

Abstract
Diverse cellular responses to external cues are controlled by a small number of signal-transduction pathways, but how the specificity of functional outcomes is achieved remains unclear. Here we describe a mechanism for signal integration based on the functional coupling of two distinct signaling pathways widely used in leukocytes: the ITAM pathway and the Jak-STAT pathway. Through the use of the receptor for interferon-γ (IFN-γR) and the ITAM adaptor Fcγ as an example, we found that IFN-γ modified responses of the phagocytic antibody receptor FcγRI (CD64) to specify cell-autonomous antimicrobial functions. Unexpectedly, we also found that in peritoneal macrophages, IFN-γR itself required tonic signaling from Fcγ through the kinase PI(3)K for the induction of a subset of IFN-γ-specific antimicrobial functions. Our findings may be generalizable to other ITAM and Jak-STAT signaling pathways and may help explain signal integration by those pathways.
Immunoreceptor tyrosine-based activation motifs (ITAMs) and ITAM-like adaptors are some of the most widely used conserved signaling modules in leukocytes1. The same ITAM is found in the cytoplasmic tail of signaling adaptors associated with all immunoreceptors, such as the T cell antigen receptor (TCR), B cell antigen receptor (BCR), natural killer (NK) cell receptor or receptor for the Fc fragment of immunoglobulins (FcR). However, in different cell types, ITAM-associated receptors regulate distinct functions as diverse as proliferation, apoptosis, degranulation, cytotoxicity or phagocytosis. In addition, even in the same cell type, a single ITAM-associated receptor can induce distinct functions when triggered in different stimulating conditions. For example, FcγR can induce functional programs involved in the killing of microbes, the presentation of antigen to CD4+ or CD8+ T cells, inflammation or tissue repair. The appropriate response therefore probably reflects the tissue state (i.e., the context in which the ITAM-relayed signal has been received).
The Jak-STAT cytokine-receptor-signaling pathway is another highly conserved pathway that regulates a large number of distinct leukocyte functions2. In response to a specific cytokine, various cell types activate an identical signaling pathway, yet each elicits a distinct, cell type – specific response3,4. However, the basis of the signaling specificity of both the ITAM signaling module and the Jak-STAT signaling module is poorly understood. All ITAMs engage the same general signaling pathway in all cells: activated immunoreceptors recruit Src kinases to phosphorylate tyrosine residues in the ITAM and generate docking sites for the binding and activation of the signaling kinase Syk or Zap70, recruitment of adaptors, and signal transmission via the MEK, NF-κB, PI(3)K and NFAT pathways1. Similarly, all activated cytokine receptors use four receptor-associated kinases of the Jak family (Tyk2, Jak1, Jak2 and Jak3) to activate seven transcription factors of the STAT family2. Thus, the question arises of how a receptor containing a single signaling module produces distinct outcomes in response to diverse signals, particularly if such a module is shared among diverse families of receptors.
ITAM adaptors such as Fcγ, DAP12, CD3ζ, immunoglobulin α-chain and immunoglobulin β-chain were initially identified as signaling modules used exclusively by immunoreceptors (for example, FcR, TCR and BCR), with which they constitutively associate and traffic to the cell surface5. However, growing evidence suggests that ITAM and ITAM-like adaptors do participate in additional signaling pathways beyond immunoreceptors6,7. In bones, DAP12 is required by the cytokine RANKL and receptor M-CSFR to induce osteoclast differentiation8,9; in NK cells, the ITAM-like adaptor DAP10 is required by the receptor for interleukin 15 (IL-15R) to induce cytotoxic responses10; in basophils, the Fcγ is required by IL-3R to induce IL-4 production11. Such observations suggest that in addition to being coupled to the canonical Src-ITAM-Syk (or Zap70) signaling pathway, ITAM adaptors may be functionally coupled to diverse receptors that engage unrelated signaling pathways, such as the Jak-STAT or TRAF–NF-κB pathway.
Here we explored the hypothesis that ITAM adaptors are functionally coupled to the Jak-STAT pathway to provide a mechanism for a basic digital logic ‘AND’ gate in context-dependent signaling in leukocytes12. An ‘AND’ gate indicates that a specific output emerges only if both input signals are ‘on’ in the same space and time window. We provide proof-of-principle evidence for our hypothesis and describe a structural and functional ‘collaboration’ between the receptor for the cytokine interferon-γ (IFN-γR)13 and the ITAM module of the phagocytic antibody receptor FcγRI (CD64)14 in specifying the cell-intrinsic antimicrobial functions of phagocytes.
RESULTS
IFN-γR colocalizes with the FcγRI signalosome in phagocytes
IFN-γ regulates the antimicrobial functions of macrophages and dendritic cells (DCs) in part by upregulating the expression of FcγRI and its ITAM adaptor Fcγ15. We confirmed upregulation of the expression of FcγRI components in mouse bone marrow (BM) macrophages after prolonged stimulation with IFN-γ (24 h) but not after short-term stimulation with IFN-γ (0.5 h) in vitro (Fig. 1a). This established that IFN-γ was able to control FcγRI-restricted functions indirectly by controlling the expression of this phagocytic receptor.
Figure 1.
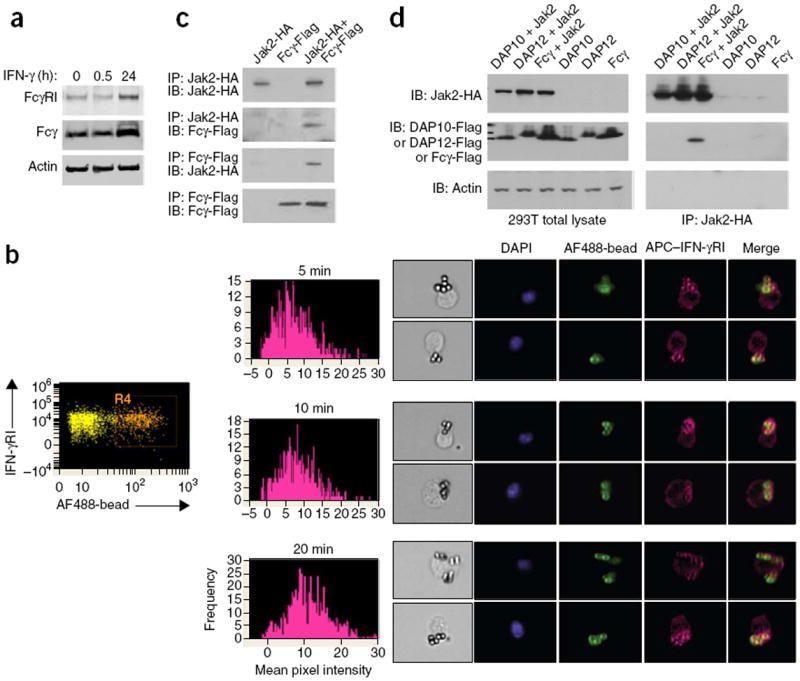
IFN-γR is coupled with the FcγRI signalosome in phagocytes. (a) Immunoblot analysis of FcγRI and Fcγ in total lysates of BM macrophages stimulated for 0, 0.5 or 24 h (above lanes) with IFN-γ; actin serves as a loading control throughout. (b) ImageStream analysis of the recruitment of allophycocyanin (APC)-labeled IFN-γRI to the phagocytic cup in BM DCs (n = 5,000 per condition) stimulated for 5, 10 or 20 min (above plots, middle) with rabbit IgG–opsonized Alexa Fluor 488–labeled latex beads (AF488-bead): far left, gating of cells that had taken up beads; middle, mean pixel intensity of IFN-γRI staining around the bead; right, images with an intensity of >10. Original magnification (right), ×40. DAPI, DNA intercalating dye. (c) Immunoblot analysis of hemagglutinintagged Jak2 (Jak2-HA) and Flag-tagged Fcγ (Fcγ-Flag) coimmunoprecipitated from lysates of 293T cells transiently overexpressing those two proteins (separately or together; above lanes) with a cotransfection efficiency of >80%. (d) Immunoblot analysis of hemagglutinin-tagged Jak2 coimmunoprecipitated with Flag-tagged DAP12, DAP10 or Fcγ from lysates (right) of 293T cells transiently overexpressing various combinations of proteins (above lanes); left, similar immunoblot analysis of total cell lysates without immunoprecipitation. Data are representative of three (a) four (c) or two (d) experiments, or three total experiments (two ImageStream; one immunofluorescence) with similar results (b).
To determine whether IFN-γR and FcγRI can directly regulate each other’s signaling, we monitored the cellular localization of IFN-γR relative to that of FcγRI during FcγR-mediated phagocytosis. We stimulated mouse BM macrophages and DCs with sheep red blood cells (SRBCs) opsonized with mouse immunoglobulin G2a (IgG2a; the ligand for the high-affinity receptor FcγRI) (called ‘SRBC (O)’ here) or fluorescent latex beads opsonized with rabbit IgG (the ligand for all FcγRs). We tracked the cellular localization of IFN-γR with an antibody to IFN-γRI (one of its components) and visualized this through immunofluorescence (for BM macrophages) or ImageStream technology (for BM DCs). ImageStream technology allows the unbiased, large-scale statistical analysis of molecular interactions in cells in suspension not only through the collection of data in a flow path similar to classical flow cytometry but also by the simultaneous collection of visual fluorescent images of every cell in the flow. Both analyses revealed that IFN-γRI was localized together with the phagocytic cup during FcγRI-mediated phagocytosis in wild-type BM DCs (Fig. 1b) and in wild-type, FcγRII-deficient and FcγRIII-deficient BM macrophages (Supplementary Fig. 1a,b). We did not see localization of IFN-γRI with the phagocytic cup in control experiments in which we stimulated BM macrophages lacking all FcγRs (Fcγ-deficient BM macrophages) with SRBC (O) or stimulated wild-type macrophages with nonopsonized SRBCs (SRBC (N)) or when we used isotype-matched control antibody for staining (Supplementary Fig. 1a,b). Recruitment of IFN-γRI to the phagocytic cup occurred within minutes of FcγRI ligation regardless of whether IFN-γ was present or not (Supplementary Fig. 1a,b), and this occurred with kinetics that resembled those of the recruitment of actin to FcγR phagosomes upon IgG-mediated activation (Supplementary Fig. 1c).
In human monocytes, FcγRI is reported to localize in larger membrane clusters proposed to be lipid rafts16. Similarly, ImageStream analysis showed that in BM DCs (Supplementary Fig. 1d) and BM macrophages (data not shown), FcγRI was preclustered into membrane microdomains and that some of the cellular IFN-γRI was present in the same microdomains as well. Antibody to IFN-γRI or antibody to FcγRI did not detect those proteins in IFN-γRI-deficient mice or Fcγ-deficient mice, respectively (Supplementary Fig. 1e,f), which indicated the specificity of the reagents used for staining. Thus, IFN-γR induced FcγRI expression and was prepositioned in cells of the innate immune system in a manner that could enable the two receptors to functionally interact during receptor engagement and signaling (Supplementary Fig.1g).
To understand the factors that mediated the FcγRI–IFN-γR coupling, we cloned a Flag-tagged form of the FcγRI-associated ITAM adaptor Fcγ and a hemagglutinin-tagged form of the IFN-γR-associated kinase Jak2 and transiently coexpressed those proteins in 293T human embryonic kidney cells. Reciprocal coimmunoprecipitation of those proteins revealed the presence of Jak2 and Fcγ in the same complex (Fig. 1c). We did not detect interactions between Jak2 and the other two related adaptors (DAP12 or DAP10) under similar conditions (Fig. 1d). Thus, different Jak kinases might potentially be found in complexes with different ITAMs, an idea in agreement with published findings that IL-15R-associated Jak3 functionally ‘collaborates’ with DAP10 in NK cells10, while IFN-αR, which uses Jak1 (not Jak2), functionally ‘collaborates’ with DAP12, not Fcγ, in macrophages17.
FcγRI and IFN-γR have independent canonical signaling
Although IFN-γR was recruited to the FcγR phagosome, we investigated whether the two receptors were able to engage their canonical signaling pathways independently of each other. We detected intact expression and canonical signaling of IFN-γR in Fcγ-deficient BM macrophages stimulated with IFN-γ (Fig. 2a) and, similarly, intact expression and canonical signaling of FcγRI in IFN-γRI-deficient BM macrophages stimulated with SRBC (O) (Fig. 2b). That was consistent with data demonstrating that Jak2 was not phosphorylated upon activation of Fcγ downstream of its cognate immunoreceptors FcγRI and dectin-2 (refs. 18-20) (Fig. 2c) and that the rate of phagocytosis was unchanged (Fig. 2d) in BM macrophages stimulated with IFN-γ at the time of or 60 min before phagocytosis (i.e., when FcγRI expression was still intact; Fig. 1a). Thus, the canonical IFN-γR and FcγRI signaling pathways were independent of each other.
Figure 2.
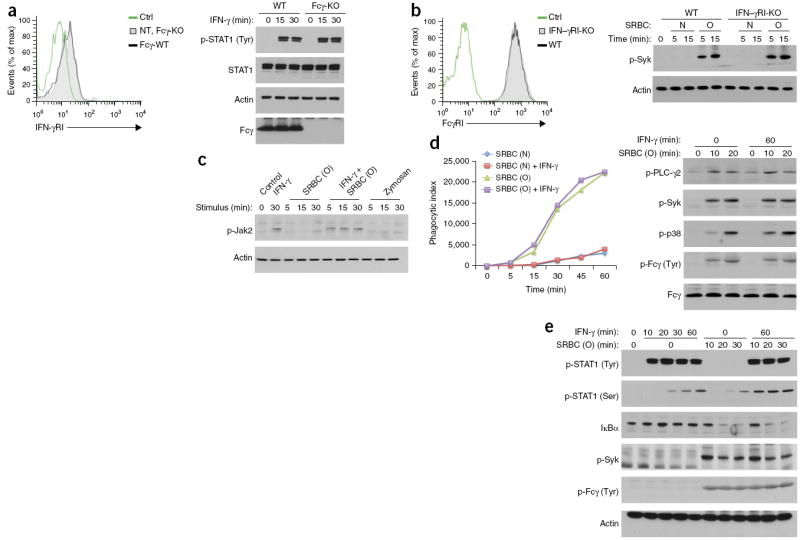
FcγR and IFN-γR are dispensable for each other’s expression and canonical signaling. (a,b) IFN-γRI expression on untransduced (NT) Fcγ-deficient (Fcγ-KO) BM macrophages and Fcγ-deficient BM macrophages reconstituted retrovirally with wild-type Fcγ (Fcγ-WT) (a), and FcγRI expression on IFN-γRI-deficient (IFN-γRI-KO) or wild-type B6 (WT) BM macrophages (b), assessed by flow cytometry (left). Ctrl, unstained cells. Right, immunoblot analysis of tyrosine-phosphorylated STAT1 (p-STAT1(Tyr)) and total STAT1, as well as Fcγ, in wild-type B6 and Fcγ-deficient BM macrophages stimulated for various times (above lanes) with IFN-γ (a), and of phosphorylated Syk (p-Syk) in BM macrophages (as at left) stimulated for various times (above lanes) with SRBC (O) or SRBC (N) (b). (c) Immunoblot analysis of phosphorylated (p-) Jak2 in wild-type (B6) BM macrophages left unstimulated (Control) or stimulated for various times (above lanes) with IFN-γ or SRBC (O) alone or in combination, or with zymosan. (d) Immunoblot analysis of phosphorylated PLC-γ2, Syk and p38, as well as tyrosine-phosphorylated Fcγ (p-Fcγ (Tyr); identified by detection of bands of the same molecular size with antibody to phosphorylated tyrosine and antibody to Fcγ) in wild-type (B6) BM macrophages stimulated for 0 or 60 min (top) with IFN-γ and for 0, 10 or 20 min (above lanes) with SRBC (O) (right), and phagocytosis of red fluorescent lipophilic dye PKH26–labeled SRBC (O) or SRBC (N) by wild-type (B6) BM macrophages in a nonsynchronized assay in the presence or absence of IFN-γ added 1 h before SRBCs (key), assessed by flow cytometry and presented as the phagocytic index (left). (e) Immunoblot analysis of STAT phosphorylated at tyrosine (p-STAT1 (Tyr)) or serine (p-STAT1 (Ser)), total IκBα, phosphorylated Syk (p-Syk) and tyrosinephosphorylated Fcγ (as in d) in wild-type (B6) BM macrophages stimulated for 0–60 min (top) with IFN-γ and for 0–30 min (above lanes) with SRBC (O) in a synchronized assay. Data are representative of four (a, left, c,e), three (a,b, right), two (b, left) or five (d) experiments with similar results.
Cytokine receptors and other signaling pathways (such as the TLR, TCR and BCR)21-25 can engage in cross-talk through the phosphorylation of Ser727 in the carboxy-terminal transactivation domain of STAT1 (refs. 26-28). That phosphorylation event recruits chromatin-modifying complexes to some STAT1- regulated genes. We found that both SRBC (O) and IFN-γ induced phosphorylation of STAT1 at Ser727 in BM macrophages and that the phosphorylation was also further enhanced when the two signals were delivered together (Fig. 2e and Supplementary Fig. 2a). FcγRI and IFN-γR activated Ser727 independently of each other and probably used different pathways to do so26,27 (Supplementary Fig. 2b–d). Thus, even though FcγRI and IFN-γR seemed to have independent signaling pathways, they were able to converge on at least one critical signal integration point.
Part of the IFN-γ-regulated gene program requires Fcγ
Most leukocytes express IFN-γR and, hence, all are responsive to IFN-γ. However, during infection, each leukocyte subset elicits cell type–specific responses to IFN-γ28. To investigate whether coupling to Fcγ in macrophages influenced IFN-γ-induced responses qualitatively, we stimulated wild-type and Fcγ-deficient peritoneal macrophages with IFN-γ and analyzed gene expression 0–6 h later by quantitative PCR. As expected from the signaling data reported above, many IFN-γ-regulated genes, such as Icam1, Cd40 and Gbp5, were induced normally in Fcγ-deficient macrophages (data not shown). However, the known IFN-γ-regulated gene Nos2, which encodes inducible nitric oxide synthase (iNOS), an enzyme critical for antimicrobial and antitumor functions29-32, was poorly induced in Fcγ-deficient macrophages in response to stimulation with IFN-γ on either a C57BL/6 (B6) background or a BALB/c background (Fig. 3a,b). The lack of Nos2 induction was not due to defective macrophage development in the Fcγ-deficient mice (Supplementary Fig. 3a), delayed kinetics of Nos2 transcription (Supplementary Fig. 3b) or unresponsiveness of the Nos2 promoter because stimulation with type I interferon induced Nos2 normally in Fcγ-deficient macrophages (Fig. 3c). The last result was consistent with reports showing that signaling via type I interferons in macrophages uses DAP12, not Fcγ, for some specific functions17. Notably, we detected normal expression of Irgm1, which encodes the antimicrobial effector LRG47 (ref. 33), in Fcγ-deficient macrophages (Fig. 3a); hence, not all IFN-γ-inducible antimicrobial responses were dependent on Fcγ expression. Finally, IL-4 did not require Fcγ for induction of the expression of arginase I, an enzyme that competes with iNOS for the common substrate arginine to elicit the alternative, M2 differentiation of macrophages (Supplementary Fig. 3c). Thus, the induction of Nos2 in response to IFN-γ in peritoneal macrophages required expression of the ITAM adaptor Fcγ.
Figure 3.

Induction of subset of IFN-γ-regulated genes in peritoneal macrophages requires expression of Fcγ. (a,b) Quantitative PCR analysis of Nos2 and Irgm1 mRNA in peritoneal macrophages obtained from naive wild-type (WT) or Fcγ-deficient (Fcγ-KO) mice (B6 or BALB/c strain in b (key)) and stimulated for 0, 3 or 6 h (below) with IFN-γ; results are presented relative to that of an unstimulated wild-type replicate, set as 1. (c) Quantitative PCR analysis of Nos2 and Irf1 mRNA in wild-type or Fcγ-deficient peritoneal macrophages stimulated for 0, 3 or 6 h (below) with IFN-α (presented as in a,b). (d) Quantitative PCR analysis (bottom) of Nos2 mRNA in Fcγ-deficient macrophages transduced with empty vector (EV) or vector encoding wild-type Fcγ or various Fcγ mutants (top) and stimulated for 0, 3 or 6 h (below graph) with IFN-γ; results are presented relative to those of unstimulated macrophages transduced with vector encoding the D11A Fcγ mutant, set as 1. Top right, expression and signaling of FcγRI by cells transduced with the mutants at left. (e) Expression of FcγRI, FcγRII-FcγRIII and IFN-γRI (middle and right) in Fcγ-deficient BM left untransduced (No Tx) or retrovirally transduced as in d (left margin), assessed by flow cytometry, with transduction (Fcγ expression) assessed based on expression of a human CD2 marker (far left; numbers in plots indicate percent human CD2–positive (hCD2+) cells). Isotype, isotype-matched control antibody. (f) Cluster analysis of gene expression upregulated (red) or downregulated (green) in wild-type or Fcγ-deficient peritoneal macrophages left unstimulated (US) or stimulated for 6 h with IFN-γ, relative to that in wild-type unstimulated macrophages; all genes with a difference in expression of over 1.5-fold were clustered. Numbers along left margin indicate gene clusters; letters along right margin indicate larger groups defined by similar gene-expression patterns (with different expression patterns (A–G; colors as above) or similar expression patterns (D and H) in wild-type and Fcγ-deficient macrophages). (g) Summary of the results in f. (h) Quantitative PCR analysis confirmation of results for selected genes of interest from f, presented relative to those of wild-type unstimulated cells, set as 1 (except Ifi202b, presented relative to results for Fcγ-deficient unstimulated cells because of its low expression in wild-type cells). Data are representative of five (a), two (b,c,e) or seven (d) experiments with similar results (mean and s.d.) or one experiment with duplicate samples (f–h), with independent validation of results (h).
We next investigated which motifs of the ITAM were relevant for the functional interaction with IFN-γR. We generated Fcγ mutants with disruption of the ITAM residues required for signaling (Y47A and Y58A) or the transmembrane region (D11A and L21A). The two transmembrane residues are required for the constitutive association between Fcγ and its cognate immunoreceptors FcγR, FcαR and FcεR and, hence, for proper trafficking and surface expression of both the adaptor and its ‘client’ receptors5,11. Retroviral transduction of the Fcγ-mutant proteins into Fcγ-deficient macrophages revealed that only wild-type Fcγ restored IFN-γ-induced Nos2 transcription, but neither the Fcγ transmembrane mutants (D11A or L21A) nor the Fcγ ITAM mutants (Y47A or Y58A) did so (Fig. 3d). This indicated that the induction of Nos2 by IFN-γ required an intact Fcγ ITAM and the proper surface expression of Fcγ or one of its other ‘client’ receptors. Notably, while neither the Fcγ transmembrane mutants (Supplementary Fig. 3d,e) nor their cognate partners FcγRI, FcγRII and FcγRIII (Fig. 3e) reached the cell surface, IFN-γR expression was normal in all conditions (Fig. 3e), which indicated that IFN-γR probably did not constitutively associate with Fcγ for proper trafficking yet somehow required Fcγ-dependent FcγR signaling from the plasma membrane for robust induction of Nos2.
To determine whether the ITAM module was required for the induction of a broader category of IFN-γ-regulated genes, we analyzed the responses of wild-type and Fcγ-deficient peritoneal macrophages to stimulation for 6 h with IFN-γ by array-based gene-expression analysis. Cluster analysis (Fig. 3f,g and Supplementary Fig. 3f) and gene-ontology analyses (Supplementary Table 1) revealed a group of IFN-γ-regulated genes that encode defense-response molecules whose expression was Fcγ dependent. For example, Ifi205, which encodes a member of the AIM2-like double-stranded DNA–sensing PYHIN family of proteins34, was one of those genes (Fig. 3f,g, group A) whose induction by IFN-γ was completely defective in Fcγ-deficient macrophages, similar to Nos2 (Fig. 3h). In contrast, another member of this family, Ifi202b, which encodes a competitive inhibitor of the AIM2 sensor of double-stranded DNA35, was among the genes whose expression was higher in resting Fcγ-deficient macrophages than in their wild-type counterparts and was further upregulated by stimulation with IFN-γ while remaining very low to undetectable in wild-type peritoneal macrophages under these experimental conditions (Fig. 3f,h, cluster 4, group C). Thus, the appropriate expression of a group of IFN-γ-regulated antimicrobial sensors and effectors was conditional on a functional Fcγ-ITAM module in peritoneal macrophages.
To determine if the data reported thus far represented an example of a more general concept of a role for the ITAM module in specifying cytokine-receptor functions, we analyzed gene expression in IL-4- stimulated Fcγ-deficient peritoneal macrophages by microarray. The data revealed that a subset of the IL-4-induced responses in macrophages was dependent on an intact ITAM module, yet the functional gene signatures were distinct from those induced by IFN-γ (Supplementary Fig. 3f–i and Supplementary Table 2). For example, after stimulation with IL-4, Fcγ-deficient macrophages were hyper-responsive in inducing genes encoding molecules that specify and characterize the M2 macrophage fate (for example, Klf4 and Clec10a (which encodes Mgl1); Supplementary Fig. 3g, cluster 5, group C). About half of the genes with different expression patterns in wild-type macrophages versus Fcγ-deficient macrophages were shared by the two microarrays analyzing cells stimulated with IFN-γ and IL-4 (Supplementary Fig. 3j). In large part, they involved genes dys-regulated in resting Fcγ-deficient macrophages before any stimulation (for example, Ifi202b; Supplementary Fig. 3i,j and Supplementary Table 3). These results indicated that an intact Fcγ-ITAM module, and probably other ITAM adaptors, may have a more general role than expected in affecting cytokine-induced responses.
Part of the IFN-γ-program requires tonic Fcγ-PI(3)K signals
The data presented thus far were consistent with one of two models: IFN-γR might access ITAM via inducible association with a classical ITAM-associated immunoreceptors such as FcγRI, or cytokine receptors might use and activate Fcγ as signaling adaptor directly and independently of the immunoreceptor (Supplementary Fig. 4a). In the former case, the immunoreceptor complex would interact functionally with the cytokine receptor to provide an accessory function that the cytokine receptor requires for part of its gene program but cannot elicit it by itself. To distinguish between two models, we investigated whether FcγRI was able to provide any signal in resting peritoneal macrophages. FcγRI is a high-affinity Fc receptor and is constitutively occupied on macrophages by monomeric IgG (probably its high-affinity ligand IgG2a) in vivo36,37. FcγRI ligated by IgG could provide tonic signaling through its Fcγ-ITAM module38. Here we defined ‘tonic signal’ as the signal generated by FcγRI bound to IgG (either free IgG or IgG in complex with monovalent antigen) that did not cause receptor crosslinking, distinct from an ‘induced signal’, which was generated by antigen-IgG immunocomplexes and caused crosslinking of FcγR.
Flow cytometry of freshly isolated wild-type peritoneal macrophages stained with F(ab′)2 antibody fragment to mouse IgG revealed that FcγR was constitutively occupied with serum IgG and, more specifically, with IgG2a (Fig. 4a). Because of the stable occupancy of FcγRs in vivo by mouse serum IgG and their variable occupancy in vitro by low-affinity bovine IgG in BM macrophage–differentiation cultures (depending on the commercial source of serum; data not shown), we used freshly isolated peritoneal macrophages for all analyses of tonic ITAM signaling. We used BM macrophages for the analysis of Fcγ transduction described above because of the low number and poor transduction efficiency of peritoneal macrophages.
Figure 4.
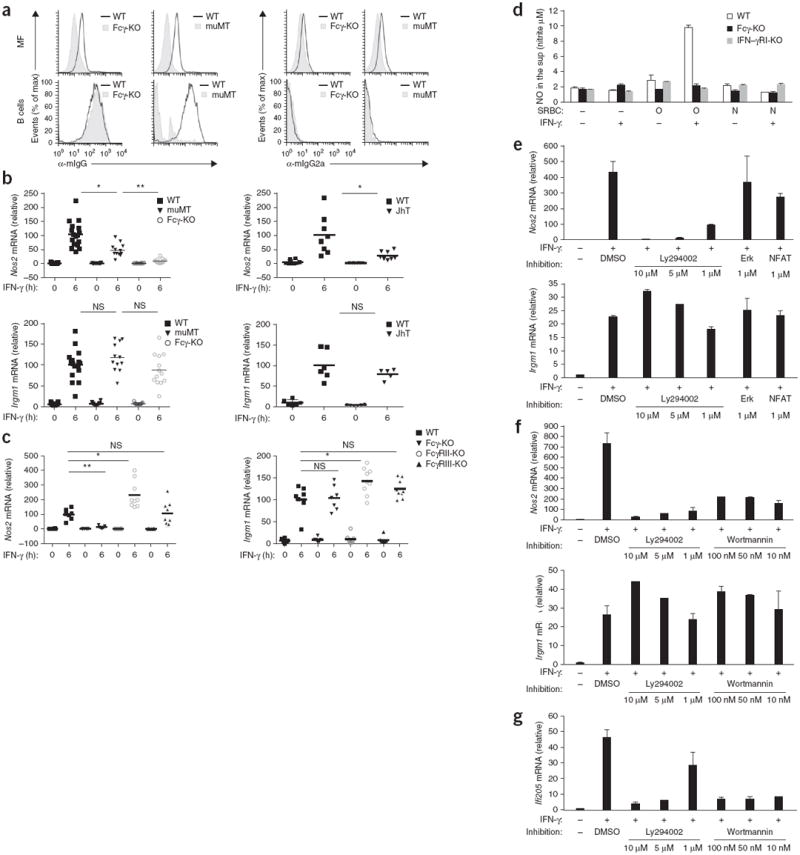
Induction of a subset of IFN-γ-regulated genes in peritoneal macrophages requires tonic PI(3)K signaling through FcγR and Fcγ. (a) Surface staining of IgG (with fluorescein isothiocyanate–conjugated F(ab′)2 fragment directed against the heavy and light chain of mouse IgG (α-mIgG)) and IgG2a (with fluorescein isothiocyanate–conjugated antibody to mouse IgG2a (α-mIgG2a)) on freshly isolated wild-type, Fcγ-deficient and muMT peritoneal cells identified as macrophages (MF; top row; F4/80+CD11b+) or B cells (bottom row; B220+), assessed by flow cytometry after blockade of nonspecific binding to FcRs. B cells serve as controls because the naive BCR will be detected with the antibody at left but not with the specific antibody at right. (b,c) Quantitative PCR analysis of Nos2 and Irgm1 mRNA in wild-type (b,c; B6 or BALB/c), muMT (b), JhT (b), Fcγ-deficient (b,c), FcγRII-deficient (c) and FcγRIII-deficient (c) peritoneal macrophages stimulated for 0 or 6 h (horizontal axis) with IFN-γ; pooled results are presented relative to the average result of IFN-γ-stimulated wild-type samples, set as 100%. Each symbol represents an individual mouse; small horizontal lines indicate the mean. NS, not significant; *P < 0.05 and **P < 0.005 (two-tailed unpaired t-test). (d) Griess assay of the production of nitric oxide (NO) by BM macrophages stimulated for 16 h without SRBCs (′) or with SRBC (O) or SRBC (N) in the presence (+) or absence (−) of IFN-γ. (e–g) Quantitative PCR analysis of Nos2, Irgm1 and Ifi205 mRNA in peritoneal macrophages left untreated (far left) or pretreated for 30 min (e,g) or 60 min (f) with the vehicle dimethyl sulfoxide (DMSO) or an inhibitor of PI(3)K (Ly294002 or Wortmannin), NFAT and Erk, then left unstimulated (−) or stimulated (+) for 6 h with IFN-γ; results are presented relative to those of an unstimulated sample, set as 1. Data are representative of two experiments with similar results (a,e–g; mean and s.d. in e–g), two (b; JhT) or three (b,c; all other strains) experiments with similar results, or five experiments with similar results (d; mean and s.d.).
To determine whether FcγR-bound serum IgG generated tonic signaling through FcγRI, we obtained peritoneal macrophages from IgG-deficient mice (i.e., muMT mice and JhT mice) and stimulated the cells for 6 h with IFN-γ. These mice have normal FcγRs but do not have circulating serum IgG to engage those receptors constitutively. As a ‘readout’, we used Nos2 as an example of a STAT- and ITAM-dependent gene transcription (Fig. 3a) and Irgm1 as an example of STAT-dependent but ITAM-independent gene transcription (Fig. 3a). Our data revealed that when stimulated with IFN-γ, IgG-deficient mice failed to properly induce Nos2 transcription but properly induced Irgm1 transcription (Fig. 4b), which suggested that tonic signaling through IgG-occupied FcγRs was, at least in part, the component required for the IFN-γR-mediated induction of Nos2.
To identify which FcγR most probably generated the tonic signal, we stimulated Fcγ-deficient, FcγRII-deficient and FcγRIII-deficient peritoneal macrophages with IFN-γ. Our data showed that tonic ITAM signaling was Fcγ dependent and was probably mediated via the high-affinity receptor FcγRI (and/or FcγRIV), not FcγRIII, and was inhibited by the immunoreceptor tyrosine-based inhibitory motif–containing receptor FcγRII (Fig. 4c). Full crosslinking of FcγRI with an antigen (SRBC (O)) delivered simultaneously with IFN-γ (which mimicked the conditions found during infections when both antigen and IFN-γ are present) was required for synergistic upregulation of full transcription of Nos2 mRNA to a level that produced detectable amounts of nitric oxide in the macrophage culture (Fig. 4d).
To identify the nature of the tonic signaling provided by FcγRI, we stimulated peritoneal macrophages with IFN-γ in the presence of inhibitors of NFAT, PI(3)K and MEK at concentrations determined to be effective (Supplementary Fig. 4c,d). Those pathways are activated by ITAMs in leukocytes. Only inhibition of PI(3)K completely eliminated transcription of the ITAM-dependent genes Nos2 and Ifi205 but not the ITAM-independent control gene Irgm1 in peritoneal macrophages (Fig. 4e–g). The NF-κB inhibitor Bay11 also completely blocked the PI(3)K pathway in peritoneal macrophages at all concentrations tested (Supplementary Fig. 4d) and hence blocked induction of the PI(3)K-dependent genes Nos2 and Ifi205 but not the PI(3)K-independent control gene Irgm1 in macrophages (Supplementary Fig. 4e). Thus, we could not use Bay11 to specifically assess the role of NF-κB pathway in tonic ITAM signaling in macrophages. As expected, the control ‘pan-Jak’ inhibitor blocked transcription of both Nos2 and Irgm1 (Supplementary Fig. 4f). Collectively, these experiments suggested that the ITAM module generated tonic PI(3)K signaling and that this signaling provided a permissive state for the induction of specific IFN-γ-dependent antimicrobial functions in peritoneal macrophages.
Early IFN-γ-mediated control of Listeria infection requires Fcγ
To determine whether the findings reported above had physiological consequences for IFN-γ-dependent antimicrobial defenses in vivo, we used infection with Listeria monocytogenes as a model. Before the production of antibodies to L. monocytogenes in vivo, early control of L. monocytogenes replication relies in part on IFN-γ-induced, iNOS-dependent killing of the bacteria in macrophage phagolysosomes before the bacteria is able to escape into the cytosol and before the killing of infected cells and systemic bacterial spread39. While antimicrobial functions elicited by IFN-γ limited bacterial loads in wild-type mice by 72 h after infection, that protection was lower in Fcγ-deficient mice as well as in control IFN-γRI-deficient mice, despite the even higher serum concentrations of IFN-γ in those two strains than in wild-type mice (Fig. 5 and Supplementary Fig. 5). These data suggested a physiological requirement for Fcγ expression and tonic ITAM signaling in IFN-γ- and iNOS-induced early bacterial clearance, even in conditions in which bacteria were not opsonized with IgG.
Figure 5.
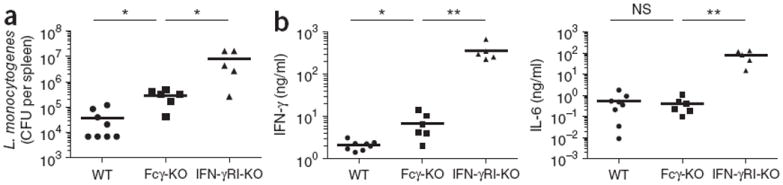
Early IFN-γ-mediated control of a b infection with L. monocytogenes in vivo requires Fcγ. (a) Bacterial load in the spleen of wild-type (B6) mice (n = 8), Fcγ-deficient mice (n = 6) and IFN-γRI-deficient mice (n = 6) 72 h after intraperitoneal infection with 2.5 × 105 L. monocytogenes, presented as colony-forming units (CFU) per spleen. (b) Enzyme-linked immunosorbent assay of IFN-γ and IL-6 in serum from mice as in a. Each symbol represents an individual mouse; small horizontal lines indicate the mean. One IFN-γRI-deficient mouse succumbed to infection. NS, not significant; *P < 0.05 and **P < 0.005 (two-tailed unpaired t-test). Data are pooled from two experiments with identical results.
‘Coincidence detection’ by IFN-γR and FcγRI
Given the possibility that IFN-γR and FcγRI could form a functional complex on the surface of phagocytic cells, we next determined if signaling through IFN-γR modulated FcγRI-triggered responses during IgG2a-mediated phagocytosis. Because such responses rely on crosslinking of the FcγR and not on tonic Fcγ signaling, we used BM macrophages that we stimulated in vitro for 6 h through the IFN-γR or FcγRI alone or through both receptors and analyzed global gene expression and gene ontology (Supplementary Fig. 6a,b and Supplementary Table 4). We used IgG2a-opsonized SRBCs to specifically target FcγRI (Supplementary Fig. 6c–f). As expected, stimulation with IFN-γ alone induced genes encoding molecules involved in preparing macrophages for antimicrobial defense, such as pathogen sensing and uptake (Fcgr1, Fcgr4, Nod1 and Myd88), macrophage maturation (Cd86 and Cd83) and antigen presentation (Tap1, Tap2, Cd74, Psmb9 (also known as Lmp2), Plmb8 (also known as Lmp7) and Ciita) (Fig. 6a, primed state I). Stimulation with FcγRI alone induced genes encoding chemokines (Cxcl1 and Cxcl2) that would probably recruit other phagocytes and genes encoding molecules involved in adaptation to oxidative stress (Prdxn1 and Srxn1), presumably triggered by heme present in SRBCs (Fig. 6a, primed state II). The engagement of FcγRI in the context of IFN-γR signaling, a scenario that would mimic an in vivo infection, resulted in a response that was not just the sum of the two individual receptor programs; instead, this synergistically induced a unique set of genes that were either not induced or were modestly induced by either stimulus alone (Fig. 6b,c). In addition to Nos2, that set included several other genes encoding molecules in the reactive nitrogen and oxygen pathway (Ass1, Nos2 and Sod2), as well as genes encoding molecules involved in regulating phagosomal trafficking and remodeling (Cdc42ep2 and Gja1 (also known as Connexin43)) and antimicrobial functions (Gbp5, Nos2, Ass1 and Gpd2), many of which are reported to be part of a unique phagosomal proteome in IFN-γ-stimulated macrophages40. The synergistic gene program also included genes encoding molecules required for the priming of cytotoxic functions of NK cells and T cells (Il27, Il15, Il15ra and Slamf7), as well as genes encoding molecules involved in tissue repair (Hbegf, Plaur and F10) and leukocyte homing (Ccl5 and Cxcl16) (Fig. 6b,c). Thus, during phagocytosis, IFN-γR and FcγRI acted like ‘coincidence detection receptors’, meaning that certain functions were elicited only when both signals were delivered in the same window of time and space12.
Figure 6.
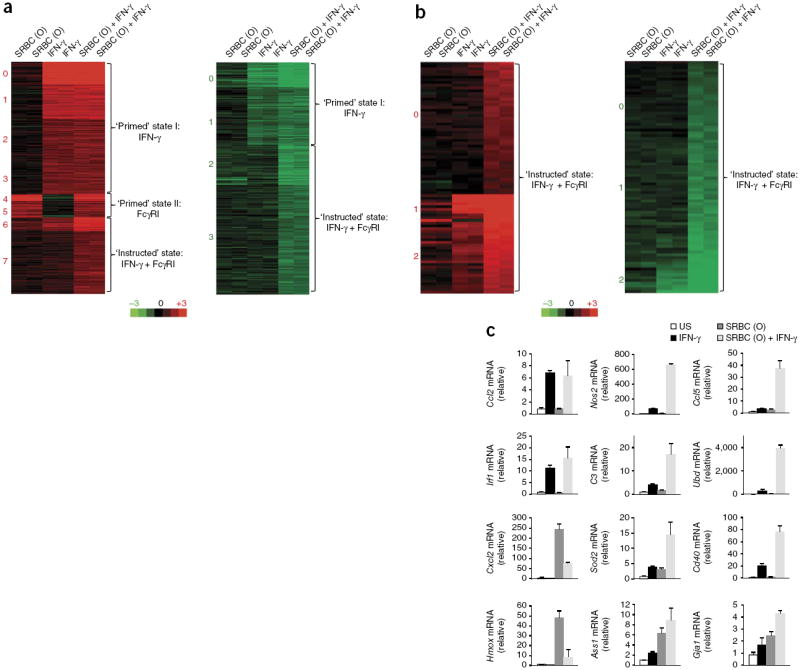
IFN-γR and FcγRI function by the ‘coincidence detection principle’ to induce a specific transcriptional program in phagocytes. (a) Cluster analysis (by k-means) of gene expression (colors as in Fig. 3f) in wild-type (B6) BM macrophages stimulated for 6 h with IFN-γ or SRBC (O) or both (above plots), for all genes with a change in expression of twofold or more relative to that of unstimulated cells, with a P value of <0.05. (b) Cluster analysis of gene expression in wild-type macrophages stimulated as in a, for genes with a change in expression of 1.5-fold after stimulation with IFN-γ plus SRBC (O) relative to that of unstimulated cells, followed by classification of those genes as synergistically induced or suppressed, by a change in expression in response to stimulation with IFN-γ plus SRBC (O) of 1.5-fold above that in response to stimulation with IFN-γ or SRBC (O) alone. (c) Quantitative PCR analysis confirming the microarray data in a, for genes upregulated by IFN-γ or SRBC (O) (left) or synergistically upregulated by both stimuli together (middle and right); results are presented relative to those of an unstimulated sample, set as 1. Data are representative of one experiment with duplicate samples (a–c), with independent result validation (c); mean and s.d.
DISCUSSION
We have described here structural and functional cooperation between IFN-γR and the ITAM signaling module of the the high-affinity antibody receptor FcγRI at the plasma membrane of macrophages, an association with functional consequences for the biology of both receptors. First, FcγRI constitutively occupied by serum IgG in vivo generated tonic Fcγ ITAM signaling in peritoneal macrophages. That tonic ITAM signaling through PI(3)K was required for the IFN-γR-triggered induction of a subset of genes encoding antimicrobial molecules in those macrophages (for example, Nos2 and Ifi205) and molecules involved in the control of acute infection by L. monocytogenes in vivo. Second, functional receptor coupling allowed IFN-γR to modify FcγRI responses during IgG-mediated crosslinking and phagocytosis in vitro. Thus, FcγRI and IFN-γR acted as a ‘coincidence detection system’, whereby unique gene-expression programs were elicited only when both activating signals were detected together at the same time by the same cells, which allowed the induction of cell-autonomous antimicrobial functions.
Tonic signaling through ITAM-associated immunoreceptors seems to be critical for the function of T cells, B cells and, as shown here, macrophages. The exact nature of the tonic signaling remains incompletely understood and its function may vary between different cell types. We propose that tonically stimulated ITAM modules provide an accessory function to cytokines or other signals that cannot otherwise elicit such functions by themselves. Such accessory signals could be the production of signaling intermediates, such as intracellular Ca2+, phosphatidylinositol-4,5-bisphosphate and phosphatidylinositol-3,4,5-trisphosphate, which are involved in multiple signaling events but can be induced only by a few signaling modules, such as ITAMs. In this context, ITAMs deliver a permissive signal for cellular activation without specifying cellular responses but allow the cytokines to do so. The PI(3)K pathway may serve as a common permissive signal delivered by many different ITAMs and ITAM-like modules: in B cells, PI(3)K transmits a key tonic signal produced by the ITAM-associated BCR that is required for the survival and proliferation of B cells41,42; we found here that in peritoneal macrophages, PI(3)K transmitted a key tonic signal from the ITAM-associated receptor FcγRI that was required for IFN-γ-induced antimicrobial responses; and in basophils, we have found that PI(3)K transmitted the tonic signal produced by an as-yet-unidentified Fcγ-associated receptor required for allergic responses (R.K.R., J.S.B. and R.M., data not shown). Finally, the PI(3)K pathway is also the main signal mediator downstream of many ITAM-like adaptors, such as DAP10 (ref. 43), that are associated with NK cell receptors. The PI(3)K pathway is activated locally at the cell membrane by the recruitment of PI(3)K subunits to an active ITAM site, which could potentially explain why cytokine receptors (such as IFN-γR) need to be recruited to the immunoreceptor site to access the pathway. In Fcγ-deficient cells, other PI(3)K activators, such as growth factors, did not compensate for the ITAM-derived permissive signal required for IFN-γ signaling, which suggests that spatially restricted, compartment- specific activation of PI(3)K might indeed be a requirement.
The colocalization of cytokine receptors and ITAM-containing immunoreceptors also allows cytokines to modify immunoreceptor responses and to make them conditional on the cytokine milieu present at the time of activation. Cytokine signaling is often thought of as an independent module that integrates its information with that emanating from immunoreceptors at the promoters of appropriate genes. While that is certainly correct in many cases, our data suggest that in addition, some cytokine receptors can also localize together with ITAM-containing receptors at the cell membrane and potentially ‘complete’ otherwise incomplete information generated by ITAM signaling alone. This would allow functionally related receptors to not only induce individual responses when only one stimulus is present but also function as a ‘coincidence detection system’ to induce an overlapping and distinct response when both signals are present.
The extent of dependence on signals from the other receptor could vary in different situations. Activation of the immunoreceptor NKG2D, which elicits the cytotoxic function of NK cells44, is completely dependent on the coexpression of the cytokine IL-15 and the NKG2D ligand Rae1 on target cells marked for killing10. In our study here, stimulation by FcγRI and uptake of cargo happened independently of cytokines, but only the detection of both FcγRI signaling and IFN-γR signaling together triggered additional transcriptional programs for the induction of antimicrobial functions in cells that had engulfed opsonized cargo. The latter event is probably important during both primary immune responses and secondary immune responses, when FcγRI is activated either by acute-phase proteins, such as pentraxins45, or antigen-specific IgG2a37, while IFN-γR is activated by cytokines secreted by innate-like lymphocytes and innate lymphoid cells46 or by antigen-specific T cells, respectively. Once FcγR has engulfed its cargo, phagocytes become unresponsive to subsequent stimulation by IFN-γ47-49; that is consistent with the idea that IFN-γ modifies FcγR function when present before or at the time of FcγR activation but is redundant once the phagocytic cargo has been taken up and the appropriate functional programs have already been initiated. Crosslinking of FcγR by high-density immunocomplexes, such as those seen in the final stages of successful immune responses, has been shown to generate IL-10-producing ‘regulatory macrophages’, presumably to dampen the response and limit further inflammation50; this suggests that functional ‘collaboration’ between IFN-γR and FcγR is spatially and temporally restricted.
The mechanics of receptor coupling remain unclear. Unlike immunoreceptors (such as the BCR, TCR, FcR and NKG2D)5, cytokine receptors do not carry a charged residue (or residues) in the transmembrane region to allow their constitutive coupling with the conserved charged residue in transmembrane region of the ITAM adaptor. We propose the existence of dedicated adaptors that could be either cytosolic proteins or transmembrane proteins, which are dynamically recruited and released to facilitate coupling of non-immunoreceptors to ITAM adaptors and to accommodate any changes in the cellular microenvironment. Finally, the mechanisms of receptor cooperation described here for FcγRI and IFN-γR are probably a common theme for other ITAM-containing receptors and cytokine receptors. The functional coupling between these receptor classes allows context-dependent responses in leukocytes.
ONLINE METHODS
Mice
Mice were maintained at Yale Animal Resources Center in accordance with the approved guidelines by the Institutional Animal Care and Use Committee at Yale. BALB/c and C57BL/6 (B6) mice were from the National Cancer Institute or Jackson Laboratories. BALB/c Fcγ-deficient, BALB/c JhT, 129 and 129 STAT1-deficient mice were from Taconic. B6 Fcγ-deficient, B6 IFN-γRI-deficient, B6 FcγRII-deficient, B6 FcγRIII-deficient and B6 muMT (B6 Ighm) mice were from Jackson Laboratories.
Reagents and antibodies
The following reagents (from vendors in parentheses) were used: Ack lysing buffer (Lonza), FBS (Benchmark), recombinant mouse IL- 4, recombinant mouse IFN-α (carrier-free; R&D Systems), recombinant mouse IFN-γ (Peprotech), Lipofectamine 2000, OVA–Alexa Fluor 488 (Invitrogen), RNA-Bee (Tel-TesT), RNeasy (Qiagen), SMART MMLV RT (Clontech), SYBR Green QPCR mix (Quanta), protease inhibitor tablets, Ly294002 (Sigma and Calbiochem), PKH26 (Sigma), 3-μm white Polybead Carboxylate Microspheres (beads; PolySciences), PolyLink Protein Coupling Kit for COOH Microspheres (PolySciences), EZ-Link Sulfo-NHS-Biotinylation Kit (Pierce), fresh SRBCs (Lampire Biological), hybridoma producing mouse IgG2a antibody to SRBCs (TIB111; American Type Culture Collection), ECL (Amersham/GE Healthcare), cyclosporine A, PD98059, Wortmannin, BAY 11-7082, Jak inhibitor I (Calbiochem), TMB Substrate for ELISA (BD Pharmingen), horseradish peroxidase–conjugated TrueBlot (for the detection of proteins after immunoprecipitation; eBioscience). Phosphatase-inhibitor mix (sodium orthovanadate, sodium fluoride and sodium glycerol phosphate) and Griess reagent (reagent A (1% sulfanilamide in 2.5% H3PO4) and reagent B (0.1% wt/v naphthylethylenediamine dihydrochloride in 2.5% H3PO4)) were made in house. Modified retroviral vector pMSCV.hCD2 and vectors MIGR2 and pMSCV.IRES-GFP were provided by D. Stetson. Antibodies were as follows: anti-Fcγ (06-727; Upstate Cell Signaling), antibody to phosphorylated tyrosine (4G10; Upstate Cell Signaling), anti-hemagglutinin (05-904; Millipore), anti-ovalbumin (AB1225; Millipore), antibody to phosphorylated PLC-γ2 (3874S; Cell Signaling Technologies), antibody to phosphorylated Syk (2711S; Cell Signaling Technologies), antibody to tyrosine-phosphorylated STAT1 (9167S; Cell Signaling Technologies), antibody to serine-phosphorylated STAT1 (9177; Cell Signaling Technologies), anti-STAT1 (9172; Cell Signaling Technologies), antibody to phosphorylated Jak1 (3331; Cell Signaling Technologies), antibody to phosphorylated Jak2 (3776; Cell Signaling Technologies), antibody to phosphorylated p38 (9216S; Cell Signaling Technologies), antibody to phosphorylated Akt (9271; Cell Signaling Technologies), anti-β-actin (loading control for immunoblot analysis; A5441; Sigma), rabbit polyclonal anti-Flag (F7425; Sigma), phalloidin (high-affinity F-actin probe for ImageStream; A22287; Invitrogen), anti-FcγRI (for immunoblot analysis; sc-7642; Santa Cruz), anti-IkBα (for immunoblot analysis; sc-371; Santa Cruz), anti-IFN-γRI (for immunofluorescence and ImageStream analysis of fixed-permeabilized cells; C20; sc-700; Santa Cruz), Alexa Fluor 647–anti-rabbit IgG (for immunofluorescence; A-21246; Life Technologies), biotin–anti-IFN-γRI (for ImageStream surface staining of unfixed cells and for flow cytomtery: 2E2; 550482; BD Pharmingen; and 13-1191-82; eBioscence), anti-FcγRI (for ImageStream surface staining of unfixed cells and for flow cytomtery; 558539; BD Pharmingen), phycoerythrin-conjugated antibody to human CD2 (555327; BD Pharmingen), anti-FcγRII-FcγIII (Fc Block (or anti-CD16/32); 553142; BD Pharmingen; and 14-0161-86; eBioscience), anti-CD11b (12-0112-83; eBioscience), anti-B220 (553093; BD Pharmingen), anti-F4/80 (17-4801-82; eBioscience), fluorescein isothiocyanate–anti–mouse IgG2a (553390; BD Pharmingen), streptavidin-phycoerythrin (554061; BD Pharmingen), streptavidin-allophycocyanin (17-4317-82; eBioscience), streptavidin–phycoerythrin–Texas Red (551487; BD Pharmingen) F(ab′)2 antibody to mouse IgG (specific for the heavy and light chains 715-096-150; Jackson ImmunoResearch), horseradish peroxidase–conjugated anti-rabbit IgG (111-035-144; Jackson ImmunoResearch), horseradish peroxidase–conjugated anti-mouse IgG (115-035-146; Jackson ImmunoResearch), horseradish peroxidase–conjugated anti-goat IgG (205-035-108; Jackson ImmunoResearch), biotin-conjugated armenian hamster IgG (isotype-matched control antibody; 13-4888-81; eBioscience), rat IgG2b (isotype-matched control antibody; 16-4031-85; eBioscience), rabbit IgG (I8140; Sigma) and normal rabbit serum (011-000-120; Jackson ImmunoReserach).
RNA extraction and quantitative PCR analysis
BM macrophages or peritoneal macrophages were plated at a density of 0.5 × 106 cells per 0.5 ml per well in 24-well non-TC dishes in macrophage-replating medium (described below) 1 d before the experiment (except for cells derived from IgG-deficient mice, described below) and then stimulated. Total RNA was extracted from BM macrophages in 0.5 ml RNA-Bee and was extracted from peritoneal macrophages with RNEasy according to the manufacturer’s instruction. RNA was reverse-transcribed with an oligo(dT) primer and SMART MMLV RT. cDNA was analyzed by quantitative PCR with SYBR Green QPCR Mix on an MX300P Stratagene System (primers, Supplementary Fig. 6b) and the comparative quantification MXPro software. Primers were designed to amplify mRNA-specific sequences and, where possible, were placed on two separate exons spanning long intron in between. Expression was normalized to the expression of RPL13a or HPRT in the same sample and was calibrated to the expression in unstimulated wild-type samples, set as 1.
Immunoblot analysis and immunoprecipitation
BM macrophages were replated at a density of 1 × 106 cells per ml per well in 12-well non-TC dishes in macrophage-replating medium 1 d before experiment. Cells were stimulated with 10–20 ng/ml IFN-γ or SRBCs (SRBC/macrophage ratio, 20:1) or both. Reactions were stopped with PBS at 4 °C, then cells were washed and lysed directly in 1× SDS loading buffer containing protease-inhibitor ‘cocktail’ and phosphatase-inhibitor ‘cocktail’ (17.5 mM β-glycerophosphate, 20 mM NaF and 1 mM sodium orthovanadate). Lysates were boiled for 5 min at 100 °C, then proteins were separated by SDS PAGE on 4–12% gradient gels, transferred to PVDF membranes and analyzed with the appropriate antibodies (identified above). All primary antibodies incubations were done overnight at 4 °C. For immunoprecipitation, 5 × 106 293T cells were lysed for 30 min at 4 °C in 0.5 ml of 1% TNT buffer (consisting of 50 mM Tris, pH7.5, 150 mM NaCl, 1% Triton X-100, and complete protease and phosphatase inhibitors). Nuclei were spun down, then 10 μl of the supernatant was retained as the input control, mixed with 5× SDS sample buffer (125 mM Tris, pH 6.8, 10 % SDS, 50 % glycerol, 0.06% bromophenol blue and 2% β-mercaptoethanol in water) and boiled. The rest of the supernatant was incubated for 2 h at 4°C (with gentle rocking) with 1–2 μg/ml anti-hemagglutinin or anti-Flag (identified above). Protein G beads (30 μl) were added for another 1 h. Beads were then spun and washed five times with washing buffer (0.1% TNT buffer). Proteins were eluted with 2× SDS buffer and analyzed by immunoblot with the appropriate antibodies (identified above) and horseradish peroxidase–conjugated TrueBlot.
Flow cytometry
BM macrophages were lifted from non-TC plates with 5 mM EDTA in PBS at 4 °C and were washed in flow cytometry buffer (2% FBS and 0.1% NaN3 in PBS). Fresh peritoneal exudate cells were collected as described elsewhere in this section. Cells were incubated for 15–60 min with anti-CD16/32 (Fc Block) before being stained for 1 h on ice with the appropriate conjugated antibodies at a dilution of 1:100 (primary antibody) and for 30 min on ice with secondary antibody at a dilution of 1:400 (all identified above). Cells were washed, fixed with 2% PFA and analyzed with a FACSCalibur (BD Biosciences) and FloJo software (TreeStar).
Peritoneal macrophage isolation
Peritoneal macrophages were isolated from naive (dead) mice by injection into peritoneum of 5 ml of 3% FBS in PBS at 4 °C, with a 27-gauge needle. The peritoneum was massaged gently for ~60 s, and peritoneal liquid was aspirated with a 22-gauege needle into a prechilled tube on ice to prevent adherence of macrophages to the plastic. Cells were plated overnight in 24-well TC dishes into ‘complete’ RPMI medium (RPMI medium containing sodium pyruvate (1 mM), HEPES buffer (25 mM), penicillin-streptomycin (100 U/ml penicillin and 100 μg/ml streptomycin), l-glutamine (2 mM) and 2-mercaptoethanol (50 μM)) with 10% FBS and 15% L929 mouse fibroblast–conditioned medium containing macrophage colony-stimulating factor, to allow macrophage adherence. When cells were collected from IgG-deficient mice (muMT or JhT), macrophages were allowed to adhere to plastic only for 2 h to limit any potential binding of serum IgG to unoccupied FcγRs during overnight culture. Floating cells (B cells, T cells, etc.) were discarded, then fresh prewarmed medium was added over the macrophages and cells were stimulated and analyzed.
BM-derived macrophages and DC cultures
After lysis of red blood cells, total BM cells were plated at a density of 6 × 106 cells per 15-cm Petri dish in 20 ml macrophage growth medium (complete RPMI medium with 20% FBS, 30% L929-conditioned medium containing macrophage colony-stimulating factor). Cells were fed with additional 20 ml of growth medium on day 4. On day 7, cells were lifted with 5 mM EDTA in PBS at 4 °C and were replated in 12- or 24-well non-TC dishes (1 × 106 cells per ml per well (12-well) or 0.5 × 106 cells per 0.5 ml per well (24-well)) in macrophages replating media (complete RPMI, 10% FBS, 15% L929-conditioned medium containing macrophage colony-stimulating factor). For DC, BM cells were grown in DC growth media (complete RPMI medium with 10% FBS, 2% L929-conditioned medium containing granulocyte-macrophage colony-stimulating factor) at a density of 1 × 106 cells per ml per well of a 12-well plate. Cells were fed on day 2 and day 4 and were used on day 5 without replating.
DNA constructs, transfection and retroviral transduction
The wild-type Fcγ sequence was isolated by PCR from basophil cDNA. Wild-type and mutant Fcγ tagged with Flag at the amino terminus was generated and transduced into the BM as described11 (R.K.R., J.S.B. and R.M., data not shown), except that BM cells were transduced on days 1 and 2 after isolation and then were differentiated into BM macrophages. The Jak2 construct was from Open Byosystems, sequence encoding the hemagglutinin tag at the carboxyl terminus was introduced by PCR and the construct cloned into vector pMSCV.IRES-GFP. 293T cells were transiently cotransfected with Lipofectamine 2000 containing vector encoding DAP10-, DAP12- or Fcg-Flag proteins10, with twice that amount (in micrograms) of vector encoding Jak2-HA or the corresponding empty vector. Cells were incubated for 24 h with DNA-containing supernatants, then the medium was replaced and cells were incubated for additional 24 h. Only in those experiments in which transfection efficiency was over 80% (to ensure sufficient numbers of double-transfected cells), 293T cells were lysed in 1% TNT buffer, and Flag- and hemagglutinin-tagged proteins were coimmunoprecipitated with the appropriate antibodies.
Microarray analysis
An eight-sample Mouse Ref-8 v2.0 Illumina array was used for comparison of global changes in gene expression in peritoneal and BM macrophages under various conditions. Cells were stimulated for 6 h with 10 ng/ml IFN-γ, 10 ng/ml IL4 and/or SRBC (O) (SRBC/macrophage ratio, 10:1). Data-processing procedures are outlined in Supplementary Figure legends as well as in all Supplementary Tables. Genes were clustered by k-means in Gene Cluster 3.0 software and were visualized with Java TreeView.
Phagocytosis assay
BM macrophages were replated at a density of 1 × 106 cells per ml per well of a 12-well non-TC dish in macrophage-replating medium 1 d before the experiment. On the day of the experiment, fresh SRBCs were labeled with PKH26 according to manufacturer’s instructions, except that 100 × 106 SRBC were used per 1 ml of labeling reaction. SRBCs were opsonized for 1–2 h at room temperature (with gentle rocking) in 1 ml of supernatants of the TIB111 mouse B cell hybridoma (American Type Culture Collection) containing IgG2a antibody to SRBC. Control SRBCs were kept in 1 ml of DMEM containing 10% FBS. SRBCs were always used fresh within 2–3 weeks of purchase. Glutardehyde-fixed SRBCs were not used because they showed substantial nonspecific uptake independently of FcγR. SRBCs were typically shipped as a 10% solution and were kept at that density for the opsonization step (for example, 100 × 106 cells were labeled in 1 ml TIB111-conditioned medium). Opsonized cells were washed in PBS for the removal of unbound antibody, then were suspended at a density of 100 × 106 cells in 100 μl of serum-free RPMI medium and were kept on ice. For stimulation, SRBCs were vortexed twice for 15 s each and added at an SRBC/macrophages ratio of 10:1 or 20:1. For nonsynchronized assays, SRBCs were simply added into macrophage-containing wells. For synchronized assays, SRBCs were spun over adherent macrophages for 5 min at 4 °C at 1,500 r.p.m. and then were moved to 37 °C to initiate signaling. Similar data were obtained in both assays. At the end of stimulation, macrophage medium was aspirated, cells were washed gently with PBS at 4 °C, with ACK lysis buffer added for 1–2 min at room temperature for lysis of SRBCs that had not been phagocytosed, and cells were washed once again with PBS. Macrophages were lifted with 5 mM EDTA and 0.1% NaN3 in PBS at 4 °C, then were fixed in 2% PFA and analyzed by flow cytometry. Phagocytic index was calculated as mean fluorescence intensity multiplied by the frequency of PKH26+ cells.
ImageStream and immunofluorescence
For ImageStream analysis, BM DCs were preferred because BM macrophages lose their native morphology when acquired in suspension. Findings were reproduced by immunofluorescence of BM macrophages as well. For the appropriate (in this case, outward) orientation of antibody Fc-regions, ovalbumin–Alexa Fluor 488 was first covalently coupled to 3-μm COOH-modified latex beads with PolyLink coupling reagents according to the manufacturer’s instruction. Beads were then opsonized for 1 h at room temperature (with gentle rocking) with rabbit IgG antibody to ovalbumin. DCs were kept in suspension, and nonsynchronized phagocytosis was initiated at 37 °C with IgG-opsonized beads (Alexa Fluor 488) at a DC/bead ratio of 1:10. IFN-γ was not added to cultures at this time. Reactions were stopped after 5, 10 and 20 min with ice-cold PBS. Cells were washed with PBS, fixed with 2% PFA, permeabilized with BDPermWash and stained with DAPI (4,6-diamidino-2-phenylindole) and phalloidin (Alexa Fluor 647) or anti-IFN-γRI (C-20) biotinylated with an EZ-Link Sulfo-NHS-Biotinylation Kit, plus streptavidin-allophycocyanin.
After being stained, cells were washed and fixed and then were analyzed at a magnification of ×40 on ImageStream flow cytometer (Amnis) and The IDEAS analysis software (Amnis). Single-color controls were used for the creation of a compensation matrix that was applied to all files to correct for spectral cross-talk. Positive cutoff values were calculated on the basis of the background ‘bright detail similarity’ (BDS) of IFN-γRI and an irrelevant signal (for example, side scatter). For phagocytosis assays, the internalization feature was first used for calculation of the ratio of the intensity of Alexa Fluor 488 (bead signal) inside the cell to the total intensity of Alexa Fluor 488 outside the cell. Higher internalization scores indicate a greater concentration of Alexa Fluor 488–labeled beads inside the cell. The BDS feature of IDEAS was then used for measurement of the spatial correlation between the fluorescent signal emanating from internalized Alexa Fluor 488–labeled beads and that of Alexa Fluor 647–phalloidin or allophycocyanin–labeled IFN-γRI. BDS is the log-transformed Pearson’s correlation coefficient of the localized bright spots with a radius of three pixels or less within the masked area in the two input images. The higher the BDS score, the higher the level of colocalization. For actin, images with an internalization score of >3 and a BDS score of >2 are presented. For IFN-γRI analysis (Fig. 1b), an additional one-pixel region was included into the analysis region to create ‘dilated mask’ around the bead, which was then used for measurement of the mean intensity score of IFNγ-RI signal around the bead. That score reflected the recruitment of IFN-γRI to the plasma membrane surrounding the Alexa Fluor 488–labeled bead, and the enrichment in signal relative to that of the IFN-γRI signal in the rest of the cell was calculated. That dilated mask was used because IFN-γRI wraps around the bead rather than over the bead, so simple BDS-colocalization scoring could miss that signal. Images with an intensity score of >10 for allophycocyanin–IFN-γRI around the Alexa Fluor 488–labeled beads are presented.
For experiments showing surface staining of DCs (Supplementary Fig. 1d), cells were stained with both biotin–anti-IFN-γRI (2E2) and anti- FcγRI (X54-5/7.1) without fixation with PFA or permeabilization for a better staining signal, as these antibody clones do not work well on previously fixed and permeabilized cells. After being stained, cells were washed and fixed and then were analyzed with an ImageStream. Staining specificity was determined with an isotype-matched control antibody and with Fcγ-deficient and IFN-γRI-deficient macrophages.
For imaging of BM macrophages, immunofluorescence was used because they are adherent cells on uncoated microscope coverslips. BM macrophages from wild-type and various Fcγ-deficient mice were allowed to adhere overnight, then were ‘fed’ IgG2a-opsonised SRBCs (TIB111 for anti-SRBC) for 15 min in nonsynchronized phagocytosis assay in the presence or absence of IFN-γ. Cells were washed once, then were fixed for 10 min with 4% PFA, permeabilized for 1 h at room temperature with BDPermWash containing 10% FBS plus Fc Block (for blockade of nonspecific binding) and then were probed for 1 h at room temperature with anti-IFN-γRI (C-20), followed by incubation for 30 min with Alexa Fluor 647–anti-rabbit. Images were visualized with 100× magnification on immunofluorescence microscope Zeiss Axioplan 2 and were analyzed using Axiovision 4.8. Because the uptake of SRBC (O) in Fcγ-deficient BM macrophageand the uptake of SRBC (N) in wild-type BM macrophages are both extremely low, arrows were added to images (Supplementary Fig. 1a,b) to indicate the location of few phagocytosed SRBC.
Griess assay
BM macrophages were replated at a density of 1 × 106 cells per 1 ml per well of a 12-well non-TC dish in macrophage-replating medium 1 d before experiments. Cells were stimulated for 16–24 h with 10 ng/ml IFN-γ with or without SRBC (O) or SRBC (N) (SRBC/macrophage ratio, 20:1). At the end of the assay, 50 µl of undiluted supernatant was transferred into flat-bottomed, clear, 96-well plates and that was mixed with 50 μl Griess reagent A and 50 μl reagent B. Absorbance at 550 nm was measured immediately in microplate reader. A standard curve was created with serial dilutions of NaNO2 from 200 μM.
L. monocytogenes infection
Wild-type (B6), Fcγ-deficient and IFN-γRI-deficient mice were infected intraperitoneally with 2.5 × 105 L. monocytogenes strain 10403S. After 72 h, spleens and serum were collected. Serum was used for measurement of cytokines by enzyme-linked immunosorbent assay. Splenocytes were lysed for 10 min in 200 μl 0.1% Triton X-100 in PBS, and lysates transferred to 800 μl PBS. For each time point, serial dilutions were made (1:10 into 1 ml PBS), and 100 μl of each dilution were plated onto Luria-Bertani plates containing streptomycin (100 μg/ml). Bacteria were grown overnight and bacterial colonies were counted.
Statistics and general methods
Where indicated, a two-tailed unpaired t-test was used for calculation of significance. Researchers were not ‘blinded’ to sample identity, and age and sex-matched littermates were used for all experiments.
Supplementary Material
Acknowledgments
We thank D. Stetson (University of Washington, Seattle) for modified retroviral vector pMSCV.hCD2 and vectors MIGR2 and pMSCV.IRES-GFP; members of the Medzhitov laboratory and S. Joyce for discussions and reading of the manuscript; and C. Annicelli and S. Cronin for assistance with animal work. Supported by the Howard Hughes Medical Institute (R.M. and J.S.B.), the US National Institutes of Health (1R56AI087725-01 AI046688, AI055502, AI089771 and DK071754 to R.M.) and the Damon Runyon Cancer Research Foundation (DRG-1968-08 to J.S.B.).
Footnotes
AUTHOR CONTRIBUTIONS
J.S.B. and R.M. designed research and wrote manuscript; J.S.B. did and analyzed experiments; R.K.R. and J.S.B. cloned and initially characterized Fcγ and its mutants; I.B. and J.S.B. did in vitro L. monocytogenes infection experiments; and R.A.D. analyzed and interpreted ImageStream data.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
Accession codes. GEO: microarray data, GSE54824.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Humphrey MB, Lanier LL, Nakamura MC. Role of ITAM-containing adapter proteins and their receptors in the immune system and bone. Immunol Rev. 2005;208:50–65. doi: 10.1111/j.0105-2896.2005.00325.x. [DOI] [PubMed] [Google Scholar]
- 2.Stark GR, Darnell JE., Jr The JAK-STAT pathway at twenty. Immunity. 2012;36:503–514. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nathan C, Sporn M. Cytokines in context. J Cell Biol. 1991;113:981–986. doi: 10.1083/jcb.113.5.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–2629. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- 5.Call ME, Wucherpfennig KW. Common themes in the assembly and architecture of activating immune receptors. Nat Rev Immunol. 2007;7:841–850. doi: 10.1038/nri2186. [DOI] [PubMed] [Google Scholar]
- 6.Bezbradica JS, Medzhitov R. Role of ITAM signaling module in signal integration. Curr Opin Immunol. 2012;24:58–66. doi: 10.1016/j.coi.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 7.Hamerman JA, Ni M, Killebrew JR, Chu CL, Lowell CA. The expanding roles of ITAM adapters FcRγ and DAP12 in myeloid cells. Immunol Rev. 2009;232:42–58. doi: 10.1111/j.1600-065X.2009.00841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koga T, et al. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature. 2004;428:758–763. doi: 10.1038/nature02444. [DOI] [PubMed] [Google Scholar]
- 9.Zou W, Reeve JL, Liu Y, Teitelbaum SL, Ross FP. DAP12 couples c-Fms activation to the osteoclast cytoskeleton by recruitment of Syk. Mol Cell. 2008;31:422–431. doi: 10.1016/j.molcel.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horng T, Bezbradica JS, Medzhitov R. NKG2D signaling is coupled to the interleukin 15 receptor signaling pathway. Nat Immunol. 2007;8:1345–1352. doi: 10.1038/ni1524. [DOI] [PubMed] [Google Scholar]
- 11.Hida S, et al. Fc receptor γ-chain, a constitutive component of the IL-3 receptor, is required for IL-3-induced IL-4 production in basophils. Nat Immunol. 2009;10:214–222. doi: 10.1038/ni.1686. [DOI] [PubMed] [Google Scholar]
- 12.Vivier E, Malissen B. Innate and adaptive immunity: specificities and signaling hierarchies revisited. Nat Immunol. 2005;6:17–21. doi: 10.1038/ni1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-γ: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 14.Nimmerjahn F, Ravetch JV. Fcγ receptors: old friends and new family members. Immunity. 2006;24:19–28. doi: 10.1016/j.immuni.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 15.Pearse RN, Feinman R, Ravetch JV. Characterization of the promoter of the human gene encoding the high-affinity IgG receptor: transcriptional induction by γ-interferon is mediated through common DNA response elements. Proc Natl Acad Sci USA. 1991;88:11305–11309. doi: 10.1073/pnas.88.24.11305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beekman JM, van der Linden JA, van de Winkel JG, Leusen JH. FcγRI (CD64) resides constitutively in lipid rafts. Immunol Lett. 2008;116:149–155. doi: 10.1016/j.imlet.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 17.Wang L, et al. ‘Tuning’ of type I interferon-induced Jak-STAT1 signaling by calcium-dependent kinases in macrophages. Nat Immunol. 2008;9:186–193. doi: 10.1038/ni1548. [DOI] [PubMed] [Google Scholar]
- 18.Sato K, et al. Dectin-2 is a pattern recognition receptor for fungi that couples with the Fc receptor γ chain to induce innate immune responses. J Biol Chem. 2006;281:38854–38866. doi: 10.1074/jbc.M606542200. [DOI] [PubMed] [Google Scholar]
- 19.Agarwal A, Salem P, Robbins KC. Involvement of p72syk, a protein-tyrosine kinase, in Fcγ receptor signaling. J Biol Chem. 1993;268:15900–15905. [PubMed] [Google Scholar]
- 20.Crowley MT, et al. A critical role for Syk in signal transduction and phagocytosis mediated by Fcγ receptors on macrophages. J Exp Med. 1997;186:1027–1039. doi: 10.1084/jem.186.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gamero AM, Larner AC. Signaling via the T cell antigen receptor induces phosphorylation of Stat1 on serine 727. J Biol Chem. 2000;275:16574–16578. doi: 10.1074/jbc.M910149199. [DOI] [PubMed] [Google Scholar]
- 22.Xu W, Nair JS, Malhotra A, Zhang JJ. B cell antigen receptor signaling enhances IFN-γ-induced Stat1 target gene expression through calcium mobilization and activation of multiple serine kinase pathways. J Interferon Cytokine Res. 2005;25:113–124. doi: 10.1089/jir.2005.25.113. [DOI] [PubMed] [Google Scholar]
- 23.Kovarik P, Stoiber D, Novy M, Decker T. Stat1 combines signals derived from IFN-γ and LPS receptors during macrophage activation. EMBO J. 1998;17:3660–3668. doi: 10.1093/emboj/17.13.3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farlik M, et al. Nonconventional initiation complex assembly by STAT and NF-κB transcription factors regulates nitric oxide synthase expression. Immunity. 2010;33:25–34. doi: 10.1016/j.immuni.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wen Z, Zhong Z, Darnell JE., Jr Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 26.Nair JS, et al. Requirement of Ca2+ and CaMKII for Stat1 Ser-727 phosphorylation in response to IFN-γ. Proc Natl Acad Sci USA. 2002;99:5971–5976. doi: 10.1073/pnas.052159099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kovarik P, et al. Stress-induced phosphorylation of STAT1 at Ser727 requires p38 mitogen-activated protein kinase whereas IFN-γ uses a different signaling pathway. Proc Natl Acad Sci USA. 1999;96:13956–13961. doi: 10.1073/pnas.96.24.13956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Boxel-Dezaire AH, Stark GR. Cell type-specific signaling in response to interferon-γ. Curr Top Microbiol Immunol. 2007;316:119–154. doi: 10.1007/978-3-540-71329-6_7. [DOI] [PubMed] [Google Scholar]
- 29.MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 30.Kleinert H, Schwarz PM, Forstermann U. Regulation of the expression of inducible nitric oxide synthase. Biol Chem. 2003;384:1343–1364. doi: 10.1515/BC.2003.152. [DOI] [PubMed] [Google Scholar]
- 31.Kamijo R, et al. Requirement for transcription factor IRF-1 in NO synthase induction in macrophages. Science. 1994;263:1612–1615. doi: 10.1126/science.7510419. [DOI] [PubMed] [Google Scholar]
- 32.Lowenstein CJ, et al. Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon γ and lipopolysaccharide. Proc Natl Acad Sci USA. 1993;90:9730–9734. doi: 10.1073/pnas.90.20.9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacMicking JD. Immune control of phagosomal bacteria by p47 GTPases. Curr Opin Microbiol. 2005;8:74–82. doi: 10.1016/j.mib.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 34.Schattgen SA, Fitzgerald KA. The PYHIN protein family as mediators of host defenses. Immunol Rev. 2011;243:109–118. doi: 10.1111/j.1600-065X.2011.01053.x. [DOI] [PubMed] [Google Scholar]
- 35.Roberts TL, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323:1057–1060. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- 36.Unkeless JC, Eisen HN. Binding of monomeric immunoglobulins to Fc receptors of mouse macrophages. J Exp Med. 1975;142:1520–1533. doi: 10.1084/jem.142.6.1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ioan-Facsinay A, et al. FcγRI (CD64) contributes substantially to severity of arthritis, hypersensitivity responses, and protection from bacterial infection. Immunity. 2002;16:391–402. doi: 10.1016/s1074-7613(02)00294-7. [DOI] [PubMed] [Google Scholar]
- 38.Ivashkiv LB. A signal-switch hypothesis for cross-regulation of cytokine and TLR signalling pathways. Nat Rev Immunol. 2008;8:816–822. doi: 10.1038/nri2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pamer EG. Immune responses to Listeria monocytogenes. Nat Rev Immunol. 2004;4:812–823. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- 40.Trost M, et al. The phagosomal proteome in interferon-γ-activated macrophages. Immunity. 2009;30:143–154. doi: 10.1016/j.immuni.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 41.Srinivasan L, et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell. 2009;139:573–586. doi: 10.1016/j.cell.2009.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schweighoffer E, et al. The BAFF receptor transduces survival signals by co-opting the B cell receptor signaling pathway. Immunity. 2013;38:475–488. doi: 10.1016/j.immuni.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu J, et al. An activating immunoreceptor complex formed by NKG2D and DAP10. Science. 1999;285:730–732. doi: 10.1126/science.285.5428.730. [DOI] [PubMed] [Google Scholar]
- 44.Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. 2003;3:781–790. doi: 10.1038/nri1199. [DOI] [PubMed] [Google Scholar]
- 45.Lu J, et al. Structural recognition and functional activation of FcγR by innate pentraxins. Nature. 2008;456:989–992. doi: 10.1038/nature07468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kastenmüller W, Torabi-Parizi P, Subramanian N, Lammermann T, Germain RN. A spatially-organized multicellular innate immune response in lymph nodes limits systemic pathogen spread. Cell. 2012;150:1235–1248. doi: 10.1016/j.cell.2012.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boekhoudt GH, Frazier-Jessen MR, Feldman GM. Immune complexes suppress IFN-γ signaling by activation of the FcγRI pathway. J Leukoc Biol. 2007;81:1086–1092. doi: 10.1189/jlb.0906543. [DOI] [PubMed] [Google Scholar]
- 48.Virgin HW, Wittenberg GF, Unanue ER. Immune complex effects on murine macrophages. I. Immune complexes suppress interferon-γ induction of Ia expression. J Immunol. 1985;135:3735–3743. [PubMed] [Google Scholar]
- 49.Esparza I, Green R, Schreiber RD. Inhibition of macrophage tumoricidal activity by immune complexes and altered erythrocytes. J Immunol. 1983;131:2117–2121. [PubMed] [Google Scholar]
- 50.Gallo P, Goncalves R, Mosser DM. The influence of IgG density and macrophage Fcγ receptor cross-linking on phagocytosis and IL-10 production. Immunol. 2010;133:70–77. doi: 10.1016/j.imlet.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.