

Abstract
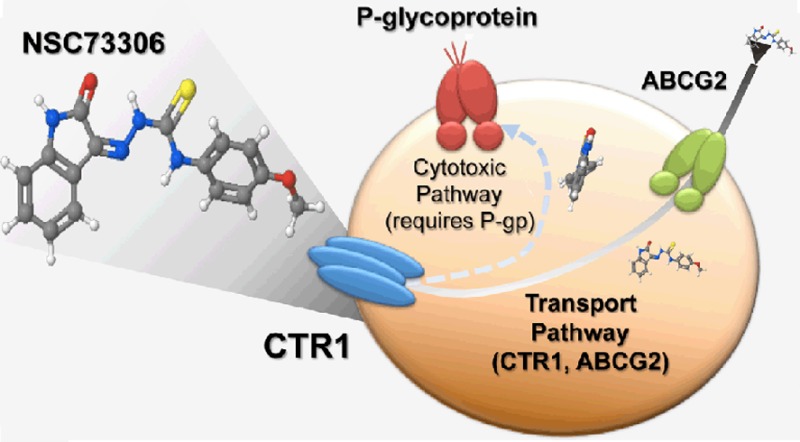
Acquired drug resistance in cancer continues to be a challenge in cancer therapy, in part due to overexpression of the drug efflux transporter P-glycoprotein (P-gp, MDR1, ABCB1). NSC73306 is a thiosemicarbazone compound that displays greater toxicity against cells expressing functional P-gp than against other cells. Here, we investigate the cellular uptake of NSC73306, and examine its interaction with P-gp and copper transporter 1 (CTR1, SLC31A1). Overexpression of P-gp sensitizes LLC-PK1 cells to NSC73306. Cisplatin (IC50 = 77 μM), cyclosporin A (IC50 = 500 μM), and verapamil (IC50 = 700 μM) inhibited cellular accumulation of [3H]NSC73306. Cellular hypertoxicity of NSC73306 to P-gp-expressing cells was inhibited by cisplatin in a dose-dependent manner. Cells transiently expressing the cisplatin uptake transporter CTR1 (SLC31A1) showed increased [3H]NSC73306 accumulation. In contrast, CTR1 knockdown decreased [3H]NSC73306 accumulation. The presence of NSC73306 reduced CTR1 levels, similar to the negative feedback of CTR1 levels by copper or cisplatin. Surprisingly, although cisplatin is a substrate of CTR1, we found that CTR1 protein was overexpressed in high-level cisplatin-resistant KB-CP20 and BEL7404-CP20 cell lines. We confirmed that the CTR1 protein was functional, as uptake of NSC73306 was increased in KB-CP20 cells compared to their drug-sensitive parental cells, and downregulation of CTR1 in KB-CP20 cells reduced [3H]NSC73306 accumulation. These results suggest that NSC73306 is a transport substrate of CTR1.
Keywords: multidrug resistance, collateral sensitivity, MDR1 sensitivity, CTR1, thiosemicarbazone
Introduction
Multidrug resistance (MDR) is one of the main causes of the failure of cancer chemotherapy. One of the major mechanisms of drug resistance is mediated by overexpression of P-glycoprotein (P-gp, ABCB1, MDR1). P-gp is a membrane-bound protein that functions as a drug efflux transporter.1,2 This protein has two halves, each containing six transmembrane domains and an ATPase domain. The drug-binding site is the primary site to interact with its substrates, and its complex structure allows structurally and functionally complex compounds to interact with it.3 Drugs interact with P-gp and stimulate ATPase activity, which energizes drug extrusion from cells, thereby reducing intracellular drug concentration.4,5 Because of the wide range of compounds recognized by P-gp, MDR cells expressing P-gp also exhibit cross-resistance to other drugs. Evidence from in vivo and in vitro studies indicates that P-gp plays an important role in drug pharmacokinetics, making it a major target to circumvent drug resistance and increase drug bioavailability.6,7
To overcome P-gp-mediated drug resistance, a variety of agents have been tested in the past 20 years.8−10 Most of the drugs were developed as P-gp inhibitors to block the function of P-glycoprotein.10 Three generations of P-gp inhibitors have been developed and have proved to be effective in vitro. These inhibitors have not shown significant effectiveness in various clinical trials.11 Multiple reasons have been suggested for these failures, including suboptimal clinical trial design, heterogeneity of patient MDR phenotypes, unexpected drug toxicity, poor pharmacokinetics of P-gp inhibitors, and the influence of endogenous P-gp or other drug transporters.11,12 Therefore, novel strategies to circumvent P-gp function in drug-resistant cancer cells are actively under development. We have focused on the effect of collateral sensitivity (CS), a phenomenon that occurs when cytotoxic drugs are more sensitive to MDR cells than the drug-sensitive, parental cells. First described in the 1950s, CS has recently emerged as a promising strategy to overcome MDR in cancer.13−17
Our laboratory identified a thiosemicarbazone named NSC73306 that is selectively toxic toward P-gp-expressing cells.18,19 Cytotoxicity studies suggest that the degree of sensitivity to NSC73306 is dependent upon the expression level of functional P-gp.18 Modulation of P-gp activity by P-gp inhibitors or downregulation of P-gp reverses hypersensitivity to NSC73306, suggesting that the cytotoxic effect of NSC73306 is linked to P-gp function.18 The molecular interactions of NSC73306 were tested with other multidrug resistance related ABC transporters, including ABCG2. NSC73306 does not specifically kill ABCG2-expressing cells, but is a potent modulator of ABCG2 by virtue of its affinity as a substrate for that efflux transporter.20
As NSC73306 can interact with other membrane proteins and metals,15,20 we hypothesized that NSC73306 might be a substrate of an unknown solute carrier (SLC) family uptake transporter. In this study, we identified compounds that could inhibit cellular accumulation of [3H]NSC73306. To achieve this goal, we established P-gp-expressing stable cell lines in LLC-PK1 porcine kidney cells.21 We found that cisplatin, a substrate transported by copper transporter 1 (CTR1, SLC31A1), is able to block NSC73306 accumulation. Our results indicate that CTR1 contributes to NSC73306 uptake.
Experimental Section
Materials
NSC73306 was obtained from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute, NIH. [3H]NSC73306 (specific activity: 40 Ci/mmol) was obtained from American Radiolabeled Chemicals (St. Louis, MO). Bodipy-verapamil, fumitremorgin C (FTC) (inhibitor of ABCG2/BCRP), and MK571 (inhibitor of multiple MRPs) were kindly provided by Dr. Suresh Ambudkar (National Cancer Institute, Bethesda, MD). (2R)-anti-5-[3-[4-(10,11-Dichloromethanodibenzo-suber-5-yl)piperazin-1-yl]-2-hydroxypropoxy] quinoline trihydrochloride (DCPQ) was kindly provided by Dr. Robert Innis (National Institute of Mental Health, Bethesda, MD). Tariquidar (XR9576) was purchased from Millennium Pharmaceuticals Inc. (Cambridge, MA). 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT), doxorubicin, verapamil, cisplatin, carboplatin, oxaliplatin, pyrilamine, quinidine, copper chloride, triethylamine, cimetidine, trimethyoprim, 3-O-methylglucose, sodium azide (NaN3), and sodium fluoride (NaF), sodium deoxycholate, and dithiothreitol (DTT) were purchased from Sigma-Aldrich (St. Louis, MO). Cyclosporin A was purchased from Calbiochem (Gibbstown, NJ). Medium 199, Dulbecco’s modified Eagle medium (DMEM), trypsin/EDTA solution, Lipofectamine2000, siRNA-Oligofectamine, and Geneticin were obtained from Life Technologies (Carlsbad, CA). Bradford reagent was purchased from Bio-Rad (Hercules, CA). Monoclonal anti-GAPDH and polyclonal anti-human CTR1 (F-190) antibodies were purchased from Santa Cruz (Santa Cruz, CA). Monoclonal mouse anti-P-gp C219 antibody was obtained from Dako (Carpinteria, CA). The anti-mouse and anti-rabbit IgG conjugated to horseradish peroxidase were purchased from Cell Signaling Technologies (Danvers, MA). ECL reagents were obtained from GE healthcare (Carpinteria, CA). The human CTR1-GFP construct was purchased from Origene (Rockville, MD). The CTR1 siRNAs and the Ctrl_AllStars_1 siRNA were purchased from Qiagen (Valencia, CA).
Cell Culture
LLC-PK1, HEK-293, and COS7 cell lines were obtained from the American Type Culture Collection (Manassas, VA). LLC-PK1 parental cells were grown at 37 °C in Medium 199 with 4.5 g/L glucose (Gibco, Carlsbad, CA) supplemented with 3% (v/v) fetal bovine serum (BioWhittaker, Walkersville, MD) with l-glutamine, penicillin, and streptomycin. The LLC-MDR1-WT and LLC-MDR1-EQ cell lines were maintained in the above medium plus 500 μg/mL Geneticin (Invitrogen, Carlsbad, CA). The parental human epidermoid carcinoma cell line KB-3-1 (a HeLa subclone) and two independent cisplatin-resistant derivatives were studied: KB-CP.5 (selected in a single step at 0.5 μg of cisplatin/mL) and KB-CP20 (selected with stepwise increases up to 20 μg of cisplatin/mL).22,23 The parental liver carcinoma cell line BEL-7404 and its cisplatin-resistant derivatives BEL-7404-CP1 (selected in a single step at 1 μg of cisplatin/mL) and BEL-7404-CP20 (selected with stepwise increases up to 20 μg of cisplatin/mL) were studied.22 With the exception of the LLC-PK1 cell lines, all cell lines were grown at 37 °C in 5% CO2, using Dulbecco’s modified Eagle medium (DMEM) with 4.5 g/L glucose, supplemented with l-glutamine, penicillin, streptomycin, and 10% fetal bovine serum.
[3H]NSC73306 Accumulation/Inhibition Assays
Experiments were done with modifications to a published method.24,25 Briefly, 2 × 105 cells were grown in each well of a 24-well culture plate (Corning, Corning, NY) for 24 h. Before initiation of the assay, cells were washed with a transport buffer containing 125 mM NaCl, 20 mM NaHCO3, 3 mM KCl, 1.8 mM CaCl2, 1 mM KH2PO4, 1.2 mM MgSO4, 10 mM d-glucose, and 10 mM HEPES (pH 7.4) and preincubated in the same buffer for 5 min. Subsequently, 200 μL of transport buffer containing [3H]NSC73306 (1 μCi/mL) and, where relevant, other small molecules was simultaneously added. To stop the experiment, drugs were removed and the cells were washed three times with ice-cold PBS. Cells were dissolved in 100 μL of 0.3 M NaOH for 3 h following neutralization with an equal volume of 0.3 M HCl. An aliquot of sample (100 μL) was taken and mixed with scintillation cocktail (10 mL) (Bio-Safe II), and the disintegrations per minute were measured using a Beckman Coulter LS-6500 liquid scintillation analyzer. Protein concentration was measured by Bradford protein assays. Except for cisplatin, verapamil, and cyclosporin A, a single concentration of drug was used. Each assay was repeated at least 3 times. For calcium-dependent transport assays, calcium was replaced with magnesium in the transport buffer. For sodium-dependent transport assays, NaCl or NaHCO3 was replaced with KCl or KHCO3 in the transport buffer.
Plasmid Transfection
Cells were seeded on a plastic Petri dish for 24 h before transfection. Transfection of pCMV6 or pCMV6-hCTR1-GFP vectors was performed by Lipofectamine2000 transfection reagent according to the manufacturer’s instructions. Briefly, DNA and Lipofectamine2000 were mixed in OPTI-MEM medium at room temperature for 30 min. The mixture was added to the cells and incubated for 24 h. Transfection efficiency was estimated by Western blot analysis using anti-CTR1 antibody.
siRNA Preparation and Transfection
Four distinct small interfering RNAs (siRNAs) were designed to target human CTR1/SLC31A1 transcripts based on predictors of on- and off-target activity (Qiagen). The sense sequences of siRNAs were as follows: Hs_SLC31A1_8, CAGCATGAGCTGCTATAGAAT; Hs_SLC31A1_9, AGCCTACTGGATTGAGGTTAA; Hs_SLC31A1_10, CCGAGAGAGCCTGCTGCGTAA; Hs_SLC31A1_7, ATCGACTTGCATTCCCACTTA. Individual siRNAs were reconstituted in nuclease-free water to achieve a 10 μM solution. The siRNAs were pooled at a 1:1:1:1 ratio prior to transfection. The Ctrl_AllStars_1 siRNA (1 nmol) negative control was reconstituted to 10 μM according to the manufacturer’s protocol. All siRNA transfection experiments were performed when the cells were >70% confluent. Briefly, prior to transfection, siRNA-Oligofectamine (Invitrogen) complexes were established according to the manufacturer’s protocol and then added to each well to achieve a final concentration of 50 nM siRNA and 0.5% Oligofectamine. After incubation at 37 °C in 5% CO2 for 20 h, medium was removed from the cells and replaced with assay medium.
Cell Growth Inhibition (MTT) Assay
Cell survival was measured by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium) assay following the published protocol.18 Briefly, cells were seeded on 96-well plates and allowed to incubate for 24 h. Drugs were then added to the cells and incubated for an additional 72 h. The remaining viable cells converted MTT to a purple formazan product, and after solubilization the absorbance at 570 nm was recorded. Cytotoxicity (IC50) was defined as the drug concentration that reduced cell viability to 50% of the untreated control. The IC50 values were calculated by GraphPad Prism 5 software (La Jolla, CA).
Western Blot Analysis
Cells grown on tissue culture plates were washed three times with ice-cold PBS and solubilized in ice-cold extraction buffer (50 mM Tris·Cl pH7.5, 150 mM NaOH, 0.5% NP-40, 1 mM DTT, 0.25% sodium deoxycholate, 1 mM EDTA pH8, 1 mM NaF, 1 mM sodium vanadate, and 1 mM PMSF). Total protein was released from the cells by repeated freeze–thawing and centrifugation (16,000 rpm, 4 °C, 5 min). In each sample, protein concentration was estimated by the Bradford method, and protein (25 μg total) was denatured and allowed to separate in 10% SDS–PAGE. The acrylamide gel was transferred to a nitrocellulose membrane using the iBlot system (Invitrogen). The membrane was first treated with blocking solution for 4 h (TBS-T supplemented with 5% nonfat dry milk) and then incubated with anti-CTR1 antibody (1:2,000) or anti-GAPDH antibody (1:25,000) in 1% TBS-T at 4 °C overnight. The following day, the membrane was washed with TBS-T, and incubated with HRP-conjugated IgG secondary antibody (1:20,000) for 90 min.
Statistical Analysis
Statistical significance of the experimental results was calculated by the two-sample t test using Prism 5 (GraphPad, La Jolla, CA). Results were considered statistically significant at P-value (p)<0.05.
Results
Cisplatin, Verapamil, and Cyclosporin A Inhibit NSC73306 Cellular Uptake
To determine the rate of initial drug uptake, LLC-PK1 (P-gp nonexpressing) and LLC-MDR1-WT (wild type P-gp expressing) cells were incubated with increasing concentrations of [3H]NSC73306 for 5 min. The initial drug accumulation rate of LLC-PK1 cells was 1.36 ± 0.2 pmol mg–1 min–1, and for P-gp-expressing LLC-MDR1-WT cells it was 1.58 ± 0.1 pmol mg–1 min–1. There was no statistically significant difference in the initial drug accumulation rates between these two cell lines (Figure S1 in the Supporting Information). This lack of difference between P-gp-expressing and parental cell lines was also confirmed in KB-3-1 and KB-V1 cells (Figure 3D). As there was no significant difference in [3H]NSC73306 accumulation between LLC-PK1 and LLC-MDR1-WT cells, we screened for small molecules that inhibit the uptake of NSC73306 into LLC-PK1 cells. LLC-PK1 cells were incubated with [3H]NSC73306 for 5 min in the presence of various compounds (Figure 1). We found that cisplatin (100 μM), cyclosporin A (1 mM), and verapamil (1 mM) significantly inhibited [3H]NSC73306 uptake (Figure 1A). However, we found that the P-gp inhibitors tariquidar (100 nM) and DCPQ (100 nM), the MRP inhibitor MK571 (50 μM), the BCRP inhibitor FTC (20 μM), and the SLC uptake transporter inhibitors pyrilamine (1 mM), quinidine (1 mM), tetraethylammonium (TEA) (1 mM), cimetidine (1 mM), and trimethoprim (1 mM) had no effect on [3H]NSC73306 uptake (Figure 1A). The P-gp substrates doxorubicin or paclitaxel also did not inhibit [3H]NSC73306 uptake. Other than drug transporter substrate/modulators, we tested whether [3H]NSC73306 uptake is dependent on sodium or calcium. Uptake studies were performed in sodium-free or calcium-free transport buffers. Again, [3H]NSC73306 uptake was not significantly affected by sodium or calcium. A significant drop in [3H]NSC73306 accumulation was observed when “cold” NSC73306 was added, suggesting saturable uptake of [3H]NSC73306.
Figure 3.

CTR1 level influences accumulation of NSC73306. (A) Transient overexpression of CTR1-GFP in COS7 cells. Cell lysates were extracted after transfection for 24 h. The presence of endogenous CTR1 and CTR1-GFP proteins was detected by immunoblotting using a polyclonal anti-CTR1 antibody. (B) Increased uptake of [3H]NSC73306 in CTR1-GFP-overexpressing cells. Accumulation of drug in control cells and CTR1-overexpressing cells was estimated. At each time point, the drug accumulation relative to time 0 was determined. (C) Knockdown of CTR1 in KB-3-1 and KB-V1 cells. Immunoblot by CTR1 antibody showing the presence of CTR1 in mock-transfected KB-3-1 and KB-V1 and CTR1 siRNA-transfected KB-3-1 and KB-V1 cells. Results of the same immunoblot with low exposure and high exposure are shown. The same blot was labeled with GAPDH antibody as a loading control. (D) Drug accumulation assay showing [3H]NSC73306 accumulation in KB-3-1 and KB-V1 with CTR1 knockdown cells. The cells were incubated with [3H]NSC73306 for 3 min, and the relative drug accumulation values with mock-transfected cells were determined. Results are mean ± SD of 3 independent experiments.
Figure 1.

Cisplatin, cyclosporin A and verapamil are inhibitors of [3H]NSC73306 uptake. (A) [3H]NSC73306 (25 pmol/mL) was incubated with LLC-PK1 cells for 5 min following cell lysis and scintillation counting. The relative [3H]NSC73306 accumulation after cells were treated with [3H]NSC73306 and DMSO was calculated. The concentrations of compounds tested are listed in the Experimental Section. Cells treated with cold NSC73306 were used as a control. (B) [3H]NSC73306 accumulation in LLC-PK1 cells with various concentrations of verapamil (up to 1 mM). (C) [3H]NSC73306 accumulation in LLC-PK1 cells treated with various concentrations of cyclosporin A (up to 1 mM). (D) Time course [3H]NSC73306 accumulation assay in the presence of cyclosporin A (0.5 mM) (black line). For control experiments, cells were treated with [3H]NSC73306 and DMSO (dashed line). Results are means ± SD of 3 independent experiments.
To characterize further the inhibitory effect of cyclosporin A, verapamil, and cisplatin, the half maximal inhibitory concentrations (IC50s) of [3H]NSC73306 cell uptake were determined. The IC50 of verapamil against [3H]NSC73306 uptake was 0.7 ± 0.1 mM (Figure 1B), and that of cyclosporin A was 0.5 ± 0.05 mM (Figure 1C). The time course of [3H]NSC73306 uptake revealed that, in the presence of cyclosporin A (0.5 mM), [3H]NSC73306 uptake was lower at all time points (compared with DMSO control) and the maximum reduction in [3H]NSC73306 uptake was 62% (Figure 1D). There were no significant differences between inhibitor-treated LLC-PK1 and LLC-MDR1-WT cells (not shown). These results showed that the IC50 values of verapamil and cyclosporin A are much higher than the amount required to inhibit P-gp function (∼20 μM), suggesting that inhibition of NSC73306 uptake by these compounds may involve other mechanisms that are yet to be identified.
Cisplatin inhibited [3H]NSC73306 uptake in a concentration-dependent manner (Figure 2A). The IC50 of cisplatin to inhibit [3H]NSC73306 uptake was 77 ± 2 μM for LLC-PK1 cells and 128 ± 40 μM for KB-3-1 cells, suggesting that this inhibitory effect is not a cell-specific effect (Figure 2A). Uptake of [3H]NSC73306 in LLC-PK1 and LLC-MDR1-WT cells in the presence and absence of cisplatin were evaluated. Coincubation of 100 μM cisplatin and [3H]NSC73306 equally reduced [3H]NSC73306 uptake in both cell lines (Figure 2B). In addition to cisplatin, a Pt-containing anticancer agent, we sought to explore whether other Pt-based compounds could share similar inhibitory effects. Both 1 mM carboplatin and 250 μM oxaliplatin had no statistically significant effect on [3H]NSC73306 uptake (Figure 2C), suggesting that the inhibitory effect of [3H]NSC73306 on platinum drug uptake is dependent on the nature of the platinum compound. Also, preincubation of cells with cisplatin, carboplatin, and oxaliplatin did not affect the IC50 (Figure 2C). The incomplete inhibition of uptake is consistent with a transporter-facilitated component of uptake, and a passive component.
Figure 2.

Inhibition of [3H]NSC73306 by platinum-based compounds. (A) Determination of IC50 of cisplatin after [3H]NSC73306 accumulation in LLC-PK1 cells and KB-3-1 cells. Cells were treated with various concentrations of cisplatin for either 5 min (LLC-PK1 cells) or 3 min (KB-3-1 cells). (B) Inhibition of accumulation of [3H]NSC73306 in LLC-PK1 and LLC-PK1-MDR1 cells. Cells were incubated with cisplatin (gray bars) or DMSO (black bars) for 5 min, and the relative [3H]NSC73306 accumulation in the DMSO-treated LLC-PK1 cells was calculated. (C) [3H]NSC73306 uptake can be inhibited by cisplatin but not carboplatin or oxaliplatin. Cells were preincubated (gray) or coincubated (black) with [3H]NSC73306 and platinum drugs. Each point in the plot represents an average cell number from 3 independent experiments.
CTR1 Influences Uptake of NSC73306
Since [3H]NSC73306 uptake and function could be modulated by cisplatin, we decided to investigate whether these two compounds share a common uptake pathway. Given cisplatin’s association with CTR1-mediated uptake,26−29 we hypothesized that CTR1 might play a role and therefore examined the effect of this Cu transporter on the cellular accumulation of NSC73306. First, we compared drug accumulation in cells that transiently overexpressed CTR1. COS7 African green monkey kidney cells were used for this experiment because they have low endogenous CTR1 expression.30 Cells were transfected with GFP-tagged human CTR1 cDNA, resulting in expression of recombinant protein (Figure 3A). In cells overexpressing CTR1, higher [3H]NSC73306 uptake was observed (Figure 3B). At 45 min, [3H]NSC73306 uptake was 36% higher in CTR1-overexpressing cells (Figure 3B). In another experiment, we examined drug accumulation in cells treated with CTR1 siRNA. We used KB-3-1 (P-gp negative) and KB-V1 (P-gp positive) human adenocarcinoma cell lines because they express human CTR1 protein (vide infra, Figure 7A). Western blot analysis confirmed that transfection of CTR1 siRNA reduced CTR1 levels in KB-3-1 and KB-V1 cells (Figure 3C). Drug uptake experiments showed that CTR1 siRNA treatment of KB-3-1 and KB-V1 cells reduced [3H]NSC73306 accumulation by approximately the same amount (Figure 3D).
Figure 7.

CTR1 level is not changed by P-gp. Immunoblots showing CTR1, P-gp, and GAPDH levels in (A) KB-3-1 and KB-V1 cells and (B) CTR1 and GAPDH levels in LLC-PK1, LLC-MDR1-WT, and LLC-MDR1-EQ cells. (C) Transient overexpression of the CTR1 gene does not change P-gp level. Protein lysate from P-gp negative KB-3-1 cells, P-gp positive KB-V1 cells, and KB-V1 cells overexpressing CTR1-GFP protein were analyzed in an immunoblot labeled with anti-P-gp antibody (C219).
Cisplatin-Resistant Cell Lines Overexpress CTR1 and Show Increased [3H]NSC73306 Uptake
We further wanted to determine if cisplatin-resistant cell lines showed cross-resistance to NSC73306, as cisplatin resistance is associated with a decrease in CTR1 protein in small cell lung cancer cells (SCLCs).28 Western blot analysis showed that the KB-CP20 cell line, which is highly resistant to cisplatin, expresses more CTR1 protein than the KB-3-1 cells (Figure 4A, left). However, cisplatin-sensitive KB-3-1 cells express amounts of CTR1 comparable to cisplatin-resistant KB-CP.5 cells. In the liver cancer cell line BEL-7404-CP20, which is highly resistant to cisplatin, CTR1 overexpression was also observed. CTR1 was not detected in the parental cell line, BEL-7404, or in its subline BEL-7404-CP1, with lower-level cisplatin resistance (Figure 4A, right).
Figure 4.

CTR1 level is elevated in cisplatin-resistant cells. (A) Immunoblot showing the level of CTR1 protein in KB-3-1, KB-CP.5, KB-CP20, BEL-7404, BEL-7404-CP1, and BEL-7404-CP20 cells. Immunoblots with GAPDH are used as loading controls. (B) [3H]NSC73306 accumulation in KB-3-1 and KB-CP20 cells. (C) [3H]NSC73306 accumulation in KB-3-1, KB-CP20, and KB-CP20 cells treated with scrambled siRNA or CTR1 siRNA. (D) Sensitivity of KB-3-1, KB-CP.5, and KB-CP20 cells to NSC73306. Cells were incubated with increasing concentrations of NSC73306 for 72 h at 37 °C. Cytotoxicity was determined by MTT assays, and the survival curves were plotted. Results are means ± SD of 3 independent experiments.
The function of CTR1 with respect to NSC73306 in cisplatin-resistant cells was examined by drug uptake assays. KB-CP20 cells showed a 2.1-fold increase in [3H]NSC73306 uptake relative to the parent line KB-3-1 (Figure 4B). To confirm that the increase in drug uptake was caused by upregulation of CTR1, we also determined the uptake of [3H]NSC73306 in KB-CP20 cells transfected with CTR1 siRNA. In CTR1 knockdown KB-CP20 cells, a significant reduction of NSC73306 uptake was observed (Figure 4C). CTR1 knockdown cells showed drug accumulation comparable to that of KB-3-1 cells. Control KB-CP20 cells transfected with scrambled siRNA also had decreased drug accumulation, suggesting that some of the reduction in NSC73306 uptake may be caused by the siRNA transfection method. Since CTR1 levels are upregulated in KB and BEL-7404 cisplatin-resistant cells, we examined whether the cellular toxicity of NSC73306 could be enhanced by the high levels of CTR1 in cisplatin-resistant cells. As noted earlier, cytotoxicity assays showed that the NSC73306 IC50 values of the KB-3-1, KB-CP.5, and KB-CP20 cells were not significantly different, suggesting that the cytotoxic effect of NSC73306 in cisplatin-resistant cells is not dependent on total intracellular NSC73306 levels (Figure 4D).
Cisplatin Reverses NSC73306 Hypertoxicity in P-gp-Expressing Cells
The correlation of P-gp expression and function with NSC73306 cytotoxicity in LLC-PK1 cells was assessed, using a wild-type P-gp-expressing stable cell line (LLC-MDR1-WT) and a functional mutant P-gp-expressing stable cell line (LLC-MDR1-EQ).31 The IC50 values of NSC73306 to LLC-PK1, LLC-MDR1-WT, and LLC-MDR1-EQ cells were 4.7 ± 2.0 μM, 0.9 ± 0.2 μM, and 4.5 ± 1.0 μM, respectively (Figure 5A). The selectivity ratio (SR) of NSC73306 killing LLC-MDR1-WT cells over parental LLC-PK1 cells was 5.4-fold (Figure 5A), which was comparable to other cell line pairs that we reported.32
Figure 5.

Sensitivity of NSC73306 requires functional P-gp and loss of NSC73306 sensitivity in LLC-MDR1-WT cells upon cisplatin treatment. (A) NSC73306 is hypersensitive to functional P-gp expressing cells. LLC-PK1 cells were treated with increasing concentrations of NSC73306 in 37 °C for 72 h following MTT assay. The IC50 values were calculated by GraphPad Prism. (B) The LLC-PK1 and LLC-MDR1-WT cells were treated with various concentrations of NSC73306. LLC-MDR1-WT cells were treated with 0, 5, 10, or 40 μM of cisplatin. The bar chart shows the EC50 for each treatment. Each point in the plot represents an average cell number from 3 independent experiments.
We further examined whether cisplatin could influence the hypertoxicity of NSC73306 in cells expressing P-gp. LLC-MDR1-WT cells were treated with increasing concentrations (5 μM, 10 μM, and 40 μM) of cisplatin for 72 h. As expected, in the absence of cisplatin, LLC-MDR1-WT cells showed ∼4-fold lower IC50 values for NSC73306 than the LLC-PK1 cells (Figure 5B). However, coincubation of cisplatin in LLC-MDR1-WT cells reversed their hypersensitivity to NSC73306 in a dose-dependent manner. In the presence of 40 μM cisplatin, the IC50 of LLC-MDR1-WT cells was ∼76% that of LLC-PK1 cells (Figure 5B).
Incubation of Cells with NSC73306 Degrades CTR1 Protein
To determine if CTR1 could be regulated by NSC73306, we analyzed CTR1 levels after drug treatment. HEK293 human embryonic kidney cells, known to express CTR1,33 were treated with increasing concentrations of NSC73306. As negative feedback mechanisms of CTR1 in response to Cu and cisplatin have been reported,34 we sought to explore whether NSC73306 could also reduce CTR1 expression. As control experiments, cells exposed to cisplatin (CDDP) for 1 h or copper (CuCl2) for 3 h produced lower CTR1 levels, as expected (Figure 6A). After 24 h, treatment with NSC73306 reduced the CTR1 level in a dose-dependent manner (Figure 6B). As GAPDH levels were slightly affected by NSC73306, the relative CTR1 level was calculated by densitometry analysis. Reduction in CTR1 levels was observed at even the lowest NSC73306 concentration tested (1 μM). In cells treated with 10 μM NSC73306, the CTR1 level was reduced by 63%, which was comparable to the effect of cisplatin (∼60% reduction). CTR1 levels in cells treated with NSC73306 for 3 h were not affected (Figure S2 in the Supporting Information).
Figure 6.

CTR1 protein is reduced by NSC73306 in a dose-dependent manner. (A) HEK293 cells were treated with different concentrations of NSC73306 at 37 °C for 24 h following cell lysis and immunoblotting. As a positive control, cells were treated with cisplatin (5 μM) and CuCl2 (100 μM) for 3 h. The presence of CTR1 was detected by polyclonal anti-CTR1 antibody. The same blot was labeled with GAPDH antibody, serving as a loading control. (B) The CTR1 level in each sample was analyzed by ImageJ software and displayed as a bar chart. Change in CTR1 level relative to control was determined by densitometry analysis. Statistical significance between NSC73306-treated cells and the control cells is shown. Results are means ± SD of 3 independent experiments.
CTR1 Expression Is Not Affected by P-gp and Vice Versa
To examine whether overexpression of P-gp modulates CTR1 levels, we tested for the presence of CTR1 in drug-resistant cell lines and recombinant cell lines. We found that CTR1 protein levels were not significantly different between KB-3-1 and KB-V1 cells, the latter expressing a high level of P-gp (Figure 7A). In LLC-PK1, LLC-MDR1-WT, and LLC-MDR1-EQ cells, CTR1 levels were also unaltered (Figure 7B). Conversely, P-gp levels did not change in KB-V1 cells overexpressing CTR1-GFP, as determined by Western blot analysis with anti-P-gp monoclonal antibody (C219) (Figure 7C).
Modulation of CTR1 Does Not Change NSC73306 Cytotoxicity in KB-3-1, KB-V1, and HEK293 cells
Since cellular CTR1 levels affect intracellular levels of NSC73306 and vice versa, we examined whether the cytotoxicity of NSC73306 could be affected by CTR1 levels. First, we examined the CTR1 level in KB cells. Transfection of KB-3-1 and KB-V1 cells with CTR1 siRNA resulted in a 25% reduction of CTR1 protein. Scrambled siRNA and CTR1 siRNA-transfected cells were incubated with NSC73306 for 72 h, and then IC50 values were determined by MTT assay. As expected, mock control and control scrambled siRNA-transfected KB-V1 cells showed higher sensitivity to NSC73306 than similarly treated KB-3-1 cells. CTR1-siRNA treatment only slightly decreased IC50 values in both KB-3-1 (from 10.9 ± 0.04 μM to 7.9 ± 0.02 μM) and KB-V1 cells (from 3.3 ± 0.04 μM to 1.7 ± 0.04 μM) (Table 1). This slight decrease might be due to the toxicity mediated by siRNA transfection. In another experiment, we sought to compare the effect of NSC73306 in KB-3-1 and HEK293 cells overexpressing CTR1. We used HEK293 cells that express low levels of P-gp instead of KB-V1 cells, because overexpression of CTR1 in KB-V1 cells resulted in cell death (not shown). Compared with the KB-3-1 cells, HEK293 cells showed higher sensitivity to NSC73306 (IC50 10.9 ± 0.04 μM vs 4.42 ± 0.03 μM), possibly due to endogenous P-gp expression. However, when compared with the mock and CTR1-GFP transfected HEK293 cells, they showed similar cytotoxicity. In addition, the IC50s were evaluated for cells incubated with NSC73306 for an extended period of time and we found no significant differences. Similarly, NSC73306 cytotoxicity was not significantly different in mock-transfected KB-3-1 cells and CTR1-GFP-transfected KB-3-1 cells (Table 1). These results indicate that while CTR1 levels affect uptake of NSC73306 (Figure 3), in the absence of P-gp, they do not correlate with toxicity of NSC73306.
Table 1. CTR1 Does Not Change Sensitivity of Cells to NSC73306a.
| cell line | mock (μM) | overexpressed CTR1 (μM) | scrambled RNA (μM) | siRNA CTR1 (μM) |
|---|---|---|---|---|
| KB-3-1 | 10.9 ± 0.04 | 10.4 ± 0.06 | 10.2 ± 0.06 | 7.9 ± 0.02 |
| KB-V1 | ND | ND | 3.3 ± 0.04 | 1.7 ± 0.04 |
| HEK-293 | 4.4 ± 0.03 | 4.7 ± 0.03 | ND | ND |
Results are mean ± SD of 3 independent experiments. Cells were incubated with increasing concentrations of NSC73306 at 37 °C for 72 h. IC50 values were determined by survival curves obtained using MTT assays. ND: not determined.
Discussion
In this work, a search was conducted for molecules that could inhibit cellular uptake of NSC73306, a compound that specifically kills P-gp-expressing cells. At physiological pH, NSC73306 is neutral, lipophilic (clogP = 2.02) and likely capable of passive diffusion. We found that its uptake in the nM range is saturable and inhibited by cisplatin, verapamil, and cyclosporin A. Therefore, we hypothesized that NSC73306 could be transported by the Cu and cisplatin uptake transporter CTR1 (verapamil and cyclosporin A are not known to be substrates of CTR1), and we show here that cellular uptake of the drug is modulated by levels of CTR1.
CTR1 is a Cu uptake transporter that mediates Cu homeostasis and is expressed in many human tissues.33,35,36 It is essential for embryonic development, and knockout of both alleles of CTR1 in the mouse produces an embryonic lethal phenotype.37,38 The functional unit of CTR1 is a homotrimeric complex with each monomer containing 190 amino acid residues, spanning the cell membrane.39,40 Electron crystallographic analysis of human CTR1 shows that the complex forms a cone-shaped structure with a narrower extracellular pore (∼8 Å) and a wider intracellular pore (∼22 Å).40 Human CTR1 has also been found to transport silver30 and regulates platinum (Pt) drug uptake (reviewed by Liu et al.).41 As such, CTR1 influences the cytotoxicity of Pt-based drugs including cisplatin, carboplatin, and oxaliplatin.28 Yeast and mammalian cells that do not express CTR1 are more resistant to Cu and Pt drugs, whereas overexpression of CTR1 increases drug uptake and increases drug sensitivity.27,29,34,42,43 Exposure of cells to cisplatin causes a rapid reduction in CTR1 protein, and this can be triggered by a low concentration of the drug.34 Although it is known that CTR1 mediates cisplatin uptake, the mechanism of Pt translocation into cells by CTR1 remains to be defined.
Our results indicate that [3H]NSC73306 uptake is inhibited by cisplatin and by cold NSC73306 in the micromolar range (77 ± 2 μM and 7.1 ± 1 μM). However, to inhibit [3H]NSC73306 uptake, ∼10-fold higher concentrations of cisplatin were required as compared to cold NSC73306, suggesting that the drug uptake pathway of NSC73306 may partially overlap with the uptake pathway of cisplatin. Also, [3H]NSC73306 uptake can be inhibited by cisplatin but not by other Pt drugs such as carboplatin and oxaliplatin. Others have reported that CTR1 is able to transport cisplatin, carboplatin, and oxaliplatin, although the kinetics of these compounds are different.27,28 There is still no consensus on exactly how Pt drugs or Cu enter the cell via CTR1. Several reports have strongly suggested that the coordination of cisplatin with CTR1 is very different from its coordination with Cu.40,43,44 Human CTR1 includes a long stretch of extracellular domain rich in methionine and histidine. Mutagenesis and truncation studies have revealed that several amino acid residues critical to regulation of Cu uptake do not affect Pt drug transport. Because of this, alternative modes of Pt drug transport by CTR1 have been proposed. Recently, Wang et al. proposed that the methionine residues of the extracellular domain of CTR1 cooperatively assist Pt drug uptake.45 The results of our drug inhibition experiments suggest that the transport of NSC73306 via CTR1 might be different from Pt or Cu transport mechanisms. The current study primarily focuses on the cellular uptake of NSC73306. Also, uptake studies and cytotoxicity studies of copper and Pt drugs are not included. Further analysis will be needed to determine the functional effects of CTR1 transport of NSC73306 and Pt drugs or Cu, and also the critical amino acids/motifs required for NSC73306 transport.
CTR1 has been known to regulate itself post-translationally in response to a sudden surge of substrates.46 Our experiments confirmed that NSC73306 could reduce CTR1 expression in a dose-dependent manner. These studies provide additional evidence that NSC73306 is a substrate of CTR1. Time-course experiments showed that the effect of NSC73306 is slower than that of cisplatin or Cu. However, the molecular mechanism of this downregulation by NSC73306 is not clear. In mammalian cells, cisplatin- and Cu-induced downregulation of CTR1 involves non-clathrin-mediated endocytosis followed by formation of ubiquitinated intermediates and degradation in the 26S proteasome.47,48 In yeast, the copper ion-sensing transcription factor Mac1p post-translationally controls the degradation of yeast CTR1, Ctr1p.49 However, our findings suggest that CTR1-mediated NSC73306 uptake in mammalian cells is regulated by a post-translational mechanism involving drug-stimulated degradation of the transporter.
This study confirms that NSC73306 requires the presence of functional P-gp in order to increase cytotoxicity to the cell. Our results demonstrate that there is no direct relationship between CTR1 expression and P-gp expression. In the LLC-MDR1-WT cells, addition of cisplatin partially reverses cell sensitivity to NSC73306. No statistical significance was found when we compared [3H]NSC73306 uptake in drug-sensitive KB-3-1 and LLC-PK1 and drug-resistant KB-V1 and LLC-MDR1-WT cell lines. These results indicate that [3H]NSC73306 import is independent of P-gp, a drug efflux transporter, and that the intracellular level of NSC73306 does not correlate with cytotoxicity. This can be understood if one considers that the [3H]NSC73306 uptake assay is done using low nM concentrations of NSC73306, while its toxicity occurs in the low μM range (a difference of 3 orders of magnitude), where uptake is likely to be diffusion-limited and not carrier dependent. This is consistent with a previous publication showing that NSC73306 interacts with the major ABC multidrug transporter ABCG2.20 Although NSC73306 alone cannot sensitize ABCG2-expressing cells, it can reduce ABCG2-mediated drug transport of other known ABCG2 substrates including mitoxantrone.20 In the current study, we found that a SLC transporter such as CTR1 could influence cellular drug uptake of NSC73306. The concentration of NSC73306 in the cell lines we tested was affected by the presence of cisplatin or a change in CTR1 protein level. However, in the absence of P-gp, CTR1 itself could not change cellular sensitivity to NSC73306. Together with ABCG2, CTR1 is involved in the drug transport pathway, but not the cell killing pathway of the drug NSC73306.
In this study, elevated CTR1 levels were found in two cell lines highly resistant to cisplatin, KB-CP20 (cervical adenocarcinoma, a HeLa subclone) and BEL-7404-CP20 (hepatoma) cells. These cells have different cellular origins, suggesting that the effect is not cell-type specific. However, we did not observe changes in CTR1 expression in cell lines only moderately resistant to cisplatin, which is in agreement with a previous study.50 Our results indicate that expression of CTR1 in cells shows three responses to cisplatin. Short-term drug exposure results in decreased CTR1 levels resulting from rapid degradation. But CTR1 levels do not vary after long-term drug exposure and selection of cisplatin-resistant cells compared with control parental cells.51,52 Western blot analysis confirmed that CTR1 levels in low-level cisplatin-resistant cells were very similar to those of the parental cells, suggesting that inhibition of drug uptake by shutting down drug transporters is replaced by another signaling mechanism, perhaps because a supply of Cu is essential. The exact mechanism that leads to higher CTR1 levels in highly cisplatin-resistant cells is not clear, but may be the result of mislocalization or a defect in post-translational modification that allows cells with such high levels of resistance to survive in cisplatin.53 We have shown that these cells accumulate less Pt than parent cells, consistent with the expected resistance phenotype, but not consistent with the expected facilitation of Pt uptake by CTR1.22 We cannot rule out the possibility that the CTR1 protein found in KB-CP20 and BEL-7404-CP20 cells is folded differently, so as to change its function, perhaps as a result of altered post-translational modification.28,54 While CTR1 levels were significantly increased in two highly cisplatin-resistant cell lines (KB-CP20 and BEL-7404-CP20), MTT assays showed that the KB-CP20 cells were not sensitized to NSC73306, in keeping with our other results showing no correlation between CTR1 levels and NSC73306 cytotoxicity.
In summary, this study demonstrates that uptake of the thiosemicarbazone NSC73306 is affected by levels of CTR1, an SLC transporter. Our data strongly suggest that, at nM concentrations, NSC73306 transport is dependent on CTR1 level and can be inhibited by the CTR1 substrate cisplatin. The connection between P-gp, CTR1, and NSC73306 provides important clues to the mechanism of P-gp sensitizing drugs that confer collateral sensitivity.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute. We thank the Developmental Therapeutics Program, Division of Cancer Treatment and Development, National Cancer Institute, for providing NSC73306. We thank George Leiman for editorial assistance.
Supporting Information Available
Figure S1 showing initial drug uptake rate of [3H]NSC73306 in LLC-PK1 cells and LLC-MDR1-WT cells. Figure S2 showing that CTR1 protein remains stable after 3 h incubation with NSC73306. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Shen D. W.; Fojo A.; Chin J. E.; Roninson I. B.; Richert N.; Pastan I.; Gottesman M. M. Human multidrug-resistant cell lines: increased mdr1 expression can precede gene amplification. Science 1986, 2324750643–5. [DOI] [PubMed] [Google Scholar]
- Fojo A. T.; Ueda K.; Slamon D. J.; Poplack D. G.; Gottesman M. M.; Pastan I. Expression of a multidrug-resistance gene in human tumors and tissues. Proc. Natl. Acad. Sci. U.S.A. 1987, 841265–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aller S. G.; Yu J.; Ward A.; Weng Y.; Chittaboina S.; Zhuo R.; Harrell P. M.; Trinh Y. T.; Zhang Q.; Urbatsch I. L.; Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 32359221718–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambudkar S. V.; Kimchi-Sarfaty C.; Sauna Z. E.; Gottesman M. M. P-glycoprotein: from genomics to mechanism. Oncogene 2003, 22477468–85. [DOI] [PubMed] [Google Scholar]
- Horio M.; Gottesman M. M.; Pastan I. ATP-dependent transport of vinblastine in vesicles from human multidrug-resistant cells. Proc. Natl. Acad. Sci. U.S.A. 1988, 85103580–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkel A. H.; Mayer U.; Wagenaar E.; Mol C. A.; van Deemter L.; Smit J. J.; van der Valk M. A.; Voordouw A. C.; Spits H.; van Tellingen O.; Zijlmans J. M.; Fibbe W. E.; Borst P. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc. Natl. Acad. Sci. U.S.A. 1997, 9484028–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horio M.; Chin K. V.; Currier S. J.; Goldenberg S.; Williams C.; Pastan I.; Gottesman M. M.; Handler J. Transepithelial transport of drugs by the multidrug transporter in cultured Madin-Darby canine kidney cell epithelia. J. Biol. Chem. 1989, 2642514880–4. [PubMed] [Google Scholar]
- Jain L.; Abraham S.; Shord S. S. The interactions of anti-cancer drugs approved in the last decade in the United States with membrane transporters. Anticancer Agents Med. Chem. 2010, 108601–16. [DOI] [PubMed] [Google Scholar]
- Nobili S.; Landini I.; Giglioni B.; Mini E. Pharmacological strategies for overcoming multidrug resistance. Curr. Drug Targets 2006, 77861–79. [DOI] [PubMed] [Google Scholar]
- Szakacs G.; Paterson J. K.; Ludwig J. A.; Booth-Genthe C.; Gottesman M. M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discovery 2006, 53219–34. [DOI] [PubMed] [Google Scholar]
- Bates S. F.; Chen C.; Robey R.; Kang M.; Figg W. D.; Fojo T. Reversal of multidrug resistance: lessons from clinical oncology. Novartis Found. Symp. 2002, 243, 83–96discussion 96–102, 180–5.. [PubMed] [Google Scholar]
- Xia C.; Smith P. G. Drug Efflux Transporters and Multidrug Resistance in Acute Leukemia: Therapeutic Impact and Novel Approaches to Mediation. Mol. Pharmacol. 2012, 8261008–21. [DOI] [PubMed] [Google Scholar]
- Bech-Hansen N. T.; Till J. E.; Ling V. Pleiotropic phenotype of colchicine-resistant CHO cells: cross-resistance and collateral sensitivity. J. Cell. Physiol. 1976, 88123–31. [DOI] [PubMed] [Google Scholar]
- Goldsborough A. S.; Handley M. D.; Dulcey A. E.; Pluchino K. M.; Kannan P.; Brimacombe K. R.; Hall M. D.; Griffiths G.; Gottesman M. M. Collateral sensitivity of multidrug-resistant cells to the orphan drug tiopronin. J. Med. Chem. 2011, 54144987–97. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Szakacs G.; Hall M. D.; Gottesman M. M.; Boumendjel A.; Kachadourian R.; Day B.; Baubichon-Cortay H.; Di Pietro A. Targeting the Achilles Heel of Multidrug-Resistant Cancer by Exploiting the Fitness Cost of Resistance. Chem. Rev. 2014, 114115753–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M. D.; Handley M. D.; Gottesman M. M. Is resistance useless? Multidrug resistance and collateral sensitivity. Trends Pharmacol. Sci. 2009, 3010546–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluchino K. M.; Hall M. D.; Goldsborough A. S.; Callaghan R.; Gottesman M. M. Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist. Updates 2012, 151–298–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig J. A.; Szakacs G.; Martin S. E.; Chu B. F.; Cardarelli C.; Sauna Z. E.; Caplen N. J.; Fales H. M.; Ambudkar S. V.; Weinstein J. N.; Gottesman M. M. Selective toxicity of NSC73306 in MDR1-positive cells as a new strategy to circumvent multidrug resistance in cancer. Cancer Res. 2006, 6694808–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M. D.; Salam N. K.; Hellawell J. L.; Fales H. M.; Kensler C. B.; Ludwig J. A.; Szakacs G.; Hibbs D. E.; Gottesman M. M. Synthesis, activity, and pharmacophore development for isatin-beta-thiosemicarbazones with selective activity toward multidrug-resistant cells. J. Med. Chem. 2009, 52103191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. P.; Shukla S.; Calcagno A. M.; Hall M. D.; Gottesman M. M.; Ambudkar S. V. Evidence for dual mode of action of a thiosemicarbazone, NSC73306: a potent substrate of the multidrug resistance linked ABCG2 transporter. Mol. Cancer Ther. 2007, 612 Part 13287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung K. L.; Pan J.; Ohnuma S.; Lund P. E.; Pixley J. N.; Kimchi-Sarfaty C.; Ambudkar S. V.; Gottesman M. M. MDR1 synonymous polymorphisms alter transporter specificity and protein stability in a stable epithelial monolayer. Cancer Res. 2014, 742598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen D. W.; Akiyama S.; Schoenlein P.; Pastan I.; Gottesman M. M. Characterisation of high-level cisplatin-resistant cell lines established from a human hepatoma cell line and human KB adenocarcinoma cells: cross-resistance and protein changes. Br. J. Cancer 1995, 714676–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleman C.; Annereau J. P.; Liang X. J.; Cardarelli C. O.; Taylor B.; Yin J. J.; Aszalos A.; Gottesman M. M. P-glycoprotein, expressed in multidrug resistant cells, is not responsible for alterations in membrane fluidity or membrane potential. Cancer Res. 2003, 63123084–91. [PubMed] [Google Scholar]
- Batist G.; Behrens B. C.; Makuch R.; Hamilton T. C.; Katki A. G.; Louie K. G.; Myers C. E.; Ozols R. F. Serial determinations of glutathione levels and glutathione-related enzyme activities in human tumor cells in vitro. Biochem. Pharmacol. 1986, 35132257–9. [DOI] [PubMed] [Google Scholar]
- Okabe M.; Szakacs G.; Reimers M. A.; Suzuki T.; Hall M. D.; Abe T.; Weinstein J. N.; Gottesman M. M. Profiling SLCO and SLC22 genes in the NCI-60 cancer cell lines to identify drug uptake transporters. Mol. Cancer Ther. 2008, 793081–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer A. K.; Samimi G.; Katano K.; Naerdemann W.; Lin X.; Safaei R.; Howell S. B. The copper influx transporter human copper transport protein 1 regulates the uptake of cisplatin in human ovarian carcinoma cells. Mol. Pharmacol. 2004, 664817–23. [DOI] [PubMed] [Google Scholar]
- Holzer A. K.; Manorek G. H.; Howell S. B. Contribution of the major copper influx transporter CTR1 to the cellular accumulation of cisplatin, carboplatin, and oxaliplatin. Mol. Pharmacol. 2006, 7041390–4. [DOI] [PubMed] [Google Scholar]
- Song I. S.; Savaraj N.; Siddik Z. H.; Liu P.; Wei Y.; Wu C. J.; Kuo M. T. Role of human copper transporter Ctr1 in the transport of platinum-based antitumor agents in cisplatin-sensitive and cisplatin-resistant cells. Mol. Cancer Ther. 2004, 3121543–9. [PubMed] [Google Scholar]
- Ishida S.; Lee J.; Thiele D. J.; Herskowitz I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. U.S.A. 2002, 992214298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertinato J.; Cheung L.; Hoque R.; Plouffe L. J. Ctr1 transports silver into mammalian cells. J. Trace Elem. Med. Biol. 2010, 243178–84. [DOI] [PubMed] [Google Scholar]
- Sauna Z. E.; Muller M.; Peng X. H.; Ambudkar S. V. Importance of the conserved Walker B glutamate residues, 556 and 1201, for the completion of the catalytic cycle of ATP hydrolysis by human P-glycoprotein (ABCB1). Biochemistry 2002, 414713989–4000. [DOI] [PubMed] [Google Scholar]
- Hall M. D.; Brimacombe K. R.; Varonka M. S.; Pluchino K. M.; Monda J. K.; Li J.; Walsh M. J.; Boxer M. B.; Warren T. H.; Fales H. M.; Gottesman M. M. Synthesis and structure-activity evaluation of isatin-beta-thiosemicarbazones with improved selective activity toward multidrug-resistant cells expressing P-glycoprotein. J. Med. Chem. 2011, 54165878–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.; Gitschier J. hCTR1: a human gene for copper uptake identified by complementation in yeast. Proc. Natl. Acad. Sci. U.S.A. 1997, 94147481–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer A. K.; Katano K.; Klomp L. W.; Howell S. B. Cisplatin rapidly down-regulates its own influx transporter hCTR1 in cultured human ovarian carcinoma cells. Clin. Cancer Res. 2004, 10196744–9. [DOI] [PubMed] [Google Scholar]
- Dancis A.; Yuan D. S.; Haile D.; Askwith C.; Eide D.; Moehle C.; Kaplan J.; Klausner R. D. Molecular characterization of a copper transport protein in S. cerevisiae: an unexpected role for copper in iron transport. Cell 1994, 762393–402. [DOI] [PubMed] [Google Scholar]
- Dancis A.; Haile D.; Yuan D. S.; Klausner R. D. The Saccharomyces cerevisiae copper transport protein (Ctr1p). Biochemical characterization, regulation by copper, and physiologic role in copper uptake. J. Biol. Chem. 1994, 2694125660–7. [PubMed] [Google Scholar]
- Kuo Y. M.; Zhou B.; Cosco D.; Gitschier J. The copper transporter CTR1 provides an essential function in mammalian embryonic development. Proc. Natl. Acad. Sci. U.S.A. 2001, 98126836–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Prohaska J. R.; Thiele D. J. Essential role for mammalian copper transporter Ctr1 in copper homeostasis and embryonic development. Proc. Natl. Acad. Sci. U.S.A. 2001, 98126842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Pena M. M.; Nose Y.; Thiele D. J. Biochemical characterization of the human copper transporter Ctr1. J. Biol. Chem. 2002, 27764380–7. [DOI] [PubMed] [Google Scholar]
- De Feo C. J.; Aller S. G.; Siluvai G. S.; Blackburn N. J.; Unger V. M. Three-dimensional structure of the human copper transporter hCTR1. Proc. Natl. Acad. Sci. U.S.A. 2009, 106114237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. J.; Lu J.; McKeage M. J. Membrane transporters as determinants of the pharmacology of platinum anticancer drugs. Curr. Cancer Drug Targets 2012, 128962–86. [DOI] [PubMed] [Google Scholar]
- Pabla N.; Murphy R. F.; Liu K.; Dong Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am. J. Physiol. 2009, 2963F505–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinani D.; Adle D. J.; Kim H.; Lee J. Distinct mechanisms for Ctr1-mediated copper and cisplatin transport. J. Biol. Chem. 2007, 2823726775–85. [DOI] [PubMed] [Google Scholar]
- Crider S. E.; Holbrook R. J.; Franz K. J. Coordination of platinum therapeutic agents to met-rich motifs of human copper transport protein1. Metallomics 2010, 2174–83. [DOI] [PubMed] [Google Scholar]
- Du X.; Wang X.; Li H.; Sun H. Comparison between copper and cisplatin transport mediated by human copper transporter 1 (hCTR1). Metallomics 2012, 47679–85. [DOI] [PubMed] [Google Scholar]
- Larson C. A.; Blair B. G.; Safaei R.; Howell S. B. The role of the mammalian copper transporter 1 in the cellular accumulation of platinum-based drugs. Mol. Pharmacol. 2009, 752324–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petris M. J.; Smith K.; Lee J.; Thiele D. J. Copper-stimulated endocytosis and degradation of the human copper transporter, hCtr1. J. Biol. Chem. 2003, 278119639–46. [DOI] [PubMed] [Google Scholar]
- Safaei R.; Maktabi M. H.; Blair B. G.; Larson C. A.; Howell S. B. Effects of the loss of Atox1 on the cellular pharmacology of cisplatin. J. Inorg. Biochem. 2009, 1033333–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonkovich J.; McKenndry R.; Shi X.; Zhu Z. Copper ion-sensing transcription factor Mac1p post-translationally controls the degradation of its target gene product Ctr1p. J. Biol. Chem. 2002, 2772723981–4. [DOI] [PubMed] [Google Scholar]
- Matsumoto S.; Tanaka T.; Kurokawa H.; Matsuno K.; Hayashida Y.; Takahashi T. Effect of copper and role of the copper transporters ATP7A and CTR1 in intracellular accumulation of cisplatin. Anticancer Res. 2007, 274B2209–16. [PubMed] [Google Scholar]
- Helleman J.; Burger H.; Hamelers I. H.; Boersma A. W.; de Kroon A. I.; Stoter G.; Nooter K. Impaired cisplatin influx in an A2780 mutant cell line: evidence for a putative, cis-configuration-specific, platinum influx transporter. Cancer Biol. Ther. 2006, 58943–9. [DOI] [PubMed] [Google Scholar]
- Yoshizawa K.; Nozaki S.; Kitahara H.; Ohara T.; Kato K.; Kawashiri S.; Yamamoto E. Copper efflux transporter (ATP7B) contributes to the acquisition of cisplatin-resistance in human oral squamous cell lines. Oncol. Rep. 2007, 184987–91. [PubMed] [Google Scholar]
- Liang X. J.; Shen D. W.; Garfield S.; Gottesman M. M. Mislocalization of membrane proteins associated with multidrug resistance in cisplatin-resistant cancer cell lines. Cancer Res. 2003, 63185909–16. [PubMed] [Google Scholar]
- Beretta G. L.; Benedetti V.; Cossa G.; Assaraf Y. G.; Bram E.; Gatti L.; Corna E.; Carenini N.; Colangelo D.; Howell S. B.; Zunino F.; Perego P. Increased levels and defective glycosylation of MRPs in ovarian carcinoma cells resistant to oxaliplatin. Biochem. Pharmacol. 2010, 7981108–17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.