Corrêa A. G.; Zarbin P. H. G. Tetrahedron Lett. 2006, 47, 5135–5137. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.