

Abstract
The endosymbiotic bacteria Wolbachia pipientis is known to infect a wide range of arthropod species yet less is known about the coevolutionary history it has with its hosts. Evidence of highly identical W. pipientis strains in evolutionary divergent hosts suggests horizontal transfer between hosts. For example, Drosophila ananassae is infected with a W. pipientis strain that is nearly identical in sequence to a strain that infects both D. simulans and D. suzukii, suggesting recent horizontal transfer among these three species. However, it is unknown whether the W. pipientis strain had recently invaded all three species or a more complex infectious dynamic underlies the horizontal transfers. Here, we have examined the coevolutionary history of D. ananassae and its resident W. pipientis to infer its period of infection. Phylogenetic analysis of D. ananassae mitochondrial DNA and W. pipientis DNA sequence diversity revealed the current W. pipientis infection is not recent. In addition, we examined the population genetics and molecular evolution of several germline stem cell (GSC) regulating genes of D. ananassae. These studies reveal significant evidence of recent and long-term positive selection at stonewall in D. ananassae, whereas pumillio showed patterns of variation consistent with only recent positive selection. Previous studies had found evidence for adaptive evolution of two key germline differentiation genes, bag of marbles (bam) and benign gonial cell neoplasm (bgcn), in D. melanogaster and D. simulans and proposed that the adaptive evolution at these two genes was driven by arms race between the host GSC and W. pipientis. However, we did not find any statistical departures from a neutral model of evolution for bam and bgcn in D. ananassae despite our new evidence that this species has been infected with W. pipientis for a period longer than the most recent infection in D. melanogaster. In the end, analyzing the GSC regulating genes individually showed two of the seven genes to have evidence of selection. However, combining the data set and fitting a specific population genetic model significant proportion of the nonsynonymous sites across the GSC regulating genes were driven to fixation by positive selection. Clearly the GSC system is under rapid evolution and potentially multiple drivers are causing the rapid evolution.
Keywords: Drosophila ananassae, Wolbachia pipientis, germline stem cell, positive selection
Introduction
Endosymbionts are organisms that reside inside its host and have effects that range from parasitism to mutualism (Dale and Moran 2006). A common mode of transmission for these symbionts involves vertical transmission where it is passed down from the host to its progeny (Werren and O’Neill 1997). As these heritable symbionts are transmitted from one cytoplasm to another, they are mainly found in female reproductive organs to ensure maximum transmission of themselves to the hosts’ offspring (Buchner 1965). Traditionally, heritable endosymbionts were thought to be mutualistic with their host because any deleterious harm that caused to its host would result in decreased transmission of the symbionts as well (Fine 1975). However, there are numerous cases of the endosymbiont being parasitic and controlling the hosts’ reproduction for its own benefit (Engelstädter and Hurst 2009). Hence, these endosymbionts that manipulate the hosts’ reproductive system are called reproductive parasites.
Reproductive parasites have a strong interest in localizing at the host reproductive tract for their own transmission because here they are predicted to be able to manipulate the host germline (Werren 2005). In the developing embryo of its host, for example, studies have shown cases where the reproductive parasites localize in developmental regions that later differentiate into reproductive tracts (Kose and Karr 1995; Ferree et al. 2005). In other cases, the reproductive parasite could directly select against uninfected germline stem cells (GSCs) for elimination and favor the transmission of infected GSCs (Werren 2005). Thus, a conflict would then arise between the host GSC and reproductive parasites.
Types of reproductive parasites can range from genetic elements, such as transposable elements (TEs) and meiotic drivers, to heritable microorganisms and organelles (Werren 2011). Wolbachia pipientis an alpha-proteobacteria is one of the most successful reproductive parasites that is estimated to infect up to 66% of all insect species (Hilgenboecker et al. 2008). The ability of W. pipientis to infect a wide range of arthropods is thought to be due to its ability to manipulate the hosts’ reproductive ability (Werren et al. 2008) and behavior (de Crespigny et al. 2006) to ensure maximum transmission through the female lineage. Evidence of W. pipientis infection is heterogeneous among Drosophila species (Mateos et al. 2006) suggesting that each host has had a unique evolutionary interactions with the reproductive parasite. However, assays of a simple absence or presence of W. pipientis are not sufficient to understand the dynamics of the infection as it ignores the duration of the W. pipientis infection. One way to examine the age of an infection is to study the population genomics of resident W. pipientis reassembled from infected host genome sequences from population samples (Richardson et al. 2012; Chrostek et al. 2013; Early and Clark 2013). However, studies sequencing only a few W. pipientis loci (rather than full genomes) failed to find sequence diversity among resident W. pipientis infecting different individuals sampled from natural populations (Guillemaud et al. 1997; James and Ballard 2000; Shoemaker et al. 2003; Dyer and Jaenike 2004), potentially due to a low rate of mutation for W. pipientis (Raychoudhury et al. 2009; Richardson et al. 2012; Early and Clark 2013). An alternative approach then is to examine the host mitochondria DNA (mtDNA) phylogeny because, like W. pipientis, it is also maternally inherited, yet accumulates substitutions at a faster rate. Thus, analysis of mtDNA would give an indirect estimate of the infectious history of W. pipientis (Hurst and Jiggins 2005). A recent W. pipientis invasion would be predicted to have eliminated the mtDNA diversity of the host due to rapid fixation in the population of the mtDNA haplotype in linkage disequilibrium with the initial female infected with W. pipientis. After the W. pipientis (and hitchhiking mtDNA) sweep through the entire population, they will both accumulate mutations returning variability to levels present prior to the W. pipientis invasion. Estimating the time to most recent common ancestor (TMRCA) of the mtDNA of a population infected with W. pipientis thus provides an indirect estimate of the time since the initiation of the most recent W. pipientis-mediated mtDNA sweep.
Interestingly within the Drosophila genus, some of the Drosophila species that have diverged millions of years ago are infected with nearly identical W. pipientis strains. For example, the genome sequence of W. pipientis Riverside strain of D. simulans (wRi) (Hoffmann et al. 1986) has very little sequence divergence from the genome sequence of W. pipientis infecting D. ananassae (wAna) (Salzberg et al. 2005) and D. suzukii (Siozios et al. 2013), suggesting recent horizontal transfers of W. pipientis among the three host species. Although vertical transmission is the main mode of transmission for W. pipientis, occasional horizontal transfer between phylogenetically distant species occurs through unknown mechanisms (Werren et al. 1995). Thus, the infectious history of W. pipientis in Drosophila is dynamic and the period of infection would be an important factor to consider when evaluating the potential for the coevolution of W. pipientis and its host.
Here, we have investigated the coevolutionary history of D. ananassae and its resident W. pipientis. We compared the TMRCA for the current W. pipientis infecting D. ananassae with those W. pipientis currently infecting D. melanogaster and D. simulans, two well-studied species that are also widely infected by W. pipientis. Additionally, we have conducted a population genetic analysis of the molecular evolution of several key GSC regulating genes in D. ananassae. Like many other reproductive parasites, W. pipientis is predicted to have an antagonistic relation with the host germline by manipulating the host GSCs. For example, the parasitic wasp Asobara tabida shows an extreme case of manipulation where the elimination of W. pipientis with antibiotics halts the formation of mature oocytes in the host (Dedeine et al. 2001). In D. melanogaster, W. pipientis is able to suppress hypomorphic sex-lethal (Starr and Cline 2002) and bag-of-marbles (bam) (Flores 2012) mutations resulting in a significant increase in the hosts’ fecundity. Furthermore, W. pipientis in the species D. mauritiana directly manipulates the host GSC regulating system, ultimately resulting in a 4-fold increase in fertility in infected individuals (Fast et al. 2011). Thus, the manipulation caused by W. pipientis could lead to selective pressure on the host GSC to resist the manipulation, resulting in a coevolutionary arms race between the host GSC and W. pipientis.
Previous studies have found two of the key GSC differentiation genes bam and benign gonial cell neoplasm (bgcn) under strong positive selection for amino acid change in D. melanogaster and D. simulans (Civetta et al. 2006; Bauer DuMont et al. 2007). Having a role in GSC differentiation for bam (McKearin and Spradling 1990) and bgcn (Gönczy et al. 1997), the evolutionary driver of selection on bam and bgcn was hypothesized to be a coevolutionary arms race between the host GSC and W. pipientis (Bauer DuMont et al. 2007). Because of the strong evidence of positive selection in bam and bgcn observed in D. melanogaster and D. simulans (Bauer DuMont et al. 2007), we examined the population genetics of bam and bgcn in D. ananassae because it is also known to be widely infected by W. pipientis (Mateos et al. 2006). We also analyzed the population genetics of five other GSC regulating genes (nanos [nos], otefin [ote], pumillio [pum], stone-wall [stwl], and female-sterile-1-Yb [Yb]) that interact downstream or upstream of bam and bgcn (Xie 2012).
Our phylogenetic results suggest that the current W. pipientis infection in D. ananassae was longer than D. melanogaster but shorter than the infections in D. simulans. As a result of this infectious period for wAna, we suggest the recent introduction of what is called the wRi strain of W. pipientis found to be rapidly spreading across several D. simulans populations within the past several decades had its origins as a horizontally transferred wAna strain that was infecting D. ananassae. Our population genetic results of D. ananassae GSC regulating gene contrast D. melanogaster and D. simulans where despite the potentially long period of infection in D. ananassae (at least longer than the current D. melanogaster infection), no significant evidence of positive selection was detected for the genes bam and bgcn whereas evidence of positive selection was observed for the genes pum and stwl. Our population genetic results thus do not support a simple W. pipientis conflict hypothesis causing adaptive evolution at bam and bgcn in all species of Drosophila, and reinforce that multiple sources of selection likely explain the heterogeneous pattern of positive selection observed for several important GSC proteins.
Results
Phylogenetics of W. pipientis in D. ananassae
The evolutionary history of the wAna strain of W. pipientis currently infecting D. ananassae populations was evaluated by examining ten wAna loci distributed evenly across the wAna genome, in 23 isofemale lines of D. ananassae from 17 different geographical locations around the world (fig. 1). Polymerase chain reaction (PCR) amplification of D. ananassae DNA from each line prior to antibiotic treatment revealed that 83% of these lines had positive evidence for W. pipientis infection (Apia77, NOU83, PPG90, and Samoa3 had negative PCR results). This suggests a worldwide prevalence of W. pipientis in the D. ananassae population. Despite the wide geographical range of wAna infection we found almost no sequence polymorphism across the approximately 7,200 bp of DNA sequenced, excluding the W. pipientis sequence from strain Samoa2 (discussed below). The only variants found were the gene mutL at site 997 and gene hcpA at site 205 both with a G/T heterozygous polymorphism for wAna that infected D. ananassae strain D38, and gene mutL at site 336 with an A/G heterozygous polymorphism for wAna that infected D. ananassae strain PNP1.
Fig. 1.

Wolbachia pipientis gene PCR results on worldwide Drosophila ananassae before (No Tet) and after (3× Tet) antibiotic treatment.(+) positive PCR result; (–) negative PCR result.
Because of prior evidence of a whole wAna genome integration into the D. ananassae chromosome (Hotopp et al. 2007), tetracycline-treated flies were also examined for wAna genes to distinguish PCR amplification of bacterial wAna genome versus the integrated genome. Almost half of the D. ananassae isofemale lines (RC102, 111DCebu, HNL0501, KMJ1, GB1, TBU146, TBU247, OGS98-K1, and VAU150) were infected with wAna based on a lack of PCR amplification of W. pipientis genes after tetracycline treatment (fig. 1). Strains for which wAna genes still amplified after tetracycline treatment (EZ104, D38, BKK13, PNP1, TB43, TI8, TBU3, and Jarkarta) presumably have an integrated wAna genome (fig. 1). However it should be noted that although treatment with tetracycline allows detection of wAna genes originating from the integrated genome, this approach cannot determine whether strains with an integrated genome also had a wAna infection. In accordance with Hotopp et al. (2007), we have also observed almost a complete absence of polymorphism between the wAna genome and the integrated wAna genome. However, our study includes a broader sample of worldwide D. ananassae population suggesting that the integrated genome has recently spread to a very wide geographic range.
The two genes with polymorphism before tetracycline treatment in D38 were still segregating as heterozygous sites in flies treated with tetracycline. This suggests that the integrated genome in this isofemale line has accumulated mutations in both genes and is now heterozygous. In the PNP1 isofemale line, tetracycline treatment reveals that the integrated gene had a variant fixed for A whereas prior to treatment, this line was polymorphic for A and G. As the reference wAna genome is fixed for G, this result suggests that PNP1 is likely to be infected with wAna that has the G variant but also has an integrated genome that had accumulated mutation and is now fixed with the A variant. These three segregating polymorphisms thus all appear to represent mutations that occurred in the integrated genome after it had inserted into the host, thus representing the D. ananassae host’s mutation rate rather than that of the infectious W. pipientis.
Drosophila ananassae isofemale line Samoa2 amplified for some but not all W. pipientis wAna PCR primers (those for mutL, ank1, ank3, and gpA did not amplify; fig. 1) suggesting a divergent strain of W. pipientis was present in this line. BLAST nucleotide search of the five MLST gene sequences from Samoa2 against nucleotide databases indicated that it did not originate from wAna but matched other W. pipientis MLST genes from different hosts with high identity (>95%). However, a completely identical sequence could not be found, suggesting that the W. pipientis infection in Samoa2 is a previously uncharacterized strain of W. pipientis. We have named this strain wAnaS for W. pipientis infecting D. ananassae Samoa2. The full genomic sequence and characteristics of wAnaS will be presented elsewhere.
D. ananassae Mitochondrial Phylogeny
The near absence of polymorphism in the approximately 7,200 bp of wAna sequence assayed is consistent with a recent invasion into the D. ananassae population. However, the lack of variability is also consistent with a very low wAna genomic mutation rate. As the both mitochondria and W. pipientis are materially inherited, variation in each will be in linkage disequilibrium. Thus, polymorphism within the faster evolving mtDNA could be used as an indirect inference for the history of W. pipientis invasion and spread within D. ananassae populations (following Hurst and Jiggins 2005). Thus, sequences from the mitochondrial genes CO1, CO2, and CytB were obtained from the same isofemale lines of D. ananassae that we had analyzed for W. pipientis.
The mtDNA phylogeny from all 23 isofemale lines was estimated by both maximum likelihood (ML) and Bayesian methods (fig. 2). The most distinguishing feature of the reconstructed D. ananassae mtDNA tree was the presence of two major clades. As all four samples from the Samoan region (Apia77, PPG90, Samoa2, and Samoa3) grouped together, we designated them as the S clade and differentiated it from the rest of the isofemale lines. The phylogeny had poor resolution in differentiating all D. ananassae mtDNA haplotypes; however, branches with the highest support were for haplotypes in isofemale lines that carried the infectious wAna (fig. 2A and B). A closer look at strains with the infectious wAna (fig. 2 strains with striped pentagon) showed relatively deep branches that clearly differentiated the wAna-infected line mtDNA haplotypes from another. Of particular note, the mtDNA phylogeny did not have a star-like topology expected after a recent selective sweep.
Fig. 2.
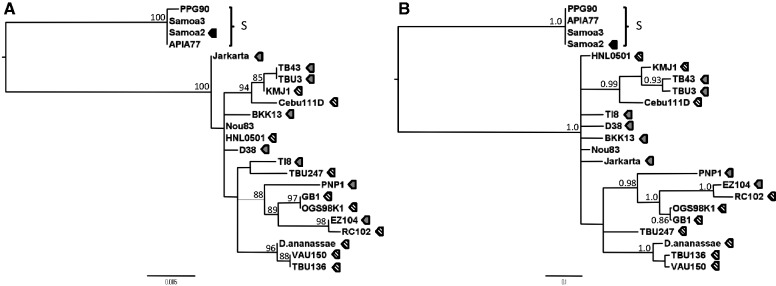
Phylogenetic reconstruction of worldwide Drosophila ananassae mitochondria using the third codon position. Genealogy was inferred using ML (A) and BI (B). The reference mitochondrial genome sequence of D. ananassae (BK006336) was included in the tree and labeled as D. ananassae. The tree was midpoint rooted. Bootstrap values of greater than 85% (A) and probability of greater than 0.85 (B) are only shown on the node of the tree. Pentagon shape filled with black color represent D. ananassae strain infected with wAnaS. Mitochondrial haplogroups that are potentially in linkage disequilibrium with wAnaS are labeled as the S group. Pentagon shape with stripes indicates D. ananassae with the infectious wAna, whereas pentagon shape filled with grey indicates D. ananassae strains with evidence of the integrated wAna genome but uncertain if they also have the infectious wAna (see text). Strains without pentagon shape have no evidence of wAnaS or wAna infection. The length of the scale bar for both trees represents the number of mutations per site.
Monophyly was not seen for the infected versus uninfected strains. For example, the mtDNA haplotypes from four uninfected isofemale lines (APIA77, NOU83, PPG90, and Samoa3) grouped with the infected line haplotypes. Although it is possible that these four lines simply have not yet been infected with W. pipientis, it is also possible that this pattern is due to the incomplete maternal transmission of W. pipientis suggested for D. melanogaster (Richardson et al. 2012; Early and Clark 2013). The mtDNA haplotypes defining the S clade grouped with high statistical support (bootstrap value 100, Bayesian probability 1.0) with the clade itself having an increased divergence compared with other haplotypes. As Samoa2 of this clade had evidence of wAnaS infection (fig. 2 strain labeled with black colored pentagon), the increase in divergence is likely to be due to a wAnaS-mediated mitochondrial sweep. Although the isofemale Apia77, PPG90, and Samoa3 did not have any evidence of W. pipientis infection (or integration), the fact that they share a virtually identical mtDNA haplotype to that in Samoa2 raises the possibility that all four lines had their mtDNA swept to fixation by a W. pipientis infection that was subsequently lost by lines Apia77, PPG90, and Samoa3.
The timing of the W. pipientis-induced mtDNA sweep through D. ananassae was inferred by estimating the TMRCA of all mtDNA haplotypes. Although we have evidence of two separate invasions of W. pipientis (wAna and wAnaS) into D. ananassae, we are unable to infer which invasion occurred first. Thus, we estimated the TMRCA of all mtDNA haplotypes to estimate the time of the first mtDNA sweep, and presumably the first infection by W. pipientis. Using the program BEAST and assuming a rate of mutation for mtDNA of 6.2 × 10−8 substitutions/site/generation (Haag-Liautard et al. 2008), we infer the median TMRCA for all D. ananassae mtDNA haplotypes at 2.1 × 105 generations, with 95% highest posterior density interval (HPD) ranging from 1.4 × 105 to 3.0 × 105 generations. As wAna infection is more prevalent worldwide (fig. 1), the TMRCA of mtDNA infected with the infectious wAna (fig. 2 strains labeled with striped pentagon) would represent the initial time point when wAna had invaded D. ananassae and spread throughout the worldwide range of this species. We cannot rule out a scenario where wAnaS was the first W. pipientis infection in D. ananassae but was incompletely replaced by wAna. In this later case, the TMRCA of mtDNA with wAna infection would represent the minimum time D. ananassae have been infected with W. pipientis. The median TMRCA for wAna-infected mtDNA was 1.1 × 105 generations (95% HPD from 6.9 × 104 to 1.5 × 105 generations).
Of interest is how the age of infection for D. ananassae compares with those of other Drosophila species. We thus used the same approach and program to estimate the TMRCA of mtDNA from D. melanogaster and D. simulans. Using only the coding sequence of the mtDNA genome obtained from the study of Ballard (2000), the median mtDNA TMRCAs were estimated at 7.9 × 104 generations (95% HPD from 5.2 × 104 to 1.1 × 105 generations) for D. melanogaster, and 8.8 × 105 generations (95% HPD from 7.5 × 105 to 1.0 × 106 generations) for D. simulans. Although our TMRCA estimates for D. melanogaster are based on only two mtDNA genomes from Ballard (2000), our estimate is similar to previous population genomic studies based on large numbers of D. melanogaster mtDNA genome sequences (Richardson et al. 2012; Early and Clark 2013). Thus, the current infection of W. pipientis in D. ananassae appears to be roughly 1.3 times older than that in D. melanogaster but only one-eighth as long as for D. simulans.
We then examined the site frequency distribution of the mtDNA as a recent selectively driven sweep caused by the spread of W. pipientis is predicted to skew the frequency distribution toward an excess of rare alleles (Simonsen et al. 1995; Fu 1997). However, after a W. pipientis-mediated mtDNA sweep, the host mtDNA polymorphism is expected to recover to its equilibrium site frequency distribution over time (Dyer and Jaenike 2004). Tests of a fit of measures of the site frequency spectrum to the expectations of an equilibrium neutral model failed to detect any skew in frequency for the full D. ananassae mtDNA data set (Tajima’s D [Tajima 1989] = −0.78, P = 0.28; Fu–Li’s D [Fu and Li 1993] = 0.53, P = 0.31). Even examination of just the mtDNA haplotypes from isofemale lines currently infected with wAna (fig. 2 strains labeled with striped pentagon) showed no departure from an equilibrium neutral model (Tajima’s D = −0.60, P = 0.30; Fu–Li’s D = −0.58, P = 0.33). These results suggest that any maternally driven selective sweep of mtDNA occurred sufficiently long enough ago so that the nucleotide site frequency spectrum has now recovered back to equilibrium levels.
Population Genetics of Several Key GSC Genes in D. ananassae
Given the potential for W. pipientis to manipulate the GSCs and reproduction of their host insect species, we examined levels and patterns of DNA sequence variation for seven GSC genes from two ancestral South East Asia populations of D. ananassae. Nucleotide polymorphism levels for these GSC genes (table 1) were generally similar to those from previous studies of D. ananassae population (Das et al. 2004; Grath et al. 2009). Additionally, overall levels of polymorphisms were similar between the two populations. Population differentiation between the two populations as measured by FST was less than that observed between an ancestral and nonancestral population of D. ananassae (Schug et al. 2007). Thus, we considered the two populations as a single population for further population genetic analysis.
Table 1.
Polymorphism and Divergence of GSC Regulating Genes in Drosophila ananassae BKK and BOG Population.
| Synonymous |
Nonsynonymous |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene | n | S | θ | π | KJC | θ | π | KJC | FST |
| bam | |||||||||
| BKK | 14 | 8 | 0.0076 | 0.0094 | 0.326 | 0.0003 | 0.0001 | 0.049 | 0.03 |
| BOG | 9 | 9 | 0.0089 | 0.0095 | 0.320 | 0.0007 | 0.0006 | 0.049 | |
| bgcn | |||||||||
| BKK | 13 | 101 | 0.0346 | 0.0368 | 0.289 | 0.0017 | 0.0018 | 0.012 | 0.10 |
| BOG | 10 | 96 | 0.0369 | 0.0375 | 0.294 | 0.0012 | 0.0012 | 0.012 | |
| nos | |||||||||
| BKK | 14 | 42 | 0.0198 | 0.0154 | 0.260 | 0.0022 | 0.0019 | 0.026 | 0.05 |
| BOG | 5 | 16 | 0.0106 | 0.0111 | 0.253 | 0.0011 | 0.0009 | 0.026 | |
| ote | |||||||||
| BKK | 14 | 64 | 0.0482 | 0.0548 | 0.244 | 0.0053 | 0.0052 | 0.033 | −0.05 |
| BOG | 4 | 44 | 0.0454 | 0.0431 | 0.226 | 0.0040 | 0.0039 | 0.034 | |
| pum | |||||||||
| BKK | 11 | 37 | 0.0267 | 0.0252 | 0.143 | 0 | 0 | 0.005 | — |
| stwl | |||||||||
| BKK | 14 | 78 | 0.0229 | 0.0226 | 0.278 | 0.0040 | 0.0031 | 0.082 | 0.12 |
| BOG | 7 | 47 | 0.0178 | 0.0176 | 0.282 | 0.0028 | 0.0023 | 0.082 | |
| Yb | |||||||||
| BKK | 10 | 103 | 0.0239 | 0.0216 | 0.470 | 0.0039 | 0.0041 | 0.156 | — |
| Average | |||||||||
| BKK | — | — | 0.0262 | 0.0266 | 0.2872 | 0.0025 | 0.0023 | 0.0520 | — |
| BOG | — | — | 0.0239 | 0.0238 | 0.2748 | 0.0020 | 0.0018 | 0.0404 | — |
Note.—n, the sample size examined; S, the number of segregating sites; Θ, polymorphism measured as Watterson’s theta; π, polymorphism measured as average pairwise differences; KJC, average nucleotide difference between D. ananassae and D. atripex with Jukes–Cantor correction; FST, fixation index for measuring genetic differentiation between the BKK and BOG population.
The average GSC gene synonymous site divergence was only slightly elevated at 28% compared with the average synonymous divergence observed between D. ananassae and D. atripex of around 20% from the study of Grath et al. (2009). Nonsynonymous divergence was elevated for bam and stwl (4.8% and 8%, respectively) which is more than twice the average nonsynonymous divergence of 2% seen between D. ananassae and D. atripex (Grath et al. 2009). For Yb, both synonymous and nonsynonymous site divergences were elevated at 35% and 12%, respectively.
The elevated divergence in synonymous sites seen in D. ananassae (table 1) raised the question of selection on synonymous sites. We examined evidence of selection on synonymous sites using a method (DuMont et al. 2004) that estimates the number of preferred and unpreferred sites and mutations that have fixed specifically along the D. ananassae lineage. Results showed significant evidence of selection on synonymous sites in the genes bgcn, ote, and stwl all in the direction of significantly favoring the fixation of preferred mutations (table 2).
Table 2.
Number of Synonymous Preference Sites and Preferred and Unpreferred Mutations Fixed along the Drosophila ananassae Lineage for Each GSC Genes.
| Gene | Number of Preferred Sites | Preferred Fixations | Number of Unpreferred Sites | Unpreferred Fixations | RP/U | FDR P Value |
|---|---|---|---|---|---|---|
| Bam | 76 | 16 | 140 | 12 | 2.46 | 0.087 |
| Bgcn | 146 | 25 | 563 | 17 | 5.67 | 5.206E-06* |
| Nos | 60 | 6 | 148 | 14 | 1.06 | 1.000 |
| Ote | 66 | 6 | 165 | 2 | 7.50 | 0.046* |
| Pum | 62 | 4 | 377 | 9 | 2.70 | 0.214 |
| Stwl | 106 | 21 | 409 | 14 | 5.79 | 2.535E-5* |
Note.—RP/U, ratio of preferred fixations over number of preferred sites to unpreferred fixations over number of unpreferred sites. FDR, false discovery rate. Significance was determined by two-tailed Fisher’s exact test. Listed P values are FDR-corrected values for multiple hypothesis comparisons.
*Significant FDR-corrected P value < 0.05.
Fay–Wu’s H (Fay and Wu 2000) was significantly negative only for the gene bgcn (table 3) indicating an excess of derived high frequency variants suggestive of a recent selective sweep. Analysis of variants using the CLSW method (Kim and Stephan 2002), only stwl and pum had a significantly better fit of a selection model versus the standard neutral model after multiple hypothesis correction. Interestingly, despite the significantly negative Fay–Wu’s H for bgcn, the CLSW method did not reject a neutral model. The ML estimate of strength of selection (2Nes) was 95.62 for pum and 143.71 for stwl, and the putative targets of selection were estimated toward the 3′-end of the coding DNA sequence (CDS) that we have physically sequenced. Using the 2Nes estimates of pum and stwl, simulations were conducted to generate 1,000 selection scenarios for each gene. The goodness-of-fit (GOF) statistics (Jensen et al. 2005), which distinguishes positive selection from population demography, for pum and stwl were not significantly different from a simulated distribution of GOF statistics from 1,000 selection scenarios. As both genes were not significantly different from a selection scenario, this suggested that the departures from an equilibrium neutral model for pum and stwl are not likely to be a false positive caused by a nonequilibrium demographic event.
Table 3.
Tests of Equilibrium Neutral Model Which Could Detect Recent Selective Sweeps.
| CLSW |
|||||
|---|---|---|---|---|---|
| Gene | FW’s H (FDR P value) | LR Value (FDR P value) | α | X | GOF-Sel (FDR P value) |
| bam | −0.925 (0.337) | 1.365 (0.518) | — | — | — |
| bgcn | −19.174 (0.016)* | 8.375 (0.087) | — | — | — |
| BKK | −12.321 (0.056) | 5.079 (0.337) | — | — | — |
| BOG | −12.267 (0.040) | 3.440 (0.518) | — | — | — |
| nos | 0.281 (0.542) | 2.203 (0.649) | — | — | — |
| ote | −3.739 (0.335) | 3.547 (0.446) | — | — | — |
| pum | 0.927 (0.518) | 5.995 (0.049)** | 95.62 | 2,399 | 61.708 (0.526) |
| stwl | −7.495 (0.047) | 13.431 (0.010)** | 143.71 | 2,178 | −81.122 (0.518) |
| BKK | −7.868 (0.049) | 8.729 (0.047)** | 42.22 | 2,408 | 64.791 (1.000) |
| BOG | −7.286 (0.051) | 7.158 (0.034)** | 59.43 | 2,092 | 139.856 (0.335) |
| Yb | 0.356 (0.567) | 2.466 (0.832) | — | — | — |
Note.—FW’s H, H statistics from Fay and Wu (2000); LR, natural log-likelihood ratio of selection versus neutrality as calculated by the method of Kim and Stephan (2002); α, the strength of selection measured by 2Nes; X, ML position of the beneficial mutation where the position is based on the Drosophila ananassae CDS; GOF-Sel, goodness of fit of the data to a selection model as calculated by the method of Jensen et al. (2005). FDR, false discovery rate. P values are FDR-corrected values for multiple hypothesis comparisons and are listed in parenthesis.
*Significant two-tailed FDR-corrected P value < 0.025; **significant one-tailed FDR-corrected P value < 0.05.
Although the FST for bgcn and stwl (table 1) was within the average FST seen across neutral and mitochondrial genes between the ancestral Bangkok (BKK), Thailand and Bogor (BOG), Indonesia populations (Schug et al. 2007, 2008), they were still the highest among the GSC regulating genes. This raises the possibility that the significant Fay–Wu’s H-test and CLSW test results for bgcn and stwl, respectively, simply reflect population structure rather than selection. The two genes were thus reanalyzed for departures from neutrality in each population (BKK and BOG) separately (table 3). In the case of stwl, both populations had a decreased strength of selection likely due to a decrease in sample size and subsequent loss in power to detect sweeps. However, for both populations the evidence of selection remained significant and the estimated position of the selective sweep was in a similar region to the combined population result. For bgcn, neither BKK nor BOG had a significant Fay–Wu’s H and CLSW test.
We used the McDonald–Kreitman (MK) test (McDonald and Kreitman 1991) to evaluate evidence of recurrent positive selection on amino acid sequences on the D. ananassae GSC regulating genes (table 4). To exclude the effect of mildly deleterious mutations that have not been purged and segregate at low frequencies in the population, polymorphisms that segregate as singletons were excluded from both synonymous and nonsynonymous sites (MK test results are qualitatively the same with singletons included, results not shown). Only stwl had a significant MK test result after multiple hypothesis corrections (table 4) and the Direction of Selection (DoS) (Stoletzki and Eyre-Walker 2011) was positive. However, as stwl had evidence of selection on synonymous site (table 2) a separate MK test was conducted by excluding synonymous changes that were potentially under selection (following Haddrill et al. 2008). Synonymous sites that had changed from its ancestral preferred codon to a preferred codon or from an ancestral unpreferred codon to an unpreferred codon were assumed to be selectively neutral as the preference of the codon has not changed. Using synonymous sites that have not changed preference as a neutral reference stwl still had a significant MK test (P = 0.007 after multiple hypothesis correction, DoS value = 0.305).
Table 4.
MK Test Result for the GSC Regulating Genes.
| Gene | Dn | Pn | Ds | Ps | DoS | FDR P Value |
|---|---|---|---|---|---|---|
| bam | 46 | 1 | 74 | 7 | 0.258 | 0.400 |
| bgcn | 28 | 10 | 160 | 83 | 0.041 | 0.517 |
| nos | 21 | 4 | 56 | 10 | −0.013 | 1.000 |
| ote | 26 | 12 | 51 | 45 | 0.127 | 0.261 |
| pum | 6 | 0 | 54 | 24 | 0.100 | 0.335 |
| stwl | 148 | 15 | 144 | 39 | 0.229 | 0.020* |
| Yb | 232 | 20 | 180 | 23 | 0.098 | 0.400 |
Note.—Dn, number of fixed nonsynonymous changes between Drosophila ananassae and D. atripex; Pn, number of nonsynonymous polymorphism within D. ananassae; Ds, number of fixed synonymous changes between D. ananassae and D. atripex; Ps, number of synonymous polymorphism within D. ananassae; DoS, Direction of Selection values (Stoletzki and Eyre-Walker 2011). FDR, false discovery rate. Significance was determined by the two-tailed Fisher’s exact test. Listed P values are FDR-corrected values for multiple hypothesis comparisons.
*Significant FDR-corrected P value < 0.05.
Long-term positive selection across the combined GSC genes was measured by estimating the proportion of fixed nonsynonymous differences that have been driven by positive selection (α). An explicit population genetic model (Eyre-Walker and Keightley 2009; Keightley and Eyre-Walker 2012) was used to estimate α and the distribution of fitness effect of nonsynonymous mutations in the GSC regulating genes. As a comparison we have estimated the same population genetic parameters in genes with sex-biased expression in D. ananassae (sex-biased genes listed in additional file 3 of the study Grath et al. 2009). Previous studies have shown that this class of genes experiences positive selection on nonsynonymous sites (Pröschel et al. 2006; Baines et al. 2008; Grath et al. 2009), and currently it is the only available D. ananassae population data set with CDS information. Although Grath et al. (2009) have estimated α for the D. ananassae sex-biased genes, the method we used incorporates a demographic model while jointly estimating α and the distribution of fitness on the nonsynonymous sites. Table 5 shows the estimate of demography, α, and the distribution of fitness with its 95% confidence intervals (CIs) for both GSC regulating genes and sex-biased genes. Both data sets show evidence of demography where the sex-biased genes indicate a population expansion whereas the GSC regulating genes a population bottleneck. However, the demographic estimates between the two data sets are not significantly different due to the overlapping CIs. For both sets of loci the predicted change in population size was estimated to be in the distant past, between 5N2 and 6.3N2 generations ago (where N2 is the current effective population size).
Table 5.
Estimates of Demography, Fraction of Adaptive Substitution, and Distribution of Fitness Effect on Nonsynonymous Sites.
| Proportion of Mutations in Different Selection Intensity |
|||||||
|---|---|---|---|---|---|---|---|
| Data Set | N2/N1 | t/N2 | α | Nes = 0–1 | Nes = 1–10 | Nes = 10–100 | Nes = 100> |
| GSC | 0.45 (0.19–6.71) | 6.28 (5–4,681.86) | 0.75 (0.45–0.85) | 0.06 (0.02–0.09) | 0.04 (0.01–0.20) | 0.07 (0.01–0.53) | 0.84 (0.20–0.96) |
| Sex-biased | 3.10 (0.35–10) | 5.00 (5–189.18) | 0.86 (0.51–0.99) | 0.02 (<0.01–0.08) | 0.10 (0.01–0.17) | 0.42 (0.10–0.85) | 0.46 (0.03–0.82) |
Note.—N2/N1, the estimated demography as the ratio of current and past population size; t/N2, the estimated time of demographic change scaled by the current population size (N2); α, proportion of nonsynonymous sites fixed by positive selection; Nes, strength of selection against newly arising mutations on nonsynonymous sites. 95% CIs are shown in parenthesis.
Estimates of α were high and significantly different from zero for GSC regulating genes where an estimated 75% of the fixed nonsynonymous sites were due to positive selection. With evidence of long-term positive selection in the gene stwl from the MK test (table 4), the majority of this 75% could be due to extensive positive selection at stwl. However, a reanalysis of the GSC data set with stwl removed revealed that α was still high at 65%. In addition, even though none of the MK test results beside stwl was significant for the other GSC regulating genes, the DoS values were still positive for most of the genes (table 4) and corroborate the high estimated α value (table 5).
The α of 75% for the total GSC data set is comparable to α in sex-biased genes that also have evidence of adaptive evolution. Despite a large proportion of the fixed nonsynonymous sites were driven by positive selection, the vast majority of newly arising deleterious mutations were strongly deleterious for both data sets (Nes > 10, where Ne is the effective population size and s is the intensity of selection against deleterious mutations). Differences in the distribution of fitness effect for the GSC regulating and sex-biased genes were seen in the extreme category where GSC regulating genes had higher proportions of its mutations in the extremely deleterious category (Nes > 100). Both data sets, however, had similar proportions of its slightly deleterious or nearly neutral mutations (0 < Nes < 1) at 6% and 2%, respectively.
Discussion
The Coevolutionary History between W. pipientis and D. ananassae
Based on our analysis of phylogeny, population genetics, and estimates of TMRCA of mtDNA, we conclude that W. pipientis has not recently invaded into D. ananassae. Schug et al. (2008) have shown that levels of FST in the mtDNA were comparable to X-linked nuclear data (Das et al. 2004). If W. pipientis had recently swept through the worldwide D. ananassae population, it would have homogenized the mtDNA haplotypes leading to reduced levels of FST. Thus the comparable levels of FST in both mtDNA and nuclear genes of D. ananassae, which is known to have high genetic structure (Vogl et al. 2003; Das et al. 2004; Schug et al. 2007), further corroborate our inference that a significant amount of evolutionary time has passed since the last W. pipientis-mediated mtDNA sweep. As theory predicts the frequency distribution of infected and uninfected mtDNA polymorphism to be similar after W. pipientis has swept through a population (Dyer and Jaenike 2004), future studies focusing on the mtDNA and nuclear polymorphisms of infected and uninfected D. ananassae would be valuable in confirming our results.
It is intriguing how W. pipientis had managed to spread through the worldwide population of D. ananassae despite the strong geographical structuring (Vogl et al. 2003; Das et al. 2004; Schug et al. 2007) and assortative mating preference (Schug et al. 2008) observed in the host. One possibility stems from the observation that the TMRCA of the worldwide mitochondria at 21,000 years (assuming ten generations per year for D. ananassae) coincides with the time when rising sea levels had begun to geographically isolate the ancestral population of D. ananassae (Das et al. 2004). This geographic event prompted the division of the ancestral populations and subsequent migration into the peripheral and South Pacific locations. We hypothesize then W. pipientis had swept through the ancestral population of D. ananassae and these infected populations subsequently colonized the peripheral and South Pacific regions ultimately leading to a worldwide infection status for D. ananassae.
The lack of polymorphism we see at the wAna genes surveyed for nine infected lines of D. ananassae might suggest a more recent sweep of wAna through the species. However, it is probably due to the low rate of mutation for W. pipientis (Raychoudhury et al. 2009; Richardson et al. 2012; Early and Clark 2013).
Other Drosophila species such as D. innubila (Dyer and Jaenike 2004), D. quinaria (Dyer et al. 2011), and D. simulans (Ballard 2004) also have evidence of long-term association with its resident W. pipientis. This contrasts with phylogenetic studies that have shown a general discordance between the arthropod infecting W. pipientis phylogeny and the host phylogeny, indicating frequent horizontal transfer and transient coevolutionary periods between W. pipientis and its arthropod hosts (Baldo et al. 2006; Baldo and Werren 2007). However, as we and others have shown, it is possible for W. pipientis to have a stable infection with its host.
D. ananassae mtDNA Phylogeny
Our mitochondrial phylogeny results contrast with those of Schug et al. (2008) who suggested the South Pacific populations (Apia, Thursday Island, and Trinity Beach) were ancestral to all other D. ananassae population. Their inferences were based on the South Pacific population having a basal mitochondrial haplotype and mate choice experiments showing strong preference of mating. We hypothesize two possible scenarios for the discrepancy: 1) With evidence of the newly identified wAnaS variant from our study, it is possible that the South Pacific population mtDNA represent divergent lineages that were in linkage disequilibrium with the wAnaS sweep; or 2) it is possible that the original D. ananassae ancestral population in central Southeast Asia (Vogl et al. 2003; Das et al. 2004; Schug et al. 2007) had an incomplete W. pipientis sweep. Here, some of these uninfected ancestral D. ananassae would have escaped the invasion and colonized regions in the South Pacific area. The basal haplotypes of the South Pacific population would then represent ancestral mitochondrial haplotypes that have not been affected by W. pipientis.
Our incomplete W. pipientis sweep hypothesis, however, may conflict with previous studies of W. pipientis invasion since an initial introduction of W. pipientis usually results in the rapid spread throughout the population, and ultimately causing high frequency of the infection (Turelli and Hoffmann 1995; Kriesner et al. 2013). If W. pipientis population had invaded the ancestral D. ananassae population, it should have led to a complete W. pipientis sweep. However, an incomplete W. pipientis sweep is possible if there are biogeographical barriers or potential host factor that confers resistance to the W. pipientis invasion (Dean et al. 2003). Both of our samples from Thursday Island and Trinity Beach have evidence of mtDNA sequences that are phylogentically related to the infected mtDNA haplotype (fig. 2). As W. pipientis-mediated mitochondrial sweeps can lead to false phylogenetic inferences (Hurst and Jiggins 2005), we conclude that the difference in mtDNA phylogeny from Schug et al. (2008) is mainly due to our analysis of D. ananassae samples that have had a W. pipientis-mediated mitochondrial sweep.
W. pipientis Has Potentially Horizontally Transferred from D. ananassae into D. simulans
Drosophila simulans is infected by several diverse strains of W. pipientis infection (Merçot and Charlat 2004) and results from previous studies have suggested that D. simulans has been infected with W. pipientis for a significant period of time (Ballard 2000, 2004; Dean and Ballard 2005). One of the W. pipientis strains infecting D. simulans, named wRi, is the most common and detected in every continent of the world (Ballard 2004). The identical mtDNA haplotypes in worldwide D. simulans stocks infected with wRi suggest a single and recent origin and spread (Hale and Hoffmann 1990; Turelli and Hoffmann 1995; Ballard 2004; Kriesner et al. 2013). The origin of the wRi variant is unknown but the high genomic similarity between wAna and wRi (Salzberg et al. 2005) has suggested recent horizontal transfer of W. pipientis between the two species. Here, our data suggest that wAna has swept through the world D. ananassae population at a more ancient time compared with the recent worldwide sweep of wRi in D. simulans (Ballard 2004). Thus, we hypothesize that the current wRi strain in D. simulans is originally a wAna variant that had horizontally transferred from D. ananassae to D. simulans. We note that D. suzukii is also infected with a W. pipientis strain highly similar to the wRi variant (Siozios et al. 2013) suggesting D. suzukii also as a potential origin of wRi. However, at least in the United States, D. suzukii was not observed in California until 2008 (Hauser 2011), which is later than the rapid wRi spreading across D. simulans populations during the late 1980s in California (Turelli and Hoffmann 1991). As this wRi from California is thought to be identical to the wRi that swept through D. simulans from Eastern Australia (Kriesner et al. 2013) and potentially the world (Hale and Hoffmann 1990; Ballard 2004), we argue that D. suzukii is not a likely candidate as the origin of wRi. As Ballard (2004) has hypothesized Ecuador as the initial location of wRi infection in D. simulans, we would predict the existence of an Ecuador D. ananassae population to harbor a wAna variant that originated the D. simulans wRi infection. Future development of polymorphic loci to differentiate within wAna and wRi diversity would help in testing our hypothesis.
The Evolution of GSC Regulating Genes in D. ananassae
We have found significant evidence of selection on synonymous sites for the genes bgcn, ote, and stwl in D. ananassae (table 2). Synonymous mutations have traditionally been assumed to be close to near neutrality. However, studies have increasingly shown significant evidence of selection on synonymous sites (e.g., DuMont et al. 2004; Hershberg and Petrov 2008). For example in the Drosophila lineage, genome-wide studies have shown significant preference in codon usage (Singh et al. 2007; DuMont et al. 2009). Unequal usage of the codon table is thought to be a result from translational accuracy (Akashi 1994), translational efficiency (Akashi 2001), or a combination of both accuracy and speed in translation (Drummond and Wilke 2008). Expression of GSC regulating genes is highly regulated where in some cases it is briefly expressed in the GSC and is immediately shut off in the differentiated cell, which is one cell diameter away from the GSC. Reflecting this tight control in expression, it is likely that bgcn, ote, and stwl have significant evidence of selection on synonymous sites.
Analysis of the seven GSC regulating genes showed evidence of recent selective sweeps at pum and stwl, and long-term selection at nonsynonymous sites for stwl. Demographic events can lead to false inferences of selection, and for D. ananassae previous studies have suggested a possibility of a demographic expansion (Das et al. 2004; Schug et al. 2007; Heled and Drummond 2008). However, effects such as population expansion are expected to affect the frequency spectrum of multiple genes toward an excess of low-frequency variants (Slatkin and Hudson 1991; Fu 1997). We argue that our results for stwl and pum are not affected severely by demography because five of the seven genes (bam, bgcn, nos, ote, and Yb) did not show deviations from neutrality. In the following, we discuss the significance of positive selection in pum and stwl, and possible drivers of selection acting on these genes.
One of the genes under positive selection, pum, is a RNA-binding protein originally identified to determine the anterior–posterior polarity in Drosophila embryos (Nusslein-Volhard et al. 1987). It is also involved in a dual role during oogenesis where it interacts with nos to retain GSC characteristics (Lin and Spradling 1997; Forbes and Lehmann 1998; Wang and Lin 2004) and interacts with brain tumor during differentiation (Harris et al. 2011). In pum, the estimated site of selection was at position 2399 of the partial D. ananassae pum CDS which corresponds to a region just outside the evolutionary conserved RNA-binding Pumillio Homology Domain (Zamore et al. 1997) (table 3). Interestingly, this region coincided with the region that harbored all the nonsynonymous-fixed differences between D. ananassae and D. atripex (table 4). Although a localized MK test specifically on the region where all the nonsynonymous-fixed differences have accumulated was not significant, a sliding window analysis of the Ka/Ks (nonsynonymous to synonymous divergence) ratio showed a spike of Ka/Ks > 1 near the location of selective sweep (results not shown). These findings suggest that the significant evidence of positive selection in pum may have been driven by the interaction between W. pipientis and D. ananassae. Why pum but not bam and bgcn were under positive selection could be explained by the difference in interaction of different strains of W. pipientis may have with its host. For example, all strains of W. pipientis are known to preferentially localize in the somatic stem cell (SSC) niche of Drosophila germarium (Frydman et al. 2006; Toomey et al. 2013). In the case of wAna however, in addition to the SSC niche, a considerable amount of concentration is also seen in the GSC niche, whereas this GSC niche accumulation is not seen for wMel (W. pipientis infecting D. melanogaster) (Toomey et al. 2013). The variation in the W. pipientis tropism suggests the possibility of varying host manipulative mechanisms for different strains of W. pipientis. Thus the GSC regulating genes in conflict with W. pipientis may differ between Drosophila species, and this highlights the value of surveying the evolution of multiple GSC regulating genes.
stwl was the only GSC regulating gene that had both recent and long-term evidence of adaptive evolution in D. ananassae. CLSW estimated the target of selection at position 2178 of the D. ananassae stwl CDS. This candidate region was not within or close to the known protein domains MADF and BESS motif of stwl (Clark and McKearin 1996). Due to its role in chromatin modification and epigenetic regulation (Maines et al. 2007; Yi et al. 2009), the selective driver for stwl could be conflict with TE. TEs are genomic parasites that cause deleterious insertions and excisions in the chromosomes, which results in the decrease in the organisms’ fitness (Slotkin and Martienssen 2007). Previous studies have suggested that the adaptive evolution of chromatin-binding proteins is caused by an arms race between the host and its TEs (Vermaak et al. 2005; Klattenhoff et al. 2009). Thus, stwl being a chromatin-binding factor may also be rapidly evolving due to a potential role in silencing TEs.
Significant Long-Term Positive Selection across the GSC Regulating Genes
When analyzed individually only two of seven GSC genes had evidence of positive selection (tables 3 and 4); however, when the GSC genes were analyzed together a large proportion of the fixed nonsynonymous changes were driven to fixation by positive selection along the D. ananassae lineage (table 5). The possibility of TEs potentially driving the evolution of stwl illustrates that other germline parasites may also be driving the evolution of the Drosophila GSCs (Werren 2011).
Alternatively the drivers of selection are not only limited to germline parasites, for example, the selective driver could be caused by factors within the GSC itself. Germline clonal experiments have shown increased competition for germline niche occupancy in GSCs with bam mutations (Jin et al. 2008), suggesting the potential for competition between GSCs. Shen et al. (2009) have shown that the gene eukaryotic initiation factor 4A is able to partially suppress bam mutant ovary phenotypes. Thus, GSC regulating genes could be evolving rapidly to outcompete the niche occupancy of its sister GSC or to suppress this competitive behavior as the most competitive GSCs from the study Jin et al. (2008) were tumorous cells.
In addition, with a recent study showing evidence of selection for competent mitochondria during oogenesis (Ma et al. 2014) mitochondrial–nuclear conflict may be another factor driving the evolution of GSC regulating genes. With the germarium as the potential site of selection (Hill et al 2014) further suggesting the possibility of a mitochondrial–nuclear conflict as a possible driver of selection.
Finally, W. pipientis may still be the driver of selection for some GSC regulating genes (Bauer DuMont et al. 2007). However, our results show the difficulty of testing this hypothesis for two reasons: 1) A present-day infection by W. pipientis is not sufficient to predict the coevolutionary history between the symbiont and its host, and 2) without the temporal information of the infection, a replacement of an old W. pipientis strain with a newer strain (Kriesner et al. 2013) can be mistaken for a novel recent W. pipientis invasion. With these caveats it would be beneficial to focus on host populations that have had a very recent W. pipientis invasion and search for signatures of recent selective sweeps in the host GSC regulating genes. Drosophila simulans would be a candidate species to examine as there are populations that have recently been invaded in California (Turelli and Hoffmann 1991) and Eastern Australia (Kriesner et al. 2013), populations that have been stably infected with W. pipientis for a long period of time (Ballard 2004), and populations that are uninfected and potentially resistant to W. pipientis (Dean et al. 2003).
Materials and Methods
Analysis of W. pipientis Genomic and D. ananassae Mitochondria Sequences
We would like to follow the convention of Lo et al. (2007) and designate all Wolbachia infecting D. ananassae as the species W. pipientis. In order for consistency with previous W. pipientis studies, we designate the W. pipientis infecting D. ananassae as wAna (Salzberg et al. 2005). Worldwide samples of isofemale D. ananassae lines were obtained from A. Kopp. The sample identifier name and location are: Apia77 (Samoa), BKK13 (Bangkok, Thailand), Cebu111D (Cebu, Philippines), D38 (India), EZ104 (Ethiopia), GB1 (Mauritius), HNL0501 (Oahu, USA), Jarkarta (Jarkarta, Indonesia), KMJ1 (Japan), NAN84 (Japan), NOU83 (Noumea, New Caledonia), OGS-98K1 (Japan), PNP1 (Phnom Pen, Cambodia), PPG90 (Pago pago, Samoa), RC102 (Rwanda), Samoa2 (Samoa), Samoa3 (Samoa), TB43 (Trinity Beach, Australia), TBU3 (Tonga), TBU136 (Tonga), TBU247 (Tonga), TI8 (Thursday Island, Australia), and Vau150 (Vava’u, Tonga). Stocks of D. atripex and D. bipectinada were obtained from the Drosophila species stock center.
Diversity of W. pipientis in D. ananassae was first examined by genotyping five conserved genes (gatB, coxA, hcpA, ftsZ, and fbpA) designed by the MLST procedure (Baldo et al. 2006). The MLST genes are mostly used for W. pipientis interspecific strain identification and have weak power in identifying intraspecific variation (e.g., Atyame et al. 2011), thus an additional four genes were also examined: DNA mismatch protein: mutL (WwAna1612), ankyrin genes: ank1 (WwAna0563) and ank3 (WwAna0805), phage-related gene: phage terminase large subunit gpA (WwAna1570). Primers for the genes mutL, ank1, ank3, and gpA were designed using the genomic sequence of wAna (Salzberg et al. 2005). Also the wsp gene traditionally used for the identification of W. pipientis was sequenced using the wsp primers designed by Baldo et al. (2005). To avoid amplification of W. pipientis genes that have integrated into the D. ananassae genome (Hotopp et al. 2007), all D. ananassae were fed 200 μg/ml of tetracycline for three generations to cure them of W. pipientis. Previous study has shown that 200 μg/ml is enough to completely cure of W. pipientis in Drosophila even after one generation of application (Osborne et al. 2012).
Conserved mitochondrial primers designed from the study Simon et al. (1994) were used to sequence the genes CO1, CO2, and CytB in all three species. DNA was extracted from adult flies using the Puregene Core Kit A DNA isolation kits (Qiagen). Sequencing was performed at Cornell University Life Sciences Core Laboratories Center (http://cores.lifesciences.cornell.edu/brcinfo/?p=about, last accessed June 2014) using ABI chemistry and Applied Biosystems Automated 3730 DNA Analyzer. All regions of the genes were sequenced at least a 2× coverage with sequence editing and assembly conducted using the program Sequencher 5.0 (Gene Codes). The D. ananassae mitochondria genome assembled from the study Montooth et al. (2009) was also incorporated into our study. Stop codons were removed and sequence alignment was conducted in the program MEGA5 using the algorithm MUSCLE. All three genes were concatenated into one supergene and only the third position of the codon was used for further phylogenetic analysis.
ML and Bayesian Inference (BI) methods of phylogenetic reconstruction on D. ananassae mitochondrial sequences were conducted using the programs RAxML7.4.2 (Stamatakis 2006) and MrBayes 3.2.1 (Ronquist et al. 2012), respectively. The user-friendly version of RAxML, raxmlGUI interface (Silvestro and Michalak 2012) was used to set the initial parameters for the ML phylogenetic trees. A general time reversible model with rate heterogeneity following a gamma distribution (GTR + G) was used for the DNA substitution matrix. The reconstruction of the phylogeny was conducted with the ML + thorough bootstrap option (-f b in RAxML) with runs = 100 and bootstrap = 1,000. This setting selects the best ML tree generated by 100 ML-based optimization from a 100 different starting tree generated by randomized Maximum Parsimony. Confidence of the tree was assessed by generating 1,000 nonparametric bootstrap runs, which were subsequently drawn on the best scoring ML tree found from the initial 100 runs. For BI phylogenetic tree, the GTR + G (lset nst = 6 rates = gamma in MrBayes) model was implemented with chains running for 5 million generations and sampling every 500 generations using the program MrBayes. At the end of the run, the standard deviation of the split frequencies was less than 0.01 and potential scale reduction factor was close to 1.0. The first 25% of the sampled generations were discarded as “burn-in” before summarizing the tree and branch length information. All trees were displayed using the program FigTree ver 1.4.0 (http://tree.bio.ed.ac.uk/software/figtree/, last accessed January 2014).
To estimate the potential time of W. pipientis invasion, we estimated the coalescent time of all D. ananassae mitochondrial haplotypes using the program BEAST 1.8.0 (Drummond et al. 2012). As mitochondria and W. pipientis are in linkage disequilibrium with each, other the initial invasion would lead to a selective sweep of mitochondrial variation from the population. Analyses were conducted assuming the HKY + G model of DNA substitution. A strict molecular clock was enforced using the D. melanogaster mitochondrial mutation rate estimated from the study Haag-Liautard et al. (2008) (6.2 × 10−8 mutations per site per fly generation). Analyses were run with 50 million generations and 25% of the initial chains were discarded as “burn-in” before analysis. The program Tracer 1.5 (http://tree.bio.ed.ac.uk/software/tracer/, last accessed January 2014) was used to check for convergence of chains and the effective sample size for all parameters was higher than 200.
Analysis of D. ananassae GSC Population Genetics
Drosophila ananassae has a high degree of population structure and the ancestral population is estimated to originate from Southeast Asia (Das et al. 2004). For the GSC population genetic analysis, D. ananassae population samples from this ancestral population (BKK and BOG) provided by M. Schug were used. For a close outgroup sequence, D. atripex provided by M. Schug was used to sequence D. atripex GSC genes using primers designed from the D. ananassae genome sequence (Assembly August 2005; http://genome.ucsc.edu/, last accessed June 2014). A third distant outgroup sequence was obtained using the genome sequence of D. bipectinada provided by the Baylor College of Medicine Human Genome Sequencing Center website (http://www.hgsc.bcm.tmc.edu, last accessed June 2014). The majority of the coding regions were sequenced for all seven genes. Large intron regions that were difficult to sequence due to indel polymorphisms were avoided. Due to incomplete annotation of the pum gene in D. ananassae, the 3′-end of the gene was only sequenced for both D. ananassae and D. atripex. In Yb, the full coding region was sequenced in D. ananassae whereas only the 5′ half of the gene could be sequenced in D. atripex. In D. bipectinada, a reliable sequence homologous to the D. ananassae Yb gene could not be found nor computationally annotated.
DNA extraction and sequencing protocols were conducted under the same protocol as previously reported in our W. pipientis and D. ananassae mitochondrial phylogeny section. Heterozygous sites were dealt by estimating the phase of the sequences using the program PHASE version 2.1 (Stephens et al. 2001). One of the haplotypes estimated by the program for each line was then randomly selected for further analysis.
Sequence alignment was conducted in the program MEGA5 (Tamura et al. 2011) using the algorithm MUSCLE (Edgar 2003). Population genetic analysis was conducted using the program DnaSP version 5 (Librado and Rozas 2009) to calculate population genetic statistics θw (Watterson’s theta), π (average pairwise difference), and K (interspecific divergence). Population structure was measured using DnaSP by calculating the population differentiation statistics FST (Lynch and Crease 1990).
Method to Detect Evidence of Selection on Synonymous Sites
Selection on synonymous sites was examined using a divergence-based method developed by DuMont et al. (2004) (cftest), which estimates the effective number of preferred and unpreferred sites and changes for a gene. Similar to the method of Nei and Gojobori (1986), which estimates the number of nonsynonymous and synonymous sites, the cftest reconstructs a parsimonious ancestral sequence which is then estimated for the effective number of preferred and unpreferred sites. Afterwards, the lineage-specific number of preferred and unpreferred fixed differences per preferred and unpreferred sites can be compared using a standard 2X2 contingency table test. Due to evidence of species within the melanogaster subgroup having conserved codon usage (Vicario et al. 2007), the codon usage table of D. melanogaster (Shields et al. 1988; Akashi 1995) was used to determine the preference of codon usage in D. ananassae.
Method to Detect Evidence of Recent Positive Selection
To detect recent evidence of selective sweeps, methods that analyze the DNA variation within a population were used. Test of neutrality using the derived site frequency spectrum (Fay and Wu’s H; Fay and Wu 2000) was conducted using DnaSP. As a proxy for the neutral frequency spectrum, only the third codon position was used as the data. Significance of the Fay–Wu’s H value was evaluated using the coalescent simulator of DnaSP under conditions of recombination. As the recombination rate of D. ananassae is not known for every gene, DnaSP was used to estimate the recombination parameter R (=4Nr) where N is the population size and r is the recombination rate per sequence. Further analysis of detecting recent selective sweep was conducted using the population genetic model of Kim and Stephan (2002) (CLSW). CLSW compares the ML estimate of a selection versus neutral model using the observed unfolded site frequency spectrum and spatial distribution of the polymorphisms. Significance of the likelihood ratio score was determined by comparing it to a simulated distribution of likelihood ratios generated from a 1,000 neutral simulations with identical mutation rate θ (4Neμ) and recombination rate R (4Ner) of the candidate gene. Candidate GSC genes that rejected the neutral model (P < 0.05 after multiple test correction) were further evaluated by the GOF test (Jensen et al. 2005). The GOF value of the candidate gene with evidence of selective sweep was compared with GOF values obtained from the 1,000 data sets generated under a selection scenario.
Methods to Detect Evidence of Long-Term Positive Selection
Deviation from neutrality was also assessed using the MK test (McDonald and Kreitman 1991). The MK test detects evidence of long-term recurrent positive selection on nonsynonymous sites by comparing the ratio of polymorphism to divergence of nonsynonymous sites to synonymous sites (neutral reference). To estimate the direction of selection, a variant of the neutrality index (Rand and Kann 1996) named DoS (Stoletzki and Eyre-Walker 2011) was used. DoS values are positive under positive selection, zero under neutrality, and negative when slightly deleterious mutations are segregating.
A second approach to detect evidence of long-term positive selection was that of Eyre-Walker and Keightley (2009), an extension from the method of Keightley and Eyre-Walker (2007) implemented in the server http://lanner.cap.ed.ac.uk/∼eang33/dfe-alpha-server.html (last accessed September 2014). This method estimates the proportion of nonsynonymous sites in the GSC regulating genes that were fixed by positive selection (α). Assuming all newly arising nonsynonymous polymorphisms to be strongly deleterious, the site frequency spectrum of the observed data can be used to estimate the fitness (distribution of fitness effect) of those newly arising deleterious mutations. A neutral reference set from the observed data (i.e., synonymous site) is used to jointly infer a past instantaneous change in the population size. Estimates from the distribution of fitness effect are then used to estimate the expected proportion of fixed difference between two species that are neutral. α is then measured as the difference seen between the expected and observed fixed differences. To exclude the difference in effective population size and consequently different evolutionary history between the autosome and X chromosome, only the autosomal genes (bam, bgcn, nos, pum, and stwl) were used for this part of the analysis. The population genetic data set from the study Grath et al. (2009) was also analyzed as a comparison. The folded site frequency spectrum for 0-fold degenerate sites was used as the selected sites whereas the 4-fold degenerate sites were used for the neutral sites. As the outgroup D. atripex was not a population data set, mutations segregating as polymorphisms within the outgroup will be falsely treated as fixed differences. To mitigate the bias in segregating polymorphisms in the outgroup sequence, the method of Keightley and Eyre-Walker (2012) was used to eliminate polymorphisms in estimates of fixed differences between the two species. CIs were obtained by 220 bootstrap sampling by locus.
Correcting for Multiple Hypothesis Comparisons
Multiple hypothesis correction was done using the false discovery rate procedure described by Benjamini and Hochberg (1995) using the P values from all analyses that required a hypothesis testing (Fisher’s exact test for cftest result; simulated distribution for Fay–Wu’s H, CLSW test, and GOF test; Fisher’s exact test for MK test).
Acknowledgments
This work was supported by the National Institute of Health grant number GM095793 to C.F.A. and Daniel A. Barbash. The authors thank Julia Goodrich with help on PCR and sequencing. They also thank Vanessa L. Bauer DuMont, Jaclyn Bubnell, Daniel Zinshteyn, and the two anonymous reviewers for their helpful discussions and comments on this manuscript.
References
- Akashi H. Synonymous codon usage in Drosophila melanogaster: natural selection and translational accuracy. Genetics. 1994;136(3):927–935. doi: 10.1093/genetics/136.3.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi H. Inferring weak selection from patterns of polymorphism and divergence at “silent” sites in Drosophila DNA. Genetics. 1995;139:1067–1076. doi: 10.1093/genetics/139.2.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi H. Gene expression and molecular evolution. Curr Opin Genet Dev. 2001;11(6):660–666. doi: 10.1016/s0959-437x(00)00250-1. [DOI] [PubMed] [Google Scholar]
- Atyame C, Delsuc F, Pasteur N, Weill M, Duron O. Diversification of Wolbachia endosymbiont in the Culex pipiens mosquito. Mol Biol Evol. 2011;28(10):2761–2772. doi: 10.1093/molbev/msr083. [DOI] [PubMed] [Google Scholar]
- Baines J, Sawyer S, Hartl D, Parsch J. Effects of X-linkage and sex-biased gene expression on the rate of adaptive protein evolution in Drosophila. Mol Biol Evol. 2008;25:1639–1650. doi: 10.1093/molbev/msn111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldo L, Dunning Hotopp J, Jolley K, Bordenstein S, Biber S, Choudhury R, Hayashi C, Maiden M, Tettelin H, Werren J. Multilocus sequence typing system for the endosymbiont Wolbachia pipientis. Appl Environ Microbiol. 2006;72:7098–7110. doi: 10.1128/AEM.00731-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldo L, Lo N, Werren J. Mosaic nature of the Wolbachia surface protein. J Bacteriol. 2005;187:5406–5418. doi: 10.1128/JB.187.15.5406-5418.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldo L, Werren JH. Revisiting Wolbachia supergroup typing based on WSP: spurious lineages and discordance with MLST. Curr Microbiol. 2007;55(1):81–87. doi: 10.1007/s00284-007-0055-8. [DOI] [PubMed] [Google Scholar]
- Ballard J. Comparative genomics of mitochondrial DNA in drosophila simulans. J Mol Evol. 2000;51(1):64–75. doi: 10.1007/s002390010067. [DOI] [PubMed] [Google Scholar]
- Ballard J. Sequential evolution of a symbiont inferred from the host: Wolbachia and Drosophila simulans. Mol Biol Evol. 2004;21(3):428–442. doi: 10.1093/molbev/msh028. [DOI] [PubMed] [Google Scholar]
- Bauer DuMont V, Flores H, Wright M, Aquadro C. Recurrent positive selection at bgcn, a key determinant of germ line differentiation, does not appear to be driven by simple coevolution with its partner protein bam. Mol Biol Evol. 2007;24:182–191. doi: 10.1093/molbev/msl141. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57(1):289–300. [Google Scholar]
- Buchner P. New York: Wiley Interscience; 1965. Endosymbiosis of animals with plant microorganisms. [Google Scholar]
- Chrostek E, Marialva M, Esteves S, Weinert L, Martinez J, Jiggins F, Teixeira L. Wolbachia variants induce differential protection to viruses in Drosophila melanogaster: a phenotypic and phylogenomic analysis. PLoS Genet. 2013;9(12):e1003896. doi: 10.1371/journal.pgen.1003896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civetta A, Rajakumar S, Brouwers B, Bacik J. Rapid evolution and gene-specific patterns of selection for three genes of spermatogenesis in Drosophila. Mol Biol Evol. 2006;23(3):655–662. doi: 10.1093/molbev/msj074. [DOI] [PubMed] [Google Scholar]
- Clark KA, McKearin DM. The Drosophila stonewall gene encodes a putative transcription factor essential for germ cell development. Development. 1996;122(3):937–950. doi: 10.1242/dev.122.3.937. [DOI] [PubMed] [Google Scholar]
- Dale C, Moran N. Molecular interactions between bacterial symbionts and their hosts. Cell. 2006;126(3):453–465. doi: 10.1016/j.cell.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Das A, Mohanty S, Stephan W. Inferring the population structure and demography of Drosophila ananassae from multilocus data. Genetics. 2004;168:1975–1985. doi: 10.1534/genetics.104.031567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Crespigny F, Pitt T, Wedell N. Increased male mating rate in Drosophila is associated with wolbachia infection. J Evol Biol. 2006;19(6):1964–1972. doi: 10.1111/j.1420-9101.2006.01143.x. [DOI] [PubMed] [Google Scholar]
- Dean MD, Ballard JW. High divergence among Drosophila simulans mitochondrial haplogroups arose in midst of long term purifying selection. Mol Phylogenet Evol. 2005;36(2):328–337. doi: 10.1016/j.ympev.2005.02.016. [DOI] [PubMed] [Google Scholar]
- Dean MD, Ballard KJ, Glass A, Ballard JW. Influence of two wolbachia strains on population structure of east African Drosophila simulans. Genetics. 2003;165(4):1959. doi: 10.1093/genetics/165.4.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedeine F, Vavre F, Fleury F, Loppin B, Hochberg M, Bouletreau M. Removing symbiotic Wolbachia bacteria specifically inhibits oogenesis in a parasitic wasp. Proc Natl Acad Sci U S A. 2001;98:6247–6252. doi: 10.1073/pnas.101304298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A, Suchard M, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29(8):1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134(2):341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuMont V, Fay J, Calabrese P, Aquadro C. DNA variability and divergence at the notch locus in Drosophila melanogaster and D. simulans: a case of accelerated synonymous site divergence. Genetics. 2004;167:171–185. doi: 10.1534/genetics.167.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuMont VL, Singh ND, Wright MH, Aquadro CF. Locus-specific decoupling of base composition evolution at synonymous sites and introns along the Drosophila melanogaster and Drosophila sechellia lineages. Genome Biol Evol. 2009;25(1):67–74. doi: 10.1093/gbe/evp008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer K, Burke C, Jaenike J. Wolbachia-mediated persistence of mtDNA from a potentially extinct species. Mol Ecol. 2011;20(13):2805–2817. doi: 10.1111/j.1365-294X.2011.05128.x. [DOI] [PubMed] [Google Scholar]
- Dyer K, Jaenike J. Evolutionarily stable infection by a male-killing endosymbiont in Drosophila innubila: molecular evidence from the host and parasite genomes. Genetics. 2004;168(3):1443–1455. doi: 10.1534/genetics.104.027854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Early AM, Clark AG. Monophyly of Wolbachia pipientis genomes within Drosophila melanogaster: geographic structuring, titre variation and host effects across five populations. Mol Ecol. 2013;22(23):5765–5778. doi: 10.1111/mec.12530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2003;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelstädter J, Hurst GD. The ecology and evolution of microbes that manipulate host reproduction. Annu Rev Ecol Evol Syst. 2009;40:127–149. [Google Scholar]
- Eyre-Walker A, Keightley P. Estimating the rate of adaptive molecular evolution in the presence of slightly deleterious mutations and population size change. Mol Biol Evol. 2009;26(9):2097–2108. doi: 10.1093/molbev/msp119. [DOI] [PubMed] [Google Scholar]
- Fast E, Toomey M, Panaram K, Desjardins D, Kolaczyk E, Frydman H. Wolbachia enhance Drosophila stem cell proliferation andtarget the germline stem cell niche. Science. 2011;334(6058):990–992. doi: 10.1126/science.1209609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay J, Wu C. Hitchhiking under positive Darwinian selection. Genetics. 2000;155(3):1405–1413. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferree P, Frydman H, Li J, Cao J, Wieschaus E, Sullivan W. Wolbachia utilizes host microtubules and dynein for anterior localization in the Drosophila oocyte. PLoS Pathog. 2005;1(2):e14. doi: 10.1371/journal.ppat.0010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine P. Vectors and vertical transmission: an epidemiologic perspective. Ann N Y Acad Sci. 1975;266:173–194. doi: 10.1111/j.1749-6632.1975.tb35099.x. [DOI] [PubMed] [Google Scholar]
- Flores HA. 2012. Evolutionary and functional analysis of the Drosophila bag of marbles gene [PhD thesis] [Ithaca (NY)]: Cornell University. [Google Scholar]
- Forbes A, Lehmann R. Nanos and pumilio have critical roles in the development and function of drosophila germline stem cells. Development. 1998;125:679–690. doi: 10.1242/dev.125.4.679. [DOI] [PubMed] [Google Scholar]
- Frydman HM, Li JM, Robson DN, Wieschaus E. Somatic stem cell niche tropism in Wolbachia. Nature. 2006;441(7092):509–512. doi: 10.1038/nature04756. [DOI] [PubMed] [Google Scholar]
- Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–925. doi: 10.1093/genetics/147.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Li W. Statistical tests of neutrality of mutations. Genetics. 1993;133(3):693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gönczy P, Matunis E, DiNardo S. Bag-of-marbles and benign gonial cell neoplasm act in the germline to restrict proliferation during Drosophila spermatogenesis. Development. 1997;124:4361–4371. doi: 10.1242/dev.124.21.4361. [DOI] [PubMed] [Google Scholar]
- Grath S, Baines J, Parsch J. Molecular evolution of sex-biased genes in the Drosophila ananassae subgroup. BMC Evol Biol. 2009;(9):291. doi: 10.1186/1471-2148-9-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemaud T, Pasteur N, Rousset F. Contrasting levels of variability between cytoplasmic genomes and incompatibility types in the mosquito Culex pipiens. Proc Biol Sci. 1997;264(1379):245–251. doi: 10.1098/rspb.1997.0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag-Liautard C, Coffey N, Houle D, Lynch M, Charlesworth B, Keightley P. Direct estimation of the mitochondrial DNA mutation rate in Drosophila melanogaster. PLoS Biol. 2008;6:e204. doi: 10.1371/journal.pbio.0060204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddrill PR, Bachtrog D, Andolfatto P. Positive and negative selection on noncoding DNA in Drosophila simulans. Mol Biol Evol. 2008;25(9):1825–1834. doi: 10.1093/molbev/msn125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale LR, Hoffmann AA. Mitochondrial DNA polymorphism and cytoplasmic incompatibility in natural populations of Drosophila simulans. Evolution. 1990;44:1383–1386. doi: 10.1111/j.1558-5646.1990.tb05241.x. [DOI] [PubMed] [Google Scholar]
- Harris RE, Pargett M, Sutcliffe C, Umulis D, Ashe HL. Brat promotes stem cell differentiation via control of a bistable switch that restricts BMP signaling. Dev Cell. 2011;20(1):72–83. doi: 10.1016/j.devcel.2010.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser M. A historic account of the invasion of Drosophila suzukii (matsumura) (Diptera: Drosophilidae) in the continental United States, with remarks on their identification. Pest Manag Sci. 2011;67(11):1352–1357. doi: 10.1002/ps.2265. [DOI] [PubMed] [Google Scholar]
- Heled J, Drummond AJ. Bayesian inference of population size history from multiple loci. BMC Evol Biol. 2008;8(1):289. doi: 10.1186/1471-2148-8-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA. Selection on codon bias. Annu Rev Genet. 2008;42:287–99. doi: 10.1146/annurev.genet.42.110807.091442. [DOI] [PubMed] [Google Scholar]
- Hilgenboecker K, Hammerstein P, Schlattmann P, Telschow A, Werren J. How many species are infected with Wolbachia? A statistical analysis of current data. FEMS Microbiol Lett. 2008;281(2):215–220. doi: 10.1111/j.1574-6968.2008.01110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill J, Chen Z, Xu H. Selective propagation of functional mitochondrial DNA during oogenesis restricts the transmission of a deleterious mitochondrial variant. Nat Genet. 2014;46(4):389–392. doi: 10.1038/ng.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann AA, Turelli M, Simmons GM. Unidirectional incompatibility between populations of Drosophila simulans. Evolution. 1986;40:692–701. doi: 10.1111/j.1558-5646.1986.tb00531.x. [DOI] [PubMed] [Google Scholar]
- Hotopp JC, Clark ME, Oliveira DC, Foster JM, Fischer P, Torres MC, Giebel JD, Kumar N, Ishmael N, Wang S, et al. Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes. Science. 2007;317:1753–1756. doi: 10.1126/science.1142490. [DOI] [PubMed] [Google Scholar]
- Hurst GD, Jiggins FM. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc Biol Sci. 2005;272:1525–1534. doi: 10.1098/rspb.2005.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James A, Ballard J. Expression of cytoplasmic incompatibility in Drosophila simulans and its impact on infection frequencies and distribution of Wolbachia pipientis. Evolution. 2000;54(5):1661–1672. doi: 10.1111/j.0014-3820.2000.tb00710.x. [DOI] [PubMed] [Google Scholar]
- Jensen J, Kim Y, DuMont V, Aquadro C, Bustamante C. Distinguishing between selective sweeps and demography using DNA polymorphism data. Genetics. 2005;170(3):1401–1410. doi: 10.1534/genetics.104.038224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, Kirilly D, Weng C, Kawase E, Song X, Smith S, Schwartz J, Xie T. Differentiation-defective stem cells outcompete normal stem cells for niche occupancy in the Drosophila ovary. Cell Stem Cell. 2008;2(1):39–49. doi: 10.1016/j.stem.2007.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley P, Eyre-Walker A. Joint inference of the distribution of fitness effects of deleterious mutations and population demography based on nucleotide polymorphism frequencies. Genetics. 2007;177(4):2251–2261. doi: 10.1534/genetics.107.080663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley P, Eyre-Walker A. Estimating the rate of adaptive molecular evolution when the evolutionary divergence between species is small. J Evol Biol. 2012;74:61–68. doi: 10.1007/s00239-012-9488-1. [DOI] [PubMed] [Google Scholar]
- Kim Y, Stephan W. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics. 2002;170:1401–1410. doi: 10.1093/genetics/160.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klattenhoff C, Xi H, Li C, Lee S, Xu J, Khurana JS, Zhang F, Schultz N, Koppetsch BS, Nowosielska A, et al. The Drosophila HP1 homolog rhino is required for transposon silencing and piRNA production by dual-strand clusters. Cell. 2009;138(6):1137–1149. doi: 10.1016/j.cell.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kose H, Karr T. Organization of Wolbachia pipientis in the Drosophila fertilized egg and embryo revealed by an anti-Wolbachia monoclonal antibody. Mech Dev. 1995;51:275–288. doi: 10.1016/0925-4773(95)00372-x. [DOI] [PubMed] [Google Scholar]
- Kriesner P, Hoffmann AA, Lee SF, Turelli M, Weeks AR. Rapid sequential spread of two Wolbachia variants in Drosophila simulans. PLoS Pathog. 2013;9(9):e1003607. doi: 10.1371/journal.ppat.1003607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Lin H, Spradling AC. A novel group of pumilio mutations affects the asymmetric division of germline stem cells in the Drosophila ovary. Development. 1997;124:2463–2476. doi: 10.1242/dev.124.12.2463. [DOI] [PubMed] [Google Scholar]
- Lo N, Paraskevopoulos C, Bourtzis K, O’Neill SL, Werren JH, Bordenstein SR, Bandi C. Taxonomic status of the intracellular bacterium Wolbachia pipientis. Int J Syst Evol Microbiol. 2007;57:654–657. doi: 10.1099/ijs.0.64515-0. [DOI] [PubMed] [Google Scholar]
- Lynch M, Crease T. The analysis of population survey data on DNA sequence variation. Mol Biol Evol. 1990;7(4):377–394. doi: 10.1093/oxfordjournals.molbev.a040607. [DOI] [PubMed] [Google Scholar]
- Ma H, Xu H, O’Farrell P. Transmission of mitochondrial mutations and action of purifying selection in Drosophila melanogaster. Nat Genet. 2014;46(4):393–397. doi: 10.1038/ng.2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maines JZ, Park JK, Williams M, McKearin DM. Stonewalling Drosophila stem cell differentiation by epigenetic controls. Development. 2007;134(8):1471–1479. doi: 10.1242/dev.02810. [DOI] [PubMed] [Google Scholar]
- Mateos M, Castrezana S, Nankivell B, Estes A, Markow T, Moran N. Heritable endosymbionts of Drosophila. Genetics. 2006;174:363–376. doi: 10.1534/genetics.106.058818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald J, Kreitman M. Adaptive protein evolution at the adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- McKearin D, Spradling A. Bag-of-marbles: A Drosophila gene required to initiate both male and female gametogenesis. Genes Dev. 1990;4:2242–2251. doi: 10.1101/gad.4.12b.2242. [DOI] [PubMed] [Google Scholar]
- Merçot H, Charlat S. Wolbachia infections in Drosophila melanogaster and D. simulans: polymorphism and levels of cytoplasmic incompatibility. Genetica. 2004;120:51–59. doi: 10.1023/b:gene.0000017629.31383.8f. [DOI] [PubMed] [Google Scholar]
- Montooth K, Abt D, Hofmann J, Rand D. Comparative genomics of Drosophila mtDNA: novel features of conservation and change across functional domains and lineages. J Mol Evol. 2009;69(1):94–114. doi: 10.1007/s00239-009-9255-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Nusslein-Volhard C, Frohnhofer HG, Lehmann R. Determination of anteroposterior polarity in Drosophila. Science. 1987;238:1675–1681. doi: 10.1126/science.3686007. [DOI] [PubMed] [Google Scholar]
- Osborne S, Iturbe-Ormaetxe I, Brownlie J, O’Neill S, Johnson K. Antiviral protection and the importance of Wolbachia density and tissue tropism in Drosophila simulans. Appl Environ Microbiol. 2012;78(19):6922–6929. doi: 10.1128/AEM.01727-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pröschel M, Zhang Z, Parsch J. Widespread adaptive evolution of Drosophila genes with sex-biased expression. Genetics. 2006;174(2):893–900. doi: 10.1534/genetics.106.058008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand DM, Kann A. Polymorphism in mitochondrial DNA: contrasts among genes from Drosophila, mice, and humans. Mol Biol Evol. 1996;13:735–748. doi: 10.1093/oxfordjournals.molbev.a025634. [DOI] [PubMed] [Google Scholar]
- Raychoudhury R, Baldo L, Oliveira D, Werren J. Modes of acquisition of Wolbachia: horizontal transfer, hybrid introgression, and codivergence in the Nasonia species complex. Evolution. 2009;63(1):165–183. doi: 10.1111/j.1558-5646.2008.00533.x. [DOI] [PubMed] [Google Scholar]
- Richardson MF, Weinert LA, Welch JJ, Linheiro RS, Magwire MM, Jiggins FM, Bergman CM. Population genomics of the Wolbachia endosymbiont in Drosophila melanogaster. PLoS Genet. 2012;8(12):e1003129. doi: 10.1371/journal.pgen.1003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg SL, Hotopp JC, Delcher AL, Pop M, Smith DR, Eisen MB, Nelson WC. Serendipitous discovery of Wolbachia genomes in multiple Drosophila species. Genome Biol. 2005;6(3):R23. doi: 10.1186/gb-2005-6-3-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug M, Smith S, Tozier-Pearce A, McEvey S. The genetic structure of Drosophila ananassae populations from Asia, Australia and Samoa. Genetics. 2007;175:1429–1440. doi: 10.1534/genetics.106.066613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug MD, Baines JF, Killon-Atwood A, Mohanty S, Das A, Grath S, Smith SG, Zargram S, McEvey SF, Stephan W. Evolution of mating isolation between populations of Drosophila ananassae. Mol Ecol. 2008;17(11):2706–2721. doi: 10.1111/j.1365-294X.2008.03770.x. [DOI] [PubMed] [Google Scholar]
- Shen R, Weng C, Yu J, Xie T. eIF4A controls germline stem cell self-renewal by directly inhibiting BAM function in the Drosophila ovary. Proc Natl Acad Sci U S A. 2009;106(28):11623–11628. doi: 10.1073/pnas.0903325106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields D, Sharp P, Higgins D, Wright F. “Silent” sites in Drosophila genes are not neutral: evidence of selection among synonymous codons. Mol Biol Evol. 1988;5(6):704–716. doi: 10.1093/oxfordjournals.molbev.a040525. [DOI] [PubMed] [Google Scholar]
- Shoemaker D, Keller G, Ross K. Effects of Wolbachia on mtDNA variation in two fire ant species. Mol Ecol. 2003;12(7):1757–1771. doi: 10.1046/j.1365-294x.2003.01864.x. [DOI] [PubMed] [Google Scholar]
- Silvestro D, Michalak I. raxmlGUI: a graphical front-end for RAxML. Org Divers Evol. 2012;12:335–337. [Google Scholar]
- Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am. 1994;87:651–701. [Google Scholar]
- Simonsen K, Churchill G, Aquadro C. Properties of statistical tests of neutrality for DNA polymorphism data. Genetics. 1995;141:413–429. doi: 10.1093/genetics/141.1.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh ND, Bauer DuMont VL, Hubisz MJ, Nielsen R, Aquadro CF. Patterns of mutation and selection at synonymous sites in Drosophila. Mol Biol Evol. 2007;24(12):2687–2697. doi: 10.1093/molbev/msm196. [DOI] [PubMed] [Google Scholar]
- Siozios S, Cestaro A, Kaur R, Pertot I, Rota-Stabelli O, Anfora G. Draft genome sequence of the Wolbachia endosymbiont of Drosophila suzukii. Genome Announc. 2013;1(1):e00032–13. doi: 10.1128/genomeA.00032-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin M, Hudson RR. Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics. 1991;129:555–562. doi: 10.1093/genetics/129.2.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin RK, Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet. 2007;8:272–285. doi: 10.1038/nrg2072. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Starr D, Cline T. A host parasite interaction rescues Drosophila oogenesis defects. Nature. 2002;418(6893):76–79. doi: 10.1038/nature00843. [DOI] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68(4):978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoletzki N, Eyre-Walker A. Estimation of the neutrality index. Mol Biol Evol. 2011;28:63–70. doi: 10.1093/molbev/msq249. [DOI] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toomey ME, Panaram K, Fast EM, Beatty C, Frydman HM. Evolutionarily conserved Wolbachia-encoded factors control pattern of stem-cell niche tropism in Drosophila ovaries and favor infection. Proc Natl Acad Sci U S A. 2013;110(26):10788–10793. doi: 10.1073/pnas.1301524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli M, Hoffmann A. Rapid spread of an inherited incompatibility factor in California Drosophila. Nature. 1991;353(6343):440–442. doi: 10.1038/353440a0. [DOI] [PubMed] [Google Scholar]
- Turelli M, Hoffmann AA. Cytoplasmic incompatibility in Drosophila simulans: dynamics and parameter estimates from natural populations. Genetics. 1995;140:1319–1338. doi: 10.1093/genetics/140.4.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermaak D, Henikoff S, Malik HS. Positive selection drives the evolution of rhino, a member of the heterochromatin protein 1 family in Drosophila. PLoS Genet. 2005;1(1):e9. doi: 10.1371/journal.pgen.0010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicario S, Moriyama E, Powell J. Codon usage in twelve species of Drosophila. BMC Evol Biol. 2007;7:226. doi: 10.1186/1471-2148-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl C, Das A, Beaumont M, Mohanty S, Stephan W. Population subdivision and molecular sequence variation: theory and analysis of Drosophila ananassae data. Genetics. 2003;165(3):1385–1395. doi: 10.1093/genetics/165.3.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Lin H. Nanos maintains germline stem cell self-renewal by preventing differentiation. Science. 2004;303:2016–2019. doi: 10.1126/science.1093983. [DOI] [PubMed] [Google Scholar]
- Werren J, Baldo L, Clark M. Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol. 2008;6:741–751. doi: 10.1038/nrmicro1969. [DOI] [PubMed] [Google Scholar]
- Werren J, Zhang W, Guo L. Evolution and phylogeny of Wolbachia: reproductive parasites of arthropods. Proc Biol Sci. 1995;261:55–63. doi: 10.1098/rspb.1995.0117. [DOI] [PubMed] [Google Scholar]
- Werren JH. Heritable microorganisms and reproductive parasitism. In: Sapp J, editor. Microbial evolution: concepts and controversies. New York: Oxford University Press; 2005. pp. 290–315. [Google Scholar]
- Werren JH. Selfish genetic elements, genetic conflict, and evolutionary innovation. Proc Natl Acad Sci U S A. 2011;108(Suppl. 2):10863–10870. doi: 10.1073/pnas.1102343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werren JH, O’Neill SL. The evolution of heritable symbionts. In: O’Neill SL, Hoffmann AA, Werren JH, editors. Influential passengers: inherited microorganisms and arthropod reproduction. New York: Oxford University Press; 1997. pp. 1–41. [Google Scholar]
- Xie T. Control of germline stem cell self-renewal and differentiation in the Drosophila ovary: concerted actions of niche signals and intrinsic factors. Wiley Interdiscip Rev Dev Biol. 2012;2:261–273. doi: 10.1002/wdev.60. [DOI] [PubMed] [Google Scholar]
- Yi X, de Vries HI, Siudeja K, Rana A, Lemstra W, Brunsting JF, Kok RM, Smulders YM, Schaefer M, Dijk F, et al. Stwl modifies chromatin compaction and is required to maintain DNA integrity in the presence of perturbed DNA replication. Mol Biol Cell. 2009;20(3):983–994. doi: 10.1091/mbc.E08-06-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamore PD, Williamson JR, Lehmann R. The pumilio protein binds RNA through a conserved domain that defines a new class of RNA-binding proteins. RNA. 1997;3:1421–1433. [PMC free article] [PubMed] [Google Scholar]