

Abstract

The flavone backbone is a well-known pharmacophore present in a number of substrates and inhibitors of various P450 enzymes. In order to find highly potent and novel P450 family I enzyme inhibitors, an acetylene group was incorporated into six different positions of flavone. The introduction of an acetylene group at certain locations of the flavone backbone lead to time-dependent inhibitors of P450 1A1. 3′-Ethynylflavone, 4′-ethynylflavone, 6-ethynylflavone, and 7-ethynylflavone (KI values of 0.035–0.056 μM) show strong time-dependent inhibition of P450 1A1, while 5-ethynylflavone (KI value of 0.51 μM) is a moderate time-dependent inhibitor of this enzyme. Meanwhile, 4′-ethynylflavone and 6-ethynylflavone are highly selective inhibitors toward this enzyme. Especially, 6-ethynylflavone possesses a Ki value of 0.035 μM for P450 1A1 177- and 15-fold lower than those for P450s 1A2 and 1B1, respectively. The docking postures observed in the computational simulations show that the orientation of the acetylene group determines its capability to react with P450s 1A1 and 1A2. Meanwhile, conformational analysis indicates that the shape of an inhibitor determines its inhibitory selectivity toward these enzymes.
Introduction
Cytochrome P450s 1A1, 1A2, and 1B1 are major P450 family I enzymes implicated in the detoxification and metabolic activation of drugs, environmental chemicals, and other xenobiotics. P450 1A2 largely metabolizes aromatic amines, whereas P450s 1A1 and 1B1 deal with the polycyclic aromatic hydrocarbons (PAHs) and polyhalogenated aromatic hydrocarbons (PHAHs).1−4 PAHs are widely distributed environmental pollutants that ubiquitously occur as the byproducts of the petroleum industry, diesel-engine exhaust, cigarette smoke, and charcoal-grilled food.5 As toxicants, they are of concern because some of these compounds have been identified as carcinogenic, mutagenic, and teratogenic agents. In vitro and in vivo experiments have shown that P450-mediated metabolism (like P450s 1A1 and 1B1) converts certain PAHs into reactive intermediates which can covalently bond to DNA and proteins, forming DNA- and protein-adducts.6,7 These adducts lead to DNA or protein damage and often induce organ toxicity and possible tumorigenicity. However, P450 1A2 is notable for its capacity to bioactivate arylamines such as 2-amino-3-methylimidazo[4,5-f]quinoline (IQ) and 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) into potent mutagenic or carcinogenic components.8 Therefore, selective inhibition of P450 family I enzymes has been selected as a molecular target for potential cancer chemoprevention.9−11
Over the years, a large number of naturally occurring and synthetic PAHs, anthraquinones, stilbenoids, coumarins, flavonoids, and alkaloids have been identified as effective inhibitors of P450 family I enzymes.12−19 In our recent study on 7-ethynylcoumarins, we have reported a highly potent mechanism-based inhibitor (MBI), 7-ethynyl-3-phenylcoumarin (7E3PC), possessing selectivity toward P450 1A1 in comparison with P450s 1A2, 2A6, and 2B1 (Figure 1).17 In this molecule, the 3-phenylcoumarin residue is the major pharmacophore, fitting into the narrow active site cavity of P450 1A1 with multiple π–π interactions, while the acetylene group could react with the enzyme, leading to its deactivation. According to these results, we incorporated the acetylene group into the flavone backbone (an analogue of 3-phenylcoumarin but much more powerful than that compound)20 to produce a series of flavone derivatives, ethynylflavones. The major points of this design could be summarized as (i) introducing the flavone backbone to enhance the inhibitory activity and (ii) modifying flavones in various positions with the ethynyl group to regulate the selectivity while keeping the mechanism-based inhibition capability.
Figure 1.
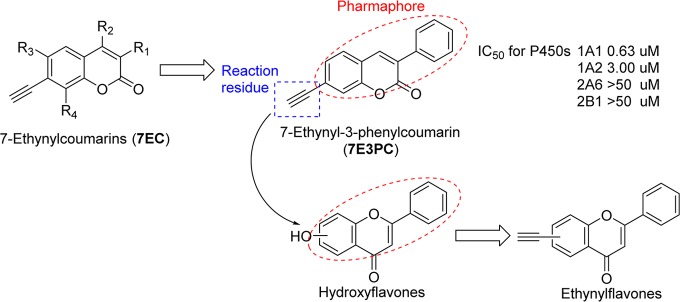
Structural optimization from ethynylcoumarins to ethynylflavones.
Materials and Methods
Chemistry
The hydroxyflavone starting materials were purchased from INDOFINE Chemical Company, Inc. (Hillsborough, NJ), and all other chemicals were purchased from Sigma-Aldrich Corporation (St. Louis, MO) and Fisher Scientific International, Inc. (Hampton, NH). Mass spectral data were collected using an Agilent 6890 GC with a 5973 MS. 1H NMR and 13C NMR spectra were recorded on a Bruker Fourier 300 MHz FT-NMR spectrometer and an Agilent 400 MHz NMR spectrometer. The data was processed using MestReNova NMR software (School of Chemistry, University of Bristol, Bristol, UK). Elemental analyses were performed by Atlantic Microlab, Inc. (Norcross, GA).
Preparation of 7-Ethynylflavone (7EF)
To a solution of 500 mg (2.1 mmol) of 7-hydroxyflavone in 10 mL of anhydrous pyridine under nitrogen atmosphere and cooling in an ice bath, 1.0 mL (5.9 mmol) of triflic anhydride was added. After stirring on ice for 2 h, the reaction mixture was quenched with 100 mL of ethyl acetate. The reaction mixture was washed with 5% KHSO4 (50 mL × 8) and saturated NaCl (50 mL × 2), dried over anhydrous MgSO4, and concentrated under vacuum to give the crude product which was recrystallized from 30 mL of anhydrous ethanol to give 545 mg (yield, 70%) of pure flavon-7-triflate as colorless crystals. GC/MS: 370 (M+, 95%), 209 ([M-CF3SO2]+, 100). 1H NMR (CDCl3, 300 HMz): δ = 8.35 (d, J = 8.7 Hz, 1H), 7.93 (m, 2H), 7.60–7.54 (m, 4H), 7.35 (dd, J = 8.7 Hz, J = 2.1 Hz, 1H), 6.87 (s, 1H). 13C NMR (CDCl3, 75 HMz): 176.89, 164.25, 165.44, 132.15, 131.00, 129.22, 128.44, 126.38, 123.71, 120.82, 118.62, 116.57, 111.59, 107.95. Flavon-7-triflate (500 mg, 1.35 mmol) was dissolved in a mixed solution of 5 mL of anhydrous pyridine and 40 mL of diisopropylamine (DIPA). To this solution, 800 mg (1.14 mmol) of bis(triphenylphosphine)palladium(II) dichloride (Pd(PPh3)2Cl2) and 60 mg (0.32 mmol) of CuI were added. After 10 min of stirring, 1.2 mL (8.43 mmol) of trimethylsilylacetylene was also added, and the reaction mixture was refluxed for 2 h. After cooling down to room temperature, the reaction mixture was concentrated to a black residue to which 100 mL of diethyl ether was added. A black precipitate formed. After filtration, the filtrate was washed with 5% KHSO4 (50 mL × 3) followed by saturated NaCl (20 mL × 2), dried over anhydrous MgSO4, and concentrated under vacuum. The crude 7-trimethylsilylethynylflavone was purified using column chromatography with petroleum ether/ethyl acetate 10:1 as the eluent to give 290 mg (yield, 68%) of silvery crystals. GC/MS: 318 (M+, 30%), 303 ([M-CH3]+, 100). 1H NMR (CDCl3, 300 HMz): δ = 8.13 (d, J = 8.4 Hz, 1H), 7.88 (m, 2H), 7.66 (d, J = 1.2 Hz, 1H), 7.50 (m, 3H), 7.45 (dd, J = 8.4 Hz, J = 1.8 Hz, 1H), 6.79 (s, 1H), 0.28 (s, 9H). 13C NMR (CDCl3, 75 HMz): 177.80, 163.52, 155.78, 131.75, 131.53, 129.09, 128.74, 128.62, 126.26, 125.62, 123.54, 121.37, 107.75, 103.12, 98.94, −0.21. To a solution of 200 mg (0.63 mmol) of 7-trimethylsilylethynylflavone in 10 mL of methanol and 10 mL of diethyl ether, 1.0 mL (1 M in methanol, 1.0 mmol) of tetrabutylammonium fluoride was added. The reaction mixture was stirred at 70 °C for 0.5 h and concentrated under vacuum. The crude product was purified using column chromatography with petroleum ether/ethyl acetate 3:1 as the eluent to produce 112 mg (yield, 72%) of 7-ethynylflavone as a yellowish powder. mp 170–173 °C. GC/MS: 246 (M+, 100%), 218 (45), 144 (30), 116 (28). 1H NMR (CDCl3, 300 HMz): δ = 8.16 (d, J = 8.1 Hz, 1H), 7.91 (m, 2H), 7.69 (d, J = 1.2 Hz, 1H), 7.53 (m, 3H), 7.49 (dd, J = 8.1 Hz, J = 1.2 Hz, 1H), 6.83 (s, 1H), 3.31 (s, 1H). 13C NMR (CDCl3, 75 HMz): 177.77, 163.72, 155.78, 131.86, 131.45, 129.13, 128.76, 127.73, 126.33, 125.82, 123.83, 121.76, 107.78, 81.97, 81.03. Anal. Calcd for C17H10O2: C, 82.91; H, 4.09; O, 12.99. Found: C, 81.91; H, 4.22.
Preparation of 2′-Ethynylflavone (2′EF)
To a solution of 500 mg (2.1 mmol) of 2′-hydroxyflavone in 15 mL of anhydrous pyridine under nitrogen atmosphere and cooling in an ice bath, 1.0 mL (5.9 mmol) of triflic anhydride was added. After stirring on ice for 1 h, the reaction mixture was transferred to a heating mantle. To this solution, 800 mg (1.14 mmol) of Pd(PPh3)2Cl2, 60 mg (0.32 mmol) of CuI, and 40 mL of DIPA were added. After 10 min of stirring, 1.2 mL (8.43 mmol) of trimethylsilylacetylene was also added, and the reaction mixture was refluxed for 2 h. After cooling down to room temperature, the reaction mixture was concentrated by vacuum to a black residue which was dissolved in a mixture of 10 mL of methanol and 10 mL of diethyl ether. To start the final step, 1.0 mL (1 M in methanol, 1.0 mmol) of tetrabutylammonium fluoride was added. The reaction mixture was stirred at 70 °C for 1.0 h and concentrated under vacuum. The residue was purified using column chromatography with petroleum ether/ethyl acetate 4:1 as the eluent to give 85 mg (yield, 16%) of 2′-ethynylflavone as yellow crystals. mp 106–108 °C. GC/MS: 246 (M+, 100%), 218 (96), 189 (92), 92 (90). 1H NMR (CDCl3, 300 HMz): δ = 8.25 (dd, J = 8.1 Hz, J = 1.8 Hz, 1 H), 7.76–7.65 (m, 3H), 7.53–7.39 (m, 4H), 6.97 (s, 1H), 3.39 (s, 1H). 13C NMR (CDCl3, 75 HMz): 177.41, 163.48, 155.94, 136.92, 131.79, 131.48, 129.84, 129.09, 126.30, 123.85, 119.50, 118.43, 107.94, 81.89, 78.37. Anal. Calcd for C17H10O2: C, 82.91; H, 4.09; O, 12.99. Found: C, 81.67; H, 4.21.
Preparation of 3′-Ethynylflavone (3′EF)
To a solution of 500 mg (2.1 mmol) of 3′-hydroxyflavone in 15 mL of anhydrous pyridine under nitrogen atmosphere and cooling in an ice bath, 1.0 mL (5.9 mmol) of triflic anhydride was added. After stirring on ice for 1 h, the reaction mixture was transferred to a heating mantle. To this solution, 800 mg (1.14 mmol) of Pd(PPh3)2Cl2, 60 mg (0.32 mmol) of CuI, and 40 mL of DIPA were added. After 10 min of stirring, 1.2 mL (8.43 mmol) of trimethylsilylacetylene was also added, and the reaction mixture was refluxed for 2 h. After cooling down to room temperature, the reaction mixture was concentrated under vacuum to a black residue which was dissolved in a mixture of 10 mL of methanol and 10 mL of diethyl ether. To start the final step, 1.0 mL (1 M in methanol, 1.0 mmol) of tetrabutylammonium fluoride was added. The reaction mixture was stirred at 70 °C for 1.0 h and concentrated under vacuum. The residue was purified using column chromatography with petroleum ether/ethyl acetate 4:1 as the eluent to give 94 mg (yield, 18%) of 3′-ethynylflavone as a brown powder. mp 129–130 °C. GC/MS: 246 (M+, 100%), 218 (55), 120 (50), 92 (60). 1H NMR (CDCl3, 400 HMz): δ = 8.24 (dd, J = 8.1 Hz, J = 1.8 Hz, 1 H), 8.12 (d, J = 8.6 Hz, 1H), 7.92 (d, J = 8.0 Hz1H), 7.72 (d, J = 8.6 Hz, 2H), 7.58 (dd, J = 8.4 Hz, J = 0.8 Hz, 1H), 7.74 (dd, J = 8.1 Hz, 1.2 Hz, 1H), 6.82 (s, 1H), 3.24 (s, 1H). 13C NMR (CDCl3, 100 HMz): 177.35, 162.20, 156.76, 132.63, 132.48, 131.66, 131.29, 129.04, 126.20, 124.60, 121.00, 118.92, 108.41, 83.35, 82.21. Anal. Calcd for C17H10O2: C, 82.91; H, 4.09; O, 12.99. Found: C, 81.33; H, 4.09.
Preparation of 4′-Ethynylflavone (4′EF)
To a solution of 500 mg (2.1 mmol) of 4-hydroxyflavone in 10 mL of anhydrous pyridine under nitrogen atmosphere and cooling in an ice bath, 1.0 mL (5.9 mmol) of triflic anhydride was added. After stirring on ice for 2 h, the reaction mixture was transferred to a heating mantle. To this solution, 800 mg (1.14 mmol) of Pd(PPh3)2Cl2, 60 mg (0.32 mmol) of CuI, and 40 mL of DIPA were added. After 10 min of stirring, 1.2 mL (8.43 mmol) of trimethylsilylacetylene was also added, and the reaction mixture was refluxed for 2 h. After cooling down to room temperature, the reaction mixture was concentrated under vacuum to a black residue which was dissolved in a mixture of 10 mL of methanol and 10 mL of diethyl ether. To start the final step, 1.0 mL (1 M in methanol, 1.0 mmol) of tetrabutylammonium fluoride was added. The reaction mixture was stirred at 70 °C for 0.5 h, and concentrated under vacuum. The residue was purified using column chromatography with petroleum ether/ethyl acetate 3:1 as the eluent to give 80 mg (yield, 15%) of 4-ethynylflavone as a brown powder. mp 164 °C decomposed. GC/MS: 246 (M+, 100%), 218 (35), 120 (35). 1H NMR (CDCl3, 300 HMz): δ = 8.24 (dd, J = 8.1 Hz, J = 1.2 Hz, 1H), 7.91 (d, J = 8.7 Hz, 2H), 7.73 (ddd, J = 8.4 Hz, J = 8.4 Hz, J = 1.5 Hz, 1H), 7.65 (d, J = 8.7 Hz, 2H), 7.58 (dd, J = 8.4 Hz, J = 0.6 Hz, 1H), 7.73 (ddd, J = 8.1 Hz, J = 8.1 Hz, J = 1.2 Hz, 1H), 6.85 (s, 1H), 3.27 (s, 1H). 13C NMR (CDCl3, 75 HMz): 178.35, 162.45, 156.21, 134.00, 133.32, 132.72, 131.82, 126.16, 125.75, 125.43, 123.89, 118.10, 107.96, 82.72, 80.16. Anal. Calcd for C17H10O2: C, 82.91; H, 4.09; O, 12.99. Found: C, 82.50; H, 4.26.
Preparation of 6-Ethynylflavone (6EF)
To a solution of 500 mg (2.1 mmol) of 6-hydroxyflavone in 10 mL of anhydrous pyridine under nitrogen atmosphere and cooling in an ice bath, 1.0 mL (5.9 mmol) of triflic anhydride was added. After stirring on ice for 1 h, the reaction mixture was transferred to a heating mantle. To this solution, 800 mg (1.14 mmol) of Pd(PPh3)2Cl2, 60 mg (0.32 mmol) of CuI, and 40 mL of DIPA were added. After 10 min of stirring, 1.2 mL (8.43 mmol) of trimethylsilylacetylene was also added, and the reaction mixture was refluxed for 2 h. After cooling down to room temperature, the reaction mixture was concentrated under vacuum to a black residue which was dissolved in a mixture of 10 mL of methanol and 10 mL of diethyl ether. To start the final step, 1.0 mL (1 M in methanol, 1.0 mmol) of tetrabutylammonium fluoride was added. The reaction mixture was stirred at 70 °C for 1.0 h and concentrated under vacuum. The residue was purified using column chromatography with petroleum ether/ethyl acetate 4:1 as the eluent to give 72 mg (yield, 14%) of 6-ethynylflavone as a blue powder. mp 154–156 °C. GC/MS: 246 (M+, 100%), 218 (15), 144 (95), 116 (60). 1H NMR (CDCl3, 300 HMz): δ = 8.34 (d, J = 1.8 Hz, 1 H), 7.93–7.89 (m, 1H), 7.78 (m, 1H), 7.55–7.51 (m, 4H), 6.82 (s, 1H), 3.14 (s, 1H). 13C NMR (CDCl3, 100 HMz): 177.41, 163.48, 155.94, 136.92, 131.79, 131.48, 129.84, 129.09, 126.30, 123.85, 119.50, 118.43, 107.94, 81.89, 78.37. Anal. Calcd for C17H10O2: C, 82.91; H, 4.09; O, 12.99. Found: C, 80.86; H, 4.53.
Preparation of 5-Ethynylflavone (5EF)
To a solution of 500 mg (2.1 mmol) of 5-hydroxyflavone in 15 mL of anhydrous pyridine under nitrogen atmosphere and cooling in an ice bath, 1.0 mL (5.9 mmol) of triflic anhydride was added. After stirring at room temperature for 3 days, the reaction mixture was quenched with 100 mL of ethyl acetate. The reaction mixture was washed with 5% KHSO4 (50 mL × 8) and saturated NaCl (50 mL × 2), dried over anhydrous MgSO4, and concentrated under vacuum to give the crude product which was recrystallized from 30 mL of anhydrous ethanol to give 560 mg (yield, 72%) of pure flavon-5-triflate as colorless crystals. GC/MS: 370 (M+, 95%), 209 ([M-CF3SO2]+, 100). 1H NMR (CDCl3, 300 HMz): δ 7.91–7.87 (m, 2H), 7.75–7.63 (m, 2H), 7.58–7.51 (m, 3H), 7.24 (d, J = 1.2 Hz, 1H), 6.79 (s, 1H). 13C NMR (CDCl3, 75 HMz): 176.00, 162.83, 157.21, 146.84, 133.35, 132.10, 130.77, 129.19, 126.32, 120.96, 119.11, 118.87, 117.84, 116.71, 108.74. Flavon-5-triflate (500 mg, 1.35 mmol) was dissolved in a mixture of 10 mL of anhydrous pyridine and 40 mL of DIPA. To this solution, 800 mg (1.14 mmol) of bis(triphenylphosphine)palladium(II) dichloride (Pd(PPh3)2Cl2) and 60 mg (0.32 mmol) of CuI were added. After 10 min of stirring, 1.2 mL (8.43 mmol) of trimethylsilylacetylene was also added, and the reaction mixture was refluxed for 2 h. After cooling down to room temperature, the reaction mixture was concentrated by vacuum to a black residue which was dissolved in a mixture of 10 mL of methanol and 10 mL of diethyl ether. To start the final step, 1.0 mL (1 M in methanol, 1.0 mmol) of tetrabutylammonium fluoride was added. The reaction mixture was stirred at 70 °C for 0.5 h and concentrated under vacuum. The residue was purified using column chromatography with petroleum ether/ethyl acetate 3:1 as the eluent to give 120 mg (37.38%) of the product. mp 157–160 °C. GC/MS: 246 (M+, 100%), 218 (40), 189 (30), 144 (30), 116 (50). 1H NMR (CDCl3, 400 HMz): δ = 8.24 (dd, J = 8.0 Hz, J = 1.2 Hz, 1H), 7.41–7.64 (m, 6H), 6.72 (s, 1H), 3.42 (s, 1H). 13C NMR (CDCl3, 100 HMz): 177.35, 162.20, 156.76, 132.63, 132.48, 131.66, 131.29, 129.04, 126.20, 124.60, 121.00, 118.92, 108.41, 83.31, 82.34. Anal. Calcd for C17H10O2: C, 82.91; H, 4.09; O, 12.99. Found: C, 82.04; H, 4.20.
Materials in Bioassays
Gentest human CYP1A1, CYP1A2, CYP1B1, and CYP2A6 supersomes and rat CYP2B1 supersomes were purchased from BD Biosciences (Franklin Lakes, NJ). d-Glucose-6-phosphate sodium salt, β-nicotinamide adenine dinucleotide phosphate sodium salt (NADP+), and glucose-6-phosphate dehydrogenase were purchased from Sigma-Aldrich Corporation. Other reagents in bioassays were purchased from Fisher Scientific International, Inc. Figures were plotted with Prism 6 (GraphPad Software, Inc., La Jolla, CA).
Fluorimetric Enzyme Inhibition Assays of P450s 1A1, 1A2, 1B1, 2A6, and 2B1
The inhibition activities of the target compounds toward P450s 1A1-, 1A2-, 1B1-, 2A6-, and 2B1-dependent reactions were tested through standard methods as previously described.21,22 These studies included P450 1A1-dependent deethylation of resorufin ethyl ether, P450 1A2-dependent demethylation of resorufin methyl ether, P450 1B1-dependent deethylation of resorufin ethyl ether, P450 2B1-dependent depentylation of resorufin pentyl ether, and P450 2A6-dependent coumarin 7-hydroxylation assays. In brief, potassium phosphate buffer (1760 μL of a 0.1 M solution, pH 7.6) was placed in a 1.0 cm quartz cuvette, and 10 μL of a 1.0 M MgCl2 solution, 10 μL of a 1.0 mM corresponding resorufin or coumarin substrate solution (final concentration of 5 μM) in dimethyl sulfoxide (DMSO), 10 μL of the microsomal P450 protein (final concentration of 1.6 nM for P450 1A1 and 5 nM for P450s 1A2, 1B1, 2A6, and 2B1), and 10 μL of an inhibitor solution in DMSO were added. For the controls, 10 μL of pure DMSO was added in place of the inhibitor solution. The reaction was initiated by the addition of 200 μL of a NADPH regenerating solution. The regenerating solution was prepared by combining 797 μL of a 0.10 M potassium phosphate buffer solution (pH 7.6), 67 μL of a 15 mM NADP+ solution in buffer, 67 μL of a 67.5 mM glucose 6-phosphate solution in buffer, and 67 μL of a 45 mM MgCl2 solution, and incubating the mixture for 5 min at 37 °C before the addition of 3 units of glucose 6-phosphate dehydrogenase/mL and a final 5 min of incubation at 37 °C. The final assay volume was 2.0 mL. The production of resorufin anion was monitored by a spectrofluorimeter (OLIS DM 45 spectrofluorimetry system) at 535 nm excitation and 585 nm emission, with a slit width of 2 nm. The production of 7-hydroxycoumarin was monitored at 338 nm excitation and 458 nm emission, with a slit width of 2 nm. The reactions were performed at 37 °C. For each inhibitor, a number of assay runs were performed using gradually diluted inhibitor solutions. At least four concentrations of each inhibitor showing 20–80% inhibition were tested.
Data Analysis
Ki Values
The initial data obtained from the above assays were a series of reaction progress curves (the time-course of product formation) in the presence of various inhibitor concentrations and in the absence of the inhibitor as the control. The Microsoft Excel program was used to fit these data (fluorescence intensity vs time) in order to obtain the parameters of the best-fit second-order curves (y = ax2 + bx + c). The coefficient b in the above second-order equation represented enzymatic activity (v). Dixon plots were used (by plotting the reciprocals of the enzymatic activity (1/v) vs inhibitor concentrations [I]) in order to determine Ki values (x-intercepts) for the inhibitors. The results based on the first 6 min of the enzymatic reactions are tabulated in Table S1 of Supporting Information. The data are represented as the mean ± SE micromolar of three independent experiments. Figure 2 plots the Ki values of the tested compounds for the inhibition of P450s 1A1, 1A2, and 1B1 as a column chart.
Figure 2.

Ki values of αNF, 7E3PC, and ethynylflavones for the inhibition of P450s 1A1, 1A2, and 1B1. For convenience, the ordinate axis was plotted on a logarithmic scale. Ki values are represented as the mean ± SE micromolar of three independent experiments. The exact Ki values are listed in Supporting Information, Table S1.
KI and Limiting kinact Values
The first-order derivatives (y = 2ax + b) of the above second-order curves (y = ax2 + bx + c) represent the enzymatic activity over time. The semilog plots of the percent relative activity (Y = log[(y/y0) × 100]) versus time demonstrate the centesimal loss of enzymatic activity with time. The linear portions of the above semilog plots were used to determine t1/2 values (the time of enzymes losing half of their activities, which equals 0.693/kinact) at various concentrations for the observed time-dependent losses of activity. To obtain KI and limiting kinact values, 1/kinact were plotted versus reciprocals of the inhibitor concentration (1/[I]) (Kitz–Wilson plots). The limiting kinact values were the abscissa intercepts of the plots, and the KI values were calculated from the ordinate intercepts (−1/KI). The KI and limiting kinact values of the time-dependent inhibitors for P450 1A1 are tabulated in Table 1.
Table 1. KI and Limiting kinact Values of Ethynylflavones for the Inhibition of P450 1A1a.
| 2′EFb | 3′EF | 4′EF | 5EF | 6EF | 7EF | |
|---|---|---|---|---|---|---|
| KI (μM) | 0.053 ± 0.007 | 0.056 ± 0.014 | 0.51 ± 0.09 | 0.043 ± 0.008 | 0.035 ± 0.012 | |
| limiting kinact (min–1) | 0.46 ± 0.02 | 0.33 ± 0.08 | 0.066 ± 0.023 | 0.33 ± 0.03 | 0.42 ± 0.04 |
The KI and limiting kinact values are represented as the mean ± SE μM of three independent experiments.
2′EF is not a time-dependent inhibitor of P450 1A1.
NADPH-Dependency Assay
All assay solution components had the same concentrations as in the above fluorimetric enzyme inhibition assays. For preincubation assays in the presence of NADPH, potassium phosphate buffer (1560 μL, pH 7.6) was placed in a 1.0 cm quartz cuvette followed by 10 μL of a 1.0 M MgCl2 solution, 10 μL of the microsomal P450 protein, 10 μL of an inhibitor solution in DMSO (the concentration leading to approximately 20% enzymatic activity inhibition), and 200 μL of a NADPH regenerating solution. The assay mixture was incubated for 5 min at 37 °C before reaction initiation by the addition of 200 μL of buffer and 10 μL of the corresponding substrate solution. For the preincubation assays in the absence of NADPH, potassium phosphate buffer (1760 μL, pH 7.6) was placed in a 1.0 cm quartz cuvette followed by 10 μL of a 1.0 M MgCl2 solution, 10 μL of the microsomal P450 protein, and 10 μL of an inhibitor solution in DMSO (the concentration leading to approximately 20% enzymatic activity inhibition). The assay mixture was incubated for 5 min at 37 °C, before reaction initiation by the addition of 200 μL of the NADPH regenerating solution and 10 μL of the corresponding substrate solution. The final assay volume for both assays was 2.0 mL. The production of P450-dependent reaction products were monitored as described above. The reactions were performed at 37 °C.
Computational Chemistry
Docking simulations of the ethynylflavones with human P450s 1A1 and 1A2 were performed using the LigandFit module in Accelrys Discovery Studio 3.5 (Accelrys, San Diego, CA). The crystal structures of human cytochrome P450s 1A1 and 1A2 in complex with α-naphthoflavone (PDB ID: 4I8V and 2HI4) are available from the Protein Data Bank (PDB).23,24 For the crystal structure of P450 1A1, subunit A was used for docking simulations. Water molecules in the crystal structures were removed, and hydrogen atoms were added to the P450 templates under the CHARMm force field. The 3D structures of ethynylflavones were built using a 3D-sketcher module in Accelrys Discovery Studio. Partial atomic charges were assigned to each atom with the Gasteiger Charge method, and energy minimization of each molecule was performed using the conjugate gradient method with CHARMm force field. The minimization was terminated when the energy gradient convergence criterion of 0.001 kcal/mol·Å was reached. To explore the binding modes of the 3D-structures of ethynylflavones to P450s 1A1 and 1A2, the docking program LigandFit was used to automatically dock the ligands into the active site cavities of the enzymes. In the docking process, the standard flexible docking protocol was performed. Ten conformers of each molecule were automatically formed, which are the best fit into the defined active site cavities.
Results
Synthesis of Ethynylflavones
Ethynylflavones were synthesized from hydroxyflavones through a similar three-step procedure described previously17 (Scheme 1). In the synthesis of 7EF, each intermediate was purified and described in detail above, as an example. In the syntheses of 2′EF, 3′EF, 4′EF, and 6EF, intermediates were not purified and rather directly used in the next steps. Thus, a simplified method was applied in the syntheses of these compounds. In the synthesis of 5EF, the first reaction is extremely slow due to the influence of an intramolecular hydrogen bond. Thus, the synthesis of 5EF was also described separately in detail. All spectra of target compounds were supplied in the Supporting Information.
Scheme 1. Synthesis and Structures of Ethynylflavones.

Reagents and conditions: (a) triflic anhydride, pyridine, 0 °C, 2 h; (b) Pd(PPh3)2Cl2, copper(I) iodide, trimethylsilylacetylene, diisopropylamine, reflux, 2 h; (c) tetrabutylammonium fluoride, methanol, 70 °C, 0.5 h.
Ethynylflavones’ Inhibition of P450s 1A1, 1A2, and 1B1
Effects of the target ethynylflavones on P450 1A1-mediated ethoxyresorufin O-deethylation (EROD), 1A2-mediated methoxyresorufin O-demethylation (MROD), and 1B1-mediated EROD were determined through fluorescence enzyme assays, and the data are shown in Figure 2. 7-Ethynyl-3-phenylcoumarin (7E3PC) and α-naphthoflavone (αNF, Scheme 1) serve as positive controls for the convenient comparison with the previous data.17,19
As is shown in Figure 2, ethynylflavones 3′EF, 4′EF, 6EF, and 7EF possess much stronger inhibitory activities toward P450 1A1 (Ki values ranging from 0.024 to 0.041 μM) than 7-ethynyl-3-phenylcoumarin (Ki value of 0.74 μM). However, 2′EF and 5EF exhibit a relatively weak inhibition of P450 1A1 (Ki, 1.64 and 1.23 μM, respectively). Time-course curves show that 3′EF, 4′EF, 5EF, 6EF, and 7EF time- and concentration-dependently inhibit P450 1A1, suggesting that they are time-dependent inhibitors (TDIs). Of these five TDIs, 3′EF and 7EF possess the highest kinact values which will be discussed below.
4′EF and 6EF have relatively weak inhibition toward P450 1A2, with the Ki values of 3.26 and 6.20 μM, respectively. While 2′EF, 3′EF, 5EF, and 7EF exhibit moderately strong inhibition of P450 1A2 (Ki values ranging from 0.23 to 0.81 μM). 2′EF is the most potent inhibitor toward P450 1A2 among the six tested compounds, with a Ki value of 0.23 μM (Figure 2).
As for P450 1B1, the ethynylflavones show small variations in their inhibition activities, with Ki values ranging from 0.13 to 0.61 μM. Interestingly, none of the compounds showed time-dependent inhibition toward P450s 1A2 and 1B1.
Effects of Ethynylflavones on P450s 2A6 and 2B1
To determine the inhibitory activities of the ethynylflavones toward P450s 2A6 and 2B1, coumarin 7-hydroxylation and pentoxyresorufin O-depentylation (PROD) assays were performed, respectively. 2′EF showed a weak inhibition toward P450 2B1 with a Ki value of 15.7 μM, while no inhibition was observed for the other compounds with concentrations under 25 μM, suggesting selectivity of the ethynylflavones toward family I over family II P450 enzymes.
3′EF, 4′EF, 5EF, 6EF, and 7EF Time- and NADPH-Dependent Inhibition of P450 1A1
The time-course curves of the tested compounds in NADPH-dependency assays are summarized in Figure 3. Among the six compounds, 3′EF, 4′EF, 5EF, 6EF, and 7EF exhibit NADPH-dependency in the inhibition toward P450 1A1, suggesting that 3′EF, 4′EF, 5EF, 6EF, and 7EF are potential mechanism-based inhibitors (MBIs) of P450 1A1. However, 2′EF does not NADPH-dependently inhibit P450 1A1. This type of assay was also performed for P450s 1A2, 1B1, 2A6, and 2B1. No time-dependent inhibition was observed for these enzymes. The kinetic parameters of TDIs 3′EF, 4′EF, 5EF, 6EF, and 7EF in the inhibition of P450 1A1 were determined through a 6 min continuous observation with various concentrations of the inhibitors. The data are listed in Table 1. 3′EF and 7EF possess the highest limiting kinact values, 0.46 ± 0.02 and 0.42 ± 0.04 min–1, respectively, suggesting that 3′EF and 7EF have very high affinity toward P450 1A1 and that their ethynyl groups tend to react with the active site of this enzyme. 5EF exhibits a relatively poor affinity toward and reaction rate with P450 1A1, with a KI value of 0.51 μM and a kinact value of 0.066 min–1.
Figure 3.

Effects of NADPH preincubation on the production of resorufin by P450 1A1 in the presence of ethynylflavones. Five-minute preincubation was applied in the presence (red triangles) and absence (blue squares) of NADPH for each test. The concentrations used for 2′EF, 3′EF, 4′EF, 5EF, 6EF, and 7EF were 0.1 μM, 0.00625 μM, 0.00625 μM, 0.1 μM, 0.00625 μM, and 0.00625 μM, respectively. For 2′EF, preincubation with NADPH exhibited a slight influence on enzymatic activity of P450 1A1. For 5EF, preincubation with NADPH caused a considerable inhibition of P450 1A1. Whereas for 3′EF, 4′EF, 6EF, and 7EF, 5 min preincubation with NADPH (0.00625 μM of inhibitor) lead to a complete inhibition of P450 1A1.
Computational Simulation of Ethynylflavones Binding to P450s 1A1 and 1A2
To investigate the interaction between target compounds and P450 enzymes 1A1 and 1A2, the docking simulations were performed with the LigandFit protocol in the Accelrys Discovery Studio described above. After 3D structure sketching and energy minimization, the standard ligand, α-naphthoflavone, as well as test compounds were docked into the active site cavities of P450s 1A1 and 1A2 in order to obtain the binding patterns. Figure 4A–E shows the docking postures of α-naphthoflavone, 2′EF, 3′EF, 5EF, and 6EF with the crystal structure of P450 1A1 (PDB ID: 4I8V). These images demonstrate that certain ethynylflavones such as 3′EF, 5EF, and 6EF dock into P450 1A1 with the acetylene group oriented toward the reactive center, heme. On the contrary, the acetylene group of 2′EF is oriented away from the heme when docked with P450 1A1. These observations explain why 3′EF, 4′EF, 5EF, 6EF, and 7EF are TDIs of P450 1A1, while 2′EF is only a competitive inhibitor of this enzyme since close interaction between the acetylene group and the heme is essential for the metabolism of these compounds by the enzyme into active intermediates which bind the protein and lead to the inactivation of the enzyme.
Figure 4.

Stereoviews of compounds αNF (A), 2′EF (B), 3′EF (C), 5EF (D), and 6EF (E) with P450 1A1. The protein is shown as ribbons (α-helix I in pink for emphasis), while the heme and ligands are shown as sticks. Selected amino acid residues around the active site cavity are labeled and exhibited as sticks. αNF is the standard ligand in the crystal structure of P450 1A1 (PDB ID: 4I8V). To compare the relative positions between αNF and the other compounds, the surface representation of αNF is shown in each image. The acetylene groups of 3′EF, 5EF, and 6EF point toward the heme, while that of 2′EF is pointing away from the heme.
Figure 5A–E shows the docking postures of α-naphthoflavone, 2′EF, 3′EF, 5EF, and 6EF with the crystal structure of P450 1A2 (PDB ID: 2HI4). As seen in these docking images, the acetylene group of all tested compounds are away from the heme, indicating that they are not TDIs of this enzyme. These docking simulation studies show the importance of relative position between the heme and the acetylene group of a TDI.
Figure 5.

Stereoviews of compounds αNF (A), 2′EF (B), 3′EF (C), 5EF (D), and 6EF (E) with P450 1A2. The protein is shown as ribbons (α-helix I in pink for emphasis), while the heme and ligands are shown as sticks. Selected amino acid residues around the active site cavity are labeled and exhibited as sticks. αNF is the standard ligand in the crystal structure of P450 1A2 (PDB ID: 2HI4). To compare the relative positions between αNF and the other compounds, the surface representation of αNF is shown in each image. The acetylene groups of all docked compounds are pointing away from the heme.
Discussion
Considering that P450 family I enzymes play an important role in carcinogenesis, discovering selective inhibitors of these enzymes is of great value. Superior to competitive inhibitors, mechanism-based inhibitors exhibit higher potency by covalent binding to the target enzymes and completely and irreversibly inactivating them. In this study, we identified five TDIs (3′EF, 4′EF, 5EF, 6EF, and 7EF) of P450 1A1. They are probably MBIs since they show time-, concentration-, and NADPH-dependent inhibition of P450 1A1, and the ethynyl functional group is a typical residue of an MBI. Some of these compounds exhibit compatible IC50 values to well-known P450 1A1 inhibitors, αNF and pyrene (both Kis around 0.04 μM).19,25 Especially, compounds 3′EF and 7EF possessing kinact values of 0.46 ± 0.02 and 0.42 ± 0.04 min–1, respectively, irreversibly inactivate half of the P450 1A1 enzymes in less than 2 min (their limiting t1/2 values are 1.52 and 1.65 min, respectively). This dramatically strong inactivation can be directly observed in the NADPH-dependency assays (Figure 3). With preincubation in the presence of NADPH for 5 min, 0.00625 μM of 3′EF or 7EF completely subvert the enzymatic activity of P450 1A1 in EROD assays.
In our projects focused on the discovery of new P450 inhibitors, the active site cavities of P450 enzymes were thoroughly investigated. In our recent studies, evidence shows that P450 1A1 has a planar long-strip active site cavity and that P450 1A2 owns a planar triangle cavity.19,20 As to the long-strip molecules 4′EF and 6EF in this study, they exhibit 85-fold and 177-fold higher selectivity toward P450 1A1 over P450 1A2, respectively (Figure 6). However, the triangle molecule 2′EF shows a 7.2-fold higher selectivity toward P450 1A2 over 1A1. These observations are consistent with the results of our previous studies.19
Figure 6.
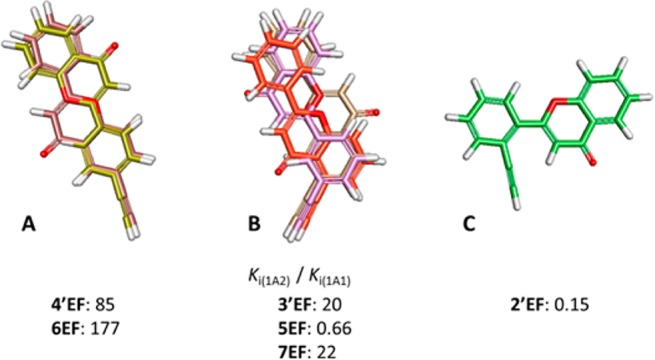
Inhibitory selectivity of ethynylflavones for P450 1A1 over P450 1A2 shows dependence on the molecular shape. (A) Superimposed 4′EF and 6EF; (B) superimposed 3′EF, 5EF, and 7EF; (C) compound 2′EF. Ki(1A2)/Ki(1A1) is the ratio of the Ki values of a compound against P450 1A2 to that of P450 1A1.
In general, the introduction of an acetylene group decreases the inhibitory activity of the flavone derivatives toward P450 1B1, compared with αNF and other flavone derivatives previously reported.19 Thus, for the compounds reported here, considerable selectivity toward P450 1A1 over P450 1B1 was observed. For example, 6EF and 7EF exhibit 15- and 18-fold higher selectivity toward P450 1A1 over P450 1B1.
The introduction of an acetylene group seems to increase the potential of the compounds for acting as inhibitors of P450s 1A1 and 1A2. At the same time, these compounds also provide us with a tool to investigate the interactions between the small molecules and the enzymes. Docking simulations in this study show that TDIs of P450 1A1 bind with the enzyme’s active site cavity with the acetylene group facing toward the heme, whereas for all the tested molecules docked into the active site cavity of P450 1A2, the acetylene group is oriented away from the heme leading to the compounds acting as competitive inhibitors of this enzyme. Because of the orientation of a TDI, its binding pattern(s) could be easily determined compared with that of other types of inhibitors. Thus, as expected, TDIs are useful tools for the investigation of enzyme–ligand interactions.
Glossary
Abbreviations
- PAHs
polycyclic aromatic hydrocarbons
- PHAH
polyhalogenated aromatic hydrocarbons (PHAHs)
- IQ
2-amino-3-methylimidazo[4,5-f]quinolone
- PhIP
2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine
- 7EC
7-ethynylcoumarin
- 7E3PC
7-ethynyl-3-phenylcoumarin
- EF
ethynylflavone
- DIPA
diisopropylamine
- Pd(PPh3)2Cl2
bis(triphenylphosphine)palladium(II) dichloride
- NADP+
β-nicotinamide adenine dinucleotide phosphate sodium salt
- DMSO
dimethyl sulfoxide
- EROD
ethoxyresorufin O-deethylation
- MROD
methoxyresorufin O-demethylation
- αNF
α-naphthoflavone
- PROD
pentoxyresorufin O-depentylation
- MBIs
mechanism-based inhibitors
- TDIs
time-dependent inhibitors
Supporting Information Available
1H and 13C NMR spectra, elemental analysis results, and activity data of all the tested compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by DoD Award W81XWH-11-1-0105. We thank NIH-MBRS SCORE (grant number S06 GM 008008) for support of the preliminary work done on this project by the Foroozesh research group. We also thank the Louisiana Cancer Research Consortium and the NIH-NIMHD grant 8G12MD007595 for their support of the Major Instrumentation at Xavier University of Louisiana. We also acknowledge the NIH-NIGMS supported RISE and MARC Programs at Xavier University of Louisiana (grant numbers 2R25GM060926-09 and 2T34GM007716) for the support of our undergraduate students working on this project.
The contents are solely the responsibility of the authors and do not necessarily represent the official views of the Louisiana Cancer Research Consortium, the DoD, or the NIH.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Nebert D. W.; Dalton T. P. (2006) The role of cytochrome P450 enzymes in endogenous signalling pathways and environmental carcinogenesis. Nat. Rev. Cancer 6, 947–960. [DOI] [PubMed] [Google Scholar]
- Shimada T.; Sugie A.; Shindo M.; Nakajima T.; Azuma E.; Hashimoto M.; Inoue K. (2003) Tissue-specific induction of cytochromes P450 1A1 and 1B1 by polycyclic aromatic hydrocarbons and polychlorinated biphenyls in engineered C57BL/6J mice of arylhydrocarbon receptor gene. Toxicol. Appl. Pharmacol. 187, 1–10. [DOI] [PubMed] [Google Scholar]
- Josephy P. D.; Evans D. H.; Williamson V.; Henry T.; Guengerich F. P. (1999) Plasmid-mediated expression of the UmuDC mutagenesis proteins in an Escherichia coli strain engineered for human cytochrome P450 1A2-catalyzed activation of aromatic amines. Mutat. Res. 429, 199–208. [DOI] [PubMed] [Google Scholar]
- Androutsopoulos V. P.; Tsatsakis A. M.; Spandidos D. A. (2009) Cytochrome P450 CYP1A1: wider roles in cancer progression and prevention. BMC Cancer 9, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z.; Dragin N.; Gálvez-Peralta M.; Jorge-Nebert L. F.; Miller M. L.; Wang B.; Nebert D. W. (2010) Organ-specific roles of CYP1A1 during detoxication of dietary benzo[a]pyrene. Mol. Pharmacol. 78, 46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkonen O.; Nebert D. W. (1982) Metabolism of polycyclic aromatic hydrocarbons: etiologic role in carcinogenesis. Pharmacol. Rev. 34, 189–222. [PubMed] [Google Scholar]
- Miller K. P.; Ramos K. S. (2001) Impact of cellular metabolism on the biological effects of benzo[a]pyrene and related hydrocarbons. Drug Metab. Rev. 33, 1–35. [DOI] [PubMed] [Google Scholar]
- Kim D.; Guengerich F. P. (2005) Cytochrome P450 activation of arylamines and heterocyclic amines. Annu. Rev. Pharmacol. Toxicol. 45, 27–49. [DOI] [PubMed] [Google Scholar]
- Kleiner H. E.; Reed M. J.; DiGiovanni J. (2003) Naturally occurring coumarins inhibit human cytochromes P450 and block benzo[a]pyrene and 7,12-dimethylbenz[a]anthracene DNA adduct formation in MCF-7 cells. Chem. Res. Toxicol. 16, 415–422. [DOI] [PubMed] [Google Scholar]
- Kleiner H. E.; Vulimiri S. V.; Starost M. F.; Reed M. J.; DiGiovanni J. (2002) Oral administration of the citrus coumarin, isopimpinellin, blocks DNA adduct formation and skin tumor initiation by 7,12-dimethylbenz[a]anthracene in SENCAR mice. Carcinogenesis 23, 1667–1675. [DOI] [PubMed] [Google Scholar]
- Kleiner H. E.; Vulimiri S. V.; Reed M. J.; Uberecken A.; DiGiovanni J. (2002) Role of cytochrome P450 1a1 and 1b1 in the metabolic activation of 7,12-dimethylbenz[a]anthracene and the effects of naturally occurring furanocoumarins on skin tumor initiation. Chem. Res. Toxicol. 15, 226–235. [DOI] [PubMed] [Google Scholar]
- Sridhar J.; Liu J.; Foroozesh M.; Klein Stevens C. L. (2012) Inhibition of cytochrome p450 enzymes by quinones and anthraquinones. Chem. Res. Toxicol. 25, 357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belluti F.; Fontana G.; Dal Bo L.; Carenini N.; Giommarelli C.; Zunino F. (2010) Design, synthesis and anticancer activities of stilbene-coumarin hybrid compounds: Identification of novel proapoptotic agents. Bioorg. Med. Chem. 18, 3543–3550. [DOI] [PubMed] [Google Scholar]
- Mikstacka R.; Rimando A. M.; Dutkiewicz Z.; Stefański T.; Sobiak S. (2012) Design, synthesis and evaluation of the inhibitory selectivity of novel trans-resveratrol analogues on human recombinant CYP1A1, CYP1A2 and CYP1B1. Bioorg. Med. Chem. 20, 5117–5126. [DOI] [PubMed] [Google Scholar]
- Shimada T.; Tanaka K.; Takenaka S.; Murayama N.; Martin M. V.; Foroozesh M. K.; Yamazaki H.; Guengerich F. P.; Komori M. (2010) Structure-function relationships of inhibition of human cytochromes P450 1A1, 1A2, 1B1, 2C9, and 3A4 by 33 flavonoid derivatives. Chem. Res. Toxicol. 23, 1921–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T.; Murajama N.; Tanaka K.; Takenaka S.; Imai Y.; Hopkins N. E.; Foroozesh M. K.; Alworth W. L.; Yamazaki H.; Guengerich F. P.; Komori M. (2008) Interaction of polycyclic aromatic hydrocarbons with human cytochrome P450 1B1 in inhibiting catalytic activity. Chem. Res. Toxicol. 21, 2313–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Nguyen T. T.; Dupart P. S.; Sridhar J.; Zhang X.; Zhu N.; Stevens C. L.; Foroozesh M. (2012) 7-Ethynylcoumarins: selective inhibitors of human cytochrome P450s 1A1 and 1A2. Chem. Res. Toxicol. 25, 1047–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Androutsopoulos V. P.; Papakyriakou A.; Vourloumis D.; Spandidos D. A. (2011) Comparative CYP1A1 and CYP1B1 substrate and inhibitor profile of dietary flavonoids. Bioorg. Med. Chem. 19, 2842–2849. [DOI] [PubMed] [Google Scholar]
- Liu J.; Taylor S. F.; Dupart P. S.; Arnold C. L.; Sridhar J.; Jiang Q.; Wang Y.; Skripnikova E. V.; Zhao M.; Foroozesh M. (2013) Pyranoflavones: a group of small-molecule probes for exploring the active site cavities of cytochrome P450 enzymes 1A1, 1A2, and 1B1. J. Med. Chem. 56, 4082–4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Sridhar J.; Foroozesh M. (2013) Cytochrome P450 family 1 inhibitors and structure-activity relationships. Molecules 18, 14470–14495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke M. D.; Thompson S.; Weaver R. J.; Wolf C. R.; Mayer R. T. (1994) Cytochrome P450 specificities of alkoxyresorufin O-dealkylation in human and rat liver. Biochem. Pharmacol. 48, 923–936. [DOI] [PubMed] [Google Scholar]
- Buters J. T.; Schiller C. D.; Chou R. C. (1993) A highly sensitive tool for the assay of cytochrome P450 enzyme activity in rat, dog and man. Direct fluorescence monitoring of the deethylation of 7-ethoxy-4-trifluoromethylcoumarin. Biochem. Pharmacol. 46, 1577–1584. [DOI] [PubMed] [Google Scholar]
- Sansen S.; Yano J. K.; Reynald R. L.; Schoch G. A.; Griffin K. J.; Stout C. D.; Johnson E. F. (2007) Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J. Biol. Chem. 282, 14348–14355. [DOI] [PubMed] [Google Scholar]
- Walsh A. A.; Szklarz G. D.; Scott E. E. (2013) Human cytochrome P450 1A1 structure and utility in understanding drug and xenobiotic metabolism. J. Biol. Chem. 288, 12932–12943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T.; Yamazaki H.; Foroozesh M.; Hopkins N. E.; Alworth W. L.; Guengerich F. P. (1998) Selectivity of polycyclic inhibitors for human cytochrome P450s 1A1, 1A2, and 1B1. Chem. Res. Toxicol. 11, 1048–1056. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.