

Abstract
The SMRT coregulator functions as a dual coactivator and corepressor for estrogen receptor-α (ERα) in a gene-specific manner, and in several studies its elevated expression correlates with poor outcome for breast cancer patients. A specific role of SMRT in breast cancer progression has not been elucidated, but SMRT knock-down limits estradiol-dependent growth of MCF-7 breast cancer cells. In this study, small-interfering RNA (siRNA) and short-hairpin RNA (shRNA) approaches were used to determine the effects of SMRT depletion on growth of ERα-positive MCF-7 and ZR-75–1 breast cancer cells, as well as the ERα-negative MDA-MB-231 breast cancer line. Depletion of SMRT inhibited growth of ERα-positive cells grown in monolayer but had no effect on growth of the ERα-negative cells. Reduced SMRT levels also negatively impacted the anchorage-independent growth of MCF-7 cells as assessed by soft agar colony formation assays. The observed growth inhibitions were due to a loss of estradiol-induced progression through the G1/S transition of the cell cycle and increased apoptosis in SMRT-depleted compared with control cells. Gene expression analyses indicated that SMRT inhibits apoptosis by a coordinated regulation of genes involved in apoptosis. Functioning as a dual coactivator for anti-apoptotic genes and corepressor for pro-apoptotic genes, SMRT can limit apoptosis. Together these data indicate that SMRT promotes breast cancer progression through multiple pathways leading to increased proliferation and decreased apoptosis.
Breast cancer remains a major health problem in the United States. In 2013, more than 230,000 women will be diagnosed with new cases of breast cancer and nearly 40,000 women are expected to die from their disease (1). Many cancers, including those of the breast, encompass gene mutations, amplifications, or deletions that can be drivers of disease progression (2). The earliest stages of breast cancer are characterized by excessive, unchecked proliferation of the breast epithelium, whereas death is ultimately caused by growth at metastatic sites (3). The majority (70%–75%) of breast cancers express estrogen receptor-α (ERα), and in these tumors it is a major driver of proliferation (4, 5). Circulating estrogens produced by the ovaries and other tissues as well as locally synthesized in breast, bind to and activate ERα leading to programs of gene expression that promote breast carcinogenesis (5–7). Treatments to block the activity of this receptor are therefore commonly used for ERα-positive tumors; these include antiestrogens and aromatase inhibitors that prevent estradiol (E2) synthesis (8, 9). With the reduction of receptor activity, breast cancer cell proliferation and consequently disease progression is inhibited.
Upon binding to ligands, ERα undergoes a conformational change that enables it to interact with coactivators and corepressors (6). These coregulators exist in large multiprotein complexes that enable them to directly or indirectly remodel chromatin by altering histone-histone and histone-DNA interactions through catalyzing the addition or removal of histone posttranslational modifications (10, 11). For example, E2 recruits coactivators with enzymatic activities (eg, histone acetyl transferase) that promote transcription of ERα target genes (12–15). Conversely, knock-down of a single coregulator can limit E2-induced transcription in a gene-specific manner (13–15). Coactivators are required for maximal growth of breast cancer cells, at least in part via their ability to stimulate E2-dependent expression of genes that promote cell proliferation (13–15). Moreover, ERα coactivators such as steroid receptor coactivator (SRC)-3 are frequently overexpressed in breast cancer, and oncogenic and driver mutations have been identified in multiple chromatin remodeling factors; collectively this demonstrates the importance of this class of proteins for disease progression (2, 12, 16, 17).
In addition to the well-known role of the silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) as a corepressor of unliganded type II nuclear receptors including retinoic acid receptor-α, SMRT can both stimulate and repress E2-dependent ERα activity in a gene-selective manner (14, 18, 19). This dual function of SMRT as a coactivator and corepressor of ERα makes it difficult to predict, a priori, whether SMRT exerts a pro- or antitumorigenic role in breast cancer. In several large studies evaluating human breast tumors, elevated SMRT protein levels correlated with poor prognosis potentially reflecting an ERα coactivator role for SMRT in breast cancer (20, 21). However, the association between higher levels of SMRT mRNA and a better outcome for untreated, lymph-node negative, ERα-positive breast cancer patients suggests a protective role for SMRT (22). The apparent discrepancy in these reports may reflect the poor correlation between SMRT mRNA and protein expression demonstrated in breast cancer cell line studies (21), but this awaits confirmation in breast tumors. In genetic studies, one nonsynonymous single nucleotide polymorphism in SMRT has been positively associated with breast cancer (23). In addition, amplification of SMRT has been detected in ductal carcinoma in situ, an early breast lesion, whereas deletion of SMRT was more common in advanced cases of mixed ductal carcinoma in situ and invasive carcinomas (24). The presence of a truncation mutation within the SMRT gene also was detected in The Cancer Genome Atlas breast cancer analyses (2). Taken together, these represent multiple reports of alterations in SMRT expression and/or function in breast cancer, and highlight the critical need for a greater understanding of the role of this coregulator in the breast under normal and pathophysiological conditions.
Previous data has shown that SMRT is required for full growth of ERα-positive MCF-7 breast cancer cells but not ERα-negative HeLa cervical cancer cells (14). This appears to be mediated, at least in part through SMRT coactivation of the ERα target gene, cyclin D1, as SMRT knock-down decreased the mRNA expression of this promoter of cell cycle progression (14, 19). Further support for SMRT as a progrowth coregulator comes from analyses of cortical progenitor cells isolated from SMRT null mouse embryos that proliferate at a lower rate than wild-type controls (25). There may also be a role for SMRT and/or other members of the SMRT corepressor complex, particularly histone deacetylase 3 (HDAC3), in regulation of mitosis. The Schizosaccharomyces pombe ortholog for SMRT, snt1, is required for cytokinesis of latrunculin A-treated cells (26, 27). Additionally, HDAC3 localizes to the mitotic spindle and promotes Aurora B kinase activity and sister chromatid cohesion (28–31), and because SMRT is an activator of HDAC3 deacetylase activity (32), there may be a role for SMRT in mitosis. However, mice with mutations in the deacetylase activation domains of SMRT and its paralog, NCoR are viable suggesting that the HDAC3-stimulating property of SMRT or NCoR is not required for growth and development (33).
The collective data indicate that SMRT may play multiple roles in promoting cell growth, and in this way contribute to poor outcomes for patients with breast cancer. Thus, the purpose of this study was to evaluate the role of SMRT in regulating the proliferation of ERα-positive breast cancer cells. Our data indicate that both the coactivator and the corepressor functions of SMRT are important for increased cell growth as SMRT increases progrowth and antiapoptotic gene expression while also inhibiting expression of proapoptotic genes. This up- and downregulation of genes by SMRT reveals how a single coregulator can function as a master regulator of multiple pathways to impact proliferation and cell viability, resulting in a dual mechanism for SMRT to promote progression of breast cancer.
Materials and Methods
Cells, hormones, and chemicals
Cells (MCF-7, MDA-MB-231, and ZR-75–1) were obtained from ATCC and maintained under recommended culture conditions. The following ER ligands were used: estradiol (E2), 4-hydroxytamoxifen (4HT), and ICI 182,780 (ICI; Tocris). All chemicals except ICI were obtained from Sigma-Aldrich.
Cell transfection
Cells were plated in 6-well multiwell dishes in growth media 24 hours prior to transfections that were conducted in Opti-MEM (Life Technologies) using Oligofectamine (Life Technologies) and 20 pmol/well small interfering RNA (siRNA) for MCF-7 and MDA-MB-231 cells or Lipofectamine RNAiMAX (Life Technologies) and 30 pmol/well siRNA for ZR-75–1 cells. After transfection, cells were fed with media supplemented with fetal bovine serum (FBS) or charcoal-stripped FBS (sFBS). The sequences of the panSMRT (siSMRT) and Sαβ2 (siSαβ) custom Select siRNA have been described (14) and the Silencer Negative Control siRNA #2 was used as the negative control (Life Technologies). To generate stable SMRT knock-down cells, MCF-7 cells were transfected using X-tremeGENE HP DNA Transfection Reagent (Roche) with pGIPz short hairpin (shRNA) vectors (Thermo Scientific Open Biosystems) encoding a nonsilencing control shRNA (ATCTCGCTTGGGCGAGAGTAAG) or a shRNA targeting SMRT (GGAATGAGCCTGAATACAA). Approximately 48 to 72 hours thereafter, cells were selected in 400 ng/mL puromycin and then maintained in 200 ng/mL puromycin.
Western blots
Western blots were performed as described previously (15). Briefly, cells were lysed in buffer [50 mM Tris HCl (pH 8.0), 400 mM NaCl, 5 mM EDTA, 1% NP40, and 0.2% Sarcosyl], and equal protein amounts were resolved on NuPAGE Novex gels (Life Technologies). For PARP analyses, adherent and nonadherent cells were lysed after the protocol provided with the anti-PARP serum (Roche) and equal lysate volumes were resolved by SDS-PAGE gel. Western blot analyses used primary antibodies for SMRT (BD Biosciences), PARP (Roche), actin or tubulin (Millipore), followed by incubation with the appropriate HRP-conjugated secondary antibodies (GE Healthcare). Amersham ECL Plus or ECL Prime (GE Healthcare) was used to visualize signals.
Growth assays
MCF-7 and ZR-75–1 cells (100,000 cells/well) and MDA-MB-231 cells (20,000 cells/well) were plated in 6-well multiwell dishes and transfected as described above. Thereafter, cells were fed with phenol red-free DMEM supplemented with 5% sFBS (MCF-7), phenol red-free RPMI supplemented with 4% sFBS (ZR-75–1), or DMEM supplemented with 10% FBS (MDA-MB-231). Ligand treatments were applied the next day, and cultures continued for 5 days with a fresh media addition after the second day. Cells were counted using a Beckman Coulter Particle Counter. For the pGIPz stable cell lines, 100,000 cells/well, cultured in phenol red-free DMEM plus 5% sFBS and 200 ng/mL puromycin, were treated with hormones to initiate the 5-day growth assay.
Anchorage-independent soft agar growth assay
Soft agar assays were conducted as previously described (34). Control pGIPz (shCtrl) and SMRT knock-down pGIPz (shSMRT) MCF-7 cells were plated (10,000 cells/well) in 0.35% SeaPlaque agarose (Lonza) on top of a base (0.7% agarose) in 6-well multiwell dishes. Minimum essential media supplemented with 5% sFBS and hormone treatments were added to the top layer and replenished every other day. After 14 days colonies were counted using a GelCount machine (Oxford Optronix).
FACS analysis
Cell cycle distribution was assessed by flow cytometry as previously described (15, 35). Briefly MCF-7 cells were transfected as described above with control- or SMRT-targeting siRNA, and then grown in phenol red-free DME containing 5% sFBS for 48 hours prior to hormone treatment for an additional 24 hours. Adherent cells were fixed with 100% ethanol and stained with 50 μg/mL propidium iodide in PBS. Flow cytometric analysis employed a Beckman Coulter EPICS SL-MCLs, and data was analyzed with WinMDI software (Scripps Research Institute).
Apoptosis assays
Cells were transfected with nontargeting or SMRT-targeting siRNA and then fed with phenol red-free DMEM plus 5% sFBS. Forty-eight hours later, cells were treated for 24 hours with vehicle, E2, 4HT, or staurosporine. For apoptosis assessment by Cell Death ELISA (Roche), adherent and floating cells were collected for measurement according to the manufacturer's recommendations. For Annexin V staining, adherent cells were harvested, resuspended in binding buffer and stained with Annexin V-FITC and PI using the Annexin V-FITC Apoptosis Detection kit protocol (Sigma). Stained cell solutions were filtered through a BD Falcon cell-strainer cap and analyzed on a BDFACS Canto II cytometer with BD FACSDiva software.
Microarray and RT-qPCR analysis
For gene expression analyses by Affymetrix Human Gene 1.0 ST array, cells were plated at 300,000 cells/well in a 6-well multiwell dish and transfected with control or SMRT-targeting siRNA. After culture in phenol-red free DME and 10% sFBS for 48 hours, vehicle or E2 was added for an additional 24 hours. Isolation of RNA was performed using an RNeasy kit (Qiagen). Data were analyzed using GenePattern from the Broad Institute (36), and differentially expressed genes were detected with a threshold of a 1.5-fold change (up or down) between control and SMRT-depleted MCF-7 cells. This gene list was analyzed using Database for Annotation, Visualization, and Integrated Discovery (DAVID) (37, 38) for gene ontology analysis, and a heatmap was generated with the GenePattern HeatMapViewer module (36). Microarray data has been deposited in Gene Expression Omnibus (GSE57935).
For RT-qPCR analyses, cells were transfected as described above and 48 hours later, treated with vehicle, E2, 4HT, or ICI for an additional 4 or 24 hours. RNA was extracted using TRIzol (Life Technologies), and cDNA was generated from 1 μg RNA using SuperScript VILO cDNA Synthesis kit (Life Technologies). Custom primers for the four genes selected for validation were designed with Primer Express software (Life Technologies; Table 1). Quantitative reverse-transcriptase PCR was performed using SYBR Green reagents on StepOnePlus or 7500 FAST Real-Time PCR System machines (Life Technologies). Analysis was conducted using ΔΔCt method and normalized to 18S rRNA.
Table 1.
Sequences of Forward and Reverse Primers Used for RT-qPCR
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Birc5 baculoviral IAP repeat containing 5 | 5′-TCTCAAGGACCACCGCATCT-3′ | 5′-CCAAGTCTGGCTCGTTCTCAGT-3′ |
| STRADB STE20-related kinase adaptor β | 5′-CCACAGGAACACTGGTAACTATAAAAAT-3′ | 5′-GTGGGATAGAATCACGGCTTTC-3′ |
| TNFSF10 tumor necrosis factor (ligand) superfamily, member 10 | 5′-TGCGTGCTGATCGTGATCTT-3′ | 5′-TTTTGGAGTACTTGTCCTGCATCT-3′ |
| XAF1 XIAP-associated factor 1 | 5′-TCGGTGTGCAGGAACTGTAAAA-3′ | 5′-CAGGAACCGCAGGCAGTAA-3′ |
Results
SMRT promotes growth of MCF-7 cells
To determine the role of SMRT in regulation of breast cancer cell growth, SMRT was depleted from ERα-positive MCF-7 cells by siRNA and growth in response to ER agonist and antagonists was assessed. In cells transfected with control siRNA, E2 elicited a 3.6-fold increase in cells in comparison to vehicle-treated cells whereas 4HT modestly increased cell number by 2.2-fold (Figure 1A). Consistent with its mixed ERα agonist/antagonist properties, 4HT reduced E2-induced growth to a level comparable to cells treated with 4HT alone. Acute siRNA-mediated SMRT knock-down reduced MCF-7 cell growth for all treatment groups, with the number of cells in the E2-treated, SMRT-depleted cultures reduced by 78% in comparison to the corresponding E2-treated control group. Likewise, SMRT depletion reduced the growth of 4HT-treated cells, and in cotreatment experiments 4HT blocked the low level of E2-stimulated cell growth. There are two major forms of SMRT expressed in MCF-7 cells, full-length SMRTα and the SMRTβ splice variant (14), and Western blot analysis confirmed that the siRNA employed for these experiments effectively reduced the expression of both (Figure 1B, left panel).
Figure 1.

Depletion of SMRT by siRNA or shRNA inhibits MCF-7 cell growth. For siRNA experiments, cells were transfected with nontargeting control siRNA (siCtrl) or siRNA targeting both SMRTα and SMRTβ (siSMRT), whereas for shRNA studies stable MCF-7 cells were generated with pGIPz vectors containing either nontargeting control shRNA (shCtrl) or shRNA targeting SMRTα and SMRTβ (shSMRT). A, The day after siRNA transfection, MCF-7 cells were treated with vehicle (0.1% ethanol), 1 nM E2, 100 nM 4HT or a combination of E2 and 4HT, and 5 days thereafter cell number was determined by Beckman Coulter Particle Counter. B, Representative Western blot for SMRT knock-down achieved by siRNA transfection (left) or by shRNA in the pGIPz stable MCF-7 cell lines (right). C, The shCtrl and shSMRT cells were treated with vehicle, 1 nM E2, 100 nM 4HT, or 100 nM ICI 182,780 (ICI) and counted 5 days later using a Beckman Coulter Particle Counter. D, The growth of shCtrl and shSMRT cells were assessed by soft agar assays after 14 days of hormone treatments using Oxford Optronix GelCount. Results are average ± SEM of three experiments normalized to respective E2-treated control groups. Statistical analyses were conducted by Student's t test; #, P < .05, versus vehicle treatment within the same siRNA or shRNA group and *, P < .05 versus the corresponding ligand treatment in the siCtrl or shCtrl groups.
Western blot analysis was used to screen shRNAs corresponding to regions of SMRT distinct from the siRNA target sequence (data not shown); this enabled an independent assessment of the effect of SMRT depletion on cell growth to ensure the above results were not due to off target effects. Stable cell lines were generated by transfecting MCF-7 cells with pGIPz vectors expressing either a control (shCtrl) or the SMRT (shSMRT) shRNA able to achieve the most effective SMRT depletion. Western blot analysis verified the effective depletion of all isoforms of SMRT in shSMRT cells (Figure 1B, right panel). This chronic shRNA-mediated SMRT depletion inhibited growth of E2-treated cultures grown in monolayer as well as vehicle-, 4HT- and ICI-treated cells (Figure 1C). The relative agonist activity of 4HT in the control shRNA line was low and not enhanced in the SMRT depleted cells, and in neither control nor SMRT shRNA cell lines was the pure ERα antagonist, ICI 182,780 (ICI), able to stimulate cell growth.
These stable cell lines were used to determine if SMRT promotes anchorage-independent growth of MCF-7 cells in soft agar colony formation assays. Vehicle and E2-treated colony formation were decreased in shSMRT compared with shCtrl cells indicating that anchorage-independent growth is enhanced by SMRT expression (Figure 1D). Neither 4HT nor ICI promoted colony formation of either shCtrl or shSMRT cells. Taken together, the ability of SMRT depletion to inhibit E2-dependent growth of MCF-7 cells grown as monolayers or in soft agar indicates that SMRT is required for proliferation of these ERα-positive breast cancer cells.
To determine if SMRT can impact growth independently of functional ERα, the effect of SMRT depletion was assessed in MCF-7 cells treated with a high dose (500 nM) of the ICI pure antiestrogen to block ERα function. As expected in the vehicle-exposed cells, E2 induces growth of control transfected (siCtrl) cells and acute depletion of SMRT inhibited growth compared with siCtrl cells (Figure 2). In the presence of high dose ICI, E2 did not stimulate growth of siCtrl cells indicating that the ERα pathway is effectively blocked. Knock-down of SMRT in ICI-treated cells results in a significant reduction in MCF-7 cell growth in comparison to siCtrl cells treated with antiestrogen, and indicates that SMRT also can regulate MCF-7 breast cancer cell growth through an ERα-independent pathway.
Figure 2.
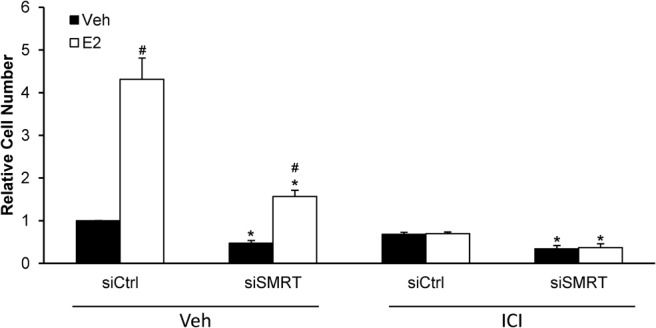
Impact of SMRT depletion on growth of antiestrogen-treated MCF-7 cells. Cells were transfected with nontargeting control (siCtrl) or SMRT-specific (siSMRT) siRNA and treated with vehicle or 500 nM ICI 182,780 (ICI) to block ERα activity. The next day cells were treated with vehicle or 1 nM E2 and after 5 days of culture, harvested for cell number determinations. Data presented is average ± SEM of three experiments normalized to vehicle-treated siCtrl cells. Statistical analyses were performed with Student's t test; #, P < .05 versus vehicle treatment within the same siRNA group and *, P < .05 versus the corresponding ligand treatment in the siCtrl group.
SMRT regulates proliferation of other breast cancer cells
The impact of SMRT depletion on growth of ZR-75–1 cells, another ERα-positive breast cancer cell line, was examined to determine if its ability to regulate growth extends to other breast cancer cells. In ZR-75–1 cells transfected with the control siRNA, E2 induced a 2.6-fold increase in cell number whereas 4HT induced a modest 1.2-fold increase in comparison to the vehicle-treated condition; ICI treatment was without effect (Figure 3A). Acute siRNA knock-down of SMRT resulted in growth inhibition for all treatments compared with control cells, as exemplified by a 22% decrease in the E2-induced growth of the SMRT-depleted versus control cells. Western blot analysis verified the efficient siRNA-mediated depletion of SMRT for these cells (Figure 3B). This indicates that this coregulator is required for maximal growth of ZR-75–1 cells and argues that SMRT promotion of cell proliferation is not unique to MCF-7 cells.
Figure 3.

Effect of SMRT knock-down on growth of ERα-positive ZR-75–1 cells and ERα-negative MDA-MB-231 cells. A, ZR-75–1 cells were transfected with nontargeting control (siCtrl) or SMRT-specific (siSMRT) siRNA and 24 hours later treated with vehicle, 1 nM E2, 100 nM 4HT, or 100 nM ICI. Cell growth was determined after 5 days of culture. Cell numbers are normalized to E2-treated siCtrl cells (n = 3). B, Representative Western blot of SMRT (top) expression for ZR-75–1 cells treated with siRNA for control (-) and SMRT (+). Actin is shown as a loading control (bottom). C, Representative Western blot of SMRT knock-down in MDA-MB-231 cells (left) and Western blot comparing levels of SMRT across cell lines grown in media supplemented with 10% FBS indicates there are variable levels of total SMRT and SMRT isoforms in the cell lines examined (right). D, MDA-MB-231 cells were transfected with control (siCtrl) or SMRT (siSMRT) siRNA, and 5 days thereafter, cell numbers were assessed. Cell numbers are normalized to siCtrl cells (n = 7). Numerical data are presented as the average ± SEM. Statistical analyses were by Student's t test; #, P < .05 versus vehicle treatment within the same siRNA group and *, P < .05 versus the corresponding ligand treatment in the siCtrl group.
Many breast cancers are ERα-negative, and to determine if SMRT can regulate the proliferation of breast cancer cells lacking this transcription factor, an experiment was conducted in ERα-negative MDA-MB-231 cells. After transfection with the SMRT siRNA, western blot analysis indicated effective reduction of SMRT protein in comparison to the control cells (Figure 3C, left). The overall levels of SMRT protein were significantly lower in MDA-MB-231 cells than in MCF-7 and ZR-75–1 cells (Figure 3C, right); the significance of this is unknown. SMRT depletion had no effect on growth of MDA-MB-231 cells (Figure 3D), consistent with a prior report (39).
Cell cycle analysis of SMRT depleted MCF-7 cells
Flow cytometry was used to compare cell cycle distribution between control and SMRT-depleted MCF-7 cells. Transfected cells were synchronized in the G1 phase of the cell cycle, with at least 70% of the siCtrl and the siSMRT vehicle-treated cells accumulating in this phase (Figure 4). In siCtrl cells, treatment with E2 resulted in a reduction in the G1 population to 47% and an increase in the population of S/G2-M cells from 25% to 49%. However, the majority (65%) of the E2-treated, SMRT-depleted cells remained in the G1 phase of the cell cycle with a corresponding small increase in S/G2-M cells (15→23%) indicating that loss of SMRT expression negatively impacts the E2-induced progression of cells from G1 to subsequent phases of the cell cycle. Additionally there was a nearly 3-fold hormone-independent increase in the subG0 population of the siSMRT cells (12.5% for vehicle) compared with siCtrl cells (4.5% for vehicle).
Figure 4.
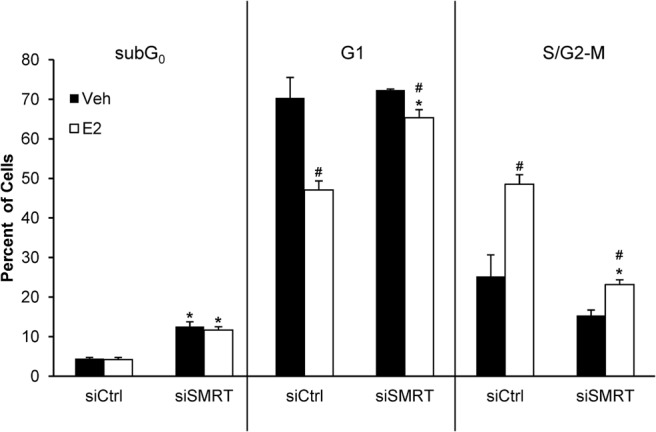
Impact of SMRT depletion on cell cycle distribution of MCF-7 cells. Cells were transfected with nontargeting control (siCtrl) or SMRT-specific (siSMRT) siRNA and allowed to accumulate in G0 for 48 hours in 5% sFBS. Thereafter cells were treated with vehicle or 1 nM E2 for 24 hours, then fixed and analyzed for DNA content by staining with propidium iodide followed by flow-cytometry using a Beckman Coulter EPICS XL-MCLs to determine the percentage of cells in the subG0 (left), G1 (middle), and S/G2-M (right) phases of cell cycle. Results shown are average ± SEM (n = 3). Statistical analyses were performed by Student's t test; #, P < .05 versus vehicle treatment within the same siRNA group and *, P < .05 versus the corresponding vehicle or E2 treatment in the siCtrl group.
SMRT depletion increased apoptosis
The increase in the subG0 population suggested increased apoptosis and multiple assays were conducted to address this possibility. The Cell Death ELISA measures nucleosomal DNA, a hallmark of apoptosis. Knock-down of SMRT resulted in a 2.1-fold hormone-independent elevation of nucleosomal DNA, indicating increased apoptosis in SMRT-depleted cells (Figure 5A). As a second, independent measurement of apoptosis, levels of annexin V binding, indicative of the disruption of the plasma membrane that occurs during apoptosis, were assessed by flow cytometry. Depletion of SMRT increased annexin V staining by 2.5-fold in comparison to vehicle-treated control cells (Figure 5B). Finally, to determine if apoptosis was mediated by a caspase pathway, cleaved PARP protein was evaluated. Elevated levels of cleaved PARP were first detected 72 hours after siRNA-mediated knock-down of SMRT expression, and this was further increased at the 96-hour timepoint (Figure 5C). In contrast, minimal PARP cleaved products were detected for the control-transfected cells. Staurosporine was used as a positive control for apoptosis in each assay, and in all cases the expected increases were observed (Figures 5, A–C). The effect of SMRT depletion on induction of apoptosis was not cell-type specific because SMRT knock-down in ZR-75–1 cells also increased levels of cleaved PARP (Figure 5D). Moreover, the impact of SMRT depletion on apoptosis in MCF-7 cells was not unique to the panSMRT siRNA as a second, independent SMRT siRNA, Sαβ2, also was able to effectively increase cleaved PARP (Figure 5E). As expected, this siSαβ effectively decreased SMRT expression as well as E2-dependent growth of MCF-7 cells (Figure 5, F and G). Together these data, using multiple cell types and different siRNAs, show that SMRT knock-down results in an E2-independent increase in apoptosis versus control breast cancer cells.
Figure 5.

Depletion of SMRT from MCF-7 cells increases apoptosis. Cells were transfected with nontargeting control (siCtrl) or SMRT-specific (siSMRT) siRNA, and after 48 hours were treated with vehicle, 1 nM E2, 100 nM 4HT, or 1 μM staurosporine (positive control) for 24 hours, as indicated. Cells were harvested and the extent of apoptosis was assessed by (A) Cell Death ELISA and (B) annexin V staining. C, MCF-7 cells transfected with control (-) or SMRT (+) siRNA were harvested 48, 72, or 96 hours after transfection for analysis of PARP cleavage by Western blot analysis (top). Tubulin (bottom) is shown as a loading control. Shown is a representative blot (n = 4). D, ZR-75–1 cells were transfected with siCtrl (-) or siSMRT (+) siRNA and harvested 96 hours thereafter for analysis of PARP cleavage by Western blot. MCF-7 cells were transfected with siCtrl (-) or siSαβ (+) siRNA and collected 96 hours thereafter for assessment of (E) cleaved PARP and (F) knock-down of SMRT expression by Western blot. Shown are representative blots of at least 3. G, MCF-7 cells were transfected with siCtrl or siSαβ siRNA and 24 hours after treated with vehicle or 1 nM E2. After 5 days cells were harvested for evaluation of cell growth. Numerical data are presented as average ± SEM (n = 3). Statistical analyses were performed by Student's t test, #, P < .05 versus siCtrl vehicle and *, P < .05 versus respective siCtrl treatment.
Differential expression of genes regulating apoptosis in control- versus SMRT-depleted cells
Gene ontology analysis of microarray data performed by DAVID revealed that genes involved in cell cycle regulation are differentially expressed between control and SMRT-depleted cells (Supplemental Table 1) and this corresponds well with the FACS analysis data indicating a defect in the E2-induced G1-S transition. One such cell cycle regulator was cyclin D1, an ERα regulated gene (40). SMRT binds to the cyclin D1 gene and positively regulates E2-dependent expression of this proproliferative cell cycle regulator (14, 19). Less expectedly, DAVID gene ontology analysis revealed enrichment for a group of genes (Figure 6A) involved in apoptosis (2, 37, 38). Two antiapoptotic genes, Birc5 and STRADB and two proapoptotic genes, TNFSF10 and XAF1 were selected for validation by RT-qPCR (41–45). Birc5 mRNA was induced by 24 hours of E2 treatment and to a lesser extent by 4HT whereas the pure antiestrogen ICI inhibited its expression, consistent with Birc5 being a known ERα-target gene (41, 42); hormonal regulation of Birc5 mRNA by ER ligands was not detected at 4 hours (Figure 6B, top and bottom). For both time points, SMRT knock-down significantly reduced Birc5 mRNA levels for vehicle, 4HT and ICI groups, and although E2-induced levels were approximately 25% lower than the corresponding control for the 24-hour timepoint, this did not reach statistical significance. Expression of the antiapoptotic gene, STRADB, was not induced by 24-hour E2 treatment although there is a minor but significant induction of STRADB mRNA by 4HT (Figure 6C). In response to SMRT knock-down, STRADB mRNA levels were approximately 20% lower for all treatment groups, and notably rather than the small induction of STRADB mRNA by 4HT observed for control cells, levels of STRADB mRNA were reduced by approximately 18% in 4HT-treated cells with SMRT depletion. The proapoptotic XAF1 gene that was not regulated by any of the tested ERα ligands, was upregulated by SMRT depletion as shown by the 1.6-fold and 2.3-fold increase in XAF1 mRNA for control versus SMRT-depleted cells exposed to vehicle for 4 and 24 hours, respectively (Figure 6D, top and bottom.). Similarly expression of the TNFSF10 gene, which was modestly induced by 4HT and ICI treatment in control cells, was increased by SMRT depletion for all treatment groups, as exemplified by a 2.1-fold increase in TNFSF10 mRNA for vehicle-treated cells (Figure 6E). Together these anti- and proapoptotic genes demonstrate that SMRT is acting as a coactivator and corepressor, respectively, in a gene-specific manner to transcriptionally regulate genes that control apoptosis pathways.
Figure 6.

Depletion of SMRT alters the expression of genes involved in apoptosis. MCF-7 cells were transfected with nontargeting control (siCtrl) or SMRT-specific (siSMRT) siRNA, and 48 hours later were treated with vehicle (-) or 1 nM E2 (+) prior to assessment of RNA expression. A, Heatmap of genes involved in apoptosis that are differentially regulated in SMRT depleted versus control cells after 24 hours of vehicle or E2 treatment. Quantitative RT-PCR analyses of mRNA levels of (B and C) antiapoptotic and (D and E) proapoptotic genes for control and SMRT depleted MCF-7 cells treated with vehicle, 1 nM E2, 100 nM 4HT, or 100 nM ICI for 4 or 24 hours, as indicated. Data presented is average ± SEM (n = 3) normalized by ΔΔCt using 18S rRNA and siCtrl vehicle as normalizers. Statistical analyses were performed by Student's t test; #, P < .05 versus vehicle treatment within the same siRNA group and *, P < .05 versus the corresponding ligand treatment in the siCtrl group.
Discussion
Aberrant control of cellular proliferation is one of the hallmarks of cancer (3), and it is therefore important to understand how transcriptional coregulators affect cell proliferation and contribute to breast cancer. Most breast cancers express ERα that when activated by E2 stimulates the expression of an array of proproliferative and antiapoptotic genes that ultimately promote tumor growth. Expression of ERα-target genes is dependent upon coactivators and corepressors recruited by the receptor, and their ability to modify chromatin to either facilitate or inhibit gene transcription. Although early studies linking corepressors such as SMRT to the antiestrogenic activity of tamoxifen suggested that elevated SMRT levels would be beneficial to patients (46), SMRT was recently shown to function as a dual coactivator and corepressor for ERα, and this raised the question if SMRT functions in an anti- or pro-proliferative capacity. The results herein demonstrate that SMRT promotes the E2-dependent proliferation of two ERα-positive breast cancer cell lines, but not ERα-negative MDA-MB-231 cells. The stimulation of cell growth by SMRT is associated with cells moving from the G1 to S phases of the cell cycle as well as inhibition of apoptosis, and together these data suggest that elevated SMRT levels contribute to a transcriptional environment that sustains proliferation.
Knock-down of SMRT decreased MCF-7 and ZR-75–1 cell numbers in growth assays. A previous cell cycle analysis study indicated that codepletion of SMRT and NCoR reduced the percentage of 1 nM E2-treated MCF-7 cells in the S/G2/M phases in comparison to controls (39), and our cell cycle analysis results with the same physiologically relevant concentration of E2 indicated that depletion of SMRT alone yields a comparable result. In contrast, SMRT depletion did not impact the distribution of cells in the S/G2/M phases in one study employing 100 nM E2 (39) whereas in another, transient SMRT knock-down increased proliferation of MCF-7 cells regardless of treatment with vehicle, 100 nM E2 or 1 μM 4HT (47). It is unknown whether these differing results are due to technical considerations such as timing or the use of very high E2 levels, or expression of only a single isoform of SMRT in the cells employed in these experiments (39, 47).
The growth inhibition of SMRT-depleted MCF-7 cells and reduction in the percentage of these cells exiting from the G1 phase after E2 treatment indicate SMRT promotes the effects of E2 on this phase of the cell cycle. Interestingly, knock-down of SRC-2 or SRC-3 coactivators also decreases the E2-induced progression of MCF-7 cells from G1 to S (15), and these coactivators as well as SMRT can stimulate E2-dependent expression of cyclin D1, a positive regulator of the G1 to S transition (14, 15, 48), likely through E2-dependent recruitment of an ERα-SMRT-SRC-3 complex to the cyclin D1 gene (19). It has been reported that SMRT expression is cell cycle-dependent with maximal expression during S phase (49), which would well place SMRT to promote the expression of genes, such as cyclin D1, that promote the E2-induced G1/S transition.
A modest ERα-independent component of growth control by SMRT was also indicated by the reduction in cell number that accompanied SMRT knock-down in ICI-treated MCF-7 cells. This was however, not observed for ERα-negative MDA-MB-231 breast cancer cells consistent with previous work indicating that codepletion of SMRT and NCoR did not alter the percent of these cells in S/G2/M (39), and the inability of SMRT knock-down to impact the growth of ERα-negative HeLa cells (14). Whether this reflects a cell-specific role for SMRT, perhaps through affecting the activity of an unknown transcription factor expressed in MCF-7 but not in MDA-MB-231 or HeLa cells, or is due to the low levels of SMRT in MDA-MB-231 versus MCF-7 cells is unknown. It was, however, somewhat surprising given the role of HDAC3 as a regulator of mitosis through deacetylation of histones and Aurora B kinase and formation of a functional mitotic spindle, as well as the localization of multiple members of the HDAC3 corepressor complex (eg, NCoR, TBL1, and TBLR1) to the mitotic spindle (28–31, 50). HDAC3 inhibitors disrupt cell growth (51), and although regulation of mitosis represents a nontranscriptional role for HDAC3 (28, 31), it would be reasonable to anticipate that SMRT and/or NCoR, as activators of HDAC3 activity, could contribute to these processes. However, the evidence for a role of these corepressors in cytokinesis and mitosis is conflicting. One study indicated that single knock-down of either SMRT or NCoR had no effect on mitosis of HeLa cells (31), whereas others report NCoR depletion led to abnormal spindle formation (28) and SMRT depletion resulted in cytokinesis failure (52). Regardless, there is no data on whether these possible effects of SMRT and/or NCoR depletion on mitosis translate into a measurable effect on the proliferation of cells over an extended growth period (eg, 5 days), and it has been suggested that the spindle phenotype of HDAC3-deficient cells may be transient (28). Whether SMRT depletion impacts mitosis or cytokinesis in breast cancer cells is the subject of ongoing investigations within our laboratory.
Cell cycle distribution and apoptosis analyses indicate that SMRT also promoted greater cell growth through inhibition of apoptosis. The apoptosis effect observed for SMRT-depleted cells was largely hormone independent, suggesting that SMRT regulation of this process was predominantly independent of ERα. That SMRT was potentially involved in control of apoptosis through regulation of gene expression was first indicated in the positive regulation of the antiapoptotic ERα target gene, Bcl-2 in MCF-7 cells (14), and with our gene ontology analysis of mRNA expression in SMRT-depleted versus control cells, it is clear that SMRT impacts a much broader array of genes to limit apoptosis. Notably, decreases in the expression of antiapoptotic genes and increases in proapoptotic gene expression noted at 52 hours post siRNA transfection (ie, 4 hours of treatment) occurred prior to the earliest detection of cleaved PARP at 72 hours, suggesting that changes in gene expression preceded apoptosis. It should also be noted, however, that some of the gene expression changes induced by SMRT depletion (eg, reduced proapoptotic BNIPL mRNA) may limit apoptosis, and the net effect of reduced SMRT expression on apoptosis undoubtedly reflects the integrated impact of the loss of this coregulator on pathways that promote and inhibit apoptosis.
A role for SMRT in regulating apoptosis may well have been expected given the ability of HDAC inhibitors that are in used as anticancer therapeutics, as well as HDAC3 knock-down to increase apoptosis (53). Indeed, the corepressor function of SMRT was recently implicated in regulating apoptotic responses in U2OS cells treated with the DNA damaging agent cisplatin through SMRT repression of Wip1 that dephosphorylates and thereby inactivates Chk2 (54). Depletion of SMRT also increased induced cell death in glioblastoma cell lines (55). With SMRT promoting the expression of a number of genes that inhibit apoptosis, including Birc5 and STRADB, our appreciation of the mechanistic roles of SMRT as an inhibitor of apoptosis is now expanded to include ERα-independent coactivation of antiapoptotic gene expression. Indeed, our data provide evidence of SMRT serving as a coactivator for antiapoptotic genes and a corepressor for proapoptotic genes, and indicate that the combined transcriptional roles of SMRT shift cells toward a relatively apoptotic-resistant state.
The abilities of breast tumor cells to proliferate and avoid apoptosis are key elements of carcinogenesis, and are often promoted through the actions of ERα and its coregulator proteins. As a positive regulator of cell cycle progression and inhibitor of apoptosis, SMRT is well positioned to coordinate multiple proliferation and survival pathways that can lead to increased tumor growth by SMRT serving as both corepressor and coactivator in a gene-specific manner. It also is possible that SMRT contributes indirectly to the growth of tumors cells by repressing the activity of transcription factors that are prodifferentiation (eg, retinoic acid receptor-α) or growth inhibitory (eg, vitamin D receptor). Whereas SMRT was originally thought to limit the activity of tamoxifen-bound ERα (46), data on its ability to contribute to the growth inhibitory effects of this antiestrogen on breast cancer cells are mixed with most studies failing to show any increase in growth after SMRT depletion (14, 39, 47, 56). Moreover, correlations between SMRT protein expression and clinical outcomes for breast cancer patients treated with tamoxifen are poor (21). In contrast, the data presented herein for SMRT promotion of breast cancer cell growth is consistent with one study demonstrating a positive correlation between nuclear SMRT expression and S-phase in human breast tumors (21), and several reports of a significant correlation between elevated SMRT protein levels and shorter time to tumor recurrence (20, 21). Taken together, this would suggest that elevated SMRT expression participates in the creation of a cellular environment in which estrogens and ERα can promote breast cancer cell proliferation, potentially through enhancing the activity of low levels of estrogens present in postmenopausal patients, as well as cell survival through the largely hormone-independent inhibition of apoptotic pathways afforded to cells by SMRT. Thus, SMRT employs both its coactivation and corepression activities to coordinately regulate cellular events that can promote the progression of breast cancer.
Acknowledgments
We thank Judy Roscoe, Cheryl Parker, and Javier Pacheco for their technical assistance, and Dr Suzanne Fuqua for her advice on soft agar assays.
The present address for S.K. is Food and Drug Administration, Center for Drug Evaluation and Research, Office of Generic Drugs, 7500 Standish Place, Rockville, Maryland 20855.
The present address for L.W. is Division of Biomedical Statistics and Informatics, Mayo Clinic, 200 first St SW, Rochester, MN 55905.
This work was supported, in part, by the Baylor College of Medicine's Genomic and RNA Profiling Core with funding from the National Institutes of Health NCI Grant (P30CA125123) and the expert assistance of Dr Lisa D. White, and the Cytometry and Cell Sorting Core with funding from the National Institutes of Health (NIAID P30AI036211, NCI P30CA125123, and NCRR S10RR024574) and the expert assistance of Joel M. Sederstrom, as well as by Public Health Service Grant DK53002 to C.L.S. from the National Institute for Diabetes, Digestive, and Kidney Diseases. S.K. was supported by a postdoctoral fellowship award (PDF 0707868) from the Susan G. Komen for the Cure Foundation, and J.K.B. received support from T32HD07165.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ERα
- estrogen receptor-α
- FBS
- fetal bovine serum
- HDAC3
- histone deacetylase 3
- sFBS
- charcoal-stripped fetal bovine serum
- SMRT
- silencing mediator of retinoic acid and thyroid hormone receptor
- SRC
- steroid receptor coactivator.
References
- 1. Siegel R, Naishadham D, Jemal A. Cancer Statistics, 2013. CA Cancer J Clin. 2013;63:11–30 [DOI] [PubMed] [Google Scholar]
- 2. Stephens PJ, Tarpey PS, Davies H, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674 [DOI] [PubMed] [Google Scholar]
- 4. Yue W, Yager JD, Wang JP, Jupe ER, Santen RJ. Estrogen receptor-dependent and independent mechanisms of breast cancer carcinogenesis. Steroids. 2013;78:161–170 [DOI] [PubMed] [Google Scholar]
- 5. Henderson BE, Ross R, Bernstein L. Estrogens as a cause of human cancer: the Richard and Hinda Rosenthal Foundation Award Lecture. Cancer Res. 1988;48:246–253 [PubMed] [Google Scholar]
- 6. Nilsson S, Mäkelä S, Treuter E, et al. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–1565 [DOI] [PubMed] [Google Scholar]
- 7. Pestell RG. New roles of cyclin D1. Am J Pathol. 2013;183:3–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maximov PY, Lee TM, Jordan VC. The discovery and development of selective estrogen receptor modulators (SERMs) for clinical practice. Curr Clin Pharmacol. 2013;8:135–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chumsri S, Howes T, Bao T, Sabnis G, Brodie A. Aromatase, aromatase inhibitors, and breast cancer. J Steroid Biochem Mol Biol. 2011;125:13–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080 [DOI] [PubMed] [Google Scholar]
- 11. Chen J, Kinyamu HK, Archer TK. Changes in attitude, changes in latitude: nuclear receptors remodeling chromatin to regulate transcription. Mol Endocrinol. 2006;20:1–13 [DOI] [PubMed] [Google Scholar]
- 12. Lonard DM, O'malley BW. Nuclear receptor coregulators: judges, juries, and executioners of cellular regulation. Molecular Cell. 2007;27:691–700 [DOI] [PubMed] [Google Scholar]
- 13. Frietze S, Lupien M, Silver PA, Brown M. CARM1 regulates estrogen-stimulated breast cancer growth through up-regulation of E2F1. Cancer Res. 2008;68:301–306 [DOI] [PubMed] [Google Scholar]
- 14. Peterson TJ, Karmakar S, Pace MC, Gao T, Smith CL. The silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) corepressor is required for full estrogen receptor alpha transcriptional activity. Mol Cell Biol. 2007;27:5933–5948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Karmakar S, Foster EA, Smith CL. Unique roles of p160 coactivators for regulation of breast cancer cell proliferation and estrogen receptor-alpha transcriptional activity. Endocrinology. 2009;150:1588–1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hudelist G, Czerwenka K, Kubista E, Marton E, Pischinger K, Singer CF. Expression of sex steroid receptors and their co-factors in normal and malignant breast tissue: AIB1 is a carcinoma-specific co-activator. Breast Cancer Res Treat. 2003;78:193–204 [DOI] [PubMed] [Google Scholar]
- 17. List HJ, Reiter R, Singh B, Wellstein A, Riegel AT. Expression of the nuclear coactivator AIB1 in normal and malignant breast tissue. Breast Cancer Res Treat. 2001;68:21–28 [DOI] [PubMed] [Google Scholar]
- 18. Privalsky ML. The role of corepressors in transcriptional regulation by nuclear hormone receptors. Annu Rev Physiol. 2004;66:315–360 [DOI] [PubMed] [Google Scholar]
- 19. Karmakar S, Gao T, Pace MC, Oesterreich S, Smith CL. Cooperative activation of cyclin D1 and progesterone receptor gene expression by the SRC-3 coactivator and SMRT corepressor. Mol Endocrinol. 2010;24:1187–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Green AR, Burney C, Granger CJ, et al. The prognostic significance of steroid receptor co-regulators in breast cancer: co-repressor NCOR2/SMRT is an independent indicator of poor outcome. Breast Cancer Res Treat. 2008;110:427–437 [DOI] [PubMed] [Google Scholar]
- 21. Smith CL, Migliaccio I, Chaubal V, et al. Elevated nuclear expression of the SMRT corepressor in breast cancer is associated with earlier tumor recurrence. Breast Cancer Res Treat. 2012;136:253–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Agthoven T, Sieuwerts AM, Veldscholte J, et al. CITED2 and NCOR2 in anti-oestrogen resistance and progression of breast cancer. Brit J Cancer. 2009;101:1824–1832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haiman CA, Garcia RR, Hsu C, et al. Screening and association testing of common coding variation in steroid hormone receptor co-activator and co-repressor genes in relation to breast cancer risk: the Multiethnic Cohort. BMC Cancer. 2009;9:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liao S, Desouki MM, Gaile DP, et al. Differential copy number aberrations in novel candidate genes associated with progression from in situ to invasive ductal carcinoma of the breast. Genes Chromosomes Cancer. 2012;51:1067–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jepsen K, Solum D, Zhou T, et al. SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature. 2007;450:415–419 [DOI] [PubMed] [Google Scholar]
- 26. Grewal C, Hickmott J, Rentas S, Karagiannis J. A conserved histone deacetylase with a role in the regulation of cytokinesis in Schizosaccharomyces pombe. Cell Div. 2012;7:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rentas S, Saberianfar R, Grewal C, et al. The SET domain protein, Set3p, promotes the reliable execution of cytokinesis in Schizosaccharomyces pombe. PLoS ONE. 2013;7:e31224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ishii S, Kurasawa Y, Wong J, Yu-Lee LY. Histone deacetylase 3 localizes to the mitotic spindle and is required for kinetochore-microtubule attachment. Proc Natl Acad Sci USA. 2008;105:4179–4184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fadri-Moskwik M, Weiderhold KN, Deeraksa A, et al. Aurora B is regulated by acetylation/deacetylation during mitosis in prostate cancer cells. FASEB J. 2012;26:4057–4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eot-Houllier G, Fulcrand G, Watanabe Y, Magnaghi-Jaulin L, Jaulin C. Histone deacetylase 3 is required for centromeric H3K4 deacetylation and sister chromatid cohesion. Genes Dev. 2008;22:2639–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li Y, Kao GD, Garcia BA, et al. A novel histone deacetylase pathway regulates mitosis by modulating aurora B kinase activity. Gene, Dev. 2006;20:2566–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guenther MG, Barak O, Lazar MA. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol. 2001;21:6091–6101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. You SH, Lim HW, Sun Z, Broache M, Won KJ, Lazar MA. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat Struct Mol Biol. 2013;20:182–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Giordano C, Cui Y, Barone I, et al. Growth factor-induced resistance to tamoxifen is associated with a mutation of estrogen receptor alpha and its phosphorylation at serine 305. Breast Cancer Res Treat. 2010;119:71–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Karmakar S, Foster EA, Blackmore JK, Smith CL. Distinctive functions of p160 steroid receptor coactivators in proliferation of an estrogen-independent, tamoxifen-resistant breast cancer cell line. Endocr Relat Cancer. 2011;18:113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38:500–501 [DOI] [PubMed] [Google Scholar]
- 37. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57 [DOI] [PubMed] [Google Scholar]
- 38. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Keeton EK, Brown M. Cell cycle progression stimulated by tamoxifen-bound estrogen receptor-alpha and promoter-specific effects in breast cancer cells deficient in N-CoR and SMRT. Mol Endocrinol. 2005;19:1543–1554 [DOI] [PubMed] [Google Scholar]
- 40. Prall OW, Rogan EM, Musgrove EA, Watts CK, Sutherland RL. c-Myc or cyclin D1 mimics estrogen effects on cyclin E-Cdk2 activation and cell cycle reentry. Mol Cell Biol. 1998;18:4499–4508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu X, Zhang X, Dhakal IB, Beggs M, Kadlubar S, Luo D. Induction of cell proliferation and survival genes by estradiol-repressed microRNAs in breast cancer cells. BMC Cancer. 2012;12:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhu J, Lu X, Hua KQ, Sun H, Yu YH, Feng YJ. Oestrogen receptor α mediates 17β-estradiol enhancement of ovarian cancer cell motility through up-regulation of survivin expression. Arch Gynecol Obstet. 2012;286:729–737 [DOI] [PubMed] [Google Scholar]
- 43. Sanna MG, da Silva Correia J, Luo Y, et al. ILPIP, a novel anti-apoptotic protein that enhances XIAP-mediated activation of JNK1 and protection against apoptosis. J Biol Chem. 2002;277:30454–30462 [DOI] [PubMed] [Google Scholar]
- 44. Wiley SR, Schooley K, Smolak PJ, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–682 [DOI] [PubMed] [Google Scholar]
- 45. Liston P, Fong WG, Kelly NL, et al. Identification of XAF1 as an antagonist of XIAP anti-Caspase activity. Nat Cell Biol. 2001;3:128–133 [DOI] [PubMed] [Google Scholar]
- 46. Smith CL, Nawaz Z, O'Malley BW. Coactivator and corepressor regulation of the agonist/antagonist activity of the mixed antiestrogen, 4-hydroxytamoxifen. Mol Endocrinol. 1997;11:657–666 [DOI] [PubMed] [Google Scholar]
- 47. Cheng X, Kao HY. G protein pathway suppressor 2 (GPS2) is a transcriptional corepressor important for estrogen receptor alpha-mediated transcriptional regulation. J Biol Chem. 2009;284:36395–36404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Planas-Silva MD, Shang Y, Donaher JL, Brown M, Weinberg RA. AIB1 enhances estrogen-dependent induction of cyclin D1 expression. Cancer Res. 2001;61:3858–3862 [PubMed] [Google Scholar]
- 49. Park EJ, Schroen DJ, Yang M, Li H, Li L, Chen JD. SMRTe, a silencing mediator for retinoid and thyroid hormone receptors-extended isoform that is more related to the nuclear receptor corepressor. Proc Natl Acad Sci USA. 1999;96:3519–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. O'Reilly MA, Gazdar AF, Clark JC, Pilot-Matias TJ, Wert SE, Hull WM, Whitsett JA. Glucocorticoids regulate surfactant protein synthesis in a pulmonary adenocarcinoma cell line. Am J Physiol. 1989;257:L385–L392 [DOI] [PubMed] [Google Scholar]
- 51. Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202 [DOI] [PubMed] [Google Scholar]
- 52. Kittler R, Pelletier L, Heninger AK, et al. Genome-scale RNAi profiling of cell division in human tissue culture cells. Nat Cell Biol. 2007;9:1401–1412 [DOI] [PubMed] [Google Scholar]
- 53. New M, Olzscha H, La Thangue NB. HDAC inhibitor-based therapies: Can we interpret the code? Mol Oncol. 2012;6:637–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Scafoglio C, Smolka M, Zhou H, Perissi V, Rosenfeld MG. The co-repressor SMRT delays DNA damage-induced caspase activation by repressing pro-apoptotic genes and modulating the dynamics of checkpoint kinase 2 activation. PLoS ONE. 2013;8:e59986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Alrfaei BM, Vemuganti R, Kuo JS. microRNA-100 targets SMRT/NCOR2, reduces proliferation, and improves survival in glioblastoma animal models. PLoS ONE. 2013;8:e80865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stanya KJ, Liu Y, Means AR, Kao HY. Cdk2 and Pin1 negatively regulate the transcriptional corepressor SMRT. J Cell Biol. 2008;183:49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]