

Deep sequencing of cytoplasmic RNA from the oocyte of the frog Xenopus reveals a population of stable lariat molecules derived from specific introns of several thousand protein-coding genes. The existence of stable intronic sequence (sis) RNAs in the cytoplasm of the oocyte, their transmission to the fertilized egg, and their persistence during early embryogenesis suggest that they might play a role in development, perhaps in regulating mRNA translation.
Keywords: intron, noncoding RNA, lariat RNA, oocyte, transcription, Xenopus
Abstract
We previously demonstrated that the oocyte nucleus (germinal vesicle or GV) of Xenopus tropicalis contains a population of stable RNA molecules derived from the introns of most expressed genes. Here we show that similar stable intronic sequence (sis) RNAs occur in the oocyte cytoplasm. About 9000 cytoplasmic sisRNAs have been identified, all of which are resistant to the exonuclease RNase R. About half have been confirmed as lariat molecules and the rest are presumed to be lariats, whereas nuclear sisRNAs are a mixture of lariat and linear molecules. Cytoplasmic sisRNAs are more abundant on a molar basis than nuclear sisRNAs and are derived from short introns, mostly under 1 kb in length. Both nuclear and cytoplasmic sisRNAs are transmitted intact to the egg at GV breakdown and persist until at least the blastula stage of embryogenesis, when zygotic transcription begins. We compared cytoplasmic sisRNAs derived from orthologous genes of X. tropicalis and X. laevis, and found that the specific introns from which sisRNAs are derived are not conserved. The existence of sisRNAs in the cytoplasm of the oocyte, their transmission to the fertilized egg, and their persistence during early embryogenesis suggest that they might play a regulatory role in mRNA translation.
INTRODUCTION
Most protein-coding genes in higher eukaryotes contain noncoding introns that interrupt the coding regions. Intronic sequences are excised from the primary transcript by the spliceosome (Hoskins and Moore 2012) and are released as lariats, which in most cases are degraded rapidly. The degradation pathway involves the RNA lariat debranching enzyme, Dbr1, which hydrolyzes the 2′–5′ covalent bond generated during splicing (Ruskin and Green 1985; Chapman and Boeke 1991; Ooi et al. 2001). Once linearized, intronic fragments are probably degraded by exonucleases in the nucleus (Hilleren and Parker 2003). Exceptions to this pathway occur when the intron contains noncoding RNAs, such as small nucleolar (sno) RNAs or micro RNAs (Rearick et al. 2011; Curtis et al. 2012; Yin et al. 2012).
Recently we carried out a high-throughput sequencing study of RNA from the giant oocyte nucleus or germinal vesicle (GV) of the frog Xenopus tropicalis (Gardner et al. 2012). We found many intronic sequences in the GV that were not detectable by sequencing whole-cell RNA. We showed that these intronic sequences are derived from the same strand as the mRNA and probably originate by splicing from pre-mRNA molecules. Because these sequences are stable for at least 48 h, we named them stable intronic sequence RNA or sisRNA.
In this study we show that some sisRNAs also occur in the cytoplasm of the X. tropicalis oocyte. We are sure of the cytoplasmic localization, because the GV can be removed intact from the oocyte before the cytoplasmic RNA is extracted and sequenced. Cytoplasmic sisRNAs are more abundant on a molar basis than nuclear sisRNAs. We show that cytoplasmic sisRNAs are insensitive to the exonuclease RNase R and that half, if not all, exist as lariat molecules. Cytoplasmic sisRNAs persist after fertilization of the egg until at least the mid-blastula transition, when major transcription first begins in the early embryo. Because of their cellular localization and persistence during early embryogenesis, we suggest that cytoplasmic sisRNAs could play a role in regulating translation of mRNAs.
RESULTS
sisRNA in the cytoplasm
Identification of rare cytoplasmic RNAs depends critically on the ability to isolate a sample of cytoplasm completely free of nuclear contamination. The GV can be removed from an oocyte with a pair of jewelers’ forceps in a matter of seconds (Gall and Wu 2010). Because one oocyte contains ∼1 µg of total RNA, only five to 10 enucleated oocytes are needed for RNA extraction and library preparation. Furthermore, <1% of total cellular RNA resides in the GV (Gardner et al. 2012). Thus, collection of an uncontaminated sample of cytoplasm is a trivial operation that requires only a few minutes. Experimentally, we cannot detect highly abundant nuclear RNAs in our cytoplasmic samples, although they are readily detectable in RNA from whole oocytes (Supplemental Figs. S1, S2). Thus, on both theoretical and experimental grounds, we are confident that our cytoplasmic RNA samples are not contaminated with nuclear RNA.
Purified cytoplasmic, nuclear, and whole oocyte RNA samples were depleted for rRNA and subjected to high-throughput sequencing (Fig. 1A). For each sample 60–100 million reads (100 bp) were obtained, of which ∼90% mapped to version 4.1 of the X. tropicalis genome (Hellsten et al. 2010). As in earlier experiments (Gardner et al. 2012), cytoplasmic reads mapped primarily to exons, whereas nuclear reads mapped primarily to intronic regions of protein-coding genes and to annotated nuclear RNAs (e.g., snRNAs, snoRNAs) (Supplemental Fig. S1). Roughly 30% of reads in both fractions mapped to unannotated regions of the genome and were not further analyzed.
FIGURE 1.

Characterization of cytoplasmic sisRNAs. (A) The oocyte nucleus or germinal vesicle (GV) can be removed from a defolliculated oocyte within seconds to provide a sample of cytoplasm uncontaminated with nuclear RNA. (B) IGV browser view of a typical gene bearing a cytoplasmic sisRNA. Cytoplasmic intronic reads (blue) occur in one intron of the eif4a1 gene (yellow exonic reads). The lower track (track height 100 reads) shows that the intronic reads do not cross the intron–exon borders. The yellow and blue arrows represent exonic and intronic primers used for RT–PCR. (C) RT–PCR results showing that cytoplasmic sisRNAs are derived from the sense strand and do not cross the intron–exon borders. (ivt) In vitro transcribed RNA, used as a control template. (D) Comparison of cytoplasmic, nuclear (GV), and whole oocyte RNAs from the rbm28 gene. Cytoplasmic and whole oocyte RNAs are essentially identical (arrow points to cytoplasmic sisRNA, blue reads). Nuclear sisRNAs (red reads in GV track) are not detectable in the whole oocyte sample. (E) Distribution of sisRNAs relative to intron length. The majority of cytoplasmic sisRNAs (blue) derive from shorter introns, whereas nuclear-specific sisRNAs (red) come from a wide range of longer introns.
Of special interest were ∼3% of cytoplasmic reads that mapped to introns. Two independent samples were sequenced with essentially identical results (Supplemental Fig. S3). Cytoplasmic intronic reads nearly always mapped as a single peak within a relatively short intron, with no reads crossing the exon–intron boundaries (Fig. 1B). These features suggested that the reads represented independent molecules, not retained introns, which would have reads that cross the exon–intron boundaries.
To confirm this hypothesis, we carried out RT–PCR experiments with exonic and intronic primers to test four introns (Fig. 1C). We first tested the strand specificity of the intronic sequences by using either the forward or reverse primer separately in the RT step. In all four cases only the reverse primer gave a product, showing that the intronic sequences are derived from the same strand as the adjacent exonic sequences. This observation was later confirmed for all cytoplasmic intronic sequences by sequencing the first strand cDNA of the library according to the TruSeq Stranded method from Illumina (Supplemental Fig. S4). We also tested for sequences that spanned the exon–intron boundary. RT–PCR was carried out with a combination of one exonic and one intronic primer. No products were detected when cytoplasmic RNA was used as the substrate. The same primers were able to amplify from in vitro-transcribed RNA that spanned the entire exon–intron–exon region (Fig. 1C). These experiments demonstrate that the cytoplasmic intronic sequences are not attached to the flanking exonic sequences and hence belong to separate molecules. They are, however, transcribed from the same strand as the exonic sequences and could be derived originally from a splicing event.
To test the stability of the cytoplasmic intronic sequences, we placed oocytes in actinomycin D for 18 h to inhibit pol II transcription. The efficiency of inhibition was verified by inspecting the loss of transcription loops and pol II staining on the lampbrush chromosomes. As in previous experiments with actinomycin (Callan 1986), inhibition of transcription was rapid and complete within 1–2 h. Cytoplasmic RNA was purified from treated and untreated oocytes and high-throughput sequencing was performed after rRNA depletion. We did not see any quantitative differences between treated and untreated oocytes samples, and the ratio between intronic transcripts and the mRNAs with which they are associated did not change (Supplemental Fig. S5). It is well known that cytoplasmic mRNA in the amphibian oocyte is unusually stable (Davidson 1986). We conclude that the cytoplasmic intronic sequences are also stable, at least during the 18-h period of actinomycin treatment.
In summary, intronic sequences in the cytoplasm are derived from the same strand as the mRNA with which they are associated, are independent molecules, and are stable for many hours. In these respects they are similar to sisRNAs from the nucleus and henceforth will be designated cytoplasmic sisRNAs.
Cytoplasmic sisRNAs are more abundant than nuclear sisRNAs and come from shorter introns
Whereas nuclear sisRNAs are derived from roughly half of all introns, cytoplasmic sisRNAs map to only ∼5% of introns. Thus, cytoplasmic sisRNAs represent a subset of all sisRNAs. Although derived from a limited number of introns, cytoplasmic sisRNAs are much more abundant on a molar basis than nuclear sisRNAs. This can be deduced by comparing sequence data from nuclear, cytoplasmic, and whole oocyte RNA. Cytoplasmic RNA and whole oocyte RNA are essentially identical with respect to intronic reads. In other words, cytoplasmic sisRNA reads are equally abundant in the two samples but nuclear sisRNA reads are not detectable in the whole oocyte RNA sample (Fig. 1D). On a whole oocyte basis, therefore, cytoplasmic sisRNAs are less abundant than mRNA sequences from the genes in which they are found, but they are more abundant than nuclear sisRNAs derived from the same genes.
These relationships can be assessed semiquantitatively by comparing the abundance of snRNAs and snoRNAs in the three samples (Supplemental Fig. S2). snRNA and snoRNA sequences are not detectable in the cytoplasmic fraction, attesting to their strict nuclear localization. They are by far the most abundant sequences in the nuclear sample, and they are readily detectable in RNA derived from the whole oocyte (nucleus plus cytoplasm). Thus, one can compare the abundance of a given snoRNA to nuclear sisRNAs (in the nucleus sample) and to cytoplasmic sisRNAs (in the whole oocyte sample). Such a calculation shows that cytoplasmic sisRNAs are about 10× more abundant than nuclear sisRNAs from the same gene.
Interestingly, introns that have cytoplasmic sisRNAs tend to be short, between 200 and 500 nucleotides (nt) in length. In contrast, the vast majority of nuclear sisRNAs are derived from introns that are 500–5000 nt in length (Fig. 1E). We have not detected any other characteristics at the sequence level that differ between nuclear and cytoplasmic sisRNAs.
Cytoplasmic sisRNAs are resistant to RNase R
As a first attempt to characterize cytoplasmic sisRNA molecules in more detail, we carried out 5′ and 3′ RACE experiments on selected examples. These experiments invariably failed, despite success with appropriate controls performed at the same time. Suspecting that the ends of the cytoplasmic sisRNA molecules might be protected by some modification, we treated cytoplasmic RNA with RNase R. This enzyme is a processive exonuclease that degrades linear single-stranded RNAs from the 3′ end regardless of internal secondary structure (Cheng and Deutscher 2002, 2005). It is not able to degrade circular or lariat RNA and it is most efficient when the terminal ribose is not modified.
We treated cytoplasmic RNA with RNase R or water and tested for the degradation of exonic and intronic sequences by RT–PCR. After RNase R treatment, no exonic product was amplified by RT–PCR for three different genes, whereas sisRNAs from the introns of these genes were still detectable (Fig. 2A). In vitro-transcribed sisRNAs were used as controls to show that sequence alone did not prevent the enzyme from working. Because sisRNAs are derived from introns, we strongly suspected that their resistance to RNase R was due to their lariat form; that is, failure to have been debranched after splicing.
FIGURE 2.

Cytoplasmic sisRNAs are resistant to RNase R. (A) Cytoplasmic RNA was treated with RNase R or water and then used for RT–PCR amplification of exonic and intronic sequences from three genes. Intronic, but not exonic, sequences were resistant to RNase R and could still be amplified. The same intronic sequences (ivt RNA), when derived from in vitro-transcribed RNA, were digested by RNase R and did not amplify. (B) Cytoplasmic RNA was digested with RNase R and subjected to deep sequencing. The upper track (control) shows that cytoplasmic sisRNA sequences (blue) are rare relative to the coding sequences (yellow) of the nasp gene. The lower track (RNase R treated) shows that the sisRNA sequences are resistant to the enzyme, whereas the mRNA is almost completely digested. (C) A relatively rare case in which an mRNA is resistant to RNase R digestion (lpcat4). Presumably, digestion began at the 3′ end of the mRNA but could not proceed past the middle of the last exon. (D) A circular molecule derived from two exons without the intervening intron in the gene lsm1. Such molecules could arise by “backsplicing,” as recently described for human fibroblasts (Jeck et al. 2013).
To determine whether resistance to RNase R is a characteristic of all cytoplasmic sisRNAs, we treated cytoplasmic RNA samples with RNase R or with water as a control, and carried out high-throughput sequencing. Approximately 70 million reads were obtained for each sample. Based on the RT–PCR experiment, we expected that all mRNA molecules would be sensitive to RNase R and all sisRNAs would be stable. The situation was somewhat more complex. The total fraction of raw reads that mapped to exons of protein-coding regions dropped from 55% in the control to 20% in the treated sample. Exonic sequences from most mRNAs were either completely undetectable or severely reduced in the sample treated with RNase R, implying that they had been digested efficiently by the enzyme (Fig. 2B). There were interesting exceptions. For example, about 360 mRNAs were enriched in the RNase R sample relative to the control. In most such cases there was a sudden drop in read number near the middle of the last exon at the 3′ end of the molecule (Fig. 2C). A possible explanation for such cases is the presence of a nucleotide with a modified sugar at that position. RNase R, being an exonuclease, could have digested the RNA from the 3′ end until it encountered this nucleotide. In a few cases we saw specific enrichment for two to four adjacent exons within a gene (Fig. 2D). Enrichment for numerous exonic sequences after RNase R treatment was recently reported for RNA from cultured human cells and explained as circularization of the molecules by “backsplicing” (Jeck et al. 2013). We did not explore this phenomenon further in our samples.
Conversely, the total fraction of raw reads mapping to introns increased dramatically from 5% to over 40%. Closer analysis of individual genes showed that all sisRNAs seen in the control sample (about 3000) were still represented in the RNase R sample (Fig. 2B; Supplemental Fig. S6). The patterns of specific sisRNAs on the genome browser did not change after enzyme treatment, suggesting that the molecules were not affected by RNase R. Moreover, because of the greater read-depth of intronic sequences in the treated sample, about 5000 additional sisRNAs were detected that had not been seen in the control.
Cytoplasmic sisRNAs exist as lariats without tails
Definitive evidence that cytoplasmic sisRNAs are lariats without tails comes from several sources. Examination of the average coverage of the most abundant cytoplasmic sisRNAs (top 200) shows a distinct bias toward the 5′ end of the intron (Fig. 3A). Reads map very close to the 5′ splice site but never close to the 3′ splice site, there being a gap of ∼30 nt between the last sisRNA read and the 3′ splice site. Because the splicing branch point is typically located at this distance from the 3′ splice site, it seemed likely that cytoplasmic sisRNAs are spliced lariats without the 3′ tail. Because of their resistance to RNase R, they could be either circles that have not been debranched or linearized lariats with some feature that inhibits RNase R digestion.
FIGURE 3.

Evidence that cytoplasmic sisRNAs are circles (lariats without tails). (A) Plot of average read number per nucleotide for 200 sisRNAs relative to nucleotide position within the intron. Reads abut the 5′ end of the intron, but are absent from a region of ∼30 nt next to the 3′ end. Consensus nucleotides are shown for positions near the ends of the introns. (B) Inverted reads contain sequences that cross the splicing branchpoint and can be mapped when appropriately split (Taggart et al. 2012). (C) Some inverted reads contain sequence errors or nucleotides inserted at the branchpoint (red nucleotides), presumably by the reverse transcriptase used for library production. (D) Cytoplasmic sisRNAs yield an RT–PCR product when amplified with outward-facing primers. These same primers amplify in vitro-transcribed RNA only when the branchpoint is in the interior of the molecule. See text for details of cloning and sequencing of the RT–PCR products.
To distinguish these alternatives we analyzed unmapped reads from the initial high-throughput sequencing data set. Inverted reads were split and mapped according to the method developed by Taggart et al. (2012). Many inverted reads mapped to intronic regions with a characteristic profile: One end mapped precisely to the 5′ splice site, whereas the other end mapped to a region ∼30 nt upstream of the 3′ splice site. Moreover, a variety of “mutations,” including short insertions or substitutions, were found at the junction site of the inverted reads. These mutations are shown in red in Figure 3B and the sequences of a few specific examples are listed in Figure 3C. It is well known that reverse transcriptase is unreliable when it reaches the 2′–5′ linkage at the branch point of a lariat. In most cases it stops at the branch point or it goes through but adds nontemplated nucleotides (Gao et al. 2008). We presume that the errors shown in Figure 3C were introduced in the reverse transcription step during preparation of the DNA library for high-throughput RNA sequencing. The evidence from inverted reads with mismatches at their junction suggests that cytoplasmic sisRNAs are derived by splicing, but for some reason are not debranched. Instead, they are stabilized in the form of lariats without tails.
High-throughput sequencing data sets contain many PCR artifacts, such as products derived from template switching. Such products can arise from amplification of a heterogeneous template pool with universal primers (Wang and Wang 1996; Acinas et al. 2005). To demonstrate that the inverted reads found in our amplified libraries are not sequencing artifacts but exist in the original sisRNA molecules, we carried out RT–PCR experiments on sisRNAs derived from three genes. First, we used reverse transcriptase to make cDNAs from a cytoplasmic RNA sample using random hexamers. The cDNAs were amplified by PCR using outward-facing primers (Fig. 3D, cytoplasmic RNA) and were then cloned and sequenced. The sequences mapped to the region between the 5′ end of the intron and the 3′ splice site, and there were mutations at the presumed junction site. In vitro-transcribed linear sisRNA was not amplified by this method (Fig. 3D, ivt RNA 1), consistent with the primers being specific for inverted sequences. Finally, cDNAs were generated from an inverted in vitro-transcribed RNA (Fig. 3D, ivt RNA 2). The sequenced products were similar to those produced from cytoplasmic RNA, except that no mutations were seen at the junction site. We conclude that the inverted reads seen in the high-throughput experiments were not PCR artifacts, but were generated by reverse transcription of lariats in the cytoplasmic RNA sample.
Cytoplasmic sisRNAs are protected from the debranching pathway
The existence of stable RNA lariats in the cytoplasm suggests that cytoplasmic sisRNAs avoid the lariat debranching pathway before their export from the nucleus. We first checked that the RNA lariat debranching enzyme Dbr1 is present in the mature Xenopus oocyte. Western blots of total GV and cytoplasmic extracts show that Dbr1 is present in the GV, as expected, but is not detected in the cytoplasm (Supplemental Fig. S7).
To test whether cytoplasmic sisRNAs are degradable by debranching activity in the nucleus, we added total cytoplasmic RNA from X. tropicalis to cytoplasmic and nuclear extracts from X. laevis oocytes (Fig. 4A). The extracts were made from a different species to avoid the complication of endogenous sequences in the extracts. After cytoplasmic RNA from X. tropicalis had been incubated in the X. laevis nuclear extract for 2 h at 25°, X. tropicalis mRNAs were still detectable, but sisRNAs were not. In contrast, both sisRNAs and mRNAs could be demonstrated after a similar incubation in the cytoplasmic extract (Fig. 4B). These results show that purified cytoplasmic sisRNAs are not inherently resistant to debranching activity. Presumably, they are protected in vivo from Dbr1 before they are exported from the nucleus.
FIGURE 4.
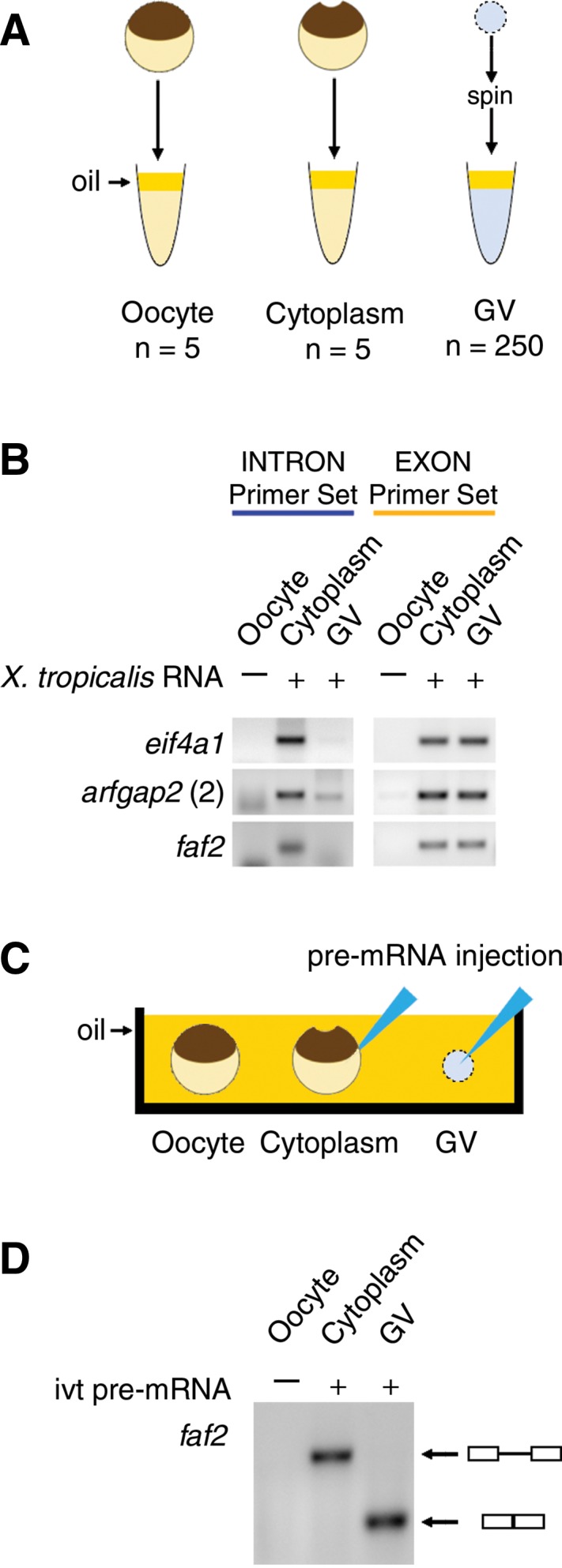
(A,B) The stability of X. tropicalis cytoplasmic RNA (1 µg) was tested by RT–PCR after incubation in X. laevis cytoplasmic or nuclear extracts. (A) Extracts of X. laevis whole oocytes, oocyte cytoplasm, and GVs were made under oil. (B) Cytoplasmic sisRNAs from X. tropicalis were degraded by a GV extract from X. laevis but not by a cytoplasmic extract (intron primer set, blue). Exonic sequences from X. tropicalis were stable for 2 h in both GV and cytoplasmic extracts (exon primer set, yellow). (C,D) Splicing activity was tested by injecting an in vitro-transcribed X. tropicalis pre-mRNA construct into the cytoplasm or GV of an X. laevis oocyte. (C) Injection of the construct was carried out under mineral oil. (D) After incubation for 2 h, an RT–PCR reaction was carried out on the cytoplasm and GV using primers from the ends of the construct. The products were run on an agarose gel and stained. Splicing occurred only in the GV, as shown by the expected smaller size of the RT–PCR product.
An intron that gives rise to an abundant cytoplasmic sisRNA can be spliced in the nucleus
Although it is well established that both major and minor splicing occur in the Xenopus oocyte nucleus (Moon et al. 2006; Mereau et al. 2007; Friend et al. 2008), it is possible that cytoplasmic sisRNAs arise by an alternative cytoplasmic pathway. To test this possibility we examined a particularly abundant cytoplasmic sisRNA derived from intron 2 of the faf2 gene of X. tropicalis. We made a splicing construct consisting of this intron along with its flanking exons. The capped and polyadenylated RNA construct was injected into an X. laevis GV or into an enucleated oocyte, both under mineral oil (Fig. 4C). Two hours later RT–PCR demonstrated that the construct had been spliced in the GV but not in the cytoplasm (Fig. 4D). This experiment demonstrates that an intron that normally gives rise to an abundant cytoplasmic sisRNA can be processed by the nuclear splicing machinery.
Some nuclear sisRNAs are also stored as lariats
In our earlier study of GV-specific RNA, we demonstrated the presence of sisRNAs derived from multiple introns of about 6700 protein-coding genes. We did not, however, assess the molecular form of these nuclear sisRNAs. To do so, we analyzed a new sample of GV RNA for branch-point reads (Taggart et al. 2012), as we did for cytoplasmic sisRNAs. We found inverted reads with mismatches at their junction that mapped to the 5′ splice site and upstream of the 3′ splice site in ∼20% of the nuclear sisRNAs (Supplemental Fig. S8A). We interpret these reads as evidence that at least some nuclear sisRNAs exist as lariats despite the presence of active Dbr1 in the GV.
To determine the stability of these lariats we reanalyzed data from an earlier actinomycin D experiment (Gardner et al. 2012). In that experiment we inhibited transcription for 15 h by incubating oocytes in actinomycin D at 20 µg/mL. We then isolated several hundred GVs, extracted the RNA, and performed high-throughput sequencing. Although that sample was not depleted for rRNA, the read-depth was sufficient to detect some inverted intronic reads. A control sample of GV RNA prepared at the same time from untreated oocytes was analyzed with similar results (Supplemental Fig. S8B). The unusual stability of these lariats implies that at least some nuclear sisRNAs are protected from Dbr1 activity and are not simply transient splicing intermediates. We have evidence from more recent RNase R experiments that the nuclear sisRNA population consists of both lariats and debranched linear molecules.
Accumulation of cytoplasmic sisRNAs during oogenesis
To assess the accumulation of cytoplasmic sisRNAs during oogenesis, we carried out high-throughput sequencing of RNA from three samples of younger oocytes selected on the basis of diameter: under 250 µm, 300–350 µm, and 350–400 µm. Because it would be technically challenging to prepare separate cytoplasm and GV fractions from these oocytes, we compared total RNA with total RNA from mature oocytes (800 µm diameter). Only the most abundant cytoplasmic sisRNAs are detectable in such samples, the majority of reads being derived from mRNA molecules. An example is shown in Figure 5A. The top track shows cytoplasmic RNA from mature oocytes, demonstrating that the sisRNA under consideration is, in fact, derived from the cytoplasm. RNA from “whole” oocytes (bottom row, 800 µm) appears essentially identical, showing that there is little or no contribution to the pattern from the GV. Total RNA from small oocytes (under 250 µm, 300–350 µm, and 350–400 µm) shows very few intronic reads, with more reads in the larger oocytes than in the smaller. A similar pattern of increasing number of reads with oocyte size was apparent when we examined other cytoplasmic sisRNAs individually (Supplemental Fig. S9).
FIGURE 5.

During oogenesis cytoplasmic sisRNAs but not snoRNAs increase in abundance relative to mRNAs. (A) Cytoplasmic sisRNAs (blue) occur in three introns of the tnpo2 gene (top track, cytoplasm from mature oocyte). During oogenesis these sisRNAs increase in abundance relative to exonic sequences (four lower tracks, total RNA from oocytes of increasing size). (B) During oogenesis two nuclear snoRNAs (green) maintain a roughly constant ratio relative to cytoplasmic exonic sequences (yellow). The arrow points to a snoRNA whose size and abundance is similar to that of the sisRNAs detected in A. The exclusively nuclear location of the snoRNA sequences is shown by their absence from cytoplasm of mature oocytes (top track).
It was not possible to gather global information on all cytoplasmic sisRNAs in these total oocyte samples. Most reads mapping to annotated introns (∼2% of the total mapped reads) were, in fact, derived from alternative splicing events. On the other hand, certain highly abundant nuclear RNAs, including snoRNAs derived from introns, were detectable in total RNA from oocytes of all sizes. Unlike cytoplasmic sisRNAs, these sequences do not change much in abundance relative to mRNAs derived from the same gene (Fig. 5B).
Cytoplasmic sisRNAs persist during early embryogenesis
In our earlier study of sisRNAs we showed by RT–PCR that three abundant sisRNAs persist in the fertilized egg and embryo until at least the blastula stage (Gardner et al. 2012). Unknown to us at that time, all three happen to be cytoplasmic sisRNAs. To gain a more complete picture of sisRNAs during early embryogenesis, we prepared total cell RNA from three stages: eggs after GV breakdown but before fertilization (white spot stage), 4-cell embryos, and early blastulae. Before high-throughput sequencing, half of each sample was digested with RNase R and half was treated with water as a control.
In the controls from the three stages we could detect all cytoplasmic sisRNAs that were evident in a sample of pure cytoplasmic RNA from mature oocytes (in Fig. 6A, cf. blue reads in “cytoplasm” with blue reads in “egg,” “4-cell,” and “early blastula”). Moreover, the mRNA to sisRNA ratio does not change across these developmental time points. That is, GV breakdown, fertilization, and early development of the embryo have little or no effect on the stability of cytoplasmic sisRNAs. After RNase R treatment of these samples, most mRNA sequences are degraded. Thus, cytoplasmic sisRNAs are dramatically increased relative to exonic sequences (blue intronic vs. orange exonic sequences in Fig. 6B for all stages). One now sees nuclear sisRNAs as well (red sequences) in the “egg,” “4-cell,” and “early blastula” stages. These results suggest that all lariat intronic sequences, both cytoplasmic and nuclear, are stable after GV breakdown until at least the blastula stage. Whether these sisRNAs are nuclear, cytoplasmic, or both during early embryogenesis is not known, since the data necessarily come from unfractionated samples.
FIGURE 6.

Persistence of sisRNAs in the early embryo. (A) A prominent cytoplasmic sisRNA in the ckap5 gene (top track, blue) persists in the egg, 4-cell, and early blastula stages. (B) The top two tracks show nuclear sisRNAs (red) in the GV and cytoplasmic sisRNAs (blue) in the cytoplasm of mature oocytes. The third track shows that cytoplasmic sisRNAs (blue) are resistant to RNase R relative to the mRNA (yellow). Note that nuclear sisRNAs are not seen in this cytoplasmic sample, attesting to its purity. The bottom three tracks show egg, 4-cell, and early blastula RNA treated with RNase R. These samples show persistence not only of cytoplasmic sisRNAs (blue) but also of nuclear sisRNAs (red) derived from GV breakdown at the time of fertilization.
Comparison of sisRNAs in X. tropicalis and X. laevis
To assess the evolutionary conservation of cytoplasmic sisRNAs we performed a deep-sequence analysis of cytoplasmic and nuclear RNA from oocytes of X. laevis. Oocytes from the two Xenopus species are similar in most respects except for size, ∼1.2-mm diameter at maturity in X. laevis compared with 0.8 mm in X. tropicalis. An important genetic difference between the two species is that X. tropicalis has a haploid chromosome number of 10, whereas X. laevis has 18. It has been known for some time that X. tropicalis is a true diploid species, but X. laevis is an ancient allotetraploid (Tymowska 1991). A recent chromosome painting analysis provides confirmation of this hypothesis (Krylov et al. 2010). Now that the X. laevis genome has been sequenced and annotated (xenbase.org), it is possible to find numerous cases of duplicate genes in this species.
We examined a number of such cases, comparing the single gene in X. tropicalis with the two orthologous copies in X. laevis. In general all three copies of a particular gene have similar intron/exon structure. Thus, it is easy to identify the “same” intron in all three genes, despite differences in their lengths and particularly in their sequences. We find cytoplasmic sisRNAs from X. laevis genes with characteristics similar to those in X. tropicalis. Specifically, the X. laevis cytoplasmic sisRNAs are found in relatively few introns compared with nuclear sisRNAs, and they are limited to shorter introns. They are resistant to RNase R and they have inverted reads with mismatches at the junction, suggesting that they are lariats. However, the probability that orthologous genes in the two species will both contain a cytoplasmic sisRNA is no greater than chance, and even when they do, different introns are involved. The same is true for the two copies of a given gene in X. laevis: When both give rise to cytoplasmic sisRNAs, different introns are involved. An example is shown in Supplemental Figure S10 for the gene cugbp1 (or celf1). In X. tropicalis the most prominent cytoplasmic sisRNA is derived from intron 12, whereas in X. laevis they come from intron 7 in cugbp1-A and intron 5 in cugbp1-B.
DISCUSSION
A population of lariat sisRNAs in the cytoplasm
The experiments reported here establish the existence of a population of intronic sequences in the oocyte cytoplasm of X. tropicalis and X. laevis. Because of their similarity to the stable intronic sequence (sis) RNAs that we previously reported from the oocyte nucleus (Gardner et al. 2012), we refer to these sequences as cytoplasmic sisRNAs. By treating samples with RNase R before sequencing, we identified about 9000 cytoplasmic sisRNAs derived from 4500 different genes. They come from specific short introns (Fig. 1E), typically only one or two per gene. Further analysis showed that many, if not all, of these molecules are lariats, which probably arise from canonical splicing events (Fig. 3). Cytoplasmic sisRNAs are stable for at least 18 h after actinomycin treatment. Furthermore, they are transmitted to the embryo at the time of GV breakdown and fertilization, and persist intact until at least the mid-blastula stage. Thus, cytoplasmic sisRNAs constitute a large class of stable lariats derived from a subset of short introns.
By comparing the abundance of molecules in nuclear, cytoplasmic, and whole oocyte RNA samples, it is possible to make rough quantitative estimates of the relative molar concentrations of mRNAs, cytoplasmic sisRNAs, and nuclear sisRNAs (Supplemental Fig. S2). On average, mRNAs are roughly 10× more abundant than cytoplasmic sisRNAs, which in turn are 10× more abundant than nuclear sisRNAs derived from the same gene.
Cytoplasmic lariats derived from introns have been described earlier (Clement et al. 1999, 2001). In the first example, an intron from the mouse T-cell receptor-β gene was shown to exist as a lariat, primarily in the nucleus but partly in the cytoplasm. In the second case, three introns from the Pem homeobox gene partitioned preferentially to the cytoplasmic fraction after subcellular fractionation. One intron was further characterized and shown to be a lariat. All three were relatively stable, with half-lives ranging from 9 to 29 min. An extensively studied example of a cytoplasmic intron is the latency-associated transcript (LAT) derived from an intron in the herpes simplex virus (for review, see Kent et al. 2003). The cytoplasmic LAT exists in a nonlinear form, presumably a lariat (Wu et al. 1996; Rodahl and Haarr 1997). A similar stable intron is derived from the murine cytomegalovirus (Kulesza and Shenk 2006). Finally, in a recent study it was shown that lariat introns accumulate in the cytoplasm of dbr1Δ yeast cells (Armakola et al. 2012). Thus, there is precedence that lariats derived from introns can be found in the cytoplasm, where they are relatively stable.
Stability of RNA classes in the oocyte
Because the ratio of nuclear to cytoplasmic volume drops dramatically during oogenesis (Hausen and Riebesell 1991), quantitative estimates of RNA classes in oocytes of different sizes could be influenced by the relative amounts of nuclear and cytoplasmic RNA. Although nascent transcripts are too low in abundance to be detected even in the smallest oocytes (with the highest nuclear to cytoplasmic volume ratio), certain highly abundant nuclear RNAs, including snoRNAs, are detectable in total RNA from oocytes of all sizes. Figure 5B shows that the ratio of snoRNA reads (from the nucleus) to mRNA reads (from the cytoplasm) remains more or less constant during oocyte development. An mRNA and its associated snoRNA are presumably transcribed in the same event, and therefore are initially equimolar in amount. The simplest way in which this ratio might remain constant is if their relative stabilities remain constant during oogenesis. Earlier estimates of mRNA stability in X. laevis oocytes suggested half-lives of >35 d (Davidson 1986). These considerations suggest that snoRNAs are similarly stable.
On the other hand, nuclear sisRNAs in the mature oocyte are present at roughly 1/100 the molar concentration of exonic sequences from the cognate gene (Gardner et al. 2012). Cytoplasmic sisRNAs are intermediate in abundance, being more abundant in the mature oocyte than nuclear sisRNAs from the same gene, but less abundant than exonic sequences. We do not know how these ratios are achieved, because they represent an equilibrium between synthesis and degradation, neither of which has been measured. In principle, both nuclear and cytoplasmic sisRNAs populations could arise because the intron degradation machinery in the GV is inefficient. Alternatively, sisRNAs could represent a subset of sequences that specifically escape the degradation machinery, either by sequestration or other means of protection. In the case of cytoplasmic sisRNAs, physical separation from the nuclear debranching enzyme is a likely component of their higher stability.
Origin and biological role of cytoplasmic sisRNAs
At present we have little or no direct information about the origin of cytoplasmic sisRNA molecules. Theoretically they could arise by cytoplasmic splicing, as has been reported for blood platelets (Denis et al. 2005; Schwertz et al. 2006; Rondina et al. 2011) and for neuronal dendrites (Khaladkar et al. 2013; Buckley et al. 2014). Lariats generated by such a mechanism might be partially processed by an exonuclease, but the resulting lariat molecules would be stable, because the cytoplasm lacks the debranching enzyme Dbr1. We consider this explanation unlikely for several reasons. First, cytoplasmic splicing would involve the excision of retained introns, whereas we do not see extensive evidence in our cytoplasmic samples for unspliced mRNAs or for retention of specific introns that might give rise to cytoplasmic sisRNAs. Second, there is well-established evidence for nuclear localization of both major and minor splicing in the Xenopus oocyte (Moon et al. 2006; Mereau et al. 2007; Friend et al. 2008). Third, cytoplasmic sisRNAs use major splice junctions, suggesting that they are processed by conventional nuclear spliceosomes (Fig. 3A). Finally, we confirmed that a pre-mRNA construct for an abundant cytoplasmic sisRNA could be spliced in the nucleus but not in the cytoplasm (Fig. 4C,D).
We think it more likely that cytoplasmic sisRNAs arise in the GV from incompletely processed introns (Fig. 7), even though such a mechanism presents its own theoretical problems. For instance, these spliced introns must avoid the debranching enzyme Dbr1, which we have shown to be present and active in the GV. The fact that many nuclear-specific sisRNAs exist as lariats, whereas others are linear molecules, is further evidence that nuclear Dbr1 activity is somehow limited to specific intronic transcripts. These sisRNAs could be physically sequestered, so that they do not come in contact with the debranching activity, or they could be in a macromolecular complex that protects them. In situ hybridization can be used to localize introns in the GV and in the cytoplasm, and could give evidence for sequestration within specific nuclear or cytoplasmic bodies.
FIGURE 7.

Diagram of a Xenopus oocyte showing proposed origin of nuclear and cytoplasmic sisRNAs.
Cytoplasmic and nuclear sisRNAs join an ever-growing list of noncoding RNAs whose specific functions remain to be discovered (Ulitsky and Bartel 2013; van Heesch et al. 2014). A recent study identified numerous circular intronic sequences in cultured human cell lines (HeLa and H9) (Zhang et al. 2013). The investigators show by in situ hybridization that at least one of these circular RNAs, derived from an intron in ankrd52, associates with its site of transcription in the nucleus. Furthermore, it interacts with the pol II machinery and acts as a positive regulator of transcription. Many or most of these circular RNAs in human cells reside in the nucleus and thus may correspond to the sisRNAs we found in the Xenopus oocyte nucleus (Gardner et al. 2012). However, because of their cytoplasmic localization, the sisRNAs described here cannot have a direct role at their site of transcription. Without further evidence, only the most general arguments can be made about their function(s). Two features stand out. First, they are derived from a relatively small number of specific introns, implying that they are not random sequences derived from an inefficient degradation machinery. Second and more importantly, they are found in the cytoplasm weeks or even months before nuclear envelope breakdown at the time of oocyte maturation. It is well known that protein synthesis in the oocyte and fertilized egg is highly regulated (Richter 2007; Richter and Lasko 2011) and cytoplasmic sisRNAs could be one of the players in this regulation. Future experiments will focus on the precise cytological localization of sisRNAs and on the proteins with which they are associated.
MATERIALS AND METHODS
Animals, embryos, and oocytes
Female X. tropicalis of various ages were purchased from Xenopus 1. Animals were anesthetized with 0.15% tricaine methane sulfonate, and one or both ovaries were removed surgically. Pieces of ovary were cultured in OR2 medium (Wallace et al. 1973) at room temperature for up to several days. Follicle cells were removed from oocytes with collagenase (Liberase, Roche Applied Science). Success of the treatment was verified by staining with DAPI (4′, 6-diamidino-2-phenylindole) at 1 μg/mL and examining under low magnification in a fluorescence microscope (Simeoni et al. 2012). Transcription was inhibited by incubating pieces of ovary in actinomycin D (20 μg/mL) in OR2 medium. Embryos were obtained by in vitro fertilization. Females and males were primed with 100 units of human chorionic gonadotropin (HCG) the night before use. The next day, females were injected with 400 units of HCG and males with 200 units. Four hours later, females were squeezed to obtain eggs, whereas testes were removed from the males and macerated in saline. The sperm suspension was dispersed onto the freshly squeezed eggs. Eggs and embryos were manually dejellied and collected with minimal liquid in 1.5-mL Eppendorf tubes on dry ice.
Preparation of nuclear and cytoplasmic RNA fractions
GVs were manually isolated in an isotonic saline solution at pH 5.6–5.8 (83 mM KCl, 17 mM NaCl, 6.0 mM Na2HPO4, 4.0 mM KH2PO4, 1 mM MgCl2, 1.0 mM dithiothreitol, adjusted to pH 5.6–5.8 with HCl) (Gardner et al. 2012). The nuclear envelope was removed with jewelers’ forceps and the GV contents were collected in 10 mM sodium citrate and 5 mM EDTA (pH 5.0) for storage. Depending on the experiment, up to 500 GVs were collected over a period of several days. Samples of cytoplasm were obtained by removing the GV from defolliculated oocytes (Simeoni et al. 2012) and immediately placing the enucleated oocyte into a 1.5-mL Eppendorf tube on dry ice. Adequate cytoplasmic RNA could be obtained from 5–10 enucleated oocytes. RNA was extracted with TRIzol reagent (Ambion) and purified with the Direct-zol kit (Zymo Research). RNA was quantitated with a NanoDrop 2000 spectrophotometer (Thermo Scientific) and further characterized with a Bioanalyzer 2100 (Agilent).
Library preparation, sequencing, and sequence analysis
Samples were depleted of rRNA using Ribozero or Ribozero-Gold kits (Epicenter) according to the standard protocol. Subsequently, libraries were prepared using the TruSeq RNA sample preparation protocol (Illumina) or TruSeq stranded total RNA sample preparation (Illumina). Sequencing was performed on an Illumina HiSeq 2000 sequencer with either 50- or 100-bp single-end reads. Reads were aligned to the X. tropicalis genome (version 4.1) or the X. laevis genome (version 6.0) using the TopHat (version 2.0.7) and Bowtie (version 2.0.6.0) sequence alignment programs (Langmead et al. 2009; Trapnell et al. 2009). Exonic and intronic regions were quantified using BEDtools version 2.15.0 and Cufflinks version 2.0.2 (Quinlan and Hall 2010; Trapnell et al. 2010). Features with FPKM ≥2 were considered expressed. Sequence alignments were examined in the Integrative Genomics Viewer (IGV) from the Broad Institute (Robinson et al. 2011). To search for lariats or other circular RNA reads, we ran the PERL script findlariat.pl (Taggart et al. 2012). Further analysis of sisRNAs was done using BEDtools in conjunction with custom PERL scripts.
RT–PCR analysis
We performed RT–PCR against a few candidates using the one-step RT–PCR kit (Qiagen) to validate bioinformatic predictions. In vitro-transcribed RNAs were used as positive control to demonstrate the competence of the primers. To generate DNA templates for in vitro transcription, the regions of interest were amplified from genomic DNA by PCR. A T3 or T7 promoter was included in forward primers. The RNA was transcribed using T3 or T7 RNA polymerase, treated with DNase, and purified with G50 columns according to standard protocols. To confirm the orientation of selected introns, we used a single primer (either forward or reverse primer) for the RT step, followed by PCR with both sets of primers (35 cycles). For detection of lariats, total RNA was reverse-transcribed for 1 h using Episcript (Epicenter) and random hexamers. Either Episcript or SuperScript II (Life Technologies) was essential for reverse transcription through the branch point. Subsequently, PCR was performed using outward facing primer pairs (40 cycles). PCR fragments were cloned with pGEM-T Easy vector system (Promega) and sequenced to verify the specificity of these reactions. Primers used in this study are listed in Supplemental Table S1.
RNase R
rRNA-depleted samples were denatured at 65°C for 5 min and cooled on ice for 1 min. 10X RNase R buffer, RNasin (1 µL) and either 20 units of RNase R (Epicenter) or water was then added. The reaction was carried out overnight at 37°C. RT–PCR was carried out directly after the incubation, whereas libraries were made after precipitation of the RNA.
Western blots
For Western blots of the RNA lariat debranching enzyme, Dbr1, we used an affinity-purified rabbit polyclonal antibody raised against a region within amino acids 68–324 of human Dbr1 (Abcam, ab154230). The antibody was used at 1:10,000 in 1% blocking reagent (Roche). The secondary antibody was anti-rabbit IgG conjugated with horseradish peroxidase, used at a dilution of 1:20,000 in 5% nonfat dry milk. Peroxidase was detected with SuperSignal West Dura Extended Duration Substrate (Thermo Scientific).
Debranching assay
Xenopus oocyte nuclei remain physiologically active for hours when isolated under mineral oil (Paine et al. 1992; Yu et al. 2001; Deryusheva and Gall 2004). We prepared both nuclear and cytoplasmic extracts from X. laevis oocytes and tested for debranching activity. Two hundred and fifty nuclei were collected under oil and centrifuged at 20,800g for 2 min to obtain a clear nuclear extract. Cytoplasm from five oocytes was gently pipetted to obtain a cytoplasmic homogenate. Purified cytoplasmic RNA from X. tropicalis oocytes was added to the extracts, incubated for 2 h at 25°C, and purified according to the RNAqueous micro-kit protocol (Ambion).
Splicing assay
Splicing was assayed in single GVs or single enucleated oocytes of X. laevis (under oil). Each was injected with a capped and polyadenlylated RNA construct consisting of intron 2 of the faf2 gene of X. tropicalis along with its flanking exons. After incubation for 2 h at 25°C, RNA was purified using the RNAqueous micro-kit protocol (Ambion). Spliced and unspliced products were detected by RT–PCR using primers from the ends of the exons.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Nicholas Ingolia and Frederick Tan for help with sequence analysis and Allison Pinder for care in library production and sequencing. We thank Svetlana Deryusheva and other members of the Gall laboratory for technical assistance and discussion. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01 GM33397. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. J.G.G. is American Cancer Society Professor of Developmental Genetics.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.045781.114.
REFERENCES
- Acinas SG, Sarma-Rupavtarm R, Klepac-Ceraj V, Polz MF 2005. PCR-induced sequence artifacts and bias: insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl Environ Microbiol 71: 8966–8969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armakola M, Higgins MJ, Figley MD, Barmada SJ, Scarborough EA, Diaz Z, Fang X, Shorter J, Krogan NJ, Finkbeiner S, et al. 2012. Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat Genet 44: 1302–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley PT, Khaladkar M, Kim J, Eberwine J 2014. Cytoplasmic intron retention, function, splicing, and the sentinel RNA hypothesis. Wiley Interdiscip Rev RNA 5: 223–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callan HG 1986. Lampbrush chromosomes. Springer-Verlag, Berlin, Germany [Google Scholar]
- Chapman KB, Boeke JD 1991. Isolation and characterization of the gene encoding yeast debranching enzyme. Cell 65: 483–492 [DOI] [PubMed] [Google Scholar]
- Cheng ZF, Deutscher MP 2002. Purification and characterization of the Escherichia coli exoribonuclease RNase R. Comparison with RNase II. J Biol Chem 277: 21624–21629 [DOI] [PubMed] [Google Scholar]
- Cheng Z-F, Deutscher MP 2005. An important role for RNase R in mRNA decay. Mol Cell 17: 313–318 [DOI] [PubMed] [Google Scholar]
- Clement JQ, Qian L, Kaplinsky N, Wilkinson MF 1999. The stability and fate of a spliced intron from vertebrate cells. RNA 5: 206–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement JQ, Maiti S, Wilkinson MF 2001. Localization and stability of introns spliced from the Pem homeobox gene. J Biol Chem 276: 16919–16930 [DOI] [PubMed] [Google Scholar]
- Curtis HJ, Sibley CR, Wood MJ 2012. Mirtrons, an emerging class of atypical miRNA. Wiley Interdiscip Rev RNA 3: 617–632 [DOI] [PubMed] [Google Scholar]
- Davidson EH 1986. Gene activity in early development. Academic Press, Orlando, FL [Google Scholar]
- Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, Yost CC, Rubner FJ, Albertine KH, Swoboda KJ, et al. 2005. Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell 122: 379–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deryusheva S, Gall JG 2004. Dynamics of coilin in Cajal bodies of the Xenopus germinal vesicle. Proc Natl Acad Sci 101: 4810–4814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friend K, Kolev NG, Shu MD, Steitz JA 2008. Minor-class splicing occurs in the nucleus of the Xenopus oocyte. RNA 14: 1459–1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall JG, Wu Z 2010. Examining the contents of isolated Xenopus germinal vesicles. Methods 51: 45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao K, Masuda A, Matsuura T, Ohno K 2008. Human branch point consensus sequence is yUnAy. Nucleic Acids Res 36: 2257–2267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner EJ, Nizami ZF, Talbot CC Jr, Gall JG 2012. Stable intronic sequence RNA (sisRNA), a new class of noncoding RNA from the oocyte nucleus of Xenopus tropicalis. Genes Dev 26: 2550–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausen P, Riebesell M 1991. The early development of Xenopus laevis. An atlas of the histology. Springer-Verlag, Berlin, Germany [Google Scholar]
- Hellsten U, Harland RM, Gilchrist MJ, Hendrix D, Jurka J, Kapitonov V, Ovcharenko I, Putnam NH, Shu S, Taher L, et al. 2010. The genome of the Western clawed frog Xenopus tropicalis. Science 328: 633–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilleren PJ, Parker R 2003. Cytoplasmic degradation of splice-defective pre-mRNAs and intermediates. Mol Cell 12: 1453–1465 [DOI] [PubMed] [Google Scholar]
- Hoskins AA, Moore MJ 2012. The spliceosome: a flexible, reversible macromolecular machine. Trends Biochem Sci 37: 179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE 2013. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19: 141–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent JR, Kang W, Miller CG, Fraser NW 2003. Herpes simplex virus latency-associated transcript gene function. J Neurovirol 9: 285–290 [DOI] [PubMed] [Google Scholar]
- Khaladkar M, Buckley PT, Lee MT, Francis C, Eghbal MM, Chuong T, Suresh S, Kuhn B, Eberwine J, Kim J 2013. Subcellular RNA sequencing reveals broad presence of cytoplasmic intron-sequence retaining transcripts in mouse and rat neurons. PLoS One 8: e76194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krylov V, Kubickova S, Rubes J, Macha J, Tlapakova T, Seifertova E, Sebkova N 2010. Preparation of Xenopus tropicalis whole chromosome painting probes using laser microdissection and reconstruction of X. laevis tetraploid karyotype by Zoo-FISH. Chromosome Res 18: 431–439 [DOI] [PubMed] [Google Scholar]
- Kulesza CA, Shenk T 2006. Murine cytomegalovirus encodes a stable intron that facilitates persistent replication in the mouse. Proc Natl Acad Sci 103: 18302–18307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereau A, Le Sommer C, Lerivray H, Lesimple M, Hardy S 2007. Xenopus as a model to study alternative splicing in vivo. Biol Cell 99: 55–65 [DOI] [PubMed] [Google Scholar]
- Moon KH, Zhao X, Yu YT 2006. Pre-mRNA splicing in the nuclei of Xenopus oocytes. Methods Mol Biol 322: 149–163 [DOI] [PubMed] [Google Scholar]
- Ooi SL, Dann C III, Nam K, Leahy DJ, Damha MJ, Boeke JD 2001. RNA lariat debranching enzyme. Methods Enzymol 342: 233–248 [DOI] [PubMed] [Google Scholar]
- Paine PL, Johnson ME, Lau YT, Tluczek LJ, Miller DS 1992. The oocyte nucleus isolated in oil retains in vivo structure and functions. Biotechniques 13: 238–246 [PubMed] [Google Scholar]
- Quinlan AR, Hall IM 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rearick D, Prakash A, McSweeny A, Shepard SS, Fedorova L, Fedorov A 2011. Critical association of ncRNA with introns. Nucleic Acids Res 39: 2357–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD 2007. CPEB: a life in translation. Trends Biochem Sci 32: 279–285 [DOI] [PubMed] [Google Scholar]
- Richter JD, Lasko P 2011. Translational control in oocyte development. Cold Spring Harb Perspect Biol 3: a002758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodahl E, Haarr L 1997. Analysis of the 2-kilobase latency-associated transcript expressed in PC12 cells productively infected with herpes simplex virus type 1: evidence for a stable, nonlinear structure. J Virol 71: 1703–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondina MT, Schwertz H, Harris ES, Kraemer BF, Campbell RA, Mackman N, Grissom CK, Weyrich AS, Zimmerman GA 2011. The septic milieu triggers expression of spliced tissue factor mRNA in human platelets. J Thromb Haemost 9: 748–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruskin B, Green MR 1985. An RNA processing activity that debranches RNA lariats. Science 229: 135–140 [DOI] [PubMed] [Google Scholar]
- Schwertz H, Tolley ND, Foulks JM, Denis MM, Risenmay BW, Buerke M, Tilley RE, Rondina MT, Harris EM, Kraiss LW, et al. 2006. Signal-dependent splicing of tissue factor pre-mRNA modulates the thrombogenicity of human platelets. J Exp Med 203: 2433–2440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeoni I, Gilchrist MJ, Garrett N, Armisen J, Gurdon JB 2012. Widespread transcription in an amphibian oocyte relates to its reprogramming activity on transplanted somatic nuclei. Stem Cells Dev 21: 181–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taggart AJ, DeSimone AM, Shih JS, Filloux ME, Fairbrother WG 2012. Large-scale mapping of branchpoints in human pre-mRNA transcripts in vivo. Nat Struct Mol Biol 19: 719–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymowska J 1991. Polyploidy and cytogenetic variation in frogs of the genus Xenopus. In Amphibian cytogenetics and evolution (ed. Green DM, Sessions SK), pp. 259–297 Academic Press, San Diego, CA [Google Scholar]
- Ulitsky I, Bartel DP 2013. lincRNAs: genomics, evolution, and mechanisms. Cell 154: 26–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heesch S, van Iterson M, Jacobi J, Boymans S, Essers PB, de Bruijn E, Hao W, Macinnes AW, Cuppen E, Simonis M 2014. Extensive localization of long noncoding RNAs to the cytosol and mono- and polyribosomal complexes. Genome Biol 15: R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace RA, Jared DW, Dumont JN, Sega MW 1973. Protein incorporation by isolated amphibian oocytes: III. Optimum incubation conditions. J Exp Zool 184: 321–333 [DOI] [PubMed] [Google Scholar]
- Wang GC, Wang Y 1996. The frequency of chimeric molecules as a consequence of PCR co-amplification of 16S rRNA genes from different bacterial species. Microbiology 142: 1107–1114 [DOI] [PubMed] [Google Scholar]
- Wu TT, Su YH, Block TM, Taylor JM 1996. Evidence that two latency-associated transcripts of herpes simplex virus type 1 are nonlinear. J Virol 70: 5962–5967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin QF, Yang L, Zhang Y, Xiang JF, Wu YW, Carmichael GG, Chen LL 2012. Long noncoding RNAs with snoRNA ends. Mol Cell 48: 219–230 [DOI] [PubMed] [Google Scholar]
- Yu YT, Shu MD, Narayanan A, Terns RM, Terns MP, Steitz JA 2001. Internal modification of U2 small nuclear (sn)RNA occurs in nucleoli of Xenopus oocytes. J Cell Biol 152: 1279–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH, Zhu S, Yang L, Chen LL 2013. Circular intronic long noncoding RNAs. Mol Cell 51: 792–806 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.