

Abstract
The post-translational modification of proteins by electrophilic oxylipids is emerging as an important mechanism that contributes to the complexity of proteomes. Enzymatic and nonenzymatic oxidation of biological lipids results in the formation of chemically diverse electrophilic carbonyl compounds, such as 2-alkenals and 4-hydroxy alkenals, epoxides and eicosanoids with reactive cyclopentenone structures. These lipoxidation products are capable of modifying proteins. Originally considered solely as markers of oxidative insult, more recently the modifications of proteins by lipid peroxidation products are being recognized as a new mechanism of cell signaling with relevance to redox homeostasis, adaptive response and inflammatory resolution. The growing interest in protein modifications by reactive oxylipid species necessitates the availability of methods that are capable of detecting, identifying and characterizing these protein adducts in biological samples with high complexity. However, the efficient analysis of these chemically diverse proteins presents a considerable analytical challenge. We first provide an introduction into the chemistry and biological relevance of the protein adduction by electrophilic lipoxidation products. We then provide an overview of tandem mass spectrometry approaches that have been developed in recent years for the interrogation of protein modifications by electrophilic oxylipid species.
Keywords: HSAB theory, electrophilic lipoxidation products, HNE, aldehyde-reactive probe, collision induced dissociation, electron capture dissociation, electron transfer dissociation, ion mobility mass spectrometry
I. Introduction
A. Chemical diversity of protein modifications by electrophilic oxylipids
Protein modifications by reactive lipoxidation-derived aldehydes have emerged as an important subgroup of post-translational modifications (PTMs). Electrophilic lipids may act as cytotoxicants and are implicated in the pathogenesis of many diseases including atherosclerosis, spinal cord injury, stroke or myocardial infarction, and neurodegenerative diseases (Bachi, et al., 2013; Butterfield, et al., 2012; Mattson, 2009; Poli, et al., 2008a; Poli, et al., 2008b; Shi, et al., 2011). Many studies have shown that protein adducts with oxidized lipids are elevated in multiple diseases but it remains still unclear whether these protein oxylipid adducts are mediators of pathology or merely serve as markers of inflammation and oxidative stress (Higdon, et al., 2012a; LoPachin, et al., 2009; Milne, et al., 2005; Perluigi, et al., 2012; West & Marnett, 2006). More recently concepts are emerging that recognize the interrelations between redox status and protein modifications by electrophilic lipoxidation products in orchestrating antioxidant and heat shock response, adaptive response and inflammation (Diez-Dacal & Perez-Sala, 2010; Higdon, et al., 2012a; Jacobs & Marnett, 2010; West & Marnett, 2006). The structural diversity of reactive lipid peroxidation products is remarkable. We will focus in this contribution on lipid peroxidation products with α,β-unsaturated keto/aldehyde moiety as reactivity site and engage in covalent interaction with proteins to exert their biological roles. Examples of such lipoxidation-derived electrophiles are compiled in Table 1.
Table 1.
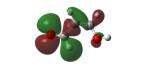
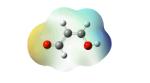
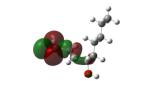
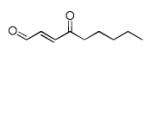
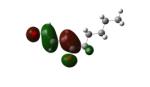
Structures of lipid aldehydes and cyclopentenone prostaglandins and their HOMO, LUMO, and maps of their electron densities in neutral states (the color gradient shows how charge is distributed in electrophiles: red color corresponds to the most negative electrostatic potential and blue indicates regions with the most positive potentials).
| Lipid Aldehydes | ||||
|---|---|---|---|---|
| Electrophile | Structure | LUMO | HOMO | Electron Density |
| Acrolein |
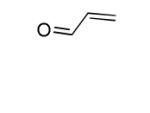
|

|

|

|
| Malon- dialdehyde MDA[1] |
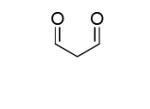
|

|
|

|
| MDA[2] |
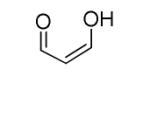
|

|
|
|
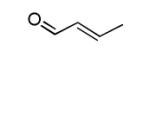
| Croton- aldehyde |
|

|
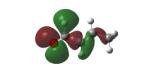
|
|
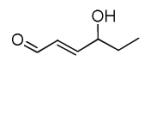
| 4-Hydroxy- 2(E)-hexenal (HHE) |
|
|
|
|
| 4-Hydroxy-2(E) nonenal (HNE) ‘s-trans’ |
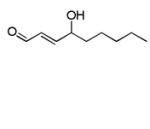
|

|
|

|
| 4-Hydroxy-2(E) nonenal (HNE) ‘s-cis’ |
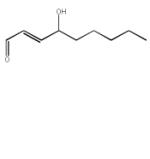
|

|
|

|
| 4-Oxo-nonenal (ONE) |
|
|

|
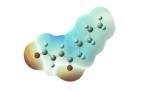
|
| 9,12-dioxo- 10(E)- dodecenoic acid (DODE) |
|

|
|

|
| Cyclopentenone Lipids | ||||
| Cyclopentenone |
|

|

|

|
| Prostaglandin A2 (PGA2) |
|

|

|

|
| PGJ2 |
|

|

|

|
| Δ12-PGJ2 |
|

|

|
|
| 15d-PGJ2 |
|
|

|

|
| Epoxide |

|

|

|

|
Tandem mass spectrometry (MS/MS) is the primary measurement technology for the detection and identification of protein modifications by electrophilic lipid oxidation products. The analytical strategies for the detection and identification of protein modifications by electrophilic lipid oxidation products are based on “bottom up approaches” meaning that peptides derived from enzymatic digestion are subjected to mass spectrometric analysis. Due to the relative low abundance of these protein modifications in biological systems, a multitude of strategies have been developed and applied that combine chemical derivatization with enrichment and separation methods. We and other have developed aldehyde/keto reactive derivatization techniques in combination with tandem mass spectrometry to decrease sample complexity prior to mass spectrometric analysis (Mirzaei & Regnier, 2006; Mirzaei, et al., 2008; Roe, et al., 2007; Ugur, et al., 2012). We pioneered the use of a hydroxyl amine-functionalized biotin tag, N’aminooxymethyl carbonylhydrazino D-biotin (aldehyde-reactive probe, ARP) for the site-specific analysis of protein modification by reactive oxylipids (Chavez, et al., 2006; Maier, et al., 2010; Wu, et al., 2011). We also developed HICAT, a hydrazine functionalized isotope-coded biotin-based affinity tag (Han, et al., 2007). More recently diverse differential quantification strategies for protein modification by electrophilic lipids based on stable-isotope labeling have become available (Han, et al., 2012; Madian, et al., 2011b; Rauniyar & Prokai, 2011). Our work and the work of others indicates that tandem mass spectrometry-based approaches for the identification and characterization of protein adducts by electrophilic lipoxidation species are indispensible for this line of research but do show limitations. Thus, we aim in this review to discuss ion activation methods that have been used for the analysis of peptide adducts of electrophilic lipoxidation species and characteristics of tandem mass spectra of peptide oxylipid adducts.
Reactive oxylipid species result from enzymatic and non-enzymatic transformation of polyunsaturated fatty acid (PUFAs), which are constituents of membrane phospholipids or lipoproteins. Non-enzymatic oxidation of PUFAs is a radical-initiated and autocatalytic process. PUFAs are highly susceptible to radical mediated oxidation due to the ease with which abstraction of the bis-allylic methylene hydrogen by other radical species can occur. The resulting conjugated pentadienyl fatty acid radical (L.) will further react with oxygen to give a dienyl peroxyl radical (LOO.). This intermediate will abstract a radial from another biomolecule, e.g. another lipid, thereby generating a lipid hydroperoxide (LOOH) and a secondary radical. The lipid hydroperoxide (LOOH) undergoes rearrangement and cleavage reactions that result in the formation of lipoxidation end products. These reactive lipoxidation products possess electrophilic properties due to their α,β-unsaturated carbonyl moieties (Esterbauer, et al., 1991; Sayre, et al., 2006). Lipid peroxidation results in the formation of several classes of reactive aldehydes including malondialdehyde, acrolein and other 2-enals, and 4-hydroxy-2-enals (Spiteller, 2005). Free-radical catalyzed oxidation of PUFAs may also lead to the formation of electrophilic eicosanoids (Stamatakis & Perez-Sala, 2006). Enzymatic oxidation of PUFAs results also in the formation of prostaglandins and electrophilic ω-3 fatty acid-derived oxo-derivatives (EFOXs) (Groeger, et al., 2010).
The chemical reactions of electrophilic species towards cellular nucleophiles are conceptually summarized in Figure 1. The interaction between α,β-unsaturated aldehyde/keto lipids with nucleophiles occurs via a 1,4-Michael-type conjugation reactions. Criteria that influence the second order reaction between an electrophilic lipid peroxidation product and the corresponding nucleophilic target are: (1) the chemical reactivity of the electrophile, (2) the reactivity of the nucleophilic site in the protein which is determined by the microenvironment which, in turn, affects the pKa of the reactive site, (3) the surface accessibility of the reactive site and (4) the pH of the intracellular compartment (Higdon, et al., 2012a; LoPachin, et al., 2012; Schopfer, et al., 2011).
Figure 1.
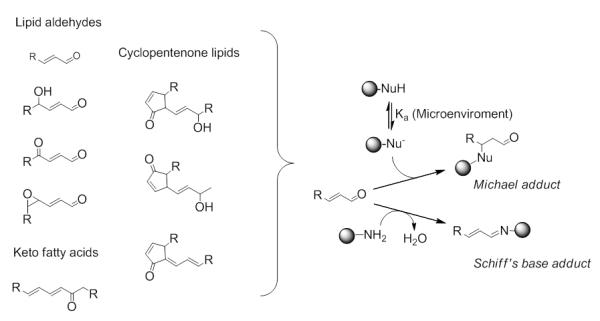
Reactions of electrophilic lipoxidation products with cellular nucleophiles. Soft electrophiles react with their α,β-unsaturated carbonyl functionality with diverse nucleophiles via 1,4-Michael-type conjugation reaction. Schiff’s base formation is predominant for hard electrophiles, such as the aldehyde group.
The chemical reactivities of α,β-unsaturated aldehydic lipids towards protein nucleophiles, such as cysteine, histidine and lysine, have been studied by thermodynamic and quantum mechanical calculations (reviewed in (LoPachin, et al., 2012)). Based on Pearson’s Hard and Soft, Acid and Bases (HSAB) theory (Pearson, 1990), LoPachin and coworkers successfully conceptually connected some cell injuries to covalent interactions of toxic electrophiles with nucleophilic sites of biologically relevant macromolecules. The initial HSAB theory (Pearson, 1990) described hardness (η) and softness (σ) of acids and bases in terms of the experimental ionization energies (EI) and electron affinities (EA) as the following: η = (EIEA)/2, and σ = 1/η. Experimental measurement of EI or EA for many reactive molecules is not always available, however, and instead calculated energies of frontier molecular orbitals (FMO) can be used within approximation of Koopmans theorem to evaluate hardness as η = (ELUMO EHOMO)/2, where LUMO stands for the lowest unoccupied MO and HOMO is the highest occupied MO. According to the FMO theory, covalent bond formation can simply be described as the frontier orbital overlap of the reacting molecules. Due to such overlap, adduct formation occurs via electron donation from nucleophile to electrophile. Therefore, the softness of an electrophile determines the ease with which the electron can be accepted. More sensitive measure of the reactivity of the electrophile can be done with introducing the electrophilicity index ω = μ2/2η (Chattaraj, et al., 2006; Parr, 1999) that combines softness (1/η) and chemical potential μ = (ELUMO+EHOMO)/2. According to the selectivity principle of the HSAB theory (LoPachin, et al., 2012; Pearson, 1990), electrophiles prefer to coordinate with nucleophiles of close softness and hardness and therefore the most stable adducts can be formed in the case of soft nucleophiles and electrophiles and weaker adducts result from the association of hard nucleophiles and electrophiles. In contrast, nucleophiles and electrophiles with very diverse softness and hardness cannot form joint complexes. The ease with which nucleophile A reacts with electrophile B is determined by its nucleophilicity index ω− = ηA(μA-μB)2/2(ηA-ηB)2 (Jaramillo, et al., 2006). This theoretical approach was used in the present work to rationalize the posttranslational modifications of proteins by electrophilic α,β-unsaturated carbonyls (see Section B, Table 1).
Although protein modifications by α,β-unsaturated lipids have originally been discussed mainly as protein modifications associated with protein damage, protein misfolding and aggregation, more recently views have emerged that consider electrophilic lipids as important and essential mediators in cell signaling cascades, eliciting response by covalent modification of proteins that are constituents of signaling pathways (Higdon, et al., 2012a; Higdon, et al., 2012b; Schopfer, et al., 2011). The formation of the electrophilic lipids will depend on the cellular context, the cellular compartment and the physiological conditions. We and others therefore propose that the chemical diversity of the reactive oxylipidome will dictate the composition of electrophile-reactive subproteomes which, in turn, govern the biological responses in a cellular, organelle and tissue-specific manner. Consequently, protein modifications by electrophilic lipids have roles in signaling and transcriptional regulatory mechanisms associated with adaptive response mechanisms and anti-inflammatory processes (Diez-Dacal & Perez-Sala, 2010; Higdon, et al., 2012a; Jacobs & Marnett, 2007; Schopfer, et al., 2011).
As producer and targets of electrophilic lipids, mitochondria have emerged as key players in electrophile-mediated cell signaling processes. Mitochondria are critically involved in cellular respiration and the regulation of the cell death pathways (Landar, et al., 2006a; Lee, et al., 2004). The inner mitochondrial membrane is rich in phospholipids with unsaturated fatty acids (in particular cardiolipins) and as such highly susceptible to lipoxidation processes (Kagan, et al., 2009). Recent works suggest that many mitochondrial membrane proteins are redox-sensitive and their activity seems to be regulated by the modification of at least some of their cysteine sulfhydryl groups. Multiple components of the electron transport chains have been described as being also modified by a diverse set of electrophilic lipids. We and others have identified key players involved in essential mitochondrial functions (e.g. bioenergetics, metabolism, and apoptosis) to be targeted and possibly regulated by electrophilic species, including the adenine nucleotide transporters (ANTs), the uncoupling proteins (UCPs) and the mitochondrial porin channels (VDACs) (Chavez, et al., 2006; Chavez, et al., 2011; Diers, et al., 2009; Guo, et al., 2011; Han, et al., 2007; Han, et al., 2012; Landar, et al., 2006b; Mailloux & Harper, 2011).
The covalent modifications of proteins by electrophilic species are emerging as important mechanisms in regulating biological system as well as possible markers of tissue damage, but techniques that are capable of studying the chemical diversity of the reactive species and the associated changes in the proteome are still insufficiently developed. Mass spectrometry has the potential to become one of the primary techniques for deciphering the complexity of subproteomes susceptible to electrophilic oxylipid species. In order to overcome limitation of current mass spectrometry methods for the comprehensive analysis of oxylipid-modified subproteomes it is necessary to understand the chemical nature of the modifications and the ion activation methods that are employed for the tandem mass spectrometry-based interrogations of oxylipid-modified peptides and proteins.
B. Computational approaches and the chemical reactivity of electrophilic lipoxidation products
To rationalize the chemical reactivity of electrophilic lipoxidation products, we have done quantum chemical calculations of electrophiles (α,β-unsaturated carbonyl derivatives and cyclopentenones) and nucleophiles in different ionization states (cysteine, histidine, arginine and two model tripeptides Ala-His-Ala and Ala-Cys-Ala) based on Gaussian-09 suite of programs (Frisch, et al., 2009). The initial conformational search of the compounds was performed via molecular mechanics (Amber and MMFF94 force fields) and molecular dynamic simulations. After selecting the thermodynamically most stable conformers, their molecular geometries were refined using density functional theory (DFT) with hybrid functional B3LYP and different basis sets depending on the complexity of a molecule from STO-3G and 3-21G to 6-31G. The optimization of molecular geometries at the final step followed by the evaluation of quantum mechanical parameters for all studied molecules was done at the UB3LYP/6-311+G(d,p) level of theory. The nucleophilicity indexes were calculated only in frame of the FMO theory, whereas the corresponding indexes of electrophiles were also determined using calculated vertical and adiabatic electron affinities (EAvert and EAad, respectively) and ionization energies (EIvert and EIad). To determine EAvert and EIvert, the energy difference of a neutral molecule and its negative or positive ions, all taken in the optimized geometry of the neutral molecule, was calculated. In the case of adiabatic EAad and EIad, the energy differences of all these species at their relaxed geometries were evaluated. An analytical frequency analysis was performed in the last case to determine zero-point energies and to confirm that optimized geometries have no imaginary frequencies and are real minima. It should be noted that both calculated HOMO and LUMO energies usually reflect the trend of EI and EA within a set of molecules correctly, however, their absolute values can differ significantly from experimental magnitudes. It is especially true for negative EAs since in this case negative ions are resonances (i.e. they are metastable species and can decay into parent neutral states via electron autodetachment). Straightforward calculations of NIs such as these in the same manner as for neutral molecules or their positively charged counterparts will most likely undergo variational collapse while applying high level of theory. One of the simple approaches to avoid such variational collapse is to use one of the stabilization methods (Hazi, 1970). In the present work, the Peyerimhoff-Simons stabilization method (Nestmann, 1985; Sobczyk, 2005) was used to determine negative vertical EAs.
Two classes of electrophilic compounds (α,β-unsaturated aldehydic lipids and cyclopentenones) were studied to determine their hardness (η), softness (σ), chemical potential (μ) and electrophilicity index (ω).The nucleophilicity indexes (ω−) of the nucleophiles (cysteine, histidine, arginine and Ala-Cys-Ala and Ala-His-Ala tripeptides) were also calculated. These quantum mechanical parameters define the reactivity of the electrophiles in their addition reactions with their biological nucleophile targets.
1. α,β-unsaturated aldehydic lipids
The calculated quantum mechanical parameters of α,β-unsaturated aldehydes in terms of the FMO theory ranked DODE (9,12-dioxo-10(E)-dodecenoic acid) and ONE (4-oxo-2(E)-nonenal) as the strongest electrophiles amongst these aldehydes with gas-phase electrophilicity indexes ω of 6.84 and 6.78, respectively (Table 2). Except for these electrophiles, the softness indexes σ, correlating with the ease of adduct formation, vary less significantly than the electrophilicity indexes ω in the case of other aldehydes (Table 2). Since ω is considered as more comprehensive measure of electrophilic reactivity (LoPachin, et al., 2012), the ranking of the aldehydes in Table 2 was performed in decreasing order of ω. The total energy difference approach, operating with theoretical EA and EI, showed essentially similar results (Table 3). The ω indexes calculated with these methods were highly correlated (r2 = 0.961 for vertical and 0.998 for adiabatic energies). The high correlation between FMO and vertical values was expected since they have similar physical meaning. Adiabatic parameters have closer relation to electron-transfer processes occurring in vivo and their high correlation with the FMO parameters is very encouraging, proving that the simpler FMO approach, which requests less calculation time and computer resources, can be used for examining similar systems in the future.
Table 2.
Calculated in terms of the FMO theory quantum mechanical parameters for electrophilic unsaturated carbonyls.
| Electrophile | ELUMO, eV | EHOMO, eV | η, eV | μ (eV) | σ, eV−1 | ω, eV |
|---|---|---|---|---|---|---|
| DODE | −3.27 | −7.54 | 2.135 | −5.405 | 0.468 | 6.84 |
| ONE | −3.24 | −7.46 | 2.11 | −5.35 | 0.474 | 6.78 |
| Acrolein | −2.24 | −7.42 | 2.59 | −4.83 | 0.386 | 4.50 |
| HNE (Cis) | −2.219 | −7.241 | 2.51 | −4.73 | 0.398 | 4.46 |
| MDA[1] | −2.08 | −7.56 | 2.74 | −4.82 | 0.365 | 4.24 |
| HHE | −2.057 | −7.26 | 2.60 | −4.659 | 0.385 | 4.17 |
| HNE (trans) | −2.04 | −7.248 | 2.60 | −4.644 | 0.384 | 4.15 |
| Crotonaldehyde | −1.91 | −7.18 | 2.635 | −4.545 | 0.380 | 3.92 |
| MDA[2] | −1.80 | −6.97 | 2.585 | −4.39 | 0.387 | 3.73 |
Table 3.
Calculated in terms of total energies quantum mechanical parameters for electrophilic unsaturated carbonyls.
| Electrophile | EAvert (EIad), eV | EIvert (EIad), (eV) | η (ηad), eV | μ (μad), eV | σ (σad), eV−1 | ω (ωad), eV |
|---|---|---|---|---|---|---|
| DODE | 1.33 (1.72) | 9.12 (9.04) | 3.895 (3.66) | −5.225 (−5.38) | 0.257 (0.273) | 3.505 (3.954) |
| ONE | 1.28 (1.63) | 9.38 (9.24) | 4.05 (3.805) | −5.33 (−5.435) | 0.247 (0.263) | 3.507 (3.88) |
| Acrolein | −0.034a (0.29) | 10.01 (9.96) | 5.02a (4.84) | −4.99a (−5.125) | 0.2a (0.21) | 2.48a (2.71) |
| HNE (cis) | 0.234 (0.54) | 9.311 (9.09) | 4.539 (4.275) | −4.773 (−4.815) | 0.22 (0.23) | 2.5 (2.71) |
| HHE | 0.032 (0.42) | 9.49 (9.34) | 4.729 (4.46) | −4.761 (−4.88) | 0.211 (0.224) | 2.4 (2.67) |
| MDA[1] | −0.73a (0.35) | 9.57a (9.44) | 5.15a (4.55) | −4.42a (−4.895) | 0.194a (0.22) | 1.9a (2.633) |
| HNE (trans) | 0.044 (0.42) | 9.35 (9.17) | 4.653 (4.375) | −4.697 (−4.795) | 0.215 (0.229) | 2.37 (2.63) |
| MDA[2] | −0.42a (0.17) | 9.47a (9.29) | 4.945a (4.56) | −4.525a (−4.73) | 0.2a (0.22) | 2.07a (2.45) |
| Crotonaldehyde | −0.27a (0.057) | 9.65a (9.55) | 4.96a (4.75) | −4.69a (−4.8) | 0.2a (0.21) | 2.217a (2.425) |
Evaluated using stabilization method (see text)
According to a recent report (LoPachin, et al., 2012), the HNE electrophile showed a significantly lower second-order reaction rate constant for a type-2 alkene reaction with cysteine at physiological pH compared to that of acrolein, although both the calculated softness and electrophilicity for the s-cis HNE conformer were found slightly higher than for acrolein. Such discrepancy between theory and experiment was explained by a steric hindrance that might be caused by the extended five-carbon alkyl tail of HNE. The present FMO theory calculations of the s-cis HNE softness and electrophilicity showed that the σ value is indeed higher compared to that of acrolein, whereas the ω value is lower (Table 2). However, the performed calculations suggested that the s-trans HNE conformer is a thermodynamically more stable isomer as its s-cis counterpart, and that the s-trans conformer is predicted to have lower values for both σ and ω. This newly established difference between softness and electrophilicity of acrolein and HNE (Table 2) can hardly explain the significant differences in their reactive rate constants (LoPachin, et al., 2012), which may point to other factors, such as steric hindrance and hemiacetal formation impacting the chemical reactivity and rate constants of these oxadiene systems under physiological conditions.
Comparison of the nucleophilicity indexes ω− for cysteine, histidine, arginine and two model peptides containing cysteine, arginine, and alanine calculated at different ionization states showed that cysteine thiolates (i.e. side chain with the deprotonated sulfhydryl group, denoted as (−1) in Table 4) are the preferential targets for α,β-unsaturated aldehydes where DODE and ONE are the softest electrophiles followed by acrolein and HNE (cis). This findings are in line with many experimental and theoretical observations (LoPachin, et al., 2012). The calculations showed reduced electron density on the β-carbon of the α,β-unsaturated carbonyl structure of the electrophiles and therefore a nucleophilic attack in a 1,4-Michael type conjugate reaction (LoPachin, et al., 2009) should likely proceed at the β-carbon. According to the calculations, the model Ala-Cys-Ala tripeptide has a slightly lower nucleophilicity index in its thiolate state than single cysteine. The thiol state (i.e. uncharged sulfhydryl group, denoted as (0) in Table 4) for both Cys and Ala-Cys-Ala is predicted to have a very low ω− value and therefore can be rejected as a nucleophile.
Table 4.
Calculated at the ub3lyp/6-311+g(d,p) level of theory the nucleophilicity indexes, ω−, of cysteine and model Ala-Cys-Ala tripeptide in their reactions with electrophilic α,β-unsaturated carbonyl compounds in different ionization states (indicated in round brackets).
| Electrophile | ω− (eV) | |||
|---|---|---|---|---|
| Cys (−1) |
Cys (0) |
Ala-Cys-Ala (−1) |
Ala-Cys-Ala (0) |
|
| DODE | 2.56 | 0.106 | 1.965 | 0.145 |
| ONE | 2.55 | 0.099 | 1.955 | 0.137 |
| Acrolein | 1.721 | 0.0306 | 1.262 | 0.051 |
| HNE (cis) | 1.726 | 0.024 | 1.264 | 0.042 |
| MDA[1] | 1.605 | 0.0284 | 1.17 | 0.047 |
| HHE | 1.62 | 0.019 | 1.177 | 0.035 |
| HNE (trans) | 1.61 | 0.018 | 1.17 | 0.034 |
| Crotonaldehyde | 1.532 | 0.0125 | 1.106 | 0.026 |
| MDA[2] | 1.483 | 0.0062 | 1.065 | 0.0166 |
The deprotonated (i.e. uncharged, denoted as (0) in Table 5) side chains of the basic amino acids His, Lys and Arg showed slightly higher ω− values compared to the cysteine’s thiol state; however, even in this case they were found to be very low (Table 5) in comparison to that of the cysteine thiolate (Table 4). This finding is in line with the experimentally determined rate constants (Doorn & Petersen, 2002) for the reaction of HNE/ONE with cysteine, basic amino acids and peptides showing much higher reactivity for cysteines. Doorn and Petersen also reported that reactivity of the basic amino acids towards ONE and HNE should be in the following order: His > Lys (>Arg for ONE) and ONE being more reactive than HNE (Doorn & Petersen, 2002). The calculated nucleophilicity indexes for the basic amino acids in the uncharged states (Table 5) are in line with the observed reactivities in their reactions with ONE and HNE. These nucleophilicity indexes, however, do not support the observed decline in reactivity as His > Lys > Arg showing very close values for all basic amino acids in uncharged states. It should be noted, however, that the discussed reactions have been performed at the physiological pH 7.4, when the relative ratio of the deprotonated (i.e. uncharged) basic amino acids to their protonated counterparts is expected to be high for His, little for Lys and very little for Arg due to the very high pKa of Lys and Arg.
Table 5.
Calculated at the ub3lyp/6-311+g(d,p) level of theory the nucleophilicity indexes, ω−, of basic amino acids histidine, lysine, arginine, and model Ala-His-Ala tripeptide in their reactions with electrophilic α,β-unsaturated carbonyl compounds in different ionization states (indicated in round brackets)
| Electrophile | ω− (eV) | |||||||
|---|---|---|---|---|---|---|---|---|
| His (0) |
His (+1) |
Ala-His-Ala (0) |
Ala-His-Ala (+1) |
Lys (0) |
Lys (+1) |
Arg (0) |
Arg (+1) |
|
| DODE | 0.164 | 0.452 | 0.167 | 0.177 | 0.186 | 0.281 | 0.184 | 0.207 |
| ONE | 0.155 | 0.474 | 0.158 | 0.191 | 0.177 | 0.298 | 0.175 | 0.221 |
| Acrolein | 0.061 | 0.552 | 0.061 | 0.259 | 0.073 | 0.366 | 0.0718 | 0.297 |
| HNE (cis) | 0.052 | 0.603 | 0.052 | 0.291 | 0.064 | 0.406 | 0.0623 | 0.33 |
| MDA[1] | 0.057 | 0.527 | 0.057 | 0.246 | 0.069 | 0.346 | 0.067 | 0.285 |
| HHE | 0.043 | 0.607 | 0.044 | 0.297 | 0.054 | 0.411 | 0.0529 | 0.338 |
| HNE (trans) | 0.042 | 0.612 | 0.043 | 0.301 | 0.053 | 0.415 | 0.0514 | 0.342 |
| Crotonaldehyde | 0.034 | 0.639 | 0.034 | 0.32 | 0.043 | 0.437 | 0.0415 | 0.363 |
| MDA[2] | 0.023 | 0.706 | 0.023 | 0.367 | 0.031 | 0.492 | 0.029 | 0.412 |
It is interesting that the calculations predicted higher ω− value for protonated His, Lys and Arg (denoted as (+1) in Table 5) than for their uncharged states. Protonated His showed, on average, larger ω− value than Lys and Arg, and this is supportive of the experimentally observed higher reactivity of His with respect to Lys and Arg (Doorn & Petersen, 2002). However, the nucleophilic indexes of the protonated states for all basic amino acids are predicted to have opposite ranking with respect to the electrophilic indexes of aldehydes and the last finding contradicts the experimental data (Doorn & Petersen, 2002). To rationalize this, we performed analysis of the HOMO responsible for the electron donation to electrophiles for protonated His, Lys and Arg (Figure 2). This analysis showed that the HOMO is mainly located at the backbone, whereas the contributions of the side chain into the HOMO were very little in the case of protonated His and none for protonated Lys and Arg. That means that the electrophilic attack can hardly occur at the side chains of the protonated His, Lys and Arg, respectively, and, therefore, basic amino acids are not very good nucleophiles for α,β-unsaturated aldehydes.
Figure 2.
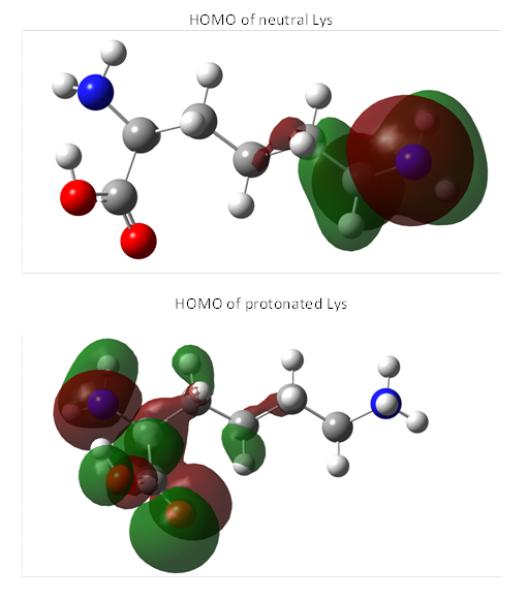
Comparison of HOMO of neutral Lys (denoted as (0) in Table 5) and HOMO of protonated Lys (denoted as (+1) in Table 5).
2. Cyclopentenone lipids
15-Deoxy-Δ-12,14-prostaglandin J2 (15d-PGJ2) was found to be the strongest electrophile in the studied series (Tables 6 and 7). However, even 15d-PGJ2 showed lower value for the electrophilicity indexes ω than DODE and ONE from the previous set of electrophiles. Nonconjugated epoxide was also considered here for comparison and according to the calculations epoxide is a poor electrophile. Other cyclopentenone lipids showed very close indexes of electrophilicity including the cyclopentenone. The last circumstance suggests that the electrophilic center for them could still be the β-carbon atom of the cyclopentonone ring like in α,β-unsaturated aldehydes. Analysis of the electron density (Table 1) confirmed a strong withdrawal effect of the carbonyl oxygen atom and reduced electron density near the β-carbon atom. Due to the rather flexible eight-carbon side arm, double carbon-carbon bonds in the side arm can hardly participate significantly into total polarizability of the molecules except for 15d-PGJ2 and Δ12-PGJ2 where double bonds can be involved in the direct conjugation with the cyclopentonone ring. The last circumstance could probably be responsible for the higher values of their electrophilicity indexes in comparison to other electrophiles of the series. According to the calculations, these molecules might in principle have additional electrophilic centers that are either the β- (Δ12-PGJ2 and 15d-PGJ2) or δ- (15d-PGJ2) carbon atoms of the eight-carbon side arm.
Table 6.
Calculated in terms of the FMO theory quantum mechanical parameters for electrophilic cyclopentenone and its derivatives (parameters for epoxide are given for comparison).
| Electrophile | ELUMO, eV | EHOMO, eV | η, eV | μ (eV) | σ, eV−1 | ω, eV |
|---|---|---|---|---|---|---|
| 15d-PGJ2 | −2.294 | −6.498 | 2.102 | −4.396 | 0.476 | 4.597 |
| Δ12-PGJ2 | −1.88 | −6.74 | 2.43 | −4.31 | 0.412 | 3.822 |
| PGA2 | −1.82 | −6.87 | 2.53 | −4.345 | 0.396 | 3.731 |
| PGJ2 | −1.77 | −6.57 | 2.4 | −4.17 | 0.417 | 3.623 |
| Cyclopentenone | −1.653 | −6.9047 | 2.626 | −4.279 | 0.381 | 3.486 |
| Epoxide | 0.296 | −7.68 | 3.988 | −3.692 | 0.251 | 1.709 |
Table 7.
Calculated in terms of total energies quantum mechanical parameters for electrophilic cyclopentenone and its derivatives (parameters for epoxide are given for comparison).
| Electrophile | EAvert (EIad), eV |
EIvert (EIad), (eV) |
η (ηad), eV | μ (μad), eV | σ (σad), eV−1 | ω (ωad), eV |
|---|---|---|---|---|---|---|
| 15d-PGJ2 | 0.73 (1.28) | 8.04 (7.774) | 3.655 (3.247) | −4.385 (−4.527) | 0.274 (0.308) | 2.63 (3.16) |
| Δ12-PGJ2 | 0.23 (1.33) | 8.35 (7.95) | 4.06 (3.31) | −4.29 (−4.64) | 0.246 (0.302) | 2.27 (3.25) |
| PGA2 | 0.153 (0.39) | 8.37 (8.211) | 4.109 (3.91) | −4.262 (−4.3) | 0.243 (0.256) | 2.21 (2.36) |
| PGJ2 | 0.14 (0.34) | 8.13 (7.833) | 3.995 (3.747) | −4.135 (−4.087) | 0.25 (0.267) | 2.14 (2.23) |
| Cyclopentenone | −0.539a (−0.133) | 9.26 (9.19) | 4.9a (4.66) | −4.36a (4.529) | 0.204a (0.215) | 1.94a (2.2) |
| Epoxide | −1.487a (−1.55) | 10.62 (10.48) | 6.05a (6.015) | −4.57a (−4.47) | 0.165a (0.166) | 1.723a (1.657) |
Evaluated using stabilization method (see text)
Similar to the unsaturated aldehydes, cyclopentenones are predicted to have high reactivity towards thiolate state of cysteines (Tables 8 and 9). Thiol states have, again, a very low nucleophilicity index both for single cysteine and tripeptide. His, Lys and Arg are predicted to be less effective nucleophiles in the reactions with cyclopentenones. Epoxide should have essentially no reaction with the His, Lys and Arg. The nucleophilic indexes of protonated His are higher than that of protonated Lys and Arg and for all of them the nucleophilic indexes are higher than that for uncharged states. As mentioned in the case of unsaturated aldehydes, these protonated states might have nucleophilic centers located on the peptide backbone rather than on the side chain.
Table 8.
Calculated at the ub3lyp/6-311+g(d,p) level of theory the nucleophilicity indexes, ω−, of cysteine and model Ala-Cys-Ala tripeptide in their reactions with electrophilic derivatives of cyclopentenone in different ionization states (indicated in round brackets). The nucleophilicity indexes of nonconjugated epoxide are shown for comparison.
| Electrophile | ω− (eV) | |||
|---|---|---|---|---|
| Cys (−1) |
Cys (0) |
Ala-Cys-Ala (−1) |
Ala-Cys-Ala (0) |
|
| 15d-PGJ2 | 1.872 | 0.0077 | 1.374 | 0.02 |
| Δ12-PGJ2 | 1.547 | 0.00398 | 1.113 | 0.013 |
| PGA2 | 1.496 | 0.00486 | 1.074 | 0.014 |
| PGJ2 | 1.491 | 0.00101 | 1.065 | 0.007 |
| Cyclopentene | 1.399 | 0.00294 | 0.996 | 0.011 |
| Epoxide | 0.657 | 0.00352 | 0.432 | 0.0004 |
Table 9.
Calculated at the ub3lyp/6-311+g(d,p) level of theory the nucleophilicity indexes, ω−, of basic amino acids histidine, lysine, arginine, and model Ala-His-Ala tripeptide in their reactions with electrophilic derivatives of cyclopentonone in different ionization states (indicated in round brackets ). The nucleophilicity indexes of nonconjugated epoxide are shown for comparison.
| Electrophile | ω− (eV) | |||||||
|---|---|---|---|---|---|---|---|---|
| His (0) |
His (+1) |
Ala-His-Ala (0) |
Ala-His-Ala (+1) |
Lys (0) |
Lys (+1) |
Arg (0) |
Arg (+1) |
|
| 15d-PGJ2 | 0.028 | 0.844 | 0.028 | 0.45 | 0.037 | 0.608 | 0.036 | 0.488 |
| Δ12-PGJ2 | 0.0188 | 0.78 | 0.0189 | 0.414 | 0.026 | 0.553 | 0.025 | 0.459 |
| PGA2 | 0.02 | 0.737 | 0.02 | 0.39 | 0.028 | 0.518 | 0.027 | 0.433 |
| PGJ2 | 0.0112 | 0.845 | 0.0113 | 0.462 | 0.017 | 0.609 | 0.0163 | 0.508 |
| Cyclo- pentene |
0.0158 | 0.737 | 0.0158 | 0.39 | 0.022 | 0.518 | 0.021 | 0.438 |
| Epoxide | 0.000003 | 0.63 | 0.000003 | 0.35 | 0.0003 | 0.44 | 0.00022 | 0.411 |
To summarize, the computational calculations provide a theoretical understanding of the chemical reactivities of electrophilic lipids to their biologically relevant nucleophilic sites in proteins (Cys, His, Lys, Arg). These calculations also emphasize that thiolate formation is a determining factor conferring nucleophilicty and, accordingly, susceptibility to electrophilic α,β-unsaturated carbonyls (Tables 4, 5 and 8). Consequently, microenvironments that lower the pKa of the unreactive thiol state of the cysteine sulfhydryl (pKa 8.3) promote the formation of the reactive thiolate state and increase the susceptibility of the site to modification by electrophilic lipids. Microenvironments that favor thiolate formation can be found, for instance, in active site pockets in which basic residues favor deprotonation of the sulfhydryl group (Martyniuk, et al., 2011; Weerapana, et al., 2010). A well documented protein target of diverse α,β-unsaturated carbonyl compound is human GAPDH which showcases multiple cysteine residues with disparate reactivities. The most susceptible target site is Cys-152, the active site cysteine with an estimated pKa of 6 caused by the pKa-lowering interaction with a nearby His residue (Martyniuk, et al., 2011). Reactive protein thiols control protein function and are targets of reversible thiol modifications (Held & Gibson, 2011). But, thiolate adduct formations are irreversible and, depending on the site of adduction, cause loss of protein function and cytotoxicity by interfering with regulatory redox modifications (Held & Gibson, 2011; Higdon, et al., 2012a; Martyniuk, et al., 2011).
II. TANDEM MASS SPECTROMETRY-BASED METHODS OF PEPTIDES MODIFIED BY ELECTROPHILIC LIPOXIDATION PRODUCTS
Tandem mass spectrometry has become the technique of choice for the detection and characterization of protein modification by electrophilic lipid peroxidation products. In tandem mass spectrometry approaches precursor ions are isolated and subsequently subjected to gas phase dissociation methods. Most tandem mass spectral studies are based on collision induced dissociation (CID). CID-MS/MS of peptide adducts of electrophilic lipids give rise to peptide backbone ions, which bond cleavage occurring at the Ccarbonyl-Npeptide amide bond leading to bn and yn-type “sequence” product ions encompassing the N- and C-terminus, respectively (Figure 3). Decarbonylation of bn-type ions leads to formation of an-type ions. In the case that the oxylipid adduct is retained on the amino acid residue during CID-MS/MS, the modified residue mass directly relates to the modification. However, in addition to the peptide backbone fragment ions, MS/MS spectra of oxylipid-modified peptides frequently show “non-sequence” product ions due to gas phase rearrangement reactions that cause loss of the oxylipid modification. The “non-sequence” product ions may serve as “diagnostic ions” but also often hamper or even sometime may preclude peptide identification and/or the localization of the modification. The type and the abundance of the product ions observed will depend on the chemical nature of the peptide precursor ion, the protonation state and the CID-MS/MS conditions used. In addition to CIDMS/MS techniques, alternative gas phase fragmentation techniques, such as electron capture dissociation (ECD) and electron transfer dissociation (ETD), have recently been explored for the site-specific analysis of peptides modified by electrophilic lipids.
Figure 3.
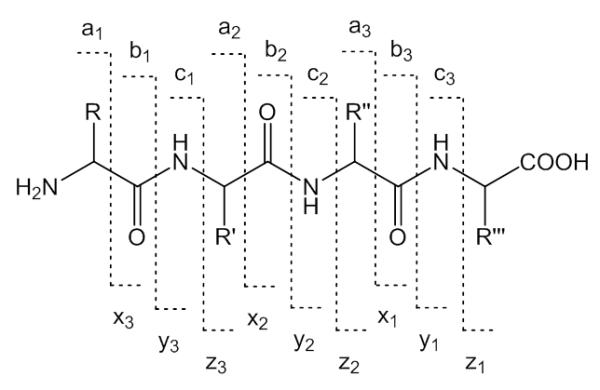
Types and nomenclature of peptide backbone fragment ions.
A. CID-MS/MS of peptide Michael adducts of electrophilic lipids
During CID experiments activation of a gas phase ions is achieved upon collision with an inert target gas (e.g. Ar or He) which leads to the conversion of the ion’s translational energy into vibrational energy. The vibrational energy is then disseminated over all modes of the ion within a picosecond timescale (≤ 10−12 s). When the vibrational energy exceeds the activation barrier of a bond dissociation of that bond occurs (McLuckey & Goeringer, 1997; Wells & McLuckey, 2005). The “mobile” proton model is usually applied for discussing peptide fragmentation mechanisms (Paizs & Suhai, 2005; Wysocki, et al., 2000). In peptide ions with mobile protons, ion activation in CID experiments leads to protonation of the amide nitrogen which weakens the amide bond and leads to dissociation under formation of the primary fragment ions; i.e. cleavages are “charge-directed”. On the other hand, in peptide ions in which the mobile proton is sequestered at the side chain of an arginine, lysine or histidine residue, more energy seems to be required to “mobilize” a proton from the basic side chains to the peptide backbone to induce “charge directed” dissociation (Dongre, 1996). For instance, in tryptic peptide ions with a C-terminal arginine, salt-bridge stabilized transition states are formed involving the Arg side-chain guanidinium, amidic or carboxylic protons (Bythell, et al., 2009).
Oxidation of lipids containing polyunsaturated fatty acids results in the formation of a plethora of electrophilic lipoxidation products. The most common group of these lipoxidation products has as characteristic feature an α,β-unsaturated carbonyl moiety. The most studied subgroup is composed of oxylipids with 2-enal structure (Esterbauer, et al., 1991; Schopfer, et al., 2011; Yin, et al., 2011). For instance, oxidation of ω-6 fatty acids, leads to formation of 4-hydroxy-2-nonenal (HNE), whereas oxidative degeneration of ω-3 fatty acids yields 4-hydroxy-2-hexenal (HHE). As outlined above, the α,β-unsaturated lipid aldehydes are soft electrophiles and form Michael-type adducts with target sites displaying suitable nucleophilicity. The notion that proteins modified by 2-enals represent a biologically active subproteome has promoted intense efforts to develop methods suitable for the detection and characterization of these PTMs. The current method of choice for the characterization of protein modifications by 2-enals is tandem mass spectrometry. The energy imparted for ion activation is depended on the instrumentation used. Low energy (10-100 eV) CID is common to quadrupole hybrid instruments (qTOFs) and linear ion trap FT mass spectrometers. The “high energy” CID regime is employed in the ToF/ToF instruments. “Very low energy” (1-20 eV) CID condition is typically used in quadrupole ion traps. The instrument design and energy regime used during CID will determine the number of collisions, the time necessary for ion activation and consequently the fragmentation pathways accessible to the precursor ions (Wells & McLuckey, 2005). Since CID resembles a “slow heating” process and is kinetically controlled the most labile bonds fragment preferentially (Wells & McLuckey, 2005).
1. CID-MS/MS of peptides with Michael-type adduction by HNE
Our laboratory and others have observed that ions of Michael-type oxylipid-modified peptides undergo preferential fragmentation via low energy rearrangement reactions which occasionally limits the generation of sequence product ions but instead results in fragment ions that stem from neutral loss of the oxylipid (Carbone, et al., 2005; Chavez, et al., 2006; Chavez, et al., 2011; Guo & Prokai, 2011; Han, et al., 2007; Rauniyar, et al., 2009; Roe, et al., 2007; Stevens, et al., 2007). For instance, CID-MS/MS spectra of peptide-HNE adducts often indicate the occurrence of retro-Michael addition (RMA) pathways that result in loss of HNE from the precursor ions (Carbone, et al., 2005; Chavez, et al., 2006; Chavez, et al., 2011; Han, et al., 2007; Rauniyar, et al., 2009; Roe, et al., 2007; Stevens, et al., 2007). Under CID MS/MS conditions peptide ions with Michael adducts on cysteine residues seem to be particular susceptible to undergo retro Michael addition dissociations (Scheme 1, Figure 4). In contrast, Michael adducts on histidine residues seem to be more likely preserved during CID experiments (Rauniyar & Prokai, 2009). The apparent higher stability of the His-adduct is also reflected by the observation of the histidine-HNE immonium ion (m/z 266); whereas the corresponding cysteine-HNE immonium ion is not observable (Figure 4). Mass spectrometric detection and identification of peptides modified by Michael adducts on lysine residues have rarely been reported, which likely relates to the limited tendency of lysine residues to form Michael adducts under physiological conditions. Lysine residues rather form Schiff base adducts (Liu, et al., 2003; Sayre, et al., 2006).
Figure 4.
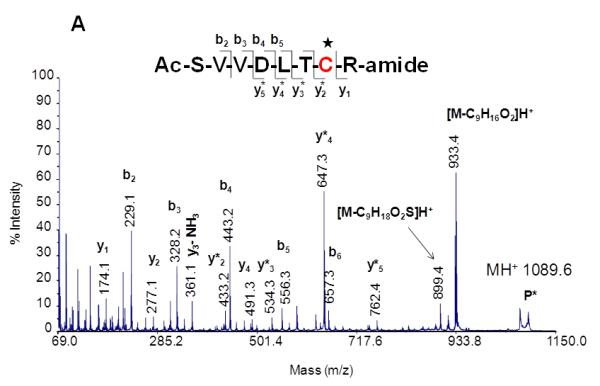
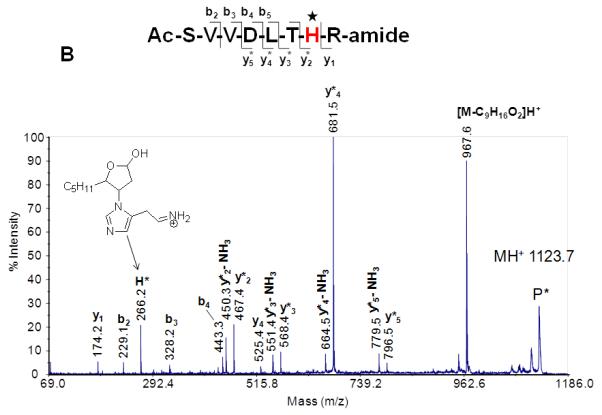
MALDI product ion spectra of peptide HNE adducts on Cys (A) and His (B). CIDMS/MS (1 keV) was performed using a MALDI ToF-ToF instrument. Note the neutral loss of the oxylipid due to retro Michael fragmentation. CID-MS/MS studies of peptides with oxylipid adduction on histidine residues yield as diagnostic product ions His-oxylipid immonium ions. Reproduced from Wu, PhD thesis (2011) with permission from Jianyong Wu, copyright 2009.
The tendency of peptide adducts of 2-enals to undergo retro Michael addition (RMA) reaction during CID-MS/MS studies is an analytical shortcoming of CID-MS/MS-based proteomics screens and hampers the development of large-scale identification and site-specific assignments of protein adducts of electrophilic lipoxidation products. Hydride reduction of the aldehyde moiety of the Michael adduct has been the preferred method to stabilize the adduct and making it amenable to CID-MS/MS studies (Scheme 2). Upon reductive stabilization (e.g. with sodium borohydride) the masses of the peptide oxylipid adduct ions and the fragment ions encompassing the modification are shifted by +2 Da (Fritz, et al., 2012; Liu, et al., 2003).
On the other hand, multistage tandem mass spectrometry-based strategies have been development that take advantage of the tendency of peptide-HNE adducts to undergo RMA. Several groups have developed data-dependent neutral loss (NL) -driven MS3 acquisition methods (Rauniyar, et al., 2009; Roe, et al., 2007). The neutral loss-driven MS3 acquisition technique is based on the detection of the neutral loss of m/z 156, 78 and 52 from single, doubly and triply protonated peptide ions, respectively, upon CID-MS/MS for triggering the MS3 experiment. A possible drawback of this method is that MS3 of the neutral loss ion does not allow location of the site of adduction. These initial studies have revealed that peptide adducts have different tendencies to undergo the retro Michael addition which is a prerequisite to utilize the NL-triggered acquisition modes (Rauniyar, et al., 2009; Stevens, et al., 2007). The disparate tendency of peptide oxylipid adducts to undergo retro Michael additions may warrant further systematic studies to foster the development of improved data-dependent acquisition protocols and bioinformatic tools.
1. CID-MS/MS of Michael adducts of electrophilic mono oxygenated derivatives of ω-3 PUFAs
Electrophilic fatty acid oxo-derivatives (EFOXs) are reactive α,β-unsaturated keto derivatives of fatty acids that are derived through enzymatic oxidation from ω-3 PUFAs. COX-2 oxidizes PUFAs to mono-hydroxy derivatives of FAs which are then further converted to EFOX derivatives by hydroxyl dehydrogenases (Serhan, et al., 2008). Groeger et al. (2010) used a combination of β-mercaptoethanol (BME) capture and RP-HPLC-MS/MS for the detection and identification of EFOX derivatives produced by activated macrophages. EFOX derivatives from the major ω-3 PUFAs, including eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) were observed after activation of RAW265.7 cells using diverse stimuli. For instance, the nanoLC-MS/MS analysis of the EFOX adduct to glutathione resulted in loss of the EFOX-D5 yielding the neutral loss ion at m/z 308 ([GSH+H]+) and adduct–specific fragment ions at m/z 420.2 and 523.3 (Figure 5). Proteomic analysis of 17-EFOX-in vitro modified GAPDH allowed localizing the site of adduction to distinct Cys- and His residues, which indicated that peptide-EFOX adducts are sufficiently stable, at least under certain CID-MS/MS conditions, to allow the unambiguous assignment of the adduction site. Shotgun proteomics studies that probe the biological significance of EFOX modifications have not yet become available.
Figure 5.
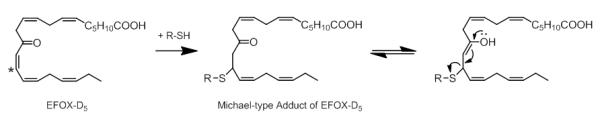
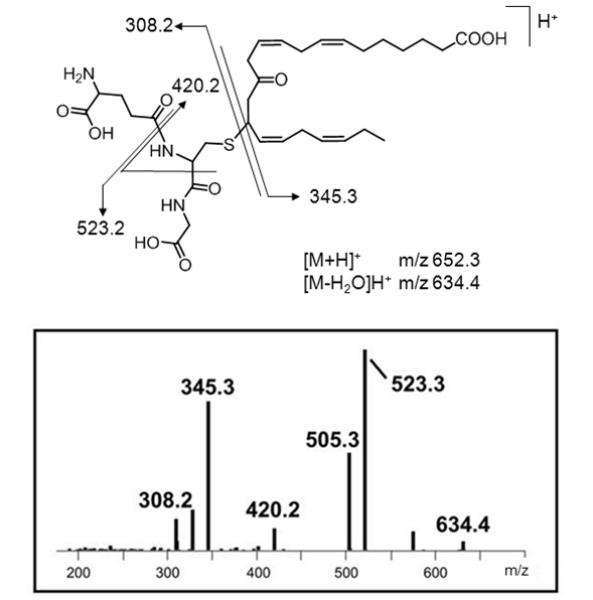
Ion trap (LCQ) CID-MS/MS spectrum of the a Michael-type glutathione adduct of 13-EFOX-D ([M+H]+ 5 m/z 652.3). The nomenclature of EFOX derivatives is based on the length of the carbon chain (i.e. docosanoids, D) and the number of the double bonds is reflected by the subscripted “x”. For instance, EFOX-D5 refers to the mono-oxygenated derivative of the 22-carbon fatty acid, docosahexaenoic acid (DHA) with a total of 5 double bonds. Retro-Michael addition results in the ions at m/z 308.2 ([GSH+H]+) and m/z 345.3 ([EFOX-D5+]H+), respectively, depending on the site of protonation. Reproduced and adapted from Groeger et al. Nat. Chem. Biol. (2010) with permission from Nature Publishing Group, copyright 2010.
2. CID-MS/MS of Michael adducts of electrophilic cyclopentenone-containing lipids
Cyclopentenone prostaglandins and isoprostanes are reactive eicosanoids that can form covalent adducts with cysteine residues in proteins through Michael-type addition reactions (Stamatakis & Perez-Sala, 2006). Cyclopentenone prostaglandins comprise a distinct group of electrophilic lipids due to their α,β-unsaturated carbonyl moiety in the prostane ring. Cyclopentenone PGs are formed from arachidonic acid (AA) via cyclooxygenase biosynthetic pathways (Musiek, et al., 2006; Uchida & Shibata, 2007). Cyclopentenone isoprostanes are structural isomers of cyclopentenone PGs and formed nonenzymatically by free radical-catalyzed generation (Hardy, et al., 2011). Examples of reactive eicosanoids are given in Figure 6A. Cyclopentenone lipids are soft electrophiles (Table 6) and Michael adduct formation has been predominantly found on cysteine residues, except for His-136 in human serum albumin due to an conducive pocket in subdomain IB (Yamaguchi, et al., 2009). Protein targets of cyclopentenone PGs have been identified and for some of these targets, site-specific modification information has become available (reviewed in (Garzon, et al., 2011)). Similarly as observed for protonated peptide 2-enal adducts, in CID-MS/MS experiments protonated peptide adducts with cyclopentenone lipids undergo fragmentation at the site of adduction due to retro Michael addition giving rise to nonsequence neutral loss ions. As an example, Figure 6B depicts the MALDI MS/MS product ion spectrum of a tryptic peptide modified by Δ12-PGJ2. Neutral loss of the Δ12-PGJ2 modification from the precursor ion results in the observation of the ion at m/z 1662 (Renedo, et al., 2007).
Figure 6.
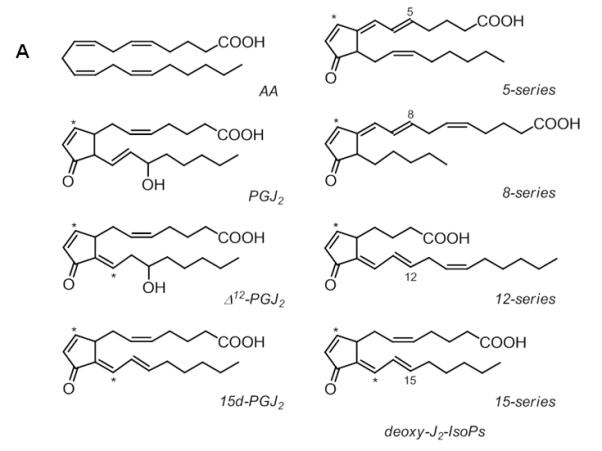
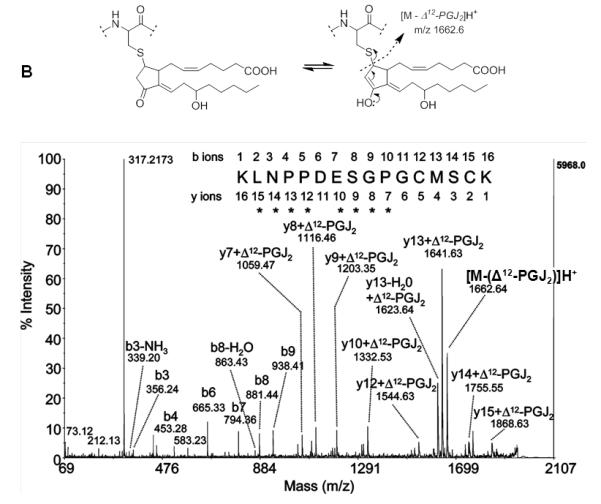
CID-MS/MS of peptide adducts with cyclopentenone lipids. A) Examples of cyclopentenone lipids derived from arachidonic acid. The asterisks mark the electrophilic carbon atom. B) MALDI CID MS/MS spectrum of the Δ12-PGJ2-modified peptide KLNPPDESGPGCMSCK with m/z 1997.2. The peptide was derived from Δ12-PGJ2-treated HRas (after digestion with trypsin). CID MS/MS analysis was performed on a MALDI MS/MS instrument with ToF-ToF optics. Michael adduct formation was confirmed by the mass increment of 334.5 which was observed for the y7 to y15 ions. The ion at m/z 1662.6 indicated retro-Michael fragmentation during MALDI ToF-ToF MS/MS. The ion at m/z 317.2 indicates charged loss of the cyclopentenone lipid associated with loss of water, [Δ12-PGJ2 – H2O]H+. Reproduced and modified from Renedo et al. (2007) with permission from American Chemical Society, copyright 2007.
B. Ion mobility-enhanced CID-MS/MS of peptide-oxylipid adducts
A challenge that we are currently attempting to address resides in the assignment of positional isomers of protein modification by lipoxidation products. One strategy that we are exploring is ion mobility-enhanced MS/MS of oxylipid-modified proteins. This strategy has been successfully applied to isomeric phosphopeptides (Thalassinos, et al., 2008). In principle, ion mobility spectrometry facilitates the separation of ions according to mass, charge, and shape. Thus, ion mobility spectrometry may enable the gas-phase separation of isobaric ions. Ions with more compact conformations will traverse the mobility separation device more quickly than those ions with extended conformations. In addition, the combination of ion mobility spectrometry and mass spectrometry will expand the separation space which, in turn, may facilitate the detection of low abundant species (Bohrer, et al., 2008; Kanu, et al., 2008).
For the analysis of peptide-oxylipids described here, we used a Waters Synapt G2 HDMS instrument equipped with an electrospray ionization source. Design principles are outlined in Figure 7A. In this instrument, a TriWaveTM section consists of three traveling wave-enabled stacked ring ion guides is is placed between a quadrupole mass filter (Q1) and an orthogonal acceleration (oa) TOF analyzer. The TriWaveTM section enables unique combinations of gas-phase ion mobility separation and fragmentation experiments. Ion activation can take place either in the trap, or in the transfer device, or in both. Precursor ions or 1st generation fragment ions can be subjected to traveling wave ion mobility spectrometry. The working principle and the design of the traveling wave ion mobility spectrometry device have been published previously by Giles et al. (Giles, et al., 2004; Giles, et al., 2011). Details on the conceptual background of classical ion mobility spectrometry and the adaption to traveling wave ion mobility spectrometry have been described elsewhere (Michaelevski, et al., 2010; Smith, 2009).
Figure 7.
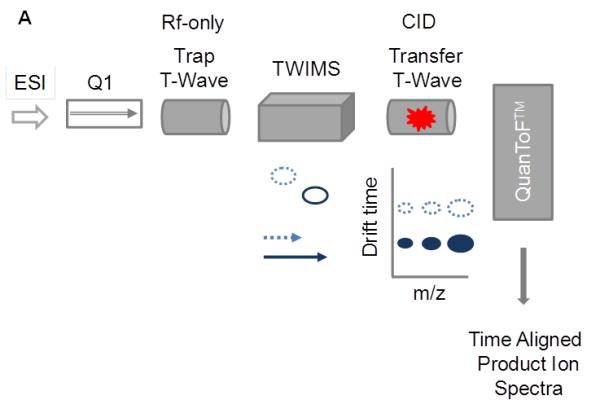
Ion mobility-enhanced CID-MS/MS for the characterization of peptide-oxylipid adducts. (A) Conceptual presentation of ion mobility-enhanced CID-MS/MS using a quadrupole traveling wave ion mobility TOF instrument (Waters Synapt G2 HDMS). In this mode, precursor ions are separated according to their ion mobilities (mass, charge and shape) in the ion mobility section of the TriWaveTM device. Ion activation is performed in the transfer region. Fragment ions generated in the transfer region are time-aligned to their precursor ions. This mode of operation is potentially useful for the unambiguous assignment of positional isomers as long as they have different interaction cross sections; v, velocity;
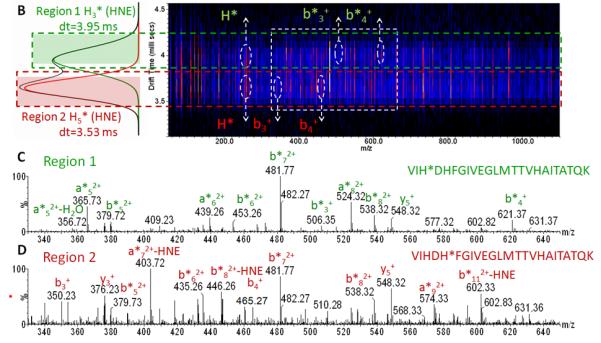
(B) IMS-CID-MS/MS analysis of positional isomers of a peptide-HNE adduct with the sequence VIH3DH5FGIVEGLM TTVH17 ox AITATQK (derived from HNE-modified GADPH by tryptic digestion). This peptide has three histidine residues as potential sites for modification by HNE. Transfer dissociation was conducted by selecting the quadruply protonated peptide with m/z 698.38 in the quadrupole device Q1, subsequently ions were separated in the TWIMS region and then subjected to collisional activation in the transfer region. The drift scope image illustrates the time-alignment of the fragment ions originating from two precursor populations with drift time distribution ~3.53 and ~3.95 ms, respectively. (C and D) The extracted tandem mass spectrum after extraction of the ion signals that are time-aligned with the respective precursors. The upper panel (C) depicts the product ion spectrum of the peptide modified on His at position 3 (His-3) by HNE and the lower panel (D) shows the same peptide but modified at His-5. (L. Huang, unpublished data).
Figure 7B illustrates the use of ion mobility-enhanced transfer fragmentation for the characterization of positional isomers of a large peptide with multiple possible sites for HNE adduction. The mono adducted peptide was derived from GAPDH after in vitro exposure to HNE, trypsination and Hz affinity-enrichment (Huang, et al., 201x). The enriched HNE-modified peptide solution was used for LC-MS in combination with ion mobility-enhanced tandem mass spectrometry. Transfer fragmentation was conducted by selecting the precursor ions using the quadrupole device Q1. Ions were then separated in the T-wave ion mobility device and ion activation was performed in the transfer region by applying elevated collision energy. The product ions were then passed on to the TOF analyzer. Figure 7B depicts the driftscope image of the ion-mobility enhanced transfer fragmentation experiment for the singly-adducted quadruply protonated peptide, VIHDHFGIVEGLMTTVHAITATQK ([M+4H]4+, m/z 698.9). The driftscope image illustrates the time-alignment of the fragment ions with their ion mobility-separated precursors. This large peptide separated into two ion populations with drift time distributions (DTDs) that differed by 0.4 ms. Fragment ions of the first DTD (region 1, dt 3.95 ms) and fragment ions of the second DTD (region 2, dt 3.53ms ) were extracted separately to obtain the product ion spectrum of each of the peptide isomers (Figures 7B and 7C). The depicted spectra show only the m/z region 330-650. The transfer fragmentation of the precursor with DTD at 3.53 ms (region 2) yielded product ions b3*, b4* that indicated modification of the His residue in position 3 by HNE (Figure 1B). Whereas, the precursor with the DTD at 3.95 ms (region 1) yielded fragment ions b3, b4 indicating the absence of the HNE modification on His-3. However, the observed mass shifts (of 156 Da) for bn fragmentation with n ≥5 pointed to HNE modification of His-5 by HNE (Figure 7C). We expect that the combination of ion mobility spectrometry with high resolution mass analyzers will become an important measurement platform for the in depth analysis of protein adducts of reactive lipids. In particular, ion mobility-enhanced tandem mass spectrometry has the potential to assist in the analysis of positional isomers enabling the unequivocal assignment of site of modification. As these in depth information on protein adduction become more readily available this knowledge may lead to an enhanced understanding of the mechanisms by which these modifications modulate protein and cell function and/or exert cytotoxicity.
III.TANDEM MASS SPECTROMETRY-BASED STRATEGIES IN CONJUNCTION WITH AFFINITY-BASED PROBES FOR PROFILING OF OXYLIPID-MODIFIED PROTEINS
Key for the successful analysis of protein modification by mass spectrometry-based approaches is an appreciation of their solution and gas-phase reactivities and an understanding of the limitation associated with diverse mass spectrometry techniques. Modification by electrophilic lipoxidation products are chemically diverse and in biological samples present only in very low abundance. Most mass spectrometry-based approaches use a combination of nano-liquid chromatography and tandem mass spectrometry (nanoLC-MS/MS). Information-dependent data acquisition modes are often used for the analysis in which the (three or five) most intense peptide ions from the eluting peptides are being submitted to gas phase fragmentation techniques. Therefore, in order to increase the likelihood to detect, identify and obtain modification site information, a collection of diverse techniques has become available for the selective enrichment of oxylipid-modified peptides and proteins enabling tandem mass spectrometry-based analyses with improved detection sensitivities which is particularly beneficial for the analysis of complex biological samples. Enrichment techniques can be implemented at the protein or at peptide-level. However, a distinct advantage of approaches that perform an enrichment step at the peptide level is that the unambiguous identification and site-specific information for the oxylipid-modification is achievable by tandem mass spectrometry. The confidence with which peptide-level identification of oxylipid-modification can be achieved benefits from the acquisition of high-resolution and high accuracy mass spectral data.
1. Chemoselective strategies that utilize hydrazine-functionalized affinity-probes
The chemoselective tagging of the aldehydic protein modification with a chemical affinity probe is the most common strategy employed for profiling of aldehydic protein modifications in complex mixtures. Current approaches are based on “bottom up” proteomics strategies and utilize small chemical probes for the targeted analysis of aldehydic PTMs to minimize interference from the high background of the proteome. The design of these probes enables the selective and covalent modification of the aldehyde group in proteins modified by direct oxidation reactions and/or adduction by α,β-unsaturated aldehydes with an affinity group. The majority of the approaches utilize hydrazide-functionalized derivatives of biotin ((Madian & Regnier, 2010; Madian, et al., 2011a; Madian, et al., 2011b; Meany, et al., 2007)). Our group explored the use of hydrazine-functionalized isotope-coded affinity probes (HICATs) for the identification and the comparative quantification of oxylipid-modified proteins in “bottom up” mass spectrometry-based proteomics approaches (Han, et al., 2007). Reductive stabilization of the hydrazone bond is beneficial for increasing the chemical stabilization of the tagged adduct. A potential drawback of using hydrazine-functionalized probes for shotgun approaches is that the CID-MS/MS spectra usually show non-peptide fragment ions that originate from the probe (Han, et al., 2007).
2. Chemoselective tagging using a hydroxylamine-functionalized affinity-probe
We introduced a hydroxylamine -functionalized biotin-containing probe, aka. aldehyde reactive probe (ARP), for the specific tagging of adducts of electrophilic lipoxidation products in complex matrices (Chavez, et al., 2006). Derivatization of the aldehyde group in oxidatively modified proteins leads to aldoxime formation (Scheme 3) and the resulting biotin-tagged peptide is amenable to enrichment and subsequent interrogation by tandem mass spectrometry. The ARP-tag introduces a mass shift of +332 Da with respect to the corresponding oxylipid-modified peptide. We used the ARP-tagging strategy for profiling protein targets of HNE exposure in THP-1 cells (Chavez, et al., 2010). The Tannenbaum group has investigated the modification of proteins by DODE, the α,β-unsaturated keto aldehyde with the highest electrophilicity (Table 2). The structure of DODE is shown in Table 1. The ARP strategy was enabled the tandem mass spectrometry-based identification of protein targets of DODE in MCF7 breast cancer cells (Slade, et al., 2010).
We employed a shotgun proteomics approach in conjunction with chemoselective tagging using ARP for the identification and characterization of HNE-adducted proteins in cardiac mitochondria (Chavez, et al., 2011). After trypsination and enrichment of ARP-tagged peptides localization of the adduction site was achieved by tandem mass spectrometry using nanoLC ESI MS/MS and nanoLC in combination with MALDI-MS/MS. An interesting finding of this work was that some of the identified protein target sites were adducted by different lipoxidation products (Chavez, et al., 2011).
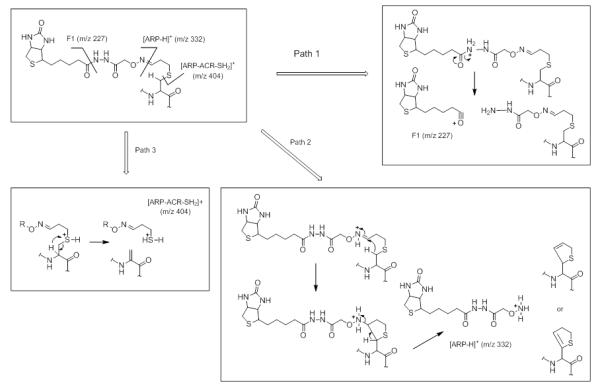
During the course of our investigations we observed that the fragmentation behavior of ARP-tagged oxylipid-peptide adducts depended to a certain degree on the CID-MS/MS conditions employed by the different instrument designs. Figure 8 shows MALDI product ion spectra of a peptide adducted on a cysteine residue by different α,β-unsaturated lipid aldehydes. MALDI ToF/ToF MS/MS (1kV) of singly protonated peptide adducts (MH+) yielded a) sequence-related bn and yn-type fragment ions that in favorable cases allowed site-specific assignment of the modification, b) HNE-His immonium ions, and c) nonsequence fragment ions that indicated loss of the ARP (−331 Da) and HNE-ARP (−469 Da) moiety, respectively. Multiply protonated CID-MS/MS spectra acquired on a qToF type instrument displayed product ions (m/z 227, 332 and 404) derived from the ARP-tag that point to the dominance of charge-directed fragmentation mechanisms (Figure 9). By contrast, analysis of multiply protonated ARP-tagged oxylipid peptide adducts by nanoLC-ESI-MS/MS using a LTQ-FT ICR MS yielded product ion spectra that revealed charge-reduced fragment ions (m/z 1443.5 and 1571.6) that originate from the “charged” loss of the ARP tag (MH+ 332) and sequential loss of “dehydrated HNE” (−138.1 Da) (Figure 10). This led us to speculate that the relatively slow activation of the precursor ion occurring in linear ion trap-type instruments promotes low energy rearrangement pathways (Figure 10). We further hypothesize that “mobile’ protons direct the fragmentation behavior in multiply protonated peptides and promote charge-directed fragmentation pathways. On the other hand, charge-directed pathways are less accessible under MALDI-ToF/ToF MS/MS conditions because the proton is sequestered at the most basic site, i.e. the C-terminal residue (Lys, Arg) in our experiments (Wang, et al., 2011; Wysocki, et al., 2000).
Figure 8.
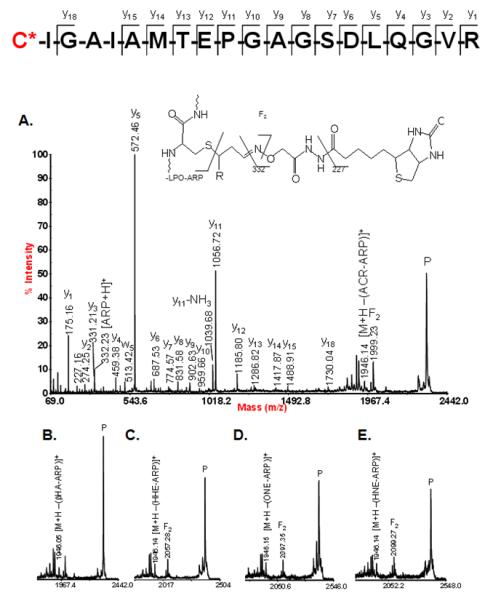
MALDI-MS/MS analysis of the mitochondrial protein acyl-CoA dehydrogenase (long-chain specific; ACADL_RAT) modified on Cys-166 with different endogenous electrophilic lipoxydation products . ARP was used for the chemoselective tagging of proteins with reactive carbonyl/keto groups. After trypsin digestion, ARP-tagged peptides were enriched using immobilized monomeric avidin beads and submitted to MALDI-CID (1keV) MS/MS analysis using an Applied Biosystem 4700 ToF-ToF instrument. “Peptide” ions as well as the ARP-derived “non-sequence” ion are annotated. The asterisks denote fragment ions that retained the lipoxidation-derived 2-alkenal modification during CID-MS/MS. Reproduced from Chavez et al (2011) with permission from Elsevier, copyright 2011.
Figure 9.
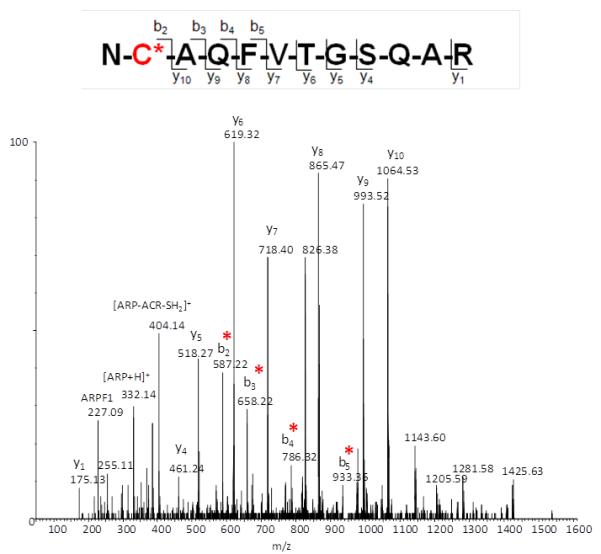
(A) QToF CID-MS/MS product ion spectrum of the ARP-tagged peptide acrolein adduct NC*AQFVTGSQAR ([M+2H]2+ ion; monoisotopic m/zcalc 825.88; accuracy Δ(m/z) = −0.01 Da). Reproduced from Chavez et al. (2011) with permission from Elsevier, copyright 2011. (B) Proposed charge-directed fragmentation pathways for multiply protonated ARP-tagged peptide acrolein adducts induced by CID-MS/MS using a qToF instrument (Waters Ultima Global).
Figure 10.
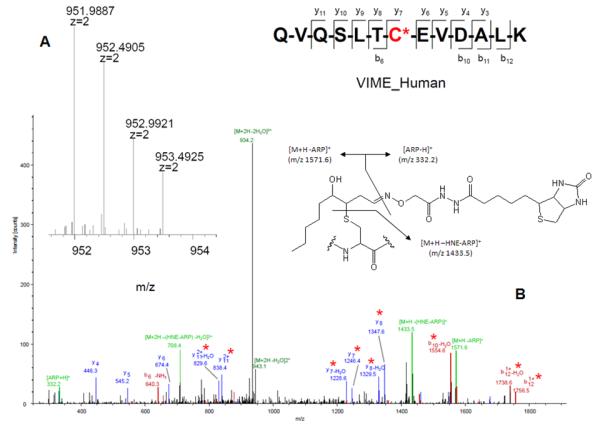
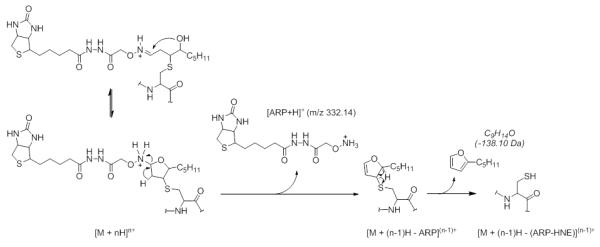
LTQ-FT mass spectrometric analysis of the ARP-tagged peptide-HNE Michael adduct QVQSLTC*EVDALK from vimentin (VIME_Human). (A) High accuracy mass measurement of the precursor ion (monoisotopic mass m/z calc 951.9869; accuracy Δ(m/z) = 2 ppm); (B) LTQ CID-MS/MS analysis of the [M+2H]2+ ion of the ARP-tagged peptide-HNE Michael adduct QVQSLTC*EVDALK; Sequence ions are denoted as bn and yn-type ions and highlighted in red and blue, respectively. “Non-sequence” ions are highlighted in green and originate from “charged loss” fragmentation of the ARP-HNE moiety. The asterisks denote fragment ions that retained the HNE adduct during CID. THP-1 cells were exposed to HNE and treated with ARP. The cell lysate was then digested and submitted to capillary LC-ESI MS/MS analysis. The CID-MS/MS analysis was conducted on a 7T LTQ-FT MS instrument (Thermo). Reproduced and adapted from Chavez et al. Supplemental Materials (2010) with permission from Elsevier, copyright 2011. D. Proposed fragmentation mechanism for ARP-tagged peptide oxylipid adducts under low energy CID-condition applied in the LTQ. Rearrangement reactions promote the “charged loss” of ARP and subsequent neutral loss of “dehydrated HNE”.
Slade et al. developed a filtered database search strategy to aid in the identification of ARP-tagged oxylipid-modified peptides (Slade, et al., 2011). An in silico filter program uses the ARP-specific fragment ions to filter the tandem mass spectral data and create a set of new datafiles that only contain the oxylipid modified peptides. Searching this curated dataset by MASCOT, a widely-used software search engine that uses tandem mass spectrometry data to identify proteins from primary sequence databases, limits the number of false positive hits and simplifies the manual inspection of the MS/MS data. As an alternative to MASCOT, Slade et al. also introduced a specialized database search engine for the identification of biotin-containing peptides without the prerequisite of defining the masses of possible oxylipid modifications (Slade, et al., 2011). This search and identification strategy identified serum proteins with diverse oxylipid modifications in a murine model of inflammation, e.g. DODE adducts were assigned to specific residues on mouse serum albumin and HNE adducts were identified on multiple murinoglobulin sites (Slade, et al., 2011).
IV. Electron-driven dissociation methods
A. Electron capture dissociation (ECD)
In electron capture dissociation (ECD) a multiply positively charged species captures a low energy electron (< 10 eV) which leads to exothermic activation and dissociation of the resultant odd electron ion (Zubarev, 1998) (Scheme 4). Because ECD causes charge reduction, multiply protonated peptides, [M+nH]n+ with n ≥ 2, are required for ECD studies. Multiply protonated peptides are also more competent in capturing a low energy electron since the electron capture cross section is proportional to the square of the ion charge (Zubarev, et al., 2000). Electron capture is not a very efficient process and therefore benefits from long interaction times. At present, most ECD-MS/MS studies in the context of protein and peptide analyses are performed in FTICR instruments (Bakhtiar & Guan, 2005; Bakhtiar & Guan, 2006; McLafferty, et al., 2001).
The ECD mechanisms is based on radical chemistry and results in the formation of cn and zn-type fragment ions (Zubarev, 2002; Zubarev, 1998). ECD processes take places in a faster timescale than internal energy distribution occurs (≤10−12 s). Consequently, bond dissociation is not kinetically controlled and, unlike in CID, low energy pathways are less likely to compete with peptide bond fragmentation. It is for this reason that ECD has been enthusiastically applied for the modification site localization of labile protein modifications (Bakhtiar & Guan, 2005; Bakhtiar & Guan, 2006; Shi, et al., 2001; Stensballe, et al., 2000). Recently, Prokai and coworkers have demonstrated that ECD studies of HNE-modified peptides yield peptide fragment ions that retain the HNE moiety resulting in the unambiguous assignment of the site of adduct formation (Figure 11) (Rauniyar, et al., 2009).
Figure 11.
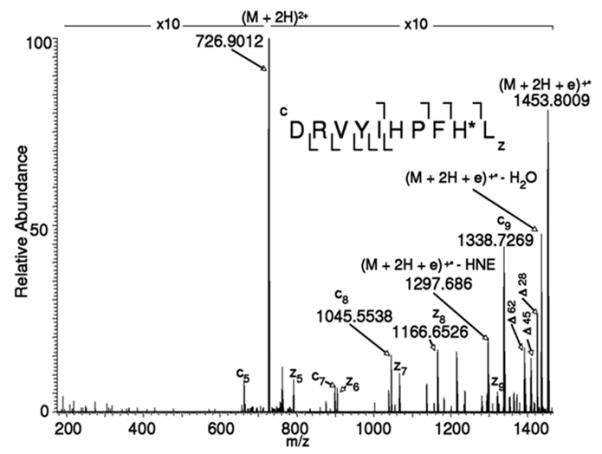
ECD-MS/MS spectrum of the (M+2H)2+ ion m/z 726.9012 of DRVYIHPFH*L. The mass difference between fragment ions c8 (m/z 1045.5538) and c9 (m/z 1338.7269) indicates that the histidine residue at position 9 is modified by HNE. Δ, markes neutral loss ions: Δ18, loss of CO; Δ45, loss of .COOH; Δ62, loss of CO2 + H2O. Reprinted from Rauniyar et al. (2009) with permission from American Chemical Society, copyright 2009.
Our laboratory has evaluated ECD-MS/MS for the analysis of ARP-tagged peptide oxylipid adducts. ECD-MS/MS analyses were conducted on a LTQ-FT MS (Figure 12). As typically observed for ECD studies, the spectra obtained for both unmodified and ARP-HNE modified peptides showed low intense fragment ions and were dominated by precursor and charge-reduced precursor ions. The ECD MS/MS spectra of the modified peptides showed usually multiple fragment ions of the ARP-lipid moiety. These non-peptide fragmentations were assigned as “charged loss” fragment ions and included ions that originated from loss of the protonated ARP tag (−331 Da) and the ARP-HNE moiety (−469 Da). Four other non-peptide ions resulted from loss of parts of the ARP tag with −129, −240, −314 and −357 Da. Despite the undesirable fragmentation of the ARP-HNE moiety and the presence of several ”charged loss” ions, the spectra allowed the assignment of c- and z-type ions to correctly decode the peptide sequence and locate the site of the HNE adduction (Tzeng & Maier, 2010).
Figure 12.
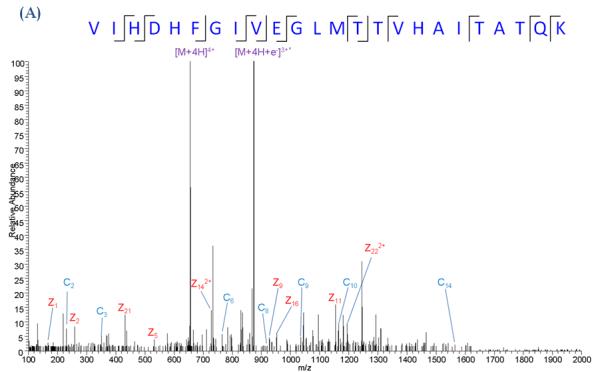
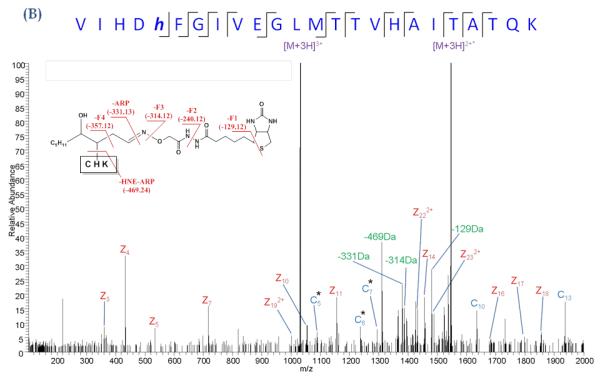
ECD MS/MS analysis of an ARP-tagged peptide-HNE Michael adduct. (A) ECDMS/MS spectrum of the quadruply protonated peptide VIHDHFGIVEGLMTTVHAITATQK (derived from GAPDH) and (B) of the same peptide but with ARP-tagged HNE modification. ECD was performed on the triply protonated peptide. Albeit ECD resulted in fragmentation of the ARP tag, ECD product ions were sufficiently generated to allow the localization of the HNE modification to the histidine residue in position 5. Asterisks denote the fragment ions that retained the ARP-HNE moiety (Tzeng & Maier, ASMS 2010).
B. Electron-transfer dissociation (ETD)
ETD mass spectrometry was first reported in 2004 by Hunt and coworkers (Syka, et al., 2004). The initial step in the ETD process involves the transfer of an electron to the multiply protonated peptide from a singly charged radical anion. Subsequent electron transfer from the radical anion to a peptide backbone amide bond generates a carbonyl radical anion that then abstracts a proton from a positively charged nitrogen in close proximity (Scheme 4). The resulting charge-reduced ion contains a radical site at the carbonyl carbon that then triggers fragmentation of the N-Cα bond to produce cn and zn- type ions (Scheme 4). Similarly to ECD, ETD results in a relative non-selective fragmentation of the N-Cα bond, while retaining labile modifications. ETD-MS/MS has been successfully used for phosphorylation site mapping (Boersema, et al., 2009; Chi, et al., 2007; Palumbo, et al., 2011), methylation site mapping (Young, et al., 2009), O-glycosylation (Perdivara, et al., 2008), and many others (Kim & Pandey, 2012; Mikesh, et al., 2006). As such, ETD represents an alternative and highly promising dissociation method for obtaining site-specific information of electrophilic oxylipid adduction. ETD seems to work most efficiently for peptide ions that have high charge-to-residue ratios (Good, et al., 2007; Molina, et al., 2007).
Very recently, ETD MS/MS data have been published for a Michael-type adduct on a cysteine residue of tryptic peptide that was derived from SIRT3 (Fritz, et al., 2012). Ion trap ETD fragmentation prevented HNE loss via retro Michael addition reaction whereas the CID MS/MS spectrum was dominated by ions that indicate loss of HNE from the triply charged peptide ion. Figure 13 depicts some of our initial experiments of using ETD (implemented in an Orbitrap XL instrument) for the analysis of HNE-modified peptides. The ETD-MS/MS spectrum shows complete series of cn- and zn-type fragment ions that allow the unambiguous assignment of the HNE adduction to the histidine residue. No ion peaks are observed that indicate loss of HNE from the precursor ion during the dissociation experiment, an essential prerequisite for establishing distinct modification sites of electrophilic lipids.
Figure 13.
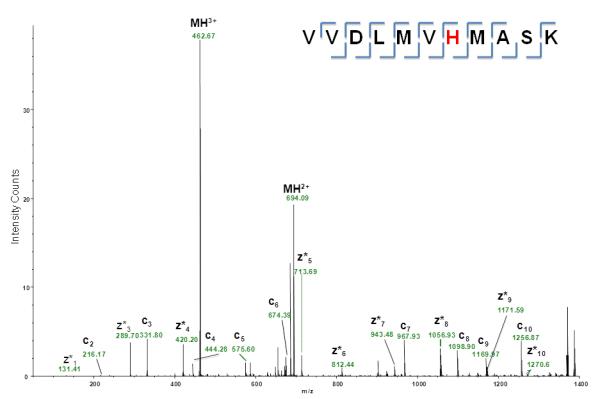
ETD MS/MS product ion spectra of the triply protonated peptide
VVDLMVH*MASK from GAPDH in vitro-modified by HNE (([M+3H]3+, m/z 462.59). Complete cn- and zn-type fragment ion series are observed. ETD did not induce retro-Michael addition of HNE. Asterisks denote the fragment ions that retained the HNE modification. The ETD-MS/MS experiment was performed with an Orbitrap XL equipped with an ETD module. Fluoranthene radical anions were generated in the CI ion source and then transferred into the linear trap (unpublished data).
V. Conclusions and Outlook
Examples of recent state-of-the-art and high-throughput proteomics strategies for the identification, quantification, characterization of carbonylation in biological samples with possible utility to clinical application have become available (Madian, et al., 2011a). Although hundreds of proteins were identified as putatively containing aldehydic protein modifications, only a relative small selection of carbonyl modifications were indeed identified and site-specific information was obtained. Most of the identified protein carbonyl modifications were identified as semialdehydic modification sites, all other modifications were formed by the addition of glycation, advanced glycation end products and by Schiff’s base formation involving glyoxal and methyl glyoxal to lysine residues. However, none of the identified sites were related to Michael-type adducts of electrophilic lipids. The failure to detect and identify the protein lipid adducts in highly complex biological mixtures reveals the shortcomings of our currently employed techniques and search engines. Loss of the modification during the CID fragmentation experiments results in unsuccessful matching of the tandem mass spectral data with database entries or in low peptide ion matching scores. The developments of new algorithms that take advantage of the non-peptide fragment ions have been shown beneficial and will need to be further extended and refined.
Key to successful large-scale proteomics strategies of oxylipid modifications will depend on chemical derivatization approaches in conjunction with enrichment techniques and/or highly efficient capture/release strategies. We believe that a prerequisite for future large scale analyses for the identification of protein targets of electrophilic lipids will depend critically on a better understanding to what extent the chemical stability and properties of Michael-type adducts of lipid peroxidation products govern the gas phase fragmention behaviors of proteolytic oxylipid-modified peptide adducts. The exploration of electron-driven MS/MS techniques for protein/peptide adducts of electrophilic lipids is promising but systematic studies of the parameters govern gas-phase fragmentation of the oxylipid-modified peptide ions are just starting to emerge (Rauniyar, et al., 2009). We foresee that tandem mass spectral techniques that are based on non-CID mechanisms will provide promising routes for the analysis of protein adducts of electrophilic lipids.
The analytical challenges associated with the detailed characterization of the interactions of electrophilic lipids with proteins in diverse cell systems are exciting and will promote the development of new innovative approaches. Deciphering the crosstalk between redox status, protein modifications by electrophilic lipids and cellular responses will advance our understanding of these electrophilic lipid protein adducts in health and disease.
Supplementary Material
Acknowledgement
We are immensely grateful to Dr. Fred Stevens for his many helpful suggestions that lead to the successful completion of the studies described and his critical comments that significantly improved this review. We would like to thank Thermo Scientific Corp. for providing access to an Orbitrap XL ETD. Work described in this contribution was supported by the National Institutes of Health Grants R01AG025372, S10RR025628 and P30ES000210. YVV is supported by the National Science Foundation under Grant No. 0924027, the National Institutes of Health under Grant No. R01RR026275 and a grant from OSU RERF.
References
- Bachi A, Dalle-Donne I, Scaloni A. Redox Proteomics: Chemical Principles, Methodological Approaches and Biological/Biomedical Promises. Chem Rev. 2013;113:596–698. doi: 10.1021/cr300073p. [DOI] [PubMed] [Google Scholar]
- Bakhtiar R, Guan Z. Electron capture dissociation mass spectrometry in characterization of post-translational modifications. Biochem Biophys Res Commun. 2005;334:1–8. doi: 10.1016/j.bbrc.2005.05.138. [DOI] [PubMed] [Google Scholar]
- Bakhtiar R, Guan Z. Electron Capture Dissociation Mass Spectrometry in Characterization of Peptides and Proteins. Biotechnol Lett. 2006;28:1047–1059. doi: 10.1007/s10529-006-9065-z. [DOI] [PubMed] [Google Scholar]
- Boersema PJ, Mohammed S, Heck AJR. Phosphopeptide fragmentation and analysis by mass spectrometry. J Mass Spectrom. 2009;44:861–878. doi: 10.1002/jms.1599. [DOI] [PubMed] [Google Scholar]
- Bohrer BC, Merenbloom SI, Koeniger SL, Hilderbrand AE, Clemmer DE. Biomolecule analysis by ion mobility spectrometry. Annu Rev Anal Chem (Palo Alto Calif) 2008;1:293–327. doi: 10.1146/annurev.anchem.1.031207.113001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Perluigi M, Reed T, Muharib T, Hughes CP, Robinson RA, Sultana R. Redox proteomics in selected neurodegenerative disorders: from its infancy to future applications. Antioxid Redox Signal. 2012;17:1610–1655. doi: 10.1089/ars.2011.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bythell BJ, Suhai S, Somogyi A, Paizs B. Proton-driven amide bond-cleavage pathways of gas-phase peptide ions lacking mobile protons. J Am Chem Soc. 2009;131:14057–14065. doi: 10.1021/ja903883z. [DOI] [PubMed] [Google Scholar]
- Carbone DL, Doorn JA, Kiebler Z, Ickes BR, Petersen DR. Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. J Pharmacol Exp Ther. 2005;315:8–15. doi: 10.1124/jpet.105.088088. [DOI] [PubMed] [Google Scholar]
- Chattaraj PK, Sarkar U, Roy DR. Electrophilicity Index. Chem Rev. 2006;106:2065–2091. doi: 10.1021/cr040109f. [DOI] [PubMed] [Google Scholar]
- Chavez J, Wu J, Han B, Chung WG, Maier CS. New role for an old probe: affinity labeling of oxylipid protein conjugates by N’-aminooxymethylcarbonylhydrazino d-biotin. Anal Chem. 2006;78:6847–6854. doi: 10.1021/ac0607257. [DOI] [PubMed] [Google Scholar]
- Chavez J, Chung WG, Miranda CL, Singhal M, Stevens JF, Maier CS. Site-specific protein adducts of 4-hydroxy-2(E)-nonenal in human THP-1 monocytic cells: protein carbonylation is diminished by ascorbic acid. Chem Res Toxicol. 2010;23:37–47. doi: 10.1021/tx9002462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez JD, Wu J, Bisson W, Maier CS. Site-specific proteomic analysis of lipoxidation adducts in cardiac mitochondria reveals chemical diversity of 2-alkenal adduction. J Proteomics. 2011;74:2417–2429. doi: 10.1016/j.jprot.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi A, Huttenhower C, Geer LY, Coon JJ, Syka JE, Bai DL, Shabanowitz J, Burke DJ, Troyanskaya OG, Hunt DF. Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc Natl Acad Sci U S A. 2007;104:2193–2198. doi: 10.1073/pnas.0607084104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diers AR, Higdon AN, Ricart KC, Johnson MS, Agarwal A, Kalyanaraman B, Landar A, Darley-Usmar VM. Mitochondrial targeting of the electrophilic lipid 15-deoxy-Delta12,14-prostaglandin J2 increases apoptotic efficacy via redox cell signalling mechanisms. Biochem J. 2009;426:31–41. doi: 10.1042/BJ20091293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez-Dacal B, Perez-Sala D. Anti-inflammatory prostanoids: focus on the interactions between electrophile signaling and resolution of inflammation. ScientificWorldJournal. 2010;10:655–675. doi: 10.1100/tsw.2010.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongre AR, Jones JL, Somogyi A, Wysocki VH. Influence of Peptide Composition, Gas-Phase Basicity, and Chemical Modification on Fragmentation Efficiency: Evidence for the Mobile Proton Model. J Am Chem Soc. 1996;118:8365–8374. [Google Scholar]
- Doorn JA, Petersen DR. Covalent modification of amino acid nucleophiles by the lipid peroxidation products 4-hydroxy-2-nonenal and 4-oxo-2-nonenal. Chem Res Toxicol. 2002;15:1445–1450. doi: 10.1021/tx025590o. [DOI] [PubMed] [Google Scholar]
- Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr., Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 09, Revision A.1. Gaussian, Inc.; Pittsburgh PA: 2009. [Google Scholar]
- Fritz KS, Kellersberger KA, Gomez JD, Petersen DR. 4-HNE adduct stability characterized by collision-induced dissociation and electron transfer dissociation mass spectrometry. Chem Res Toxicol. 2012;25:965–970. doi: 10.1021/tx300100w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon B, Oeste CL, Diez-Dacal B, Perez-Sala D. Proteomic studies on protein modification by cyclopentenone prostaglandins: expanding our view on electrophile actions. J Proteomics. 2011;74:2243–2263. doi: 10.1016/j.jprot.2011.03.028. [DOI] [PubMed] [Google Scholar]
- Giles K, Pringle SD, Worthington KR, Little D, Wildgoose JL, Bateman RH. Applications of a travelling wave-based radio-frequency-only stacked ring ion guide. Rapid Commun Mass Spectrom. 2004;18:2401–2414. doi: 10.1002/rcm.1641. [DOI] [PubMed] [Google Scholar]
- Giles K, Williams JP, Campuzano I. Enhancements in travelling wave ion mobility resolution. Rapid Commun Mass Spectrom. 2011;25:1559–1566. doi: 10.1002/rcm.5013. [DOI] [PubMed] [Google Scholar]
- Good DM, Wirtala M, McAlister GC, Coon JJ. Performance characteristics of electron transfer dissociation mass spectrometry. Mol Cell Proteomics. 2007;6:1942–1951. doi: 10.1074/mcp.M700073-MCP200. [DOI] [PubMed] [Google Scholar]
- Groeger AL, Cipollina C, Cole MP, Woodcock SR, Bonacci G, Rudolph TK, Rudolph V, Freeman BA, Schopfer FJ. Cyclooxygenase-2 generates anti-inflammatory mediators from omega-3 fatty acids. Nat Chem Biol. 2010;6:433–441. doi: 10.1038/nchembio.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Prokai-Tatrai K, Nguyen V, Rauniyar N, Ughy B, Prokai L. Protein targets for carbonylation by 4-hydroxy-2-nonenal in rat liver mitochondria. J Proteomics. 2011;74:2370–2379. doi: 10.1016/j.jprot.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Prokai L. To tag or not to tag: a comparative evaluation of immunoaffinity-labeling and tandem mass spectrometry for the identification and localization of posttranslational protein carbonylation by 4-hydroxy-2-nonenal, an end-product of lipid peroxidation. J Proteomics. 2011;74:2360–2369. doi: 10.1016/j.jprot.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, Stevens JF, Maier CS. Design, synthesis, and application of a hydrazide functionalized isotope-coded affinity tag for the quantification of oxylipid-protein conjugates. Anal Chem. 2007;79:3342–3354. doi: 10.1021/ac062262a. [DOI] [PubMed] [Google Scholar]
- Han B, Hare M, Wickramasekara S, Fang Y, Maier CS. A comparative ‘bottom up’ proteomics strategy for the site-specific identification and quantification of protein modifications by electrophilic lipids. J Proteomics. 2012;75:5724–5733. doi: 10.1016/j.jprot.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy KD, Cox BE, Milne GL, Yin H, Roberts LJ. Nonenzymatic free radical-catalyzed generation of 15-deoxy-Δ(12,14)-prostaglandin J2-like compounds (deoxy-J2-isoprostanes) in vivo. J Lipid Res. 2011;52:113–124. doi: 10.1194/jlr.M010264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazi AU, Taylor HS. Stabilization method for calculating resonance energies: model problem. Phys. Rev. A. 1970;1:1109–1120. [Google Scholar]
- Held JM, Gibson BW. Regulatory control or oxidative damage? Proteomic approaches to interrogate the role of cysteine oxidation status in biological processes. Mol Cell Proteomics. 2011;11:R111. doi: 10.1074/mcp.R111.013037. 013037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higdon A, Diers AR, Oh JY, Landar A, Darley-Usmar VM. Cell signalling by reactive lipid species: new concepts and molecular mechanisms. Biochem J. 2012a;442:453–464. doi: 10.1042/BJ20111752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higdon AN, Landar A, Barnes S, Darley-Usmar VM. The electrophile responsive proteome: integrating proteomics and lipidomics with cellular function. Antioxid Redox Signal. 2012b;17:1580–1589. doi: 10.1089/ars.2012.4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Wickramasekara S, Maier CS. Ion mobility-enhanced tandem mass spectrometry for the characterization of positional isomers of peptides modified by electrophilic lipid aldehydes. 201x. in preparation.
- Jacobs AT, Marnett LJ. Heat shock factor 1 attenuates 4-Hydroxynonenal-mediated apoptosis: critical role for heat shock protein 70 induction and stabilization of Bcl-XL. J Biol Chem. 2007;282:33412–33420. doi: 10.1074/jbc.M706799200. [DOI] [PubMed] [Google Scholar]
- Jacobs AT, Marnett LJ. Systems Analysis of Protein Modification and Cellular Responses Induced by Electrophile Stress. Acc Chem Res. 2010;43:673–683. doi: 10.1021/ar900286y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo P, Perez P, Contreras R, Tiznado W, Fuentealba P. Definition of a nucleophilicity scale. J Phys Chem A. 2006;110:8181–8187. doi: 10.1021/jp057351q. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Bayir HA, Belikova NA, Kapralov O, Tyurina YY, Tyurin VA, Jiang J, Stoyanovsky DA, Wipf P, Kochanek PM, Greenberger JS, Pitt B, Shvedova AA, Borisenko G. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Radic Biol Med. 2009;46:1439–1453. doi: 10.1016/j.freeradbiomed.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanu AB, Dwivedi P, Tam M, Matz L, Hill HH., Jr. Ion mobility-mass spectrometry. J Mass Spectrom. 2008;43:1–22. doi: 10.1002/jms.1383. [DOI] [PubMed] [Google Scholar]
- Kim M-S, Pandey A. Electron transfer dissociation mass spectrometry in proteomics. Proteomics. 2012;12:530–542. doi: 10.1002/pmic.201100517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landar A, Zmijewski JW, Dickinson DA, Le Goffe C, Johnson MS, Milne GL, Zanoni G, Vidari G, Morrow JD, Darley-Usmar VM. Interaction of electrophilic lipid oxidation products with mitochondria in endothelial cells and formation of reactive oxygen species. Am J Physiol Heart Circ Physiol. 2006a;290:H1777–1787. doi: 10.1152/ajpheart.01087.2005. [DOI] [PubMed] [Google Scholar]
- Landar A, Shiva S, Levonen AL, Oh JY, Zaragoza C, Johnson MS, Darley-Usmar VM. Induction of the permeability transition and cytochrome c release by 15-deoxy-Delta12,14-prostaglandin J2 in mitochondria. Biochem J. 2006b;394:185–195. doi: 10.1042/BJ20051259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Je JH, Kim DH, Chung SW, Zou Y, Kim ND, Ae Yoo M, Suck Baik H, Yu BP, Chung HY. Induction of endothelial apoptosis by 4-hydroxyhexenal. Eur J Biochem. 2004;271:1339–1347. doi: 10.1111/j.1432-1033.2004.04042.x. [DOI] [PubMed] [Google Scholar]
- Liu Z, Minkler PE, Sayre LM. Mass spectroscopic characterization of protein modification by 4-hydroxy-2-(E)-nonenal and 4-oxo-2-(E)-nonenal. Chem Res Toxicol. 2003;16:901–911. doi: 10.1021/tx0300030. [DOI] [PubMed] [Google Scholar]
- LoPachin RM, Gavin T, Petersen DR, Barber DS. Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: nucleophilic targets and adduct formation. Chem Res Toxicol. 2009;22:1499–1508. doi: 10.1021/tx900147g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPachin RM, Gavin T, Decaprio A, Barber DS. Application of the Hard and Soft, Acids and Bases (HSAB) theory to toxicant--target interactions. Chem Res Toxicol. 2012;25:239–251. doi: 10.1021/tx2003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madian AG, Regnier FE. Profiling carbonylated proteins in human plasma. J Proteome Res. 2010;9:1330–1343. doi: 10.1021/pr900890k. [DOI] [PubMed] [Google Scholar]
- Madian AG, Diaz-Maldonado N, Gao Q, Regnier FE. Oxidative stress induced carbonylation in human plasma. J Proteomics. 2011a;74:2395–2416. doi: 10.1016/j.jprot.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madian AG, Myracle AD, Diaz-Maldonado N, Rochelle NS, Janle EM, Regnier FE. Differential carbonylation of proteins as a function of in vivo oxidative stress. J Proteome Res. 2011b;10:3959–3972. doi: 10.1021/pr200140x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier CS, Chavez J, Wang J, Wu J. Protein adducts of aldehydic lipid peroxidation products identification and characterization of protein adducts using an aldehyde/keto-reactive probe in combination with mass spectrometry. Methods Enzymol. 2010;473:305–330. doi: 10.1016/S0076-6879(10)73016-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailloux RJ, Harper M-E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic Biol Med. 2011;51:1106–1115. doi: 10.1016/j.freeradbiomed.2011.06.022. [DOI] [PubMed] [Google Scholar]
- Martyniuk CJ, Fang B, Koomen JM, Gavin T, Zhang L, Barber DS, Lopachin RM. Molecular mechanism of glyceraldehyde-3-phosphate dehydrogenase inactivation by alpha,beta-unsaturated carbonyl derivatives. Chem Res Toxicol. 2011;24:2302–2311. doi: 10.1021/tx200437y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp Gerontol. 2009;44:625–633. doi: 10.1016/j.exger.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLafferty FW, Horn DM, Breuker K, Ge Y, Lewis MA, Cerda B, Zubarev RA, Carpenter BK. Electron capture dissociation of gaseous multiply charged ions by Fourier-transform ion cyclotron resonance. J Am Soc Mass Spectrom. 2001;12:245–249. doi: 10.1016/S1044-0305(00)00223-3. [DOI] [PubMed] [Google Scholar]
- McLuckey SA, Goeringer DE. Slow Heating Methods in Tandem Mass Spectrometry. J Mass Spectrom. 1997;32:461–474. [Google Scholar]
- Meany DL, Xie H, Thompson LV, Arriaga EA, Griffin TJ. Identification of carbonylated proteins from enriched rat skeletal muscle mitochondria using affinity chromatography-stable isotope labeling and tandem mass spectrometry. Proteomics. 2007;7:1150–1163. doi: 10.1002/pmic.200600450. [DOI] [PubMed] [Google Scholar]
- Michaelevski I, Kirshenbaum N, Sharon M. T-wave ion mobility-mass spectrometry: basic experimental procedures for protein complex analysis. J Vis Exp. 2010 doi: 10.3791/1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikesh LM, Ueberheide B, Chi A, Coon JJ, Syka JEP, Shabanowitz J, Hunt DF. The utility of ETD mass spectrometry in proteomic analysis. Biochim Biophys Acta. 2006;1764:1811–1822. doi: 10.1016/j.bbapap.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne GL, Musiek ES, Morrow JD. F2-isoprostanes as markers of oxidative stress in vivo: an overview. Biomarkers. 2005;10(Suppl 1):S10–23. doi: 10.1080/13547500500216546. [DOI] [PubMed] [Google Scholar]
- Mirzaei H, Regnier F. Enrichment of carbonylated peptides using Girard P reagent and strong cation exchange chromatography. Anal Chem. 2006;78:770–778. doi: 10.1021/ac0514220. [DOI] [PubMed] [Google Scholar]
- Mirzaei H, Baena B, Barbas C, Regnier F. Identification of oxidized proteins in rat plasma using avidin chromatography and tandem mass spectrometry. Proteomics. 2008;8:1516–1527. doi: 10.1002/pmic.200700363. [DOI] [PubMed] [Google Scholar]
- Molina H, Horn DM, Tang N, Mathivanan S, Pandey A. Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc Natl Acad Sci U S A. 2007;104:2199–2204. doi: 10.1073/pnas.0611217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiek ES, Breeding RS, Milne GL, Zanoni G, Morrow JD, McLaughlin B. Cyclopentenone isoprostanes are novel bioactive products of lipid oxidation which enhance neurodegeneration. J Neurochem. 2006;97:1301–1313. doi: 10.1111/j.1471-4159.2006.03797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestmann B, Peyerimhoff SD. Calculation of the discrete component of resonance states in negative ions by variation of nuclear charges. J Phys B: At Mol Phys. 1985;18:615–626. [Google Scholar]
- Paizs B, Suhai S. Fragmentation pathways of protonated peptides. Mass Spectrom Rev. 2005;24:508–548. doi: 10.1002/mas.20024. [DOI] [PubMed] [Google Scholar]
- Palumbo AM, Smith SA, Kalcic CL, Dantus M, Stemmer PM, Reid GE. Tandem mass spectrometry strategies for phosphoproteome analysis. Mass Spectrom Rev. 2011;30:600–625. doi: 10.1002/mas.20310. [DOI] [PubMed] [Google Scholar]
- Parr RG, Szentpály LV, Liu S. Electrophilicity Index. J Am Chem Soc. 1999;121:1922–1924. [Google Scholar]
- Pearson RG. Hard and soft acids and bases – the evolution of a chemical concept. Coord Chem Rev. 1990;100:403–425. [Google Scholar]
- Perdivara I, Petrovich R, Allinquant B, Deterding LJ, Tomer KB, Przybylski M. Elucidation of O-Glycosylation Structures of the beta-Amyloid Precursor Protein by Liquid Chromatography Mass Spectrometry Using Electron Transfer Dissociation and Collision Induced Dissociation. J Proteome Res. 2008;8:631–642. doi: 10.1021/pr800758g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perluigi M, Coccia R, Butterfield DA. 4-Hydroxy-2-Nonenal, a Reactive Product of Lipid Peroxidation, and Neurodegenerative Diseases: A Toxic Combination Illuminated by Redox Proteomics Studies. Antioxid Redox Signal. 2012 doi: 10.1089/ars.2011.4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli G, Schaur RJ, Siems WG, Leonarduzzi G. 4-hydroxynonenal: a membrane lipid oxidation product of medicinal interest. Med Res Rev. 2008a;28:569–631. doi: 10.1002/med.20117. [DOI] [PubMed] [Google Scholar]
- Poli G, Biasi F, Leonarduzzi G. 4-Hydroxynonenal-protein adducts: A reliable biomarker of lipid oxidation in liver diseases. Mol Aspects Med. 2008b;29:67–71. doi: 10.1016/j.mam.2007.09.016. [DOI] [PubMed] [Google Scholar]
- Rauniyar N, Stevens SM, Prokai-Tatrai K, Prokai L. Characterization of 4-hydroxy-2-nonenal-modified peptides by liquid chromatography-tandem mass spectrometry using data-dependent acquisition: neutral loss-driven MS3 versus neutral loss-driven electron capture dissociation. Anal Chem. 2009;81:782–789. doi: 10.1021/ac802015m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauniyar N, Prokai L. Detection and identification of 4-hydroxy-2-nonenal Schiff-base adducts along with products of Michael addition using data-dependent neutral loss-driven MS3 acquisition: method evaluation through an in vitro study on cytochrome c oxidase modifications. Proteomics. 2009;9:5188–5193. doi: 10.1002/pmic.200900116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauniyar N, Prokai L. Isotope-coded dimethyl tagging for differential quantification of posttranslational protein carbonylation by 4-hydroxy-2-nonenal, an end-product of lipid peroxidation. J Mass Spectrom. 2011;46:976–985. doi: 10.1002/jms.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renedo M, Gayarre J, Garcia-Dominguez CA, Perez-Rodriguez A, Prieto A, Canada FJ, Rojas JM, Perez-Sala D. Modification and activation of Ras proteins by electrophilic prostanoids with different structure are site-selective. Biochemistry. 2007;46:6607–6616. doi: 10.1021/bi602389p. [DOI] [PubMed] [Google Scholar]
- Roe MR, Xie H, Bandhakavi S, Griffin TJ. Proteomic mapping of 4-hydroxynonenal protein modification sites by solid-phase hydrazide chemistry and mass spectrometry. Anal Chem. 2007;79:3747–3756. doi: 10.1021/ac0617971. [DOI] [PubMed] [Google Scholar]
- Sayre LM, Lin D, Yuan Q, Zhu X, Tang X. Protein adducts generated from products of lipid oxidation: focus on HNE and one. Drug Metab Rev. 2006;38:651–675. doi: 10.1080/03602530600959508. [DOI] [PubMed] [Google Scholar]
- Schopfer FJ, Cipollina C, Freeman BA. Formation and signaling actions of electrophilic lipids. Chem Rev. 2011;111:5997–6021. doi: 10.1021/cr200131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi R, Rickett T, Sun W. Acrolein-mediated injury in nervous system trauma and diseases. Mol Nutr Food Res. 2011;55:1320–1331. doi: 10.1002/mnfr.201100217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi SD, Hemling ME, Carr SA, Horn DM, Lindh I, McLafferty FW. Phosphopeptide/phosphoprotein mapping by electron capture dissociation mass spectrometry. Anal Chem. 2001;73:19–22. doi: 10.1021/ac000703z. [DOI] [PubMed] [Google Scholar]
- Slade PG, Williams MV, Brahmbhatt V, Dash A, Wishnok JS, Tannenbaum SR. Proteins modified by the lipid peroxidation aldehyde 9,12-dioxo-10(E)-dodecenoic acid in MCF7 breast cancer cells. Chem Res Toxicol. 2010;23:557–567. doi: 10.1021/tx9002808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slade PG, Williams MV, Chiang A, Iffrig E, Tannenbaum SR, Wishnok JS. A filtered database search algorithm for endogenous serum protein carbonyl modifications in a mouse model of inflammation. Mol Cell Proteomics. 2011;10:M111. doi: 10.1074/mcp.M111.007658. 007658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D, Knapman TW, Campuzano I, Malham RW, Berryman JT, Radford SE, Ashcroft AE. Deciphering drift time measurements from travelling wave ion mobility spectrometry-mass spectrometry studies. Eur J Mass Spectrom (Chichester, Eng) 2009;15:113–130. doi: 10.1255/ejms.947. [DOI] [PubMed] [Google Scholar]
- Sobczyk M, Anusiewicz I, Berdys-Kochanska J, Sawicka A, Skurski P, Simons J. Coulomb-assisted dissociative electron attachment: Application to a model peptide. J Phys Chem A. 2005;109:250–258. doi: 10.1021/jp0463114. [DOI] [PubMed] [Google Scholar]
- Spiteller G. The relation of lipid peroxidation processes with atherogenesis: a new theory on atherogenesis. Mol Nutr Food Res. 2005;49:999–1013. doi: 10.1002/mnfr.200500055. [DOI] [PubMed] [Google Scholar]
- Stamatakis K, Perez-Sala D. Prostanoids with cyclopentenone structure as tools for the characterization of electrophilic lipid-protein interactomes. Ann N Y Acad Sci. 2006;1091:548–570. doi: 10.1196/annals.1378.096. [DOI] [PubMed] [Google Scholar]
- Stensballe A, Jensen ON, Olsen JV, Haselmann KF, Zubarev RA. Electron capture dissociation of singly and multiply phosphorylated peptides. Rapid Commun Mass Spectrom. 2000;14:1793–1800. doi: 10.1002/1097-0231(20001015)14:19<1793::AID-RCM95>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Stevens SM, Jr., Rauniyar N, Prokai L. Rapid characterization of covalent modifications to rat brain mitochondrial proteins after ex vivo exposure to 4-hydroxy-2-nonenal by liquid chromatography-tandem mass spectrometry using data-dependent and neutral loss-driven MS3 acquisition. J Mass Spectrom. 2007;42:1599–1605. doi: 10.1002/jms.1349. [DOI] [PubMed] [Google Scholar]
- Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalassinos K, Grabenauer M, Slade SE, Hilton GR, Bowers MT, Scrivens JH. Characterization of Phosphorylated Peptides Using Traveling Wave-Based and Drift Cell Ion Mobility Mass Spectrometry. Anal Chem. 2008;81:248–254. doi: 10.1021/ac801916h. [DOI] [PubMed] [Google Scholar]
- Tzeng S-C, Maier CS. 58th American Society of Mass Spectrometry Annual Conference; Salt Lake City, Utah USA. 2010. [Google Scholar]
- Uchida K, Shibata T. 15-Deoxy-Δ12,14-prostaglandin J2: An Electrophilic Trigger of Cellular Responses. Chem Res Toxicol. 2007;21:138–144. doi: 10.1021/tx700177j. [DOI] [PubMed] [Google Scholar]
- Ugur Z, Coffey CM, Gronert S. Comparing the efficiencies of hydrazide labels in the study of protein carbonylation in human serum albumin. Anal Bioanal Chem. 2012;404:1399–1411. doi: 10.1007/s00216-012-6235-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhang J, Arbogast B, Maier CS. Tandem mass spectrometric characterization of thiol peptides modified by the chemoselective cationic sulfhydryl reagent (4-iodobutyl)triphenylphosphonium--effects of a cationic thiol derivatization on peptide fragmentation. J Am Soc Mass Spectrom. 2011;22:1771–1783. doi: 10.1007/s13361-011-0192-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, Cravatt BF. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells JM, McLuckey SA. Collision-induced dissociation (CID) of peptides and proteins. Methods Enzymol. 2005;402:148–185. doi: 10.1016/S0076-6879(05)02005-7. [DOI] [PubMed] [Google Scholar]
- West JD, Marnett LJ. Endogenous Reactive Intermediates as Modulators of Cell Signaling and Cell Death. Chem Res Toxicol. 2006;19:173–194. doi: 10.1021/tx050321u. [DOI] [PubMed] [Google Scholar]
- Wu J, Stevens JF, Maier CS. Mass spectrometry-based quantification of myocardial protein adducts with acrolein in an in vivo model of oxidative stress. Mol Nutr Food Res. 2011;55:1401–1410. doi: 10.1002/mnfr.201100255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocki VH, Tsaprailis G, Smith LL, Breci LA. Mobile and localized protons: a framework for understanding peptide dissociation. J Mass Spectrom. 2000;35:1399–1406. doi: 10.1002/1096-9888(200012)35:12<1399::AID-JMS86>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S, Aldini G, Ito S, Morishita N, Shibata T, Vistoli G, Carini M, Uchida K. Delta 12-prostaglandin J2 as a product and ligand of human serum albumin: formation of an unusual covalent adduct at His146. J Am Chem Soc. 2009;132:824–832. doi: 10.1021/ja908878n. [DOI] [PubMed] [Google Scholar]
- Yin H, Xu L, Porter NA. Free radical lipid peroxidation: mechanisms and analysis. Chem Rev. 2011;111:5944–5972. doi: 10.1021/cr200084z. [DOI] [PubMed] [Google Scholar]
- Young NL, DiMaggio PA, Plazas-Mayorca MD, Baliban RC, Floudas CA, Garcia BA. High Throughput Characterization of Combinatorial Histone Codes. Mol Cell Proteomics. 2009;8:2266–2284. doi: 10.1074/mcp.M900238-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, Kruger NA, Lewis MA, Carpenter BK, McLafferty FW. Electron capture dissociation for structural characterization of multiply charged protein cations. Anal Chem. 2000;72:563–573. doi: 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- Zubarev RA, Haselmann KF, Budnik B, Kjeldsen F, Jensen F. Account:Towards an understanding of the mechanism of electron-capture dissociation: A historical perspective and modern ideas. Eur J Mass Spectrom (Chichester, Eng) 2002;8:337–349. [Google Scholar]
- Zubarev RA, Kelleher NL, McLafferty FW. Electron capture dissociation of multiply charged protein cations. J Am Chem Soc. 1998;120:3265–3266. doi: 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.