

Abstract
The self-assembly of proteins into highly ordered nanoscale architectures is a hallmark of biological systems. The sophisticated functions of these molecular machines inspire the development of methods to engineer novel self-assembling protein structures. Although there has been exciting recent progress in this area, designing multi-component protein nanomaterials with high accuracy remains an outstanding challenge. Here we address this challenge by developing a general computational method for designing protein nanomaterials in which two distinct types of subunits coassemble to a target symmetric architecture. We use the method to design five novel 24-subunit cage-like protein nanomaterials in two distinct symmetric architectures, and experimentally demonstrate that the structures of the materials are in close agreement with the computational design models. The accuracy of the method and the universe of two-component materials that it makes accessible pave the way for the construction of functional protein nanomaterials tailored to specific applications.
Introduction
The unique functional opportunities afforded by protein self-assembly range from the dynamic cellular scaffolding provided by cytoskeletal proteins to the encapsulation, protection, and delivery of viral genomes to new host cells by virus capsids. Although natural assemblies can be repurposed to perform new functions1, 2, this strategy is limited to the structures of existing proteins, which may not be suited to a given application. To overcome this limitation, methods for designing novel self-assembling proteins are of considerable interest3–6. The central challenge in designing self-assembling proteins is to encode the information necessary to direct assembly in the structures of the protein building blocks. Although the complexity and irregularity of protein structures resulted in slow initial progress in this area, advances in computational protein design algorithms and new approaches such as metal-mediated assembly have recently yielded exciting results6–16. Despite these advances, the self-assembling protein structures designed to date have been relatively simple, and continued improvements in design strategies are needed in order to enable the practical design of functional materials.
The level of structural complexity available to self-assembled nanomaterials generally increases with the number of unique molecular components used to construct the material. This is illustrated by DNA nanotechnology, in which specific and directional interactions between hundreds of distinct DNA strands allow the construction of nanoscale objects with essentially arbitrary structures17–20. In contrast, designing well-ordered multi-component protein nanomaterials has remained a significant challenge. Multiple distinct intermolecular contacts are necessary to drive the assembly of such materials3,4,8,11, 21, and programming new, geometrically precise interactions between proteins is generally difficult. Compared to homooligomers, multi-component protein nanomaterials offer several advantages: a wider range of possible structures due to their combinatorial nature, greater control over the timing of assembly, and enhanced modularity through independently addressable building blocks. Although multi-component protein assemblies have recently been generated using disulfide bonds14,22, flexible genetic linkers11,15,22, or stereotyped coiled-coil interactions to drive assembly14,15, the flexibility of these relatively minimal linkages has generally resulted in materials that are somewhat polydisperse. Most natural protein assemblies, on the other hand, are constructed from protein-protein interfaces involving many contacts distributed over large interaction surfaces that serve to precisely define the positions of the subunits relative to each other23,24. Advances in computational protein modeling and design algorithms have recently made it possible to design such interfaces25–29 and thereby direct the formation of novel self-assembling protein nanomaterials with atomic-level accuracy7,9,10, but the methods reported to date have been limited to the design of materials comprising only a single type of molecular building block. Here we expand the structural and functional range of designed protein materials with a general computational method for designing two-component coassembling protein nanomaterials with high accuracy.
Computational design method
Our method centers on encoding the information necessary to direct assembly in designed protein-protein interfaces. In addition to providing the energetic driving force for assembly, the designed interfaces also precisely define the relative orientations of the building blocks. We illustrate the method in Figure 1 using the dual tetrahedral architecture (designated here as T33) as an example. In this architecture, four copies each of two distinct, naturally trimeric building blocks are aligned at opposite poles of the three-fold symmetry axes of a tetrahedron (Figure 1a). This places one set of building blocks at the vertices of the tetrahedron and the other at the center of the faces, totaling twelve subunits of each protein. Each trimeric building block is allowed to rotate around and translate along its three-fold symmetry axis (Figure 1b); other rigid body moves are disallowed because they would lead to asymmetry. These four degrees of freedom are systematically explored during docking to identify configurations with symmetrically repeated instances of a novel inter-building block interface that is suitable for design (Figure 1c). A docking score function maximizes the number of inter-building block neighbors per residue and favors residues in highly anchored regions of the protein structure that are less likely to change conformation upon mutation of surface side chains (Figure 1d). RosettaDesign30,31 is then used to sample the identities and configurations of the side chains near the inter-building block interface, generating interfaces with features resembling those found in natural protein assemblies such as well-packed hydrophobic cores surrounded by polar rims24 (Figure 1e). The end result is a pair of new amino acid sequences, one for each building block, predicted to stabilize the modeled interface and thereby spontaneously drive assembly to the specific target configuration.
Figure 1. Overview of the computational design method.

a, The T33 architecture comprises four copies each of two distinct trimeric building blocks (green and blue) arranged with tetrahedral point group symmetry (24 total subunits; triangles indicate three-fold symmetry axes). b, Each building block has two rigid body degrees of freedom, one translational (r) and one rotational (ω), that are systematically explored during docking. c–d, The docking procedure, which is independent of the amino acid sequence of the building blocks, identifies large interfaces with high densities of contacting residues formed by well-anchored regions of the protein structure. e, Amino acid sequences are designed at the new interface to stabilize the modeled configuration and drive coassembly of the two components. f, In the T32 architecture, four trimeric (grey) and six dimeric (orange) building blocks are aligned along the three-fold and two-fold symmetry axes passing through the vertices and edges of a tetrahedron, respectively.
These docking and design procedures were implemented by extending the Rosetta software31,32 to enable the simultaneous modeling of multiple distinct symmetrically arranged protein components. The new protocol allows the different components to be arranged and moved independently according to distinct sets of symmetry operators (Extended Data Figure 1). This enables the design strategy described above to be generalized to a wide variety of symmetric architectures in which multiple symmetric building blocks are combined in geometrically specific ways3,4,21. Combining even two types of symmetry elements (as in the present study) can give rise to a large number of distinct symmetric architectures with a range of possible morphologies, including those with dihedral and cubic point group symmetries, as well as helical, layer, and space group symmetries (ref. 21 and T.O.Y., manuscript in preparation).
In this study we targeted two distinct tetrahedral architectures: the T33 architecture described above and the T32 architecture shown in Figure 1f, in which the materials are formed from four trimeric and six dimeric building blocks aligned along the three-fold and two-fold tetrahedral symmetry axes. We docked all pairwise combinations of a set of 1,161 dimeric and 200 trimeric protein building blocks of known structure in the T32 and T33 architectures (Supplementary Methods). This resulted in a large set of potential novel nanomaterials: 232,200 and 19,900 docked protein pairs, respectively, with a given pair often yielding several distinct promising docked configurations. Interface sequence design calculations were carried out on the 1,000 highest scoring docked configurations in each architecture, and the designs were evaluated based on the predicted binding energy, shape complementarity33, and size of the designed interfaces, as well as the number of buried unsatisfied hydrogen bonding groups (Supplementary Methods). After filtering on these criteria, 30 T32 and 27 T33 materials were selected for experimental characterization (Extended Data Figure 2). The 57 designs were derived from 39 distinct trimeric and 19 dimeric proteins, and contained an average of 19 amino acid mutations per pair of subunits compared to the native sequences. The designed interfaces resided mostly on elements of secondary structure, both α-helices and β-strands, with nearby loops often making minor contributions.
Screening and characterization of assembly state
Synthetic genes encoding each designed pair of proteins were cloned in tandem in a single expression vector to allow inducible co-expression in E. coli (Supplementary Methods). Polyacrylamide gel electrophoresis (PAGE) under denaturing and non-denaturing (native) conditions was used to rapidly screen the level of soluble expression and assembly state of the designed proteins in clarified cell lysates. For most of the designs, either one or both of the designed proteins was not detectable in the soluble fraction, suggesting that insoluble expression is a common failure mode for the designed materials. Given that the majority of the mutations introduced by our method are polar to hydrophobic surface mutations at the designed interfaces, it is likely that the insolubility of these designs is due to either misfolding or nonspecific aggregation of the designed protein subunits. Nevertheless, several designed protein pairs yielded single bands under non-denaturing conditions that migrated more slowly than the wild-type proteins from which they were derived, suggesting assembly to higher-order species (Extended Data Figure 3). These proteins were subcloned to introduce a hexahistidine tag at the C terminus of one of the two subunits and purified by nickel affinity chromatography and size exclusion chromatography (SEC). Five pairs of designed proteins, one T32 design (T32-28) and four T33 designs (T33-09, T33-15, T33-21, and T33-28), co-purified off of the nickel column and yielded dominant peaks at the expected size of approximately 24 subunits when analyzed by SEC (Figure 2a and Supplementary Table 1).
Figure 2. Experimental characterization of coassembly.
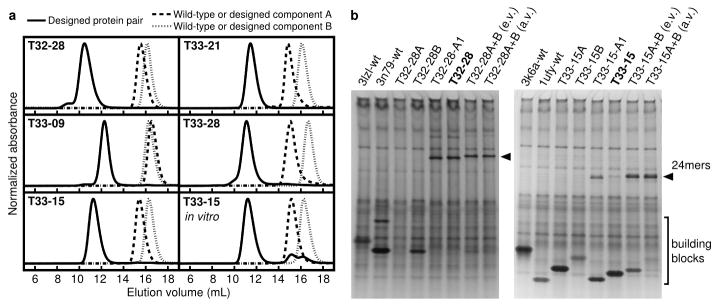
a, SEC chromatograms of the designed pairs of proteins (solid lines) and the wild-type oligomeric proteins from which they were derived (dashed and dotted lines). The co-expressed designed proteins elute at the volumes expected for the target 24-subunit nanomaterials, while the wild-type proteins elute as dimers or trimers. The T33-15 in vitro panel shows chromatograms for the individually produced and purified designed components (T33-15A and T33-15B) as well as a stoichiometric mixture of the two components. b, Native PAGE analysis of in vitro-assembled T32-28 (left panel) and T33-15 (right panel) in cell lysates. Lysates containing the co-expressed design components (lanes 5–6) contain slowly migrating species (arrows) not present in lysates containing the wild-type and individually expressed components (lanes 1–4). Mixing equal volumes (e.v.) of crude lysates containing the individual designed components yields the same assemblies (lane 7), although some unassembled building blocks remain due to unequal levels of expression (particularly for T33-15). When the differences in expression levels are accounted for by mixing adjusted volumes of lysates (a.v.), more efficient assembly is observed (lane 8).
We tested the ability of each of the five materials to assemble in vitro by expressing the two components in separate E. coli cultures and mixing them at various points after cell lysis (Extended Data Figure 3). Native PAGE revealed that in two cases (T33-15 and T32-28) the two separately expressed components efficiently assembled to the designed materials in vitro when equal volumes of cell lysates were mixed (Figure 2b, Extended Data Figure 3a, c). Adjusting the volume of each lysate in the mixture to account for differences in the level of soluble expression of the two components allowed for more quantitative assembly. In the case of T33-15, the two components of the material could also be purified independently: T33-15A and T33-15B each eluted from the SEC column as trimers in isolation. After mixing the two purified components in a 1:1 molar ratio and allowing a two hour incubation at room temperature, the mixture eluted from the SEC column as predominantly the 24mer assembly, with small amounts of residual trimeric building blocks remaining (Figure 2a). It is thus possible to control the assembly of our designed materials by simply mixing the two independently produced components.
The details of the designed interfaces for the five materials are presented in Figure 3. Qualitatively, the interfaces are similar to those in the other designs that were experimentally characterized and reflect the hypothesis underlying the design protocol: they feature well-packed and highly complementary cores of hydrophobic side chains residing mostly in elements of secondary structure, surrounded by polar side chains lining the periphery of the hydrophobic cores. The successful designs are also quantitatively similar to the other designs according to the interface metrics used to select designs for experimental characterization (predicted binding energy, shape complementarity, interface size, and number of buried unsatisfied hydrogen bonding groups; Extended Data Figure 4). The similarity of the successful and unsuccessful designs according to these structural metrics, combined with the observed insolubility of many of the designs, suggests that focusing on improving the level of soluble expression of the designed proteins could substantially improve the success rate of our approach in the future.
Figure 3. Modeled interfaces of designed two-component protein nanomaterials.

The models of the designed interfaces in each component of T32-28, T33-09, T33-15, T33-21, and T33-28 are shown at left or right, and side views of each interface as a whole are shown at center. Each image is oriented such that a vector originating at the center of the tetrahedral material and passing through the center of mass of the designed interface would pass vertically through the center of the image. The side chains of all amino acids allowed to repack and minimize during the interface design procedure are shown in stick representation. The alpha carbon atoms of positions that were mutated during design are shown as spheres, and the mutations are labeled. To highlight the morphologies of the contacting surfaces, atoms within 5 Å of the opposite building block are shown in semi-transparent surface representation. Oxygen atoms are red; nitrogen, blue; and sulfur, orange.
Structural characterization of the designed materials
Negative stain electron microscopy of the five designed materials confirmed that they assemble specifically to the target architectures (Figure 4). For each material, fields of monodisperse particles of the expected size and symmetry were observed, confirming the homogeneity of the materials suggested by SEC. Particle averaging yielded images that recapitulate features of the computational design models at low resolution. For example, class averages of T33-09 revealed roughly square or triangle-shaped structures with well-defined internal cavities that closely resemble projections calculated from the computational design model along its two-fold and three-fold axes (Figure 4, T33-09 inset). Micrographs of T33-15 assembled in vitro as described above were indistinguishable from those of co-expressed T33-15 (Figure 4 and Extended Data Figure 5), demonstrating that the same material is obtained using both methods.
Figure 4. Electron micrographs of designed two-component protein nanomaterials.

Negative stain electron micrographs for five designed materials are shown to scale (scale bar: 25 nm). The T33-15 in vitro sample was prepared by stoichiometrically mixing the independently purified components (T33-15A and T33-15B) in vitro and purifying the assembled material by SEC (see Figure 2). Micrographs of unpurified, in vitro-assembled T33-15 as well as T33-15A and T33-15B in isolation are shown in Extended Data Figure 5. For each material, two different class averages of the particles are shown in the insets (left) alongside back projections calculated from the computational design models (right).
We solved X-ray crystal structures of four of the designed materials (T32-28, T33-15, T33-21, and T33-28) to resolutions ranging from 2.1 to 4.5 Å (Figure 5 and Supplementary Tables 2 and 3). In all cases, the structures reveal that the inter-building block interfaces were designed with high accuracy: comparing a pair of chains from each structure to the computationally designed model yields backbone root mean square deviations (r.m.s.d.) between 0.5 and 1.2 Å (Figure 5 right and Extended Data Table 1). In the structures with resolutions that permit detailed analysis of side chain configurations (T33-15 and two independent crystal forms of T33-21), 87 of 113 side chains at the designed interfaces adopt the predicted conformations (Supplementary Tables 5 and 6). As intended, the designed interfaces drive the assembly of cage-like nanomaterials that closely match the computational design models: the backbone r.m.s.d. over all 24 subunits in each material range from 1.0 to 2.6 Å (Figure 5 left and Extended Data Table 1). The precise control over interface geometry offered by our method thus enables the design of two-component protein nanomaterials with diverse nanoscale features such as surfaces, pores, and internal volumes with high accuracy.
Figure 5. Crystal structures of designed two-component protein nanomaterials.

The computational design models (top) and X-ray crystal structures (bottom) are shown at left for a, T32-28, b, T33-15, c, T33-21, and d, T33-28. Views of each material are shown to scale along the 2-fold and 3-fold tetrahedral symmetry axes (scale bar: 15 nm). The r.m.s.d. values given are those between the backbone atoms in all 24 chains of the design models and crystal structures. For T33-21, r.m.s.d. values are shown for both crystal forms (images are shown for the higher resolution crystal form with backbone r.m.s.d. 2.0 Å), while the r.m.s.d. range for T33-28 derives from the four copies of the fully assembled material in the crystallographic asymmetric unit. At right, overlays of the designed interfaces in the design models (white) and crystal structures (grey, orange, green, and blue) are shown. Due to the limited resolution of the T32-28 structure, the amino acid side chains were not modeled beyond the beta carbon. For the interface overlays, the crystal structures were aligned to the design models using the backbone atoms of two subunits, one of each component.
Discussion
Due to the unique functional capabilities of self-assembling proteins, there is intense interest in engineering protein nanomaterials for applications in various fields. Most efforts to date have focused on repurposing naturally occurring protein assemblies, a strategy that is ultimately limited by the structures available and their tolerances for modification. Similarly, while directed evolution is a powerful method for protein engineering34,35 and can be used to improve, for example, the packaging capability of existing protein nanocontainers36,37, it is difficult to envision how it could accurately generate new protein nanomaterials with target structures defined at the atomic level. Our results demonstrate that computational protein design provides a general route for designing novel two-component self-assembling protein nanomaterials with high accuracy. The combinatorial nature of two-component materials greatly expands the number and variety of potential nanomaterials that can be designed. For example, in this study we used 1,361 protein building blocks to dock over 250,000 distinct protein pairs among two target architectures with tetrahedral point group symmetry, resulting in a very large set of potential nanomaterials exhibiting a variety of sizes, shapes, and arrangements of chemically and genetically addressable functional groups, loops, and termini. With continued effort to increase the success rate of protein-protein interface design and reduce the rate of designed proteins that express insolubly, it should become possible to simultaneously design multiple novel interfaces in a single material, which would enable the construction of increasingly complex materials built from more than two components.
The conceptual framework that underlies our method—symmetric docking followed by protein-protein interface design—can be generally applied to a wide variety of symmetric architectures, including those capable of forming repetitive protein arrays that extend in one, two, or three dimensions. Multi-component materials are advantageous in these extended architectures because the uncontrolled self-assembly of a single-component material inside the cell can complicate biological production5,11,21. We have shown that the two components of the designed materials T32-28 and T33-15 can be produced separately and mixed in vitro to initiate assembly of the designed structure. With new symmetric modeling algorithms capable of handling the additional degrees of freedom associated with these architectures, the accurate computational design and controllable assembly of complex, multi-component protein fibers, layers, and crystals should also be possible.
The capability to design highly homogeneous protein nanostructures with atomic-level accuracy and controllable assembly should open up new opportunities in targeted drug delivery, vaccine design, plasmonics, and other applications that can benefit from the precise patterning of matter on the sub-nanometer to hundred nanometer scale. Extending beyond static structure design, methods for incorporating the kinds of dynamic and functional behaviors observed in natural protein assemblies should make possible the design of novel protein-based molecular machines with programmable structures, dynamics, and functions.
METHODS SUMMARY
The symmetric modeling framework in Rosetta31,32 was updated to enable the modeling of multi-component symmetrical structures. A new application, tcdock, docks pairs of protein scaffolds in higher order symmetries, scoring each docked configuration according to its suitability for interface design. tcdock was used to dock all possible pairwise combinations of 200 trimeric scaffold proteins and all possible pairwise combinations of the same trimers and 1,161 dimeric proteins in the T33 and T32 symmetric architectures, respectively. New two-component protein-protein interface design protocols were used to design new amino acid sequences predicted to stabilize selected docked configurations. During the sequence design protocols, the symmetric rigid body degrees of freedom and the identities and conformations of the side chains at the inter-building block interfaces were optimized to identify low-energy sequence-structure combinations. 30 T32 and 27 T33 designs were selected for experimental characterization.
The assembly states of the designed pairs of proteins were assessed by native PAGE, and those that migrated more slowly than the wild-type scaffolds were subjected to affinity purification and SEC. The ability of the materials to assemble in vitro was investigated by independently producing the two components, mixing them at various points after cell lysis, and analyzing the mixtures by native PAGE and SEC. The materials were structurally characterized by negative stain electron microscopy including particle averaging, and at high resolution by X-ray crystallography.
Full Methods and any associated references are available in the online version of the paper.
Extended Data
Extended Data Figure 1. Comparison of one-component and multi-component symmetric fold trees.
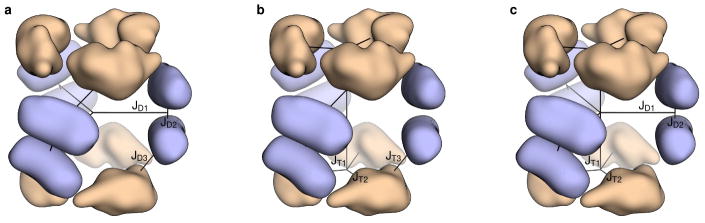
Three different symmetric fold tree representations of a D32 architecture are shown. In this architecture, two trimeric building blocks (wheat) are aligned along the three-fold rotational axes of D3 point group symmetry and three dimeric building blocks (light blue) are aligned along the two-folds. a, The dimer-centric one-component symmetry case. Rigid body degree of freedom (RB DOF, black lines) JD3 connecting the master dimer subunit to the master trimer subunit is a child of RB DOFs JD1 and JD2 controlling the master dimer subunit; in this case the positions of the trimeric subunits depend on the positions of the dimeric subunits. b, The trimer-centric one-component symmetry case. RB DOF JT3 connecting the master trimer subunit to the master dimer subunit is a child of RB DOFs JT1 and JT2 controlling the master trimer subunit; in this case the positions of the dimeric subunits depend on the positions of the trimeric subunits. c, The multi-component symmetry case. With multi-component symmetric modeling, the RB DOFs controlling the master trimer subunit (JT1 and JT2) and the master dimer subunit (JD1 and JD2) are independent. In this case the positions of the dimeric subunits do not depend on the positions of the trimeric subunits and vice versa, allowing the internal DOFs for each building block (JT2 and JD2) to be maintained while moving the building blocks independently (JT1 and JD1). See the Supplementary Methods for additional discussion.
Extended Data Figure 2. Models of the 57 designs selected for experimental characterization.

Smoothed surface representations are shown of each of the 30 T32 and 27 T33 designs. The trimeric component of each T32 design is shown in grey and the dimeric component in orange. The two different trimeric components of each T33 design are shown in blue and green. The tetrahedral two-fold and three-fold symmetry axes (black lines) are shown passing through the center of each component. Each design is named according to its symmetric architecture (T32 or T33) followed by a unique identification number. The pairs of scaffold proteins from which the designs are also indicated.
Extended Data Figure 3. Native PAGE analysis of cleared cell lysates.

Each gel contains cleared lysates pertaining to a, T32-28, b, T33-09, c, T33-15, d, T33-21, or e, T33-28. Lane 1 is from cells expressing the wild-type scaffold for component A and lane 2 the wild-type scaffold for component B. Lanes 3–4 are from cells expressing the individual design components and lanes 5–6 the co-expressed components. Lanes 7–8 are from samples mixed as crude lysates (cr.e.v or cr.a.v), while lanes 9–10 are from samples mixed as cleared lysates (cl.e.v. or cl.a.v.). Lanes 7 and 9 are from lysates mixed with equal volumes (cr.e.v. or cl.e.v.), while lanes 8 and 10 are from lysates mixed with adjusted volumes (cr.a.v. or cl.a.v.). Lane 5 is from cells expressing the C-terminally A1-tagged constructs; all other lanes are from cells expressing the C-terminally His-tagged constructs. An arrow is positioned next to each gel indicating the migration of 24-subunit assemblies and the gel regions containing unnassembled building blocks are bracketed. Each gel was stained with GelCode Blue (Thermo Scientific). Portions of the gels in a and c are also shown in Figure 2b.
Extended Data Figure 4. Structural metrics for the computational design models.

Selected metrics related to the designed interfaces are plotted for the 57 designs that were experimentally characterized, including a, the predicted binding energy measured in Rosetta Energy Units (REU), b, the surface area buried by each instance of the designed interface, c, the binding energy density (calculated as the predicted binding energy divided by the buried surface area), d, the number of buried unsatisfied polar groups at the designed interface, e, the shape complementarity of the designed interface, and f, the total number of mutations in each designed pair of proteins. Each circle represents a single design; the five successful materials are plotted as filled circles and labeled. In each plot, the designs are arranged on the x axis in order of increasing value.
Extended Data Figure 5. Electron micrographs of in vitro-assembled T33-15 (unpurified) and T33-15A and T33-15B in isolation.

Negative stain electron micrographs of independently purified T33-15 components and unpurified, in vitro-assembled T33-15 are shown to scale (scale bar: 25 nm).
Extended Data Table 1.
Root mean square deviations (r.m.s.d.) between crystal structures and design models.
| Design model | Crystal structure | Global r.m.s.d. (Å)* | Two-chain r.m.s.d. (Å)† | Contents of asymmetric unit | Structure used for superposition‡ |
|---|---|---|---|---|---|
| T32-28 | 4NWN | 2.586 | 1.246 | One cage (24 subunits) | Asymmetric unit |
| T33-15 | 4NWO | 1.433 | 0.876 | One chain of each component (2 subunits) | One cage generated from crystallographic 2- and 3-folds |
| T33-21 | 4NWP | 1.962 | 0.924 | 4 chains of each component (8 subunits) | One cage generated from one crystallographic 3-fold |
| T33-21 | 4NWQ | 1.482 | 0.765 | One chain of each component (2 subunits) | One cage generated from crystallographic 2- and 3-folds |
| T33-28 | 4NWR | 0.965 | 0.503 | Four complete cages (96 subunits) | One complete cage from the asymmetric unit |
| T33-28 | 4NWR | 0.965 | 0.548 | Four complete cages (96 subunits) | One complete cage from the asymmetric unit |
| T33-28 | 4NWR | 1.195 | 0.567 | Four complete cages (96 subunits) | One complete cage from the asymmetric unit |
| T33-28 | 4NWR | 1.212 | 0.477 | Four complete cages (96 subunits) | One complete cage from the asymmetric unit |
Global r.m.s.d. was calculated over all 24 subunits of each design model and corresponding subunits in each crystal structure.
Two-chain r.m.s.d. was calculated over chains A and B of each design model and corresponding subunits in each crystal structure.
24 subunits composing one complete cage were derived from each crystal structure as indicated and the chains renamed to match the corresponding names in the design models. In the case of T33-28, four different sets of r.m.s.d. calculations were carried out; one for each of the four cages contained in the asymmetric unit of 4NWR.
Supplementary Material
Acknowledgments
We would like to thank Dan Shi and Brent Nannenga (JFRC) for help with electron microscopy, Frank DiMaio and Rocco Moretti for assistance with software development, Per Greisen for scripts used to compare side chain conformations, Jasmine Gallaher for technical assistance, Michael Collazo for help with preliminary crystallization screening, Duilio Cascio and Michael Sawaya for help with crystallographic experiments, and Malcolm Capel, Jonathan Schuermann, and Igor Kourinov at NE-CAT beamline 24-ID-C for help with data collection. This work was supported by the Howard Hughes Medical Institute (TG and DB) and the JFRC visitor program (SG), the National Science Foundation under CHE-1332907 (DB and TOY), grants from the International AIDS Vaccine Initiative, DTRA (N00024-10-D-6318/0024), AFOSR (FA950-12-10112), and DOE (DE-SC0005155) to DB, an NIH Biotechnology Training Program award to DEM (T32GM067555), and an NSF graduate research fellowship to JBB (DGE-0718124). TOY and DEM also acknowledge support from the BER program of the DOE Office of Science.
Footnotes
Author Contributions. NPK, JBB, WS, and DB designed the research. NPK, JBB, and WS wrote program code and performed the docking and design calculations. NPK and JBB biophysically characterized the designed materials and prepared samples for structural analysis. SG characterized the designed materials by EM; SG and TG analysed EM data. DEM crystallized the designed protein materials; DEM and TOY analysed crystallographic data. NPK, JBB, and DB analysed data and wrote the manuscript. All authors discussed the results and commented on the manuscript.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
The crystal structures and structure factors for the designed materials have been deposited in the RCSB Protein Data Bank (http://www.rcsb.org/) under the accession codes 4NWN (T32-28), 4NWO (T33-15), 4NWP (T33-21, R32 crystal form), 4NWQ (T33-21, F4132 crystal form), and 4NWR (T33-28).
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding bodies.
References
- 1.Howorka S. Rationally engineering natural protein assemblies in nanobiotechnology. Curr Opin Biotechnol. 2011;22:485–491. doi: 10.1016/j.copbio.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 2.Douglas T, Young M. Viruses: making friends with old foes. Science. 2006;312:873–875. doi: 10.1126/science.1123223. [DOI] [PubMed] [Google Scholar]
- 3.Lai YT, King NP, Yeates TO. Principles for designing ordered protein assemblies. Trends Cell Biol. 2012;22:653–661. doi: 10.1016/j.tcb.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 4.King NP, Lai YT. Practical approaches to designing novel protein assemblies. Curr Opin Struct Biol. 2013;23:632–638. doi: 10.1016/j.sbi.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 5.Sinclair JC. Constructing arrays of proteins. Curr Opin Chem Biol. 2013;17:946–951. doi: 10.1016/j.cbpa.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 6.Salgado EN, Radford RJ, Tezcan FA. Metal-directed protein self-assembly. Acc Chem Res. 2010;43:661–672. doi: 10.1021/ar900273t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.King NP, et al. Computational design of self-assembling protein nanomaterials with atomic level accuracy. Science. 2012;336:1171–1174. doi: 10.1126/science.1219364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brodin JD, et al. Metal-directed, chemically tunable assembly of one-, two- and three-dimensional crystalline protein arrays. Nat Chem. 2012;4:375–382. doi: 10.1038/nchem.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lanci CJ, et al. Computational design of a protein crystal. Proc Natl Acad Sci USA. 2012;109:7304–7309. doi: 10.1073/pnas.1112595109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stranges PB, Machius M, Miley MJ, Tripathy A, Kuhlman B. Computational design of a symmetric homodimer using beta-strand assembly. Proc Natl Acad Sci USA. 2011;108:20562–20567. doi: 10.1073/pnas.1115124108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sinclair JC, Davies KM, Venien-Bryan C, Noble ME. Generation of protein lattices by fusing proteins with matching rotational symmetry. Nat Nanotechnol. 2011;6:558–562. doi: 10.1038/nnano.2011.122. [DOI] [PubMed] [Google Scholar]
- 12.Lai YT, Cascio D, Yeates TO. Structure of a 16-nm cage designed by using protein oligomers. Science. 2012;336:1129. doi: 10.1126/science.1219351. [DOI] [PubMed] [Google Scholar]
- 13.Der BS, et al. Metal-mediated affinity and orientation specificity in a computationally designed protein homodimer. J Am Chem Soc. 2012;134:375–385. doi: 10.1021/ja208015j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fletcher JM, et al. Self-assembling cages from coiled-coil peptide modules. Science. 2013;340:595–599. doi: 10.1126/science.1233936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyle AL, et al. Squaring the circle in peptide assembly: from fibers to discrete nanostructures by de novo design. J Am Chem Soc. 2012;134:15457–15467. doi: 10.1021/ja3053943. [DOI] [PubMed] [Google Scholar]
- 16.Grigoryan G, et al. Computational design of virus-like protein assemblies on carbon nanotube surfaces. Science. 2011;332:1071–1076. doi: 10.1126/science.1198841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seeman NC. Nanomaterials based on DNA. Annu Rev Biochem. 2010;79:65–87. doi: 10.1146/annurev-biochem-060308-102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rothemund PW. Folding DNA to create nanoscale shapes and patterns. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- 19.Ke Y, Ong LL, Shih WM, Yin P. Three-dimensional structures self-assembled from DNA bricks. Science. 2012;338:1177–1183. doi: 10.1126/science.1227268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han D, et al. DNA gridiron nanostructures based on four-arm junctions. Science. 2013;339:1412–1415. doi: 10.1126/science.1232252. [DOI] [PubMed] [Google Scholar]
- 21.Padilla JE, Colovos C, Yeates TO. Nanohedra: using symmetry to design self assembling protein cages, layers, crystals, and filaments. Proc Natl Acad Sci USA. 2001;98:2217–2221. doi: 10.1073/pnas.041614998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Usui K, et al. Nanoscale elongating control of the self-assembled protein filament with the cysteine-introduced building blocks. Protein Sci. 2009;18:960–969. doi: 10.1002/pro.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodsell DS, Olson AJ. Structural symmetry and protein function. Annu Rev Biophys Biomol Struct. 2000;29:105–153. doi: 10.1146/annurev.biophys.29.1.105. [DOI] [PubMed] [Google Scholar]
- 24.Janin J, Bahadur RP, Chakrabarti P. Protein-protein interaction and quaternary structure. Q Rev Biophys. 2008;41:133–180. doi: 10.1017/S0033583508004708. [DOI] [PubMed] [Google Scholar]
- 25.Huang PS, Love JJ, Mayo SL. A de novo designed protein protein interface. Protein Sci. 2007;16:2770–2774. doi: 10.1110/ps.073125207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jha RK, et al. Computational design of a PAK1 binding protein. J Mol Biol. 2010;400:257–270. doi: 10.1016/j.jmb.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karanicolas J, et al. A de novo protein binding pair by computational design and directed evolution. Mol Cell. 2011;42:250–260. doi: 10.1016/j.molcel.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fleishman SJ, et al. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science. 2011;332:816–821. doi: 10.1126/science.1202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khare SD, Fleishman SJ. Emerging themes in the computational design of novel enzymes and protein-protein interfaces. FEBS Lett. 2013;587:1147–1154. doi: 10.1016/j.febslet.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 30.Kuhlman B, Baker D. Native protein sequences are close to optimal for their structures. Proc Natl Acad Sci USA. 2000;97:10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leaver-Fay A, et al. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DiMaio F, Leaver-Fay A, Bradley P, Baker D, Andre I. Modeling symmetric macromolecular structures in Rosetta3. PLoS One. 2011;6:e20450. doi: 10.1371/journal.pone.0020450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 34.Arnold FH, Volkov AA. Directed evolution of biocatalysts. Curr Opin Chem Biol. 1999;3:54–59. doi: 10.1016/s1367-5931(99)80010-6. [DOI] [PubMed] [Google Scholar]
- 35.Jackel C, Kast P, Hilvert D. Protein design by directed evolution. Annu Rev Biophys. 2008;37:153–173. doi: 10.1146/annurev.biophys.37.032807.125832. [DOI] [PubMed] [Google Scholar]
- 36.Worsdorfer B, Pianowski Z, Hilvert D. Efficient in vitro encapsulation of protein cargo by an engineered protein container. J Am Chem Soc. 2012;134:909–911. doi: 10.1021/ja211011k. [DOI] [PubMed] [Google Scholar]
- 37.Worsdorfer B, Woycechowsky KJ, Hilvert D. Directed evolution of a protein container. Science. 2011;331:589–592. doi: 10.1126/science.1199081. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.