

Abstract
Tortuous carotid arteries are often seen in aged populations and are associated with atherosclerosis, but the underlying mechanisms to explain this preference are unclear. Artery buckling has been suggested as one potential mechanism for the development of tortuous arteries. The objective of this study, accordingly, was to determine the effect of buckling on cell proliferation and associated NF-κB activation in arteries. We developed a technique to generate buckling in porcine carotid arteries using long artery segments in organ culture without changing the pressure, flow rate, and axial stretch ratio. Using this technique, we examined the effect of buckling on arterial wall remodeling in 4-day organ culture under normal and hypertensive pressures. Cell proliferation, NF-κB p65, IκB-α, ERK1/2, and caspase-3 were detected using immunohistochemistry staining and immunoblot analysis. Our results showed that cell proliferation was elevated 5.8-fold in the buckling group under hypertensive pressure (n = 7, P < 0.01) with higher levels of NF-κB nuclear translocation and IκB-α degradation (P < 0.05 for both). Greater numbers of proliferating cells were observed on the inner curve side of the buckled arteries compared with the outer curve side (P < 0.01). NF-κB colocalized with proliferative nuclei. Computational simulations using a fluid-structure interaction model showed reduced wall stress on the inner side of buckled arteries and elevated wall stress on the outer side. We conclude that arterial buckling promotes site-specific wall remodeling with increased cell proliferation and NF-κB activation. These findings shed light on the biomechanical and molecular mechanisms of the pathogenesis of atherosclerosis in tortuous arteries.
Keywords: tortuous arteries, vascular remodeling, porcine carotid artery, mechanical stress, ex vivo organ culture, nuclear factor-κB
large- and medium-sized arteries often become tortuous in the aged population (8, 20, 40). Specifically, tortuous carotid arteries are observed in 4–45% of angiograms, with a greater prevalence in the aged population than in the young population (8, 56). Tortuous carotid arteries exhibit a higher incidence of atherosclerosis compared with nontortuous arteries (8, 20, 46), but the underlying mechanisms are poorly understood. Aging is known to promote hypertension, reduce axial tension, and increase the risk for atherosclerosis and other cardiovascular diseases (15, 31, 40, 44, 58). Our recent studies (17–19) have shown that arteries lose stability and buckle into a tortuous shape ex vivo due to hypertensive pressure, reduced axial tension, or weakened arterial wall structure. Artery buckling has been suggested as a possible mechanism leading to tortuosity (18, 20, 21). Buckling-induced curving alters the distributions of fluid shear stress and wall stress in arteries (20, 28), which could stimulate arterial wall remodeling (11, 14, 28). Therefore, it is important to determine how arteries remodel after buckling to understand the mechanism of tortuous artery development.
In addition, it is of clinical interest to determine whether artery buckling stimulates atherosclerosis, since 80% atherosclerosis in carotid and coronary arteries occurs at curving and branching areas where shear stress is lower and wall stresses are elevated (29, 39). Cell proliferation and intima hyperplasia are key initiating events in atherosclerosis development (27, 29). Previous studies from many groups, including our own, have demonstrated that altered axial stretch, shear stress, and pulse pressure stimulate cell proliferation in porcine carotid arteries (22, 24, 28, 32, 36, 59). Therefore, it is of clinical interest to investigate whether increased cell proliferation occurs in buckled arteries.
NF-κB is a key nuclear transcription factor that regulates a variety of inflammatory and proliferative gene responses in cells that contribute to arterial remodeling and atherosclerosis (2, 37, 43). NF-κB activation in endothelial cells may play an important role in the pathogenesis of atherosclerosis, especially during the early stages of disease progression (16). In addition, disturbed blood flow and acute alteration of shear stress promote arterial inflammation and wall remodeling through the induction of NF-κB expression in endothelial cells (5, 6).
Therefore, the objective of this study was to investigate cell proliferation and the associated NF-κB activation in buckled carotid arteries. We examined buckling-induced site-specific wall remodeling in curved porcine carotid arteries using a well-established organ culture model (22, 36).
MATERIALS AND METHODS
Ex vivo arterial organ culture.
Porcine common carotid arteries were obtained from farm pigs (∼100 kg) after death at a local abattoir with approval from the Texas Department of State Health Service and the University of Texas at San Antonio Institutional Biosafety Committee. Arteries were transported to our laboratory in ice-cold PBS and set up in an ex vivo perfusion organ culture system as previously described in detail (36, 59). Briefly, arteries were cleaned and mounted at their in situ length to diameter-matched stainless steel cannulae in vessel chambers filled with bath medium. Medium perfusion to the arteries was driven by a peristaltic roller pump at a controlled flow rate (160 ml/min) and pulse frequency (2.5 Hz). Perfusion and bath media consisted of cell culture medium DMEM (Sigma) supplemented with sodium bicarbonate (3.7 g/l, Sigma), l-glutamine (2 mM, Sigma), calf serum (10%, Sigma), HEPES (25 mM, GIBCO), and antibiotic-antimycotic solution (1%, Sigma). Dextran (50 g/l, Sigma) was added to the perfusion medium to adjust its viscosity close to that of human blood (4 cP). Bromodeoxyuridine (BrdU; 5 μg/ml, Roche) was added to the perfusion medium at the first day of the experiment to label the nuclei of newly proliferated cells. Flow loops were then placed in 5% CO2 incubators at 37°C.
To facilitate buckling, long arterial segments (40–60 mm) were used in the experimental groups, whereas short arterial segments (15–25 mm) were used to avoid buckling in control groups. This design was based on our previous theoretical analysis, which showed that longer segments have lower critical buckling pressure than shorter segments (17, 18). With the use of a long segment, the critical pressure of the experimental group was reduced to a level below the perfusion pressure while using a short segment increased the critical pressure of the control arteries to a level above the perfusion pressure. Consequently, when the perfusion pressure was gradually increased to set values of 100 and 200 mmHg, the long segments buckled, whereas the short segments remained straight. This approach allowed us to achieve buckling without altering the flow rate and pressure and thus to distinguish the effects of buckling from the effects of pressure or flow rate.
All arteries were maintained for up to 4 days under a physiological flow rate of 160 ml/min and a subphysiological axial stretch ratio of 1.3 to facilitate artery buckling (17, 18). The subphysiological axial stretch ratio was used to mimic the loosening of axial tension in tortuous arteries in vivo. Both buckling and control groups (without buckling) were cultured under normotensive pressure (120 ± 20 mmHg).
To further investigate the combined effects of artery buckling and hypertension, another group of arteries was cultured under hypertensive pressure (230 ± 30 mmHg). A separate control group was used for the hypertensive conditions.
The extent of artery buckling was described using the tortuosity index. During organ culture, pictures were taken with a camera and analyzed using Image-Pro Plus software. The distance between the sutures on the two ends and deflection were measured from these images (Fig. 1). The tortuosity index of the vessel was determined as follows:
| (1) |
Fig. 1.
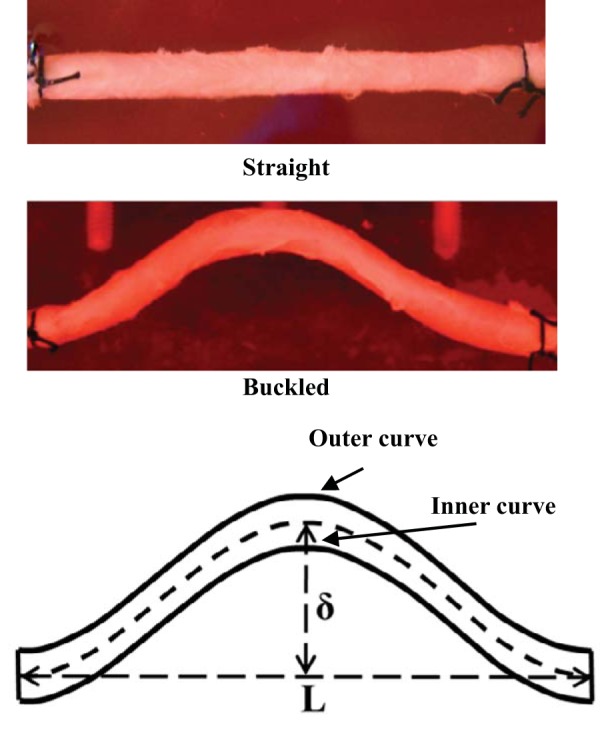
Top: photographs of a straight (control) artery and a buckled artery, both maintained in an ex vivo organ culture system for 4 days. Bottom: schematic of a buckled artery illustrating the buckling deflection (δ), end distance (L), and inner curve and outer curve sides.
where TI is the tortuosity index, δ is deflection, and L is the distance between the sutures on the two ends.
Immunohistochemistry staining.
Immunohistochemistry staining was conducted as previously described (36, 57). Arterial specimens were flushed with sterile ice-cold PBS and pressure fixed in 2% paraformaldehyde in PBS. A partial lengthwise incision was made to mark the outer curve. Fixed arteries were then transferred in 30% sucrose in PBS until processed for embedding in OCT compound.
For immunofluorescent staining, 5-μm frozen cross-sections were treated with 0.01 M citrate buffer (pH 6.0), permeabilized with 0.5% Triton X-100 in PBS, blocked with 10% FBS in PBS, and processed for labeling with the following primary antibodies: mouse anti-BrdU (catalog no. 11296736001, Roche), rabbit anti-NF-κB (catalog no. 372, Santa Cruz Biotechnology), rabbit anti-CD31 (catalog no. 28364, Abcam), and rabbit anti-α-smooth muscle actin (catalog no. 5694, Abcam). For costaining with multiple primary antibodies, the primary antibodies species were matched so that the corresponding FITC-conjugated (green) or Cy3-conjugated (red) secondary antibodies (Molecular Probes) could be used to distinguish the different primary antibodies. 4′,6-Diamidino-2-phenylindole dihydrochloride was used to counterstain nuclei.
Images (×20 and ×40 magnification) were acquired with a Qcolor3 camera under an Olympus CX41 fluorescence microscope. They were merged as multicolor images and analyzed to determine the circumferential distribution of BrdU-positive cells using Image-Pro Plus 7.0. Cell proliferation rates and NF-κB nuclear translocation rates were quantified by counting positively stained nuclei in six to nine views (3 views each for 2–3 cross-sections) and expressed as percentages of total counterstained nuclei.
Apoptosis was determined using caspase-3 immunoblotting and terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL). For TUNEL staining, frozen sections were dried and fixed as described above. After being permeabilized with 0.2% Triton X-100, sections were stained with TUNEL kit reagents (Roche Applied Science). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole dihydrochloride. Quantification of the percentage of TUNEL-positive cells was performed using a cell count under fluorescent microscopy, with images taken at ×10 magnification.
Endothelial protein extraction and immunoblot analysis for NF-κB p65, IκB-α, ERK1/2, and caspase-3 measurements.
From each artery, two tissue samples were harvested for fresh and 4-day cultured arteries. One sample was used for whole cell lysates of the arterial wall in Laemmli sample buffer (catalog no. 161-0737, Bio-Rad) to detect ERK1/2 and caspase-3 by immunoblot analysis. The other sample was used to culture endothelial cells, which were gently scraped from the arterial wall. Harvested cells were divided into two aliquots, one aliquot in Laemmli sample buffer for whole cell lysates to detect total IκB-α using Western blot analysis and another aliquot in cold PBS for nuclear extraction of NF-κB p65. The extraction was done by isolating nuclear fractions using NE-PER nuclear and cytoplasmic extraction reagents (catalog no. 78833, Thermo Fisher Scientific) as described below.
Briefly, endothelial cells collected in cold PBS were resuspended, homogenized in cytoplasmic extraction reagent (CER) I, and incubated on ice for 10 min. CER II was then added, and extracts were further incubated on ice for 1 min. Extracts were then centrifuged for 5 min, and the supernatant (cytoplasmic fraction) was transferred to a new tube. Nuclear extraction reagent (NER) was added to the nuclear pellet, and the nuclear pellet was incubated on ice for 40 min, with vortexing every 10 min. The extract was centrifuged for 10 min, and the supernatant (nuclear fraction) was transferred to a new tube. In addition, protease inhibitor cocktail (Roche) was added to the nuclear extraction fraction and whole cell lysates to prevent protein degradation. Isolated nuclear extraction fractions and whole cell lysates were stored at −80°C until immunoblot analysis.
Immunoblot analysis was conducted as previously described (57). Briefly, genomic DNA in whole cell lysates was sheered via sonication on ice three times (5 s each). Nuclear extractions and whole cell lysates were loaded onto SDS polyacrylamide gels for electrophoresis separation. The separated protein bands were blotted onto Immun-Blot polyvinylidene difluoride membranes (Bio-Rad) for detection with the following primary antibodies: rabbit anti-NF-κB p65 (catalog no. 372, Santa Cruz Biotechnology), rabbit anti-total IκB-α (catalog no. 371, Santa Cruz Biotechnology), rabbit anti-total ERK1/2 (catalog no. 4695, Cell Signaling), rabbit anti-phospho-ERK1/2 (catalog no. 4370, Cell Signaling), rabbit anti- caspase-3 (catalog no. 9665, Cell Signaling), mouse anti-β-actin (catalog no. 47778, Santa Cruz Biotechnology), and mouse anti-GAPDH (catalog no. CB1001, Millipore). Primary antibody bindings were detected with the corresponding secondary antibodies conjugated with horseradish peroxidase (Amersham GE Healthcare) and visualized using standard ECL detection (Amersham GE Healthcare). Immunoblot results were quantified using densitometry performed with ImageJ (National Institutes of Health), with values normalized to the densitometry of β-actin and GAPDH.
Arterial stress analysis using the fluid-solid interaction model.
To illustrate shear stress and wall stress in the buckled arteries, we performed computational simulations using a fluid-solid interaction (FSI) model of arteries with the mean dimensions and loads of each group shown in Table 1 using ADINA software (version 8.9, ADINA R&D). To eliminate the effect of dimensional differences between buckled and control groups, an additional simulation was done for a vessel identical to the buckled artery of the hypertensive group but half of its length to avoid buckling (“hyper-half length”).
Table 1.
Arterial segment unloaded dimensions and organ culture conditions for the experimental groups
| Normotensive |
Hypertensive |
|||
|---|---|---|---|---|
| Straight arteries | Buckled arteries | Straight arteries | Buckled arteries | |
| Sample size | 5 | 5 | 7 | 7 |
| Unloaded length, mm | 19.6 ± 2.4 | 59.8 ± 4.8 | 21.4 ± 1.8 | 39.0 ± 4.0 |
| Outer diameter, mm | 4.2 ± 0.2 | 4.6 ± 0.3 | 6.9 ± 0.3 | 6.4 ± 0.3 |
| Inner diameter, mm | 1.8 ± 0.1 | 2.0 ± 0.2 | 3.9 ± 0.2 | 3.4 ± 0.2 |
| Thickness, mm | 1.2 ± 0.2 | 1.3 ± 0.1 | 1.5 ± 0.2 | 1.5 ± 0.2 |
| Tortuosity index | 0.05 ± 0.01 | 0.12 ± 0.01* | 0.06 ± 0.01 | 0.23 ± 0.02† |
| Lumen pressure, mmHg | 120 ± 20 | 120 ± 20 | 230 ± 30 | 230 ± 30 |
| Flow rate (ml/min) | 160 | 160 | 160 | 160 |
| Axial stretch ratio | 1.3 | 1.3 | 1.3 | 1.3 |
P < 0.01 vs. normotensive straight arteries;
P < 0.01 vs. hypertensive straight arteries.
Arterial walls were modeled as a nonlinear anisotropic thick-walled model using the following Holzapfel strain-energy function (1, 25):
| (2) |
where W is the Holzapfel strain-energy function; μn and αn are the Ogden material constants; λ1, λ2, and λ3 are the principal stretch ratios in the radial, circumferential, and axial directions; k1 and k2 are the anisotropic constants; and J4 and J6 are the square of stretch ratios along the two diagonal fiber directions (J4 = J6 = λ22 sin2 β + λ32 cos2 β, where β is the angle between the fiber and the axial direction of the artery). These material constants were determined by fitting the experimental data of pressurized inflation test of a porcine carotid artery (artery 4) from a previous study (38). Values yielded from the fitting used in all simulations were α1 = 1.5, α2 = 7, α3 = 0.5, μ1 = 3,200, μ2 = 7,800, μ3 = 5,100, k1 = 15.09 kPa, k2 = 1.06, and β = 58.7°. The wall thickness was also assumed to be the same among groups. Buckling was induced by adding an imperfection of the tiny initial curvature and gradually increasing the lumen pressure as previously described (7, 60).
A steady fluid flow rate of 160 ml/min at the inlet, a pressure of 120 mmHg (normotensive) or 230 mmHg (hypertensive) at the outlet, and an initial axial stretch ratio of 1.3 were used in the simulations. The fluid was modeled as a Newtonian fluid with a specific gravity of 1.01 and viscosity of 4 cP.
Statistical analyses.
All values are presented as means ± SE. Statistical significance between means was determined using Student's t-test for two group comparisons or two-way ANOVA for multiple group comparison. The significance level was set at P < 0.05.
RESULTS
Arterial buckling was achieved under both hypertensive and normotensive pressure conditions.
Using long artery segments for the buckling group and a short segment for the control group, we were able to achieve buckling in the buckling group and avoid buckling in the control group while maintaining the same pressure, flow, and axial stretch ratio in both groups (Fig. 1). Vessel dimensions, hemodynamic conditions, and tortuosity indexes are shown in Table 1 for all groups. The tortuosity index of buckled arteries was significantly higher under hypertensive pressure (0.23 ± 0.02) compared with the normotensive pressure group (0.12 ± 0.01, P < 0.01).
Cell proliferation was increased by buckling under hypertensive pressures.
In normotensive groups, anti-BrdU staining showed similar numbers of positive cells as straight controls, indicating that buckling in the absence of elevated pressure did not stimulate proliferation (Fig. 2A). In hypertensive pressure groups, the percentage of BrdU-positive cells was significantly higher in buckled arteries (9.8 ± 1.5%) compared with straight control arteries (1.7 ± 0.3%, P < 0.01; Fig. 2B). BrdU-positive cells were present mainly in endothelial cells in the intima and smooth muscle cells in the media of the arterial wall, as confirmed by anti-CD31 staining and anti-α-actin staining (Fig. 3). Percentages of BrdU-positive cells in both intimal and medial layers were significantly higher in buckled arteries compared with the corresponding layers in straight arteries (Fig. 2C). Meanwhile, TUNEL staining and caspase-3 immunoblot results demonstrated that there were no differences in apoptotic cell death between buckled arteries and control arteries (Fig. 4). These results suggest that arterial buckling under hypertensive pressure significantly promoted cell proliferation. Furthermore, the percentage of BrdU-positive nuclei in hypertensive controls (straight arteries) was similar to that of normotensive controls (see Fig. 2), suggesting that increased pressure alone did not cause an increase in cell proliferation in the arteries.
Fig. 2.
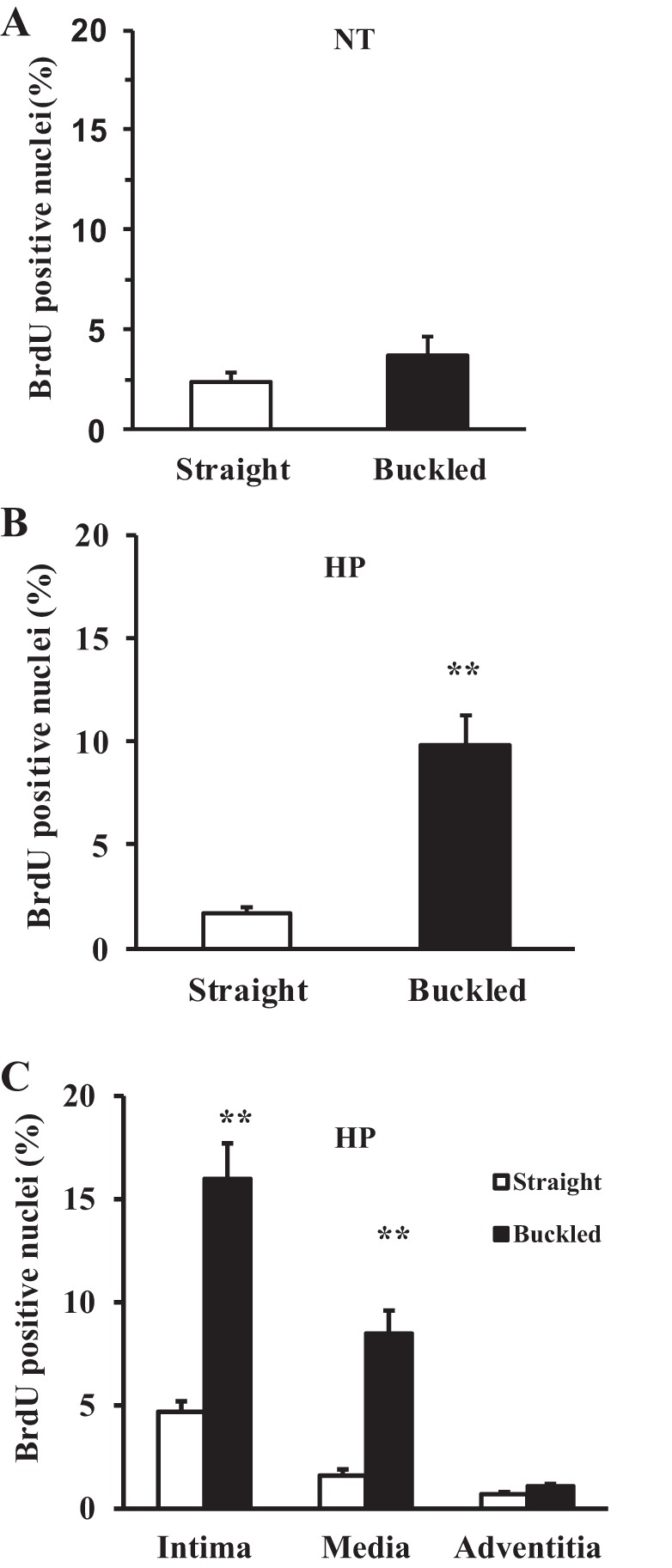
Effect of artery buckling on cell proliferation. A: comparison of the percentage of bromodeoxyuridine (BrdU)-positive cells (mean ± SE, n = 5) in straight and buckled arteries after being cultured under normotensive pressure (NT) for 4 days. B and C: comparisons of the percentage of BrdU-positive cells (mean ± SE, n = 7) in straight and buckled arteries cultured under hypertensive pressure (HP) and the corresponding BrdU-positive cells in intimal, medial, and adventitial layers, respectively. **P < 0.01.
Fig. 3.

Cross-sectional images (×40 magnification, lumens on the right) illustrating 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) nuclei counterstaining (blue) and BrdU-labeled nuclei (green fluorescence) in conjunction with CD31 staining (red) for endothelial cells (ECs) and anti-α-actin staining (red) for smooth muscle cells in straight and buckled arteries after being cultured under hypertensive pressure for 4 days.
Fig. 4.
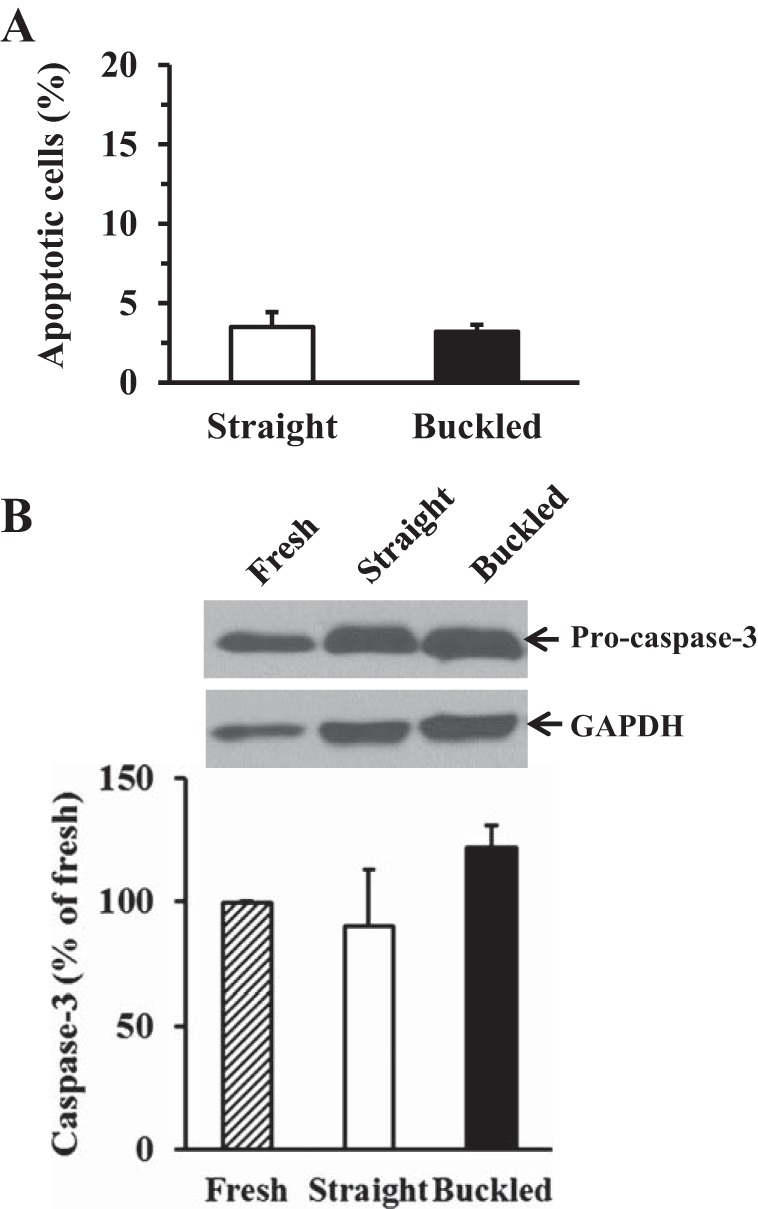
Effect of artery buckling on cell apoptosis in hypertensive arteries. A: comparison of TUNEL-positive cells (in %) between straight and buckled arteries cultured under hypertensive pressure. B: representative immunoblot images of caspase-3 and comparison of caspase-3 normalized to GAPDH among fresh, straight, and buckled arteries. Note that no significant differences in cell apoptosis were seen.
Cell proliferation was significantly higher on the inner curve side of hypertensive buckled arteries compared with the outer curve side.
BrdU-positive cells also demonstrated a nonuniform variation along the circumference in hypertensive buckled arteries (Fig. 5A). Further measurements showed that the percentage of BrdU-positive cells was more than threefold higher on the inner curve side (16.1 ± 2.5%) than on the outer curve side (4.9 ± 0.9%) of buckled arteries under hypertensive pressure (P < 0.01; Fig. 5, B and C). Both inner and outer curve sides demonstrated more cell proliferation than straight controls.
Fig. 5.
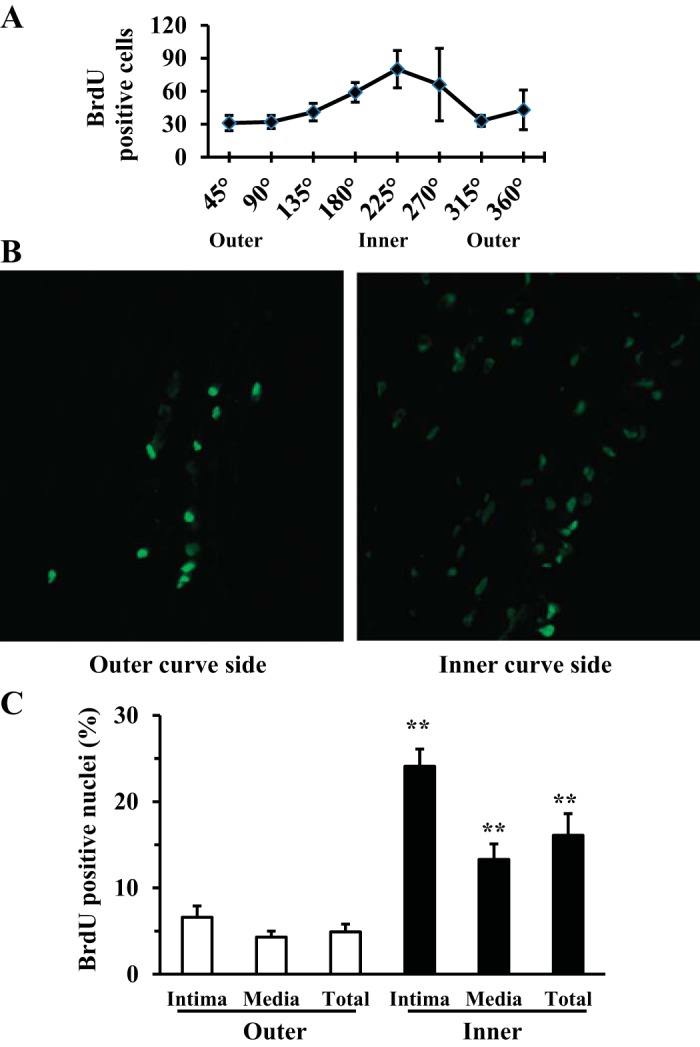
Artery buckling-induced cell proliferation was site specific in hypertensive arteries. A: the circumferential distribution of BrdU-positive cells was determined using Image-Pro Plus 7.0. B: representative fluorescent microscope images captured at the inner and outer curve sides of a buckled artery (×20 magnification; lumens on the right). C: comparison of the percentage of BrdU-positive cells of inner and outer curve sides and in intimal and medial layers. **P < 0.01 vs. the outer curve side.
NF-κB is significantly activated in hypertensive buckled arteries.
Immunohistochemical staining demonstrated that NF-κB p65 nuclear translocation occurred in both intimal and medial regions of the artery (Fig. 6). This translocation was increased significantly in endothelial cells of the intimal layer (7.0 ± 1.4% vs 2.1 ± 0.8%, P < 0.05; Fig. 6A) in buckled arteries compared with straight arteries but not in smooth muscle cells of the medial layer. Immunoblot results confirmed increased NF-κB p65 nuclear translocation in buckled arteries (Fig. 6, B and D).
Fig. 6.
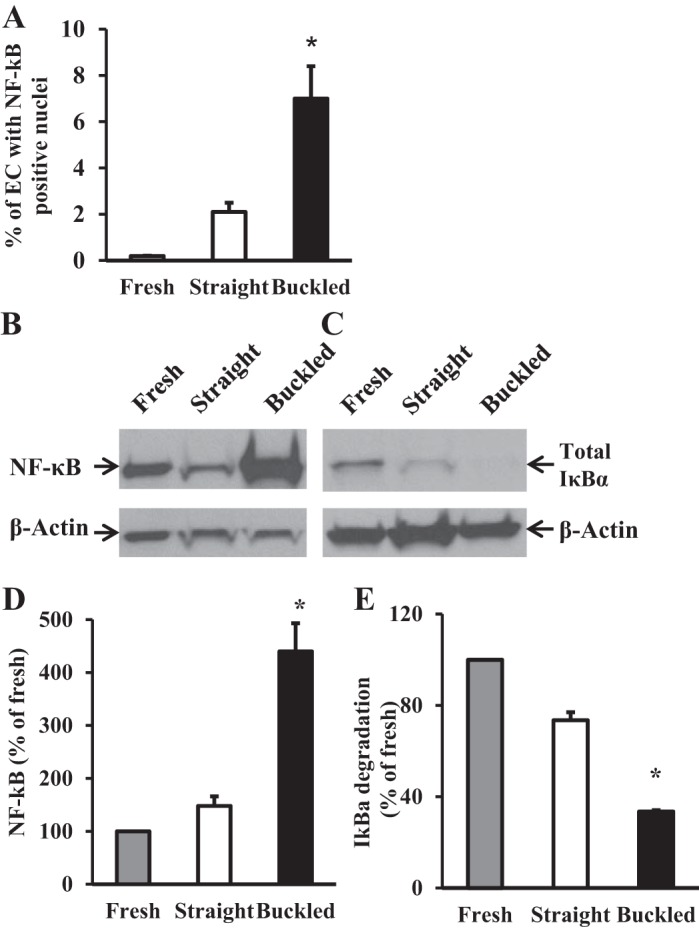
Artery buckling induced significant NF-κB activation in hypertensive arteries. A: comparison of the percentage of NF-κB p65-positive EC nuclei in fresh, straight, and buckled arteries. B and C: immunoblot images of NF-κB p65 in the EC nuclear fraction and total IκB-α protein in the EC whole cell lysate. D and E: densitometric measurement showing the significantly higher level of NF-κB p65 activation and IκB-α degradation in buckled arteries compared with both fresh and control arteries. *P < 0.05.
An additional independent indicator of NF-κB activation, IκB-α degradation, was also significantly higher in buckled arteries compared with either fresh or straight arteries (Fig. 6, C and E). Of note, NF-κB colocalized with BrdU-positive nuclei in buckled arteries (Fig. 7). Meanwhile, immunoblot analysis demonstrated no differences in ERK1/2 activation as measured by the ratio of phospho-ERK1/2 to total ERK1/2 among fresh, straight, and buckled groups (Fig. 8). Together, these results demonstrated that hypertensive arterial buckling resulted significant NF-κB activation in the intima but not ERK1/2 activation.
Fig. 7.
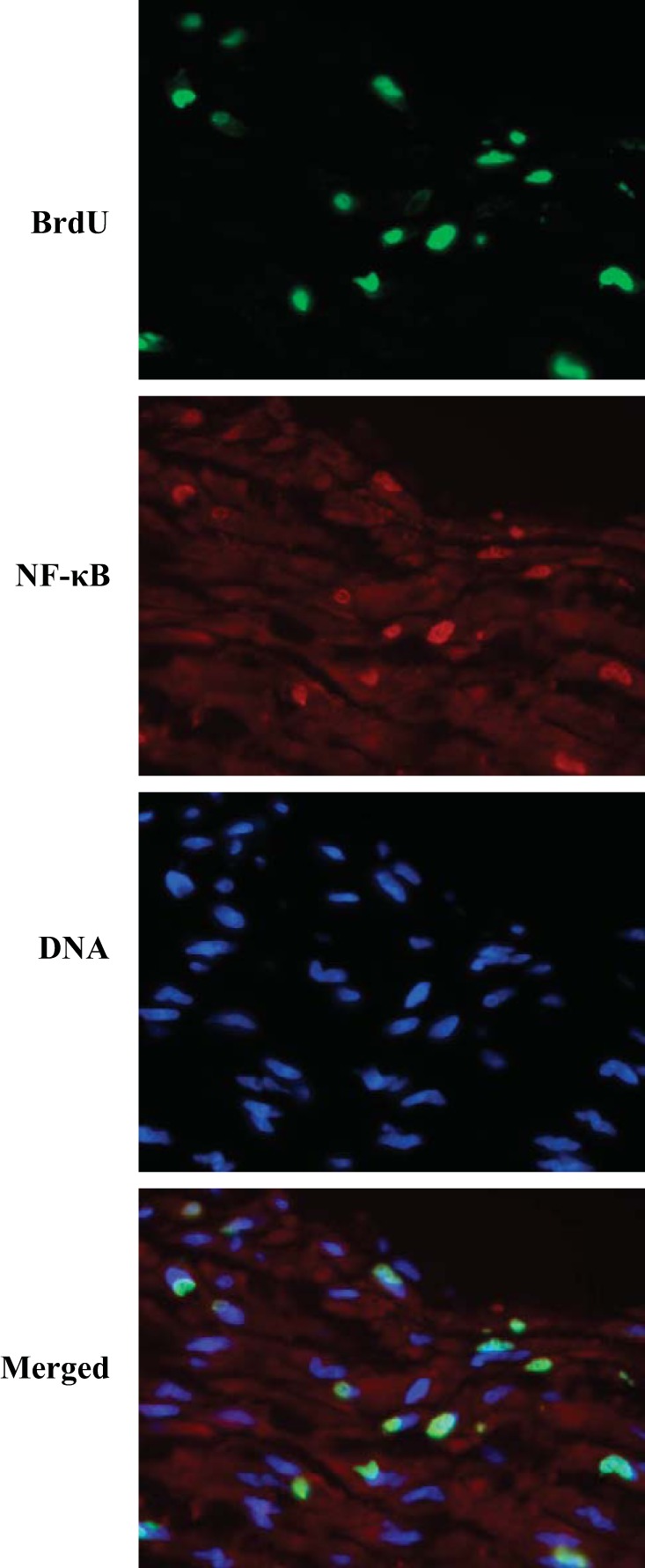
Fluorescent images of BrdU-labeled nuclei (green), NF-κB p65 activation (red), and counterstained nuclei (DNA, blue) as well as merged images. The colocalization (yellow) of NF-κB p65 and BrdU in the merged image can be seen (×40 magnification).
Fig. 8.
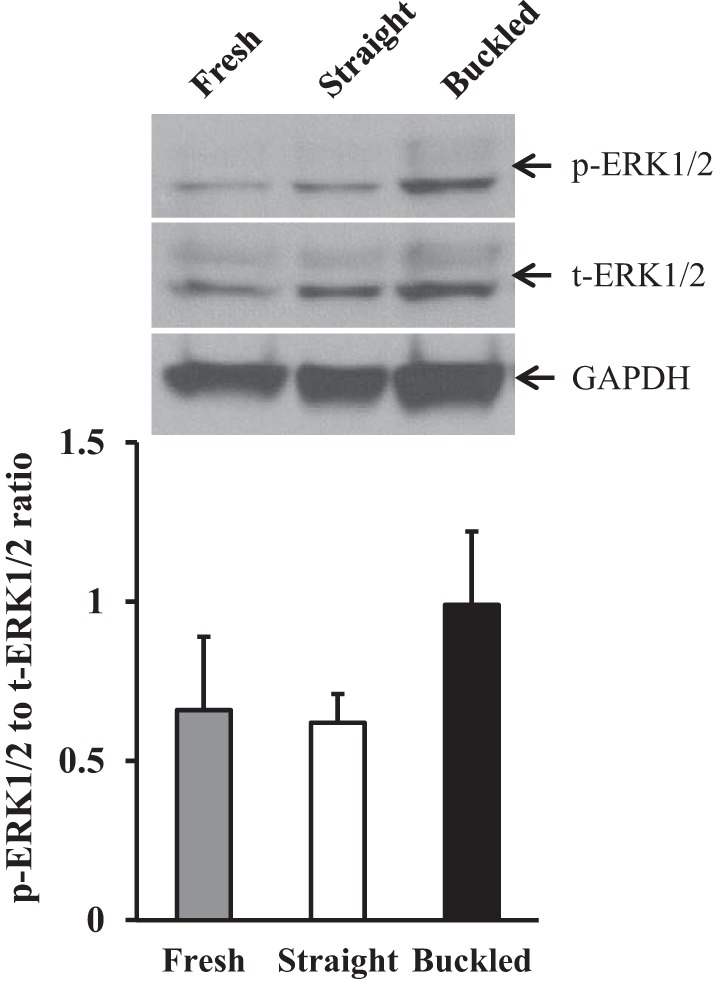
Artery buckling did not induce significant ERK1/2 activation in hypertensive arteries. A: immunoblot images of phosphorylated (p-)ERK1/2, total (t-)ERK1/2, and GAPDH. B: comparison of the ratio of p-ERK1/2 to t-ERK1/2 among the three groups (fresh, straight, and buckled arteries after being cultured for 4 days).
Wall stress and lumen shear stress in buckled arteries.
FSI simulations demonstrated that lumen shear stress, circumferential stress, and longitudinal stress became nonaxisymmetric in buckled arteries (Fig. 9), whereas straight (control) arterial segments were subjected to symmetric axial and circumferential tensile stresses and lumen shear stress. Buckling deflection created lower circumferential and longitudinal stresses on the inner curve side of the artery than on the outer curve side, especially in hypertensive arteries (Fig. 9).
Fig. 9.

Lumen shear stress and wall stress in arteries determined by fluid-solid interaction simulations. Left: velocity profile and lumen surface shear stress contours in buckled arteries under normotensive pressure and hypertensive pressure. Right: comparisons of circumferential stress and longitudinal stress in the arterial wall at the middle cross-section among the four groups. Positions 1–4 denote four equally distributed circumferential locations along the lumen surface with 1 at the outer curve and 3 at the inner curve sides of the vessel. “Hyper-half length” represents a vessel identical to the buckled vessel but half of its length to avoid buckling. See text for more details.
DISCUSSION
The present study evaluated artery buckling-induced cell proliferation and NF-κB activation using an ex vivo organ culture model. The key findings of this study were that 1) artery buckling stimulated increased cell proliferation, 2) the increase in proliferation was predominantly induced in the inner curve side under hypertensive lumen pressure, and 3) this increase in cell proliferation was associated with NF-κB activation in the arterial wall as demonstrated by increased IκB-α degradation and NF-κB p65 nuclear translocation that colocalized with BrdU-positive nuclei.
Model novelty.
Arteries could buckle and become tortuous due to hypertensive pressure and/or reduced axial strain (17, 18). Our previous model demonstrated that long arterial segments have lower critical buckling pressure (17, 18). Hence, we used long artery segments to facilitate buckling in this study. The length of control arteries (straight cylinders) was shorter, but the length does not affect the flow and wall stresses as well as biological activities in the vessels. This is because mechanical analyses showed that lumen shear stress and wall stress in cylindrical vessels are independent of vessel length (11, 28). The unique feature of this model, as we demonstrated for the first time, is that it can generate buckling in arteries without altering the pressure, flow rate, and axial stretch ratio. Thus, the observed changes were attributed specifically to the buckling. This ex vivo artery buckling model is complementary with the in vivo artery buckling model (60) and provides a valuable tool to further investigate the biomechanical and molecular mechanisms in artery tortuosity-associated atherosclerosis.
Mechanobiological mechanisms.
In the present study, our FSI simulations demonstrated that artery buckling altered circumferential stress and longitudinal stress in the arterial wall as well as lumen shear stress. Local alterations of lumen shear stress and wall stress modulate vascular cell biology and wall remodeling, leading to localized atherosclerosis in both humans and animal models (3, 45, 50). Our results showed that cell proliferation is elevated on the inner curve side of hypertensive buckled arteries, where circumferential and longitudinal stresses are lower compared with the outer curve side. This finding is consistent with previous studies (13, 26) that have demonstrated low shear stress and low longitudinal stress stimulate vascular remodeling with increased cell proliferation in animal and organ culture models. Therefore, the significant differences in cell proliferation between buckled and straight arteries and between inner and outer curved sides are likely due to a stress difference resulted from artery buckling.
Our results showed that cell proliferation in buckled arteries cultured under normotensive pressure was higher than in straight arteries, but the difference was not statistically significant. A possible explanation is that the bending deformation in buckled arteries was small under normotensive pressure and thus the stress difference between inner and outer curve sides was much less than in hypertensive arteries. In addition, the results demonstrated low cell proliferation in both normotensive and hypertensive controls with no statistical difference between them. The low cell proliferation in normotensive and hypertensive controls is consistent with our previous measurements (23), although previous measurements demonstrated some difference between the two. Our present results are also consistent with a previous report (33) showing that hypertensive pressure per se did not stimulate cell proliferation. Further statistical analysis demonstrated that there was an interaction between buckling and hypertensive pressure that causes a synergetic effect on cell proliferation.
Changes in mechanical stresses are sensed by vascular cells (endothelial cells and smooth muscle cells) through several different signaling pathways, including the NF-κB pathway (37, 43). The signal transduction involves proteins related to inflammation, proliferation, and oxidative stress, including ROS (55). Although ROS were not determined in the present study, a previous study (55) has demonstrated that NF-κB is an oxidant mechanosensitive transcription factor regulated by flow shear stress and that production and accumulation of ROS with disturbed shear stress associates with activation of NF-κB and increased DNA synthesis. Therefore, one possible mechanism for uneven remodeling in the buckled artery may be linked to ROS-mediated activation of NF-κB. Potential specific molecules targeting NF-κB may be a strategy in the prevention and treatment of tortuosity-associated atherosclerosis.
Relevance to atherosclerosis.
Previous studies (8, 47, 54) have demonstrated a strong association between arterial tortuosity and increased atherosclerotic lesions. It has been shown that curved regions of coronary arteries have more lesions than straight regions (54) and that inner sides of curved segments of femoral arteries were more atherosclerotic than outer sides, whereas both were more atherosclerotic than straight segments (47). Consistent with the literature, our present results demonstrated that arterial buckling increased cell proliferation that was significantly higher at the inner curve side, suggesting that buckling led to site-specific remodeling with elevated cell proliferation that might be associated with atherosclerosis. Previously, Van Epps and colleagues (53) have reported that arterial segments exposed to cyclic flexure significantly increased cell apoptosis and macromolecular permeability in a similar ex vivo organ culture system. However, they did not observe an increase in cell proliferation. The difference may be due to differences in the arteries (femoral vs. carotid), loading conditions (flexure under cyclic axial stretch vs. buckling under pulsatile pressure at fixed axial stretch), and duration of the experiments (12 h vs. 4 days).
In addition, Stein et al. (48) demonstrated that cyclic flexion increased lumen wall stress of the inner curve, which contributed to the progression of atherosclerotic plaques in human coronary arteries. Using magnetic resonance images and FSI model analysis, Tang et al. (51) recently demonstrated cyclic bending significantly increased maximum stress in atherosclerotic plaques. Our present results showed a similar pattern of stress change with increased cell proliferation. Taken together, the data suggest that buckling-induced stress could be a mechanism for the high prevalence of atherosclerosis in tortuous arteries, but further studies are needed.
While alterations in wall stress and shear stress in buckled arteries could be a possible mechanism leading to atherosclerosis, the development of atherosclerosis could potentiate wall stress and shear stress effects, leading to an increased level of buckling. It is generally believed that long-standing hypertension and atherosclerosis can cause elongation of arteries that reduces axial stretch, resulting in buckling of the vessels (40, 42, 49, 56). Clinical observations showed that atherosclerotic arteries tend to be more tortuous than other arteries and that tortuosity influences the development of atherosclerosis (8, 46). The two-way interaction between atherosclerosis and buckling stresses may be a possible biomechanical mechanism for the progression of atherosclerosis in the tortuous artery.
Relevance to aging.
Clinical studies (8, 42) have shown that carotid artery buckling and tortuosity are highly associated with hypertension, atherosclerosis, and aging. Individuals aged 50 yr or older have a higher prevalence of artery carotid artery buckling or kinking, and women have a greater incidence of artery buckling than men (8, 49, 56). It is well known that aging affects arterial structure and function (31, 40). Arterial wall stiffness increases with aging due to increases in collagen content and cross-linking, a high collagen-to-elastin ratio, increased medial wall thickness, and vascular calcification (9, 10, 12, 30, 31). In addition, it has been reported that sex affects the mechanical properties, biochemical contents, and wall structure of arteries (41). Our recent theoretical analysis showed that structure and mechanical stiffness of the arterial wall directly elevate the critical buckling pressure, and thus the aged-related changes and sex differences in vascular structure and mechanical properties could be a possible explanation for age-related artery buckling and its sex differences.
In addition, axial tension in arteries reduces with the loss of elastin in aged animals and humans (34, 40). Both reduced axial tension and elastin degradation with aging decrease arterial wall stiffness and can trigger artery buckling (4, 17, 35). Here, we showed that artery buckling could trigger arterial wall remodeling. This remodeling could be a mechanism that lead to tortuosity and the associated atherosclerosis observed in carotid arteries in the aged population (8).
There have been many reports of buckling of carotid arteries, which describes the tortuous shape of arteries under in vivo blood flow and pressure, since the 1930s (20, 49, 52). Here, we studied the buckling of arteries under lumen pressure and flow, in which the curved shape resulted from the buckling deformation. There are naturally curved vessels, such as the arch of aorta, where the curved shape is natural (the curved shape remains after the pressure load is removed). It will be interesting to study site-specific remodeling in the arch of aorta with aging in the future to compare remodeling in natural and buckling-induced curved vessels. In addition, it would be interesting to determine whether cell proliferation and NF-κB activation are increased in human arteries.
In conclusion, our results demonstrated that artery buckling stimulates cell proliferation possibly through a NF-κB-mediated mechanism. Buckling-induced cell proliferation indicates that buckling may contribute to the atherosclerosis observed in tortuous arteries. Further studies are needed to determine the longer-term effects of artery buckling and the adaptation process.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute (NHLBI) Grant HL-095852. It was also partially supported by NHLBI Grant HL-075360 and HHSN 268201000036C (N01-HV-00244) for the San Antonio Cardiovascular Proteomics Center, by Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development Award 5I01BX000505, and by National Science Foundation CAREER Award 0644646.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: Y.X. and D.H. performed experiments; Y.X., S.S.K., and H.-C.H. analyzed data; Y.X., M.L.L., and H.-C.H. interpreted results of experiments; Y.X. prepared figures; Y.X. and H.-C.H. drafted manuscript; Y.X., D.H., M.L.L., and H.-C.H. edited and revised manuscript; Y.X., D.H., S.S.K., M.L.L., and H.-C.H. approved final version of manuscript; H.-C.H. conception and design of research.
ACKNOWLEDGMENTS
The authors thank Granzins Meat Market (New Braunfels, TX), Watark (Poth, TX), and the Pathology laboratory at The University of Texas Health Science Center at San Antonio for help in this work. The authors also thank Dr. Qin Liu for help in this work.
REFERENCES
- 1.Bathe KJ. ADINA Theory and Modeling Guide Volume III: ADINA CFD & FSI. Watertown, MA: ADINA R&D, 2012 [Google Scholar]
- 2.Brand K, Page S, Rogler G, Bartsch A, Brandl R, Knuechel R, Page M, Kaltschmidt C, Baeuerle PA, Neumeier D. Activated transcription factor nuclear factor-κB is present in the atherosclerotic lesion. J Clin Invest 97: 1715–1722, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev 91: 327–387, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chow MJ, Mondonedo JR, Johnson VM, Zhang Y. Progressive structural and biomechanical changes in elastin degraded aorta. Biomech Model Mechanobiol 12: 361–372, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cuhlmann S, Van der Heiden K, Saliba D, Tremoleda JL, Khalil M, Zakkar M, Chaudhury H, Luong le A, Mason JC, Udalova I, Gsell W, Jones H, Haskard DO, Krams R, Evans PC. Disturbed blood flow induces RelA expression via c-Jun N-terminal kinase 1: a novel mode of NF-κB regulation that promotes arterial inflammation. Circ Res 108: 950–959, 2011 [DOI] [PubMed] [Google Scholar]
- 6.Dai G, Kaazempur-Mofrad MR, Natarajan S, Zhang Y, Vaughn S, Blackman BR, Kamm RD, Garcia-Cardena G, Gimbrone MA., Jr Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. Proc Natl Acad Sci USA 101: 14871–14876, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Datir P, Lee AY, Lamm SD, Han HC. Effects of geometric variations on the buckling of arteries. Int J Appl Mech 3: 385–406, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Del Corso L, Moruzzo D, Conte B, Agelli M, Romanelli AM, Pastine F, Protti M, Pentimone F, Baggiani G. Tortuosity, kinking, and coiling of the carotid artery: expression of atherosclerosis or aging? Angiology 49: 361–371, 1998 [DOI] [PubMed] [Google Scholar]
- 9.Dinardo CL, Venturini G, Zhou EH, Watanabe IS, Campos LC, Dariolli R, da Motta-Leal-Filho JM, Carvalho VM, Cardozo KH, Krieger JE, Alencar AM, Pereira AC. Variation of mechanical properties and quantitative proteomics of VSMC along the arterial tree. Am J Physiol Heart Circ Physiol 306: H505–H516, 2014 [DOI] [PubMed] [Google Scholar]
- 10.Dinenno FA, Jones PP, Seals DR, Tanaka H. Age-associated arterial wall thickening is related to elevations in sympathetic activity in healthy humans. Am J Physiol Heart Circ Physiol 278: H1205–H1210, 2000 [DOI] [PubMed] [Google Scholar]
- 11.Fung YC. Biomechanics: Motion, Flow Stress, and Growth. New York: Springer, 1990 [Google Scholar]
- 12.Gaballa MA, Jacob CT, Raya TE, Liu J, Simon B, Goldman S. Large artery remodeling during aging: biaxial passive and active stiffness. Hypertension 32: 437–443, 1998 [DOI] [PubMed] [Google Scholar]
- 13.Gambillara V, Thacher T, Silacci P, Stergiopulos N. Effects of reduced cyclic stretch on vascular smooth muscle cell function of pig carotids perfused ex vivo. Am J Hypertens 21: 425–431, 2008 [DOI] [PubMed] [Google Scholar]
- 14.Gleason RL, Humphrey JD. Effects of a sustained extension on arterial growth and remodeling: a theoretical study. J Biomech 38: 1255–1261, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Grimes KM, Voorhees A, Chiao YA, Han HC, Lindsey ML, Buffenstein R. Cardiac function of the naked mole-rat: ecophysiological responses to working underground. Am J Physiol Heart Circ Physiol 306: H730–H737, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-κB signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci USA 97: 9052–9057, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han HC. A biomechanical model of artery buckling. J Biomech 40: 3672–3678, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han HC. Blood vessel buckling within soft surrounding tissue generates tortuosity. J Biomech 42: 2797–2801, 2009 [DOI] [PubMed] [Google Scholar]
- 19.Han HC. Nonlinear buckling of blood vessels: a theoretical study. J Biomech 41: 2708–2713, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Han HC. Twisted blood vessels: symptoms, etiology and biomechanical mechanisms. J Vasc Res 49: 185–197, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han HC, Chesnutt JK, Garcia JR, Liu Q, Wen Q. Artery buckling: new phenotypes, models, and applications. Ann Biomed Eng 41: 1399–1410, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han HC, Ku DN, Vito RP. Arterial wall adaptation under elevated longitudinal stretch in organ culture. Ann Biomed Eng 31: 403–411, 2003 [DOI] [PubMed] [Google Scholar]
- 23.Han HC, Marita S, Ku DN. Changes of opening angle in hypertensive and hypotensive arteries in 3-day organ culture. J Biomech 39: 2410–2418, 2006 [DOI] [PubMed] [Google Scholar]
- 24.Hayman DM, Xiao Y, Yao Q, Jiang Z, Lindsey ML, Han HC. Alterations in pulse pressure affect artery function. Cell Mol Bioeng 5: 474–487, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holzapfel G, Gasser T, Ogden R. A new constitutive framework for arterial wall mechanics and a comparative study of material models. J Elasticity 61: 1–48, 2000 [Google Scholar]
- 26.Jackson ZS, Dajnowiec D, Gotlieb AI, Langille BL. Partial off-loading of longitudinal tension induces arterial tortuosity. Arterioscler Thromb Vasc Biol 25: 957–962, 2005 [DOI] [PubMed] [Google Scholar]
- 27.Jongstra-Bilen J, Haidari M, Zhu SN, Chen M, Guha D, Cybulsky MI. Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J Exp Med 203: 2073–2083, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ku DN. Blood flow in arteries. Ann Rev Fluid Mech 29: 399–434, 1997 [Google Scholar]
- 29.Ku DN, Giddens DP, Zarins CK, Glagov S. Pulsatile flow and atherosclerosis in the human carotid bifurcation. Positive correlation between plaque location and low oscillating shear stress. Arteriosclerosis 5: 293–302, 1985 [DOI] [PubMed] [Google Scholar]
- 30.Lakatta EG. Central arterial aging and the epidemic of systolic hypertension and atherosclerosis. J Am Soc Hypertens 1: 302–340, 2007 [DOI] [PubMed] [Google Scholar]
- 31.Lakatta EG, Mitchell JH, Pomerance A, Rowe GG. Human aging: changes in structure and function. J Am Coll Cardiol 10: 42A–47A, 1987 [DOI] [PubMed] [Google Scholar]
- 32.Lawrence AR, Gooch KJ. Differences in transmural pressure and axial loading ex vivo affect arterial remodeling and material properties. J Biomech Eng 131: 101009, 2009 [DOI] [PubMed] [Google Scholar]
- 33.Lawrence AR, Gooch KJ. Transmural pressure and axial loading interactively regulate arterial remodeling ex vivo. Am J Physiol Heart Circ Physiol 297: H475–H484, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Learoyd BM, Taylor MG. Alterations with age in the viscoelastic properties of human arterial walls. Circ Res 18: 278–292, 1966 [DOI] [PubMed] [Google Scholar]
- 35.Lee AY, Han B, Lamm SD, Fierro CA, Han HC. Effects of elastin degradation and surrounding matrix support on artery stability. Am J Physiol Heart Circ Physiol 302: H873–H884, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee YU, Luo J, Sprague E, Han HC. Comparison of artery organ culture and co-culture models for studying endothelial cell migration and its effect on smooth muscle cell proliferation and migration. Ann Biomed Eng 38: 801–812, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lemarie CA, Tharaux PL, Lehoux S. Extracellular matrix alterations in hypertensive vascular remodeling. J Mol Cell Cardiol 48: 433–439, 2010 [DOI] [PubMed] [Google Scholar]
- 38.Liu Q, Wen Q, Mottahedi M, Han HC. Artery buckling analysis using four-fiber wall model. J Biomech; 10.1016/j.jbiomech.2014.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McDaniel MC, Galbraith EM, Jeroudi AM, Kashlan OR, Eshtehardi P, Suo J, Dhawan S, Voeltz M, Devireddy C, Oshinski J, Harrison DG, Giddens DP, Samady H. Localization of culprit lesions in coronary arteries of patients with ST-segment elevation myocardial infarctions: relation to bifurcations and curvatures. Am Heart J 161: 508–515, 2011 [DOI] [PubMed] [Google Scholar]
- 40.Nichols WW, O'Rourke MF. McDonald's Blood Flow in Arteries: Theoretical, Experimental, and Clinical Principles (4th ed.). London: Arnold, 1998, chapt. 16 [Google Scholar]
- 41.Ozolanta I, Tetere G, Purinya B, Kasyanov V. Changes in the mechanical properties, biochemical contents and wall structure of the human coronary arteries with age and sex. Med Eng Phys 20: 523–533, 1998 [DOI] [PubMed] [Google Scholar]
- 42.Pancera P, Ribul M, Presciuttini B, Lechi A. Prevalence of carotid artery kinking in 590 consecutive subjects evaluated by Echocolordoppler. Is there a correlation with arterial hypertension? J Intern Med 248: 7–12, 2000 [DOI] [PubMed] [Google Scholar]
- 43.Partridge J, Carlsen H, Enesa K, Chaudhury H, Zakkar M, Luong L, Kinderlerer A, Johns M, Blomhoff R, Mason JC, Haskard DO, Evans PC. Laminar shear stress acts as a switch to regulate divergent functions of NF-κB in endothelial cells. FASEB J 21: 3553–3561, 2007 [DOI] [PubMed] [Google Scholar]
- 44.Pursnani S, Diener-West M, Sharrett AR. The effect of aging on the association between coronary heart disease risk factors and carotid intima media thickness: An analysis of the Atherosclerosis Risk in Communities (ARIC) cohort. Atherosclerosis 233: 441–446, 2014 [DOI] [PubMed] [Google Scholar]
- 45.Samady H, Eshtehardi P, McDaniel MC, Suo J, Dhawan SS, Maynard C, Timmins LH, Quyyumi AA, Giddens DP. Coronary artery wall shear stress is associated with progression and transformation of atherosclerotic plaque and arterial remodeling in patients with coronary artery disease. Circulation 124: 779–788, 2011 [DOI] [PubMed] [Google Scholar]
- 46.Smedby O, Bergstrand L. Tortuosity and atherosclerosis in the femoral artery: what is cause and what is effect? Ann Biomed Eng 24: 474–480, 1996 [DOI] [PubMed] [Google Scholar]
- 47.Smedby O, Johansson J, Molgaard J, Olsson AG, Walldius G, Erikson U. Predilection of atherosclerosis for the inner curvature in the femoral artery. A digitized angiography study. Arterioscler Thromb Vasc Biol 15: 912–917, 1995 [DOI] [PubMed] [Google Scholar]
- 48.Stein PD, Hamid MS, Shivkumar K, Davis TP, Khaja F, Henry JW. Effects of cyclic flexion of coronary arteries on progression of atherosclerosis. Am J Cardiol 73: 431–437, 1994 [DOI] [PubMed] [Google Scholar]
- 49.Subramanyam BR, Horii SC. Sonographic demonstration of buckling of the great vessels of the neck. Am J Roentgenol 142: 1111–1113, 1984 [DOI] [PubMed] [Google Scholar]
- 50.Suo J, Ferrara DE, Sorescu D, Guldberg RE, Taylor WR, Giddens DP. Hemodynamic shear stresses in mouse aortas: implications for atherogenesis. Arterioscler Thromb Vasc Biol 27: 346–351, 2007 [DOI] [PubMed] [Google Scholar]
- 51.Tang D, Yang C, Kobayashi S, Zheng J, Woodard PK, Teng Z, Billiar K, Bach R, Ku DN. 3D MRI-based anisotropic FSI models with cyclic bending for human coronary atherosclerotic plaque mechanical analysis. J Biomech Eng 131: 061010, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Torrens RA, Horton BT. Buckling of the right common carotid artery in hypertension. Ann Intern Med 12: 688–691, 1938 [Google Scholar]
- 53.Van Epps JS, Chew DW, Vorp DA. Effects of cyclic flexure on endothelial permeability and apoptosis in arterial segments perfused ex vivo. J Biomech Eng 131: 101005, 2009 [DOI] [PubMed] [Google Scholar]
- 54.Wahle A, Lopez JJ, Olszewski ME, Vigmostad SC, Chandran KB, Rossen JD, Sonka M. Plaque development, vessel curvature, and wall shear stress in coronary arteries assessed by X-ray angiography and intravascular ultrasound. Med Image Anal 10: 615–631, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wei Z, Costa K, Al-Mehdi AB, Dodia C, Muzykantov V, Fisher AB. Simulated ischemia in flow-adapted endothelial cells leads to generation of reactive oxygen species and cell signaling. Circ Res 85: 682–689, 1999 [DOI] [PubMed] [Google Scholar]
- 56.Weibel J, Fields WS. Tortuosity, coiling, and kinking of the internal carotid artery. I. Etiology and radiographic anatomy. Neurology 15: 7–18, 1965 [DOI] [PubMed] [Google Scholar]
- 57.Xiao Y, Zhong Y, Su H, Zhou Z, Chiao P, Zhong G. NF-κB activation is not required for Chlamydia trachomatis inhibition of host epithelial cell apoptosis. J Immunol 174: 1701–1708, 2005 [DOI] [PubMed] [Google Scholar]
- 58.Yabluchanskiy A, Ma Y, Chiao YA, Lopez EF, Voorhees AP, Toba H, Hall ME, Han HC, Lindsey ML, Jin YF. Cardiac aging is initiated by matrix metalloproteinase-9 mediated endothelial dysfunction. Am J Physiol Heart Circ Physiol 306: H1398–H1407, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yao Q, Hayman DM, Dai Q, Lindsey ML, Han HC. Alterations of pulse pressure stimulate arterial wall matrix remodeling. J Biomech Eng 131: 101011, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang J, Liu Q, Han HC. An in vivo rat model of artery buckling for studying wall remodeling. Ann Biomed Eng. In press [DOI] [PMC free article] [PubMed] [Google Scholar]