

Abstract
Protein-tyrosine phosphatase 1B (PTP1B) is a physiological regulator of glucose homeostasis and energy balance. However, the role of PTP1B in pancreatic endocrine function remains largely unknown. To investigate the metabolic role of pancreatic PTP1B, we generated mice with pancreas PTP1B deletion (panc-PTP1B KO). Mice were fed regular chow or a high-fat diet, and metabolic parameters, insulin secretion and glucose tolerance were determined. On regular chow, panc-PTP1B KO and control mice exhibited comparable glucose tolerance whereas aged panc-PTP1B KO exhibited mild glucose intolerance. Furthermore, high-fat feeding promoted earlier impairment of glucose tolerance and attenuated glucose-stimulated insulin secretion in panc-PTP1B KO mice. The secretory defect in glucose-stimulated insulin secretion was recapitulated in primary islets ex vivo, suggesting that the effects were likely cell-autonomous. At the molecular level, PTP1B deficiency in vivo enhanced basal and glucose-stimulated tyrosyl phosphorylation of EphA5 in islets. Consistently, PTP1B overexpression in the glucose-responsive MIN6 β-cell line attenuated EphA5 tyrosyl phosphorylation, and substrate trapping identified EphA5 as a PTP1B substrate. In summary, these studies identify a novel role for PTP1B in pancreatic endocrine function.
Type 2 diabetes is one of the most prevalent metabolic diseases and is characterized by hyperinsulinemia, insulin resistance, and pancreatic β-cell dysfunction (1, 2). Pancreatic islets regulate the insulin secretory response that is critical for maintaining glucose homeostasis. Impaired insulin secretion caused by loss and/or dysfunction of β-cells is observed in most forms of type 2 diabetes and is considered essential for the manifestation of the disease (3, 4). β-Cells respond to fluctuations in blood glucose level with the regulated secretion of insulin. Communication between β-cells ensures suppression of basal insulin secretion and enhancement of glucose-stimulated insulin secretion (GSIS) (5). This enables the secretion of low amounts of insulin during starvation and higher levels during feeding. However, the molecular mechanisms that underlie communication between β-cells remain largely unknown.
Tyrosyl phosphorylation is an important regulator of insulin secretion and action and is controlled by the dynamic and opposing actions of protein tyrosine kinases and protein-tyrosine phosphatases (PTPs). Eph receptor tyrosine kinases and their plasma membrane-bound ephrin ligands play multiple roles including regulation of cell-cell communication (6). Ephs and ephrins are divided into A and B subclasses, with most EphAs binding to ephrin As (A1–A5) and most EphBs binding to ephrin Bs (B1–B3) (6). A unique feature of the Eph-ephrin system is bidirectional signaling that originates from the Eph receptor (forward signaling) and ephrin ligands (reverse signaling). Importantly, EphA-ephrin A signaling is critical for β-cell communication and insulin secretion (7). EphA5 signaling inhibits whereas ephrin A5 signaling promotes GSIS. Of note, EphA5 tyrosyl phosphorylation decreases after glucose stimulation and alleviates the inhibitory effects on insulin secretion (7). Thus, under basal conditions, EphA5 signaling inhibits insulin secretion, but upon glucose stimulation, dephosphorylation of EphA5 attenuates its signaling and favors ephrin A5 signaling, leading to insulin secretion. However, the identity of the phosphatases that regulate EphA5 signaling in β-cells remains to be determined.
PTP1B is a ubiquitously expressed phosphatase that is localized on the cytoplasmic face of the endoplasmic reticulum (ER) via a hydrophobic C-terminal targeting sequence (8–10). Insights into the physiological functions of PTP1B have emerged from studies in mice with global PTP1B deficiency. Whole-body PTP1B knockout (KO) mice exhibit improved systemic insulin sensitivity and enhanced glucose tolerance and are resistant to high-fat diet (HFD)-induced obesity (11–14). The metabolic role of PTP1B in liver, muscle, adipose, and brain has been reported (15–19), but its function in the pancreas remains largely unknown. Previously, we showed with a compound KO mouse model that PTP1B global deficiency could mitigate the severe diabetes caused by insulin receptor substrate 2 (IRS2) deletion. In this model, PTP1B deficiency increased islet area, enhanced glucose tolerance, and delayed diabetes (20). More recently, we demonstrated that PTP1B modulates ER stress signaling in the glucose-responsive MIN6 β-cell line, and alterations in pancreatic PTP1B expression may serve as an adaptive response for the mitigation of chronic ER stress (21). In the current study, we assessed the physiological role of PTP1B using the Cre-loxP approach to delete PTP1B in the pancreas. Metabolic changes, insulin secretion, and systemic glucose homeostasis were determined in chow-fed vs HFD-fed control and pancreatic PTP1B KO mice, and the underlying molecular mechanism was investigated.
Materials and Methods
Mouse studies
PTP1B-floxed (PTP1Bfl/fl) mice on a 129Sv/J × C57BL/6J background were generated previously (18). Pdx1-Cre mice on C57BL/6J background were provided by Dr D. Melton (Harvard University) (22). All mice studied were age-matched and maintained on a 12-hours light, 12- hour dark cycle in a temperature-controlled facility, with free access to water and food. Mice were fed standard laboratory chow (Purina Lab chow 5001) at weaning and, in a separate cohort, switched to an HFD (60% kcal from fat) (D12492; Research Diets) at 4 weeks of age. Genotyping for the PTP1B floxed allele and for the presence of Cre was performed by PCR using DNA extracted from tails as previously described (18). All mouse studies were conducted according to federal guidelines and were approved by the Institutional Animal Care and Use Committee at University of California Davis.
Metabolic measurements
Glucose was measured in blood collected from the tail using a glucometer (Home Aide Diagnostics). Insulin concentration in serum and primary islets culture media was determined by an ultrasensitive mouse insulin ELISA kit (Crystal Chem). Fed glucose measurements were performed between 7:00 and 9:00 am and, where indicated, from mice fasted for 12 hours. For insulin tolerance tests (ITTs), mice were fasted for 4 hours and injected ip with 0.8 to 1 mU/g human insulin (HumulinR; Eli Lilly). Blood glucose values were measured before and at 15, 30, 60, and 90 minutes after injection. For ip glucose tolerance tests (GTTs), overnight-fasted mice were injected with 20% d-glucose (2 mg/g body weight), and blood glucose was measured before and at 15, 30, 60, and 120 minutes after injection. In vivo GSIS was performed on overnight-fasted mice injected with 20% d-glucose (3 mg/g body weight), and tail blood was collected before and at 2, 5, 10, 20, and 30 minutes after injection, blood insulin, and C-peptide concentrations were determined using ELISA and RIA kits (Linco Research), respectively. For ex vivo GSIS, primary islets from pancreas PTP1B (panc-PTP1B) KO and control mice were isolated using the intra-ductal collagenase method (23). After overnight culture, 20 size-matched islets were hand-picked and starved for 1 hour in Krebs-Ringer buffer (KRB) containing 2mM glucose as described (7). Islets were then incubated in Krebs-Ringer buffer containing 3mM, 7mM, 11mM, and 16.7mM glucose for 30, 60, and 120 minutes. Media were collected for insulin measurement and islets lysed to determine total protein content. Insulin levels were normalized to islet protein content.
Cell culture and immunostaining
MIN6 β-cells were maintained in DMEM containing 25mM glucose, 15% fetal bovine serum, 50 U/mL penicillin, and 50 μg/mL streptomycin. For overexpression studies, MIN6 β-cells were infected with retrovirus (pWZL) expressing wild-type (WT) human PTP1B (hPTP1B), and hygromycin-resistant pools of cells were generated. The control MIN6 β-cell line was generated from cells infected with empty (pWZL) vector. For immunofluorescence, MIN6 β-cells expressing hPTP1B were fixed using methanol at −20°C for 15 minutes and then incubated with EphA5 (1:100; Santa Cruz Biotechnology) and hPTP1B (FG6, 1:200; Abcam) antibodies. Fluorophore-conjugated secondary antibodies (1:400; Jackson Immunoresearch) were used to visualize EphA5 and PTP1B, and images were acquired using an Olympus FluoView FV1000 confocal microscope and FV10-ASW software (Olympus). For immunohistochemistry, pancreata from control and panc-PTP1B KO mice were fixed in Z-fix (Fisher Scientific) and embedded in paraffin. Sections were stained with insulin (1:100; Zymed Laboratories), mouse PTP1B (1:100; Millipore), and EphA5 (1:100; Santa Cruz Biotechnology) antibodies overnight at 4°C. Detection was performed with appropriate fluorophore-conjugated secondary antibodies (1:200; Jackson Immunoresearch) and visualized using Olympus BX51 microscope. Islet size and number were determined by using ImageJ software (National Institutes of Health, http://rsb.info.nih.gov/ij/).
Biochemical analyses
Pancreatic islets and MIN6 β-cells were lysed using a radioimmunoprecipitation assay buffer (10mM Tris-HCl [pH: 7.4], 150mM NaCl, 0.1% sodium dodecyl sulfate, 1% Triton X-100, 1% sodium deoxycholate, 5mM EDTA, 1mM NaF, 1mM sodium orthovanadate, and protease inhibitors). For substrate-trapping experiments, cells were lysed in 1% Nonidet P-40 buffer with a protease inhibitor cocktail (without sodium orthovanadate) as previously described (24–26). Lysates were cleared by centrifugation at 13 000 rpm for 10 minutes and protein concentrations determined using a bicinchoninic acid protein assay kit (Pierce Chemical). Proteins (500 μg) were subjected to immunoprecipitation using EphA5 antibodies (Santa Cruz Biotechnology), and immune complexes were collected on protein G-Sepharose beads (GE Healthcare) and washed with lysis buffer. Proteins were resolved by SDS-PAGE and transferred to polyvinylidene difluoride membranes. Immunoblotting of lysates and immunoprecipitates was performed with antibodies for phosphotyrosine (4G10) (1:5000; Millipore), EphA5 (1:500), mouse PTP1B (1:1000), and hPTP1B (1:2000). After incubation with appropriate secondary antibodies, proteins were visualized using enhanced chemiluminescence (ECL; Amersham Biosciences). Pixel intensities of immunoreactive bands were quantified using ImageQuant software (Amersham Biosciences).
Statistical analyses
Data are expressed as means ± SEM. Repeated-measures ANOVA was used to analyze multiple comparisons, ie, ITTs and GTTs. Single data point comparisons were performed using Tukey-Kramer Honest Significant Difference analyses, both analyses were performed using the JMP program (SAS Institute). Differences were considered significant at P ≤ .05 and highly significant at P ≤ .01.
Results
Generation of panc-PTP1B KO mice
To determine the metabolic role of pancreatic PTP1B, we crossed PTP1Bfl/fl mice with those expressing Cre recombinase under the control of pancreatic and duodenal homeobox 1 (Pdx1) promoter to generate mice lacking PTP1B in the (endocrine and exocrine) pancreas (22). PTP1Bfl/fl mice (controls) and panc-PTP1B KO appeared normal and did not display gross defects in the pancreas. Immunoblot analyses of primary islets revealed a significant reduction in PTP1B protein expression in panc-PTP1B KO compared with control islets (Figure 1A). Comparable expression of PTP1B was observed in the pancreas of WT and PTP1Bfl/fl (control) mice (data not shown). Expression of the closely related T-cell PTP (27) and another phosphatase that is implicated in β-cell function, Src homology phosphatase 2 (28), was not significantly altered upon PTP1B deletion. Importantly, PTP1B expression was unchanged in peripheral insulin-responsive tissues (liver, muscle, and fat) or the brain (Figure 1A). Immunostaining pancreata sections from control mice revealed PTP1B expression that was stronger in islets compared with the surrounding exocrine tissue (Figure 1B). PTP1B staining was exclusively cytosolic, consistent with its localization on the ER (8). Consistent with biochemical data, PTP1B staining was significantly decreased in panc-PTP1B KO islets, confirming deletion. Together, these data demonstrate that PTP1B is expressed in islets and indicate that the knockout approach results in efficient deletion of PTP1B in islets, enabling the determination of its metabolic role in the endocrine pancreas.
Figure 1.
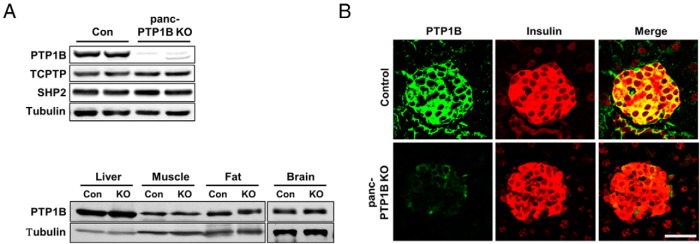
PTP1B deletion in the pancreas. A, Upper panel, Immunoblots of PTP1B expression in primary islets from PTP1Bfl/fl (control [Con]) and panc-PTP1B KO female mice fed an HFD at 28 weeks of age. Lysates were also probed for T-cell PTP (TCPTP), Src homology phosphatase 2 (SHP2), and tubulin. Lower panel, Immunoblots of PTP1B expression in liver, muscle, fat, and brain of control and panc-PTP1B KO mice. Lysates were also probed for tubulin. B, Confocal images of pancreas sections from female control and panc-PTP1B KO mice fed regular chow at 20 weeks of age stained for PTP1B (green) and insulin (red) and merged. Scale bar, 100 μm.
High-fat feeding impairs glucose tolerance in mice with pancreatic PTP1B deletion
To investigate the physiological effects of pancreatic PTP1B deletion, we measured body weights of female and male panc-PTP1B KO and control mice fed regular chow and an HFD. As expected, mice fed an HFD gained significantly more weight compared with their chow-fed littermates, and control and panc-PTP1B KO mice exhibited comparable body weights on either diet (Figure 2, A and B). This suggests that any differences in glucose tolerance are primary and not due to body weight alterations. Fed and fasted glucose and insulin concentrations were comparable between panc-PTP1B KO and control mice fed regular chow and HFD (Supplemental Table 1 and Supplemental Figure 1, A and B). To directly assess the effects of pancreatic PTP1B deletion on glucose homeostasis, we determined the ability of mice to dispose of a glucose load during GTTs. Control and panc-PTP1B KO mice of both genders exhibited comparable glucose tolerance when fed regular chow (Figure 2, C–F). However, when fed an HFD, panc-PTP1B KO mice exhibited a reduced ability to dispose of glucose load when compared with controls (Figure 2, C–F). Glucose intolerance was observed in both sexes but was more pronounced in females; hence, they were used in subsequent studies. To determine whether differences in peripheral insulin sensitivity contribute to the glucose intolerance of panc-PTP1B KO mice, we performed ITTs. Insulin sensitivity was comparable between the 2 groups when fed regular chow and an HFD (Figure 2, G and H). It is worth noting that when glucose levels were analyzed as percent change from basal, comparable insulin sensitivity was also observed (Supplemental Figure 1, C and D). Thus, the glucose intolerance in panc-PTP1B KO mice was not caused by differential insulin sensitivity. Comparable results were obtained in an independent cohort of mice (Supplemental Figure 1, E–G). Of note, old (72 weeks) panc-PTP1B KO mice fed regular chow exhibited attenuated glucose tolerance compared with controls without significant alterations in peripheral insulin sensitivity (Supplemental Figure 1, H and I). This suggests that the effects of aging resemble that of high-fat feeding in panc-PTP1B KO mice. Together, these data demonstrate that high-fat feeding and/or aging promote glucose intolerance in panc-PTP1B KO mice.
Figure 2.

Impaired glucose tolerance in mice with pancreatic PTP1B deletion fed an HFD. Body weights of female (A) and male (B) control (Con) and panc-PTP1B KO mice fed regular chow (full lines) and an HFD (dotted lines). GTTs in female (C and D) and male (E and F) control and panc-PTP1B KO mice fed regular chow and HFD at 8 (C and E) and 24 (D and F) weeks of age. ITTs in female (G) and male (H) control and panc-PTP1B KO mice at 24 weeks of age. Data are presented as mean ± SEM; n = 6–9. *, P ≤ .05; **, P ≤ .01 control vs panc-PTP1B KO mice.
Pancreatic PTP1B deficiency attenuates GSIS
To determine whether the impaired glucose tolerance in panc-PTP1B KO mice is caused by defects in insulin secretion, we evaluated insulin secretory responses to glucose in vivo in mice fed regular chow and HFD. GSIS occurs in 2 phases, a rapid initial phase (∼2–3 minutes after glucose in mice) and a prolonged phase that lasts up to 30 minutes. Accordingly, we determined basal insulin and GSIS in control and panc-PTP1B KO mice during the initial and prolonged phases. Data are presented as absolute level of circulating insulin as well as relative increment from basal because it is an adequate index of an insulin response to a glucose stimulus (29–31). The panc-PTP1B KO mice exhibited a trend for increased basal insulin but did not reach statistical significance (Figure 3, A and B). When fed an HFD, control mice exhibited a robust increase in insulin secretion 2 minutes after glucose injection, and insulin concentrations remained high at 30 minutes, indicating a second-phase response (Figure 3, A and B). In contrast, panc-PTP1B KO mice exhibited attenuated initial and prolonged phases of GSIS compared with controls. To rule out differential insulin clearance, serum C-peptide (a surrogate marker for insulin secretion) concentrations were measured in the same cohort of mice (Figure 3C). A comparable pattern of C-peptide was observed relative to insulin, suggesting no differences in insulin clearance. Similar findings were observed in an independent cohort of mice fed an HFD (Supplemental Figure 2, A and B). To examine whether altered GSIS in panc-PTP1B KO mice was due to decreased islet mass, we immunostained pancreas sections for insulin and quantified insulin-positive stained areas. No significant differences in islets size and number were observed between panc-PTP1B KO and control mice (Supplemental Figure 3A). In addition, pancreas weights were comparable between groups when fed regular chow or HFD. On regular chow, pancreas weight of control and panc-PTP1B KO female mice (7 months) were 0.35 ± 0.03 vs 0.33 ± 0.05 g, respectively (P = .66). When fed an HFD, the pancreas weight of control and panc-PTP1B KO female mice (7 months) were 0.41 ± 0.06 vs 0.45 ± 0.06 g, respectively (P = .67). When fed regular chow, control and panc-PTP1B KO mice exhibited comparable GSIS (Supplemental Figure 2C) in line with their comparable glucose tolerance. On the other hand, old panc-PTP1B KO mice fed regular chow exhibited attenuated GSIS compared with controls consistent with their attenuated glucose tolerance (Supplemental Figure 2D). Together, these data indicate that high-fat feeding and/or aging lead to attenuated GSIS in panc-PTP1B KO mice.
Figure 3.

PTP1B deficiency attenuates GSIS in vivo and ex vivo in isolated islets. A and B, In vivo GSIS. Insulin secretion at 2 and 5 minutes (A) and 10, 20, and 30 minutes (B) after ip injection of glucose in female control (Con) and panc-PTP1B KO mice fed an HFD at 11 (A) and 24 (B) weeks of age. Fold change from basal is shown on the left and circulating insulin concentrations on the right. C, C-peptide concentrations were determined in the same mice at 10 weeks of age and presented as fold change from basal and absolute concentration. D, Primary islets from control and panc-PTP1B KO mice (n = 3 per genotype) fed an HFD at 16 weeks of age were treated with increasing concentrations of glucose (Gluc) for 30, 60, and 120 minutes. Insulin concentrations were normalized to islet protein content, and representative data are shown as mean ± SEM (n = 3). *, Significant differences between groups; #, significant differences between basal and glucose-stimulated condition within a group.
To ascertain that attenuated GSIS in panc-PTP1B KO mice fed an HFD was due to PTP1B deficiency in islets and not in other pancreatic cell types and/or secondary to neural effects and secreted factors, we evaluated insulin secretion in isolated primary islets ex vivo. Primary islets from control and panc-PTP1B KO mice fed an HFD were isolated and cultured as described in Materials and Methods. Size-matched islets were subjected to step-up glucose stimulation for different periods. Control islets exhibited a significant dose- and time-dependent GSIS, whereas islets from panc-PTP1B KO mice exhibited significantly lower GSIS at 60 and 120 minutes (Figure 3D). Additionally, panc-PTP1B KO islets exhibited a comparable response to glucagon-like peptide-1 as controls (Supplemental Figure 3B), suggesting a preserved glucagon-like peptide-1 response in KO primary islets. These data are consistent with observations in vivo and suggest that PTP1B deficiency modulates GSIS. Collectively, these findings indicate that attenuated GSIS in panc-PTP1B KO mice is likely due to a cell autonomous defect, although the effect of other factors cannot be completely excluded.
PTP1B regulates EphA5 tyrosyl phosphorylation in β-cells
EphA5 signaling is critical for β-cell communication and insulin secretion (7), and pharmacological inhibition of Eph receptors enhances GSIS from mouse and human islets (32). In addition, PTP1B regulates EphA3 function and trafficking (33) and EphA2 tyrosyl phosphorylation (34). Immunostaining of EphA5 in pancreata revealed expression in islets as previously reported (7) (Figure 4A and Supplemental Figure 4). To determine whether PTP1B and EphA5 colocalize, and because currently available antibodies preclude costaining in mouse pancreatic sections, we generated MIN6 β-cells that overexpress hPTP1B. hPTP1B exhibited reticular staining that is characteristic of its ER localization and revealed significant colocalization with endogenous EphA5 (Figure 4B). To establish whether PTP1B regulates EphA5 signaling, EphA5 tyrosyl phosphorylation was evaluated in primary islets from control and panc-PTP1B KO mice fed an HFD. Primary islets from control and panc-PTP1B KO mice fed an HFD were isolated, cultured overnight, and stimulated with glucose and EphA5 tyrosyl phosphorylation was determined. EphA5 immunoprecipitates revealed elevated basal and glucose-stimulated EphA5 tyrosyl phosphorylation in panc-PTP1B KO islets compared with controls (Figure 4C). In addition, and consistent with a previous report (7), control and panc-PTP1B KO islets exhibited higher EphA5 tyrosyl phosphorylation under basal than glucose-stimulated conditions. Enhanced EphA5 tyrosyl phosphorylation in panc-PTP1B KO mice did not lead to significant alterations in ERK1/2 or AKT phosphorylation (Figure 4D). To determine whether enhanced EphA5 tyrosyl phosphorylation in panc-PTP1B KO islets is cell autonomous, we used MIN6 β-cells overexpressing hPTP1B. Basal and glucose-stimulated EphA5 tyrosyl phosphorylation was reduced in MIN6 β-cells overexpressing hPTP1B compared with controls (Figure 4E).
Figure 4.

PTP1B regulates EphA5 tyrosyl phosphorylation in islets and MIN6 β-cells. A, Confocal images of pancreas sections stained for EphA5 (green) and insulin (red) in control female mice fed regular chow for 3 months. Boxed areas are magnified in the insets. Scale bar, 100 μm. B, Confocal images of MIN6 β-cells stained for hPTP1B (green) and endogenous EphA5 (red). C, Primary islets from control (Con) and panc-PTP1B KO mice fed an HFD were starved then stimulated with 25mM glucose for 10 minutes. EphA5 immunoprecipitates were immunoblotted for phosphotyrosine (pTyr) and EphA5. Bar graph represents normalized data from 3 mice per group. Data are presented as mean ± SEM. *, Significant differences between groups; #, significant differences between basal and glucose-stimulated conditions within a group. D, Islet lysates were immunoblotted for pERK, ERK, Akt, and Akt. Data are presented as mean ± SEM; n = 4. E, EphA5 tyrosyl phosphorylation in MIN6 β-cells overexpressing hPTP1B. Cell lysates were immunoblotted for hPTP1B, mPTP1B, and tubulin (top panel). EphA5 was immunoprecipitated from control and MIN6 β-cells overexpressing hPTP1B under basal (0) and glucose-stimulated (5 and 10 minutes) conditions and then immunoblotted for pTyr and EphA5. Data of pEphA5/EphA5 are presented relative to control (0) and as mean ± SEM; n = 3. *, Significant differences between groups; #, significant differences between basal and glucose-stimulated conditions within a group. F, MIN6 β-cells were transfected with WT hPTP1B and substrate-trapping mutant D/A. Cells were starved and then stimulated with glucose (25mM) and lysed as indicated in Materials and Methods with and without vanadate treatment. hPTP1B was immunoprecipitated and then immunoblotted for endogenous EphA5 and hPTP1B. Abbreviations: IB, immunoblot; IP, immunoprecipitation.
The regulation of EphA5 tyrosyl phosphorylation by PTP1B prompted us to test whether EphA5 is a direct PTP1B substrate in β-cells. WT PTP1B has a high catalytic constant, rendering its interaction with substrates difficult to detect (35). However, steady-state interaction of PTP1B with its substrates can be enhanced using the substrate-trapping mutant (PTP1B D181A [D/A]) that forms stable complexes with tyrosine-phosphorylated substrates and was successfully used to identify PTP1B substrates (10, 36–38). WT hPTP1B and D/A were transiently expressed in MIN6 β-cells, and hPTP1B was immunoprecipitated and co-association with EphA5 was determined under basal and glucose-stimulated conditions (Figure 4F). As expected, no significant association was detected using WT hPTP1B, but co-association was observed with hPTP1B D/A under basal and glucose-stimulated conditions. Importantly, treatment with pervanadate, a strong inhibitor of tyrosine phosphatases that oxidizes the essential cysteinyl residue in the catalytic center of the enzymes (39), disrupted PTP1B D/A-EphA5 interactions (Figure 4F). This indicated that the association was consistent with enzyme-substrate interaction that is mediated by the active site cysteinyl residue of PTP1B. Together, findings in primary islets and MIN6 β-cells indicate that EphA5 is a potential target of PTP1B in the endocrine pancreas.
Discussion
Deciphering the molecular mechanisms that regulate pancreatic β-cell communication and insulin secretion is a prerequisite for treating type 2 diabetes. The metabolic role of pancreatic PTP1B in β-cell function has heretofore remained largely unexplored. In the current study, we used gene-KO and biochemical approaches to demonstrate a role for PTP1B in pancreas endocrine function. Pancreatic PTP1B deficiency impaired glucose tolerance and attenuated GSIS in mice fed an HFD and in old mice fed regular chow. We also demonstrated that PTP1B regulates EphA5 tyrosyl phosphorylation in islets and in MIN6 β-cells and identified EphA5 as a potential PTP1B substrate in the endocrine pancreas. Collectively, these findings provide new insights into pancreatic PTP1B function and regulation of glucose homeostasis.
Mice with pancreatic PTP1B deficiency exhibited comparable glucose tolerance to controls. However, when subjected to a robust challenge (such as prolonged high-fat feeding or aging) pancreatic PTP1B deficiency impaired glucose tolerance and attenuated GSIS. Tissue-specific deletion of PTP1B helped define its physiological function in the brain (18), muscle (17), liver (15), and adipose tissue (19). To determine the precise pancreas-specific function of PTP1B, we used Pdx1-Cre transgenic mice to generate pancreas PTP1B KO mice. Although some studies indicate that Pdx1-Cre transgenic lines exhibit expression of Cre recombinase in select regions of the brain including the hypothalamus (41, 42) we observed efficient and selective PTP1B deletion only in the pancreas without changes in expression in other tissues including the brain. Although we recognize that immunoblotting of whole-brain lysates is not adequate to detect selected deletion in the hypothalamus, the comparable gain in body weights in the panc-PTP1B KO and controls both on chow and HFD suggests this issue is not a confounding factor in our studies. This is especially relevant given that PTP1B deletion in brain and hypothalamus has profound effects on body weight (18, 43), a feature that is not observed in our studies. Thus, panc-PTP1B KO mice provide a useful platform to dissect the metabolic functions of pancreatic PTP1B. When challenged with high-fat feeding, panc-PTP1B KO mice exhibit glucose intolerance that was independent of altered insulin sensitivity in the periphery and was due, in large part, to attenuated GSIS. Importantly, the defect in GSIS was likely cell autonomous because it was recapitulated in isolated primary islets although we cannot completely rule out effects secondary to non–β-cells. These findings were not expected given the salutary metabolic effects of whole-body PTP1B deficiency (11, 12) as well as its deletion in IRS2 KO mice (20). However, the phenotype of whole-body PTP1B KO mice is the net effect of PTP1B ablation in various tissues. In addition, the beneficial metabolic effects of PTP1B deletion in IRS2 KO mice are in the context of disrupted insulin signaling in the KO mice, and the enhanced islet area in compound PTP1B/IRS2 KO mice is likely due to improved insulin signaling. Finally, the current findings are in contrast to adenoviral-mediated knockdown of PTP1B in rat islets that demonstrates improved GSIS under conditions of high-fat feeding (44). The reason for the discrepancy with our studies is unclear and could be due, in part, to the different methodologies and/or effects of non–β-cells. Our findings that pancreatic PTP1B deletion is influenced by diet and age are relevant to observations in humans who become more susceptible to develop glucose intolerance and overt diabetes when they age or consume a high-calorie diet. We also observed that the phenotypes in female panc-PTP1B KO mice were more pronounced compared with males. Indeed, gender-related effects on insulin secretion have been reported, and the loss of the transcription factor aryl hydrocarbon receptor nuclear translocator (ARNT) in islets has been shown to lead to severe insulin secretory defects in female mice (45). In addition, in G protein-coupled receptor 39 KO mice, only female mice exhibit significant reduction in insulin secretion upon glucose challenge (46).
The current studies identify EphA5 as a potential PTP1B substrate in the endocrine pancreas. PTP1B colocalized with EphA5 in MIN6 β-cells and in islets, and PTP1B deficiency increased basal and glucose-stimulated EphA5 tyrosyl phosphorylation. This led to enhanced EphA5 signaling and attenuated GSIS as previously reported (7). Despite the enhanced basal EphA5 tyrosyl phosphorylation in panc-PTP1B KO, basal insulin secretion was not reduced, in agreement with previous studies (7). Moreover, substrate trapping demonstrated a direct association between EphA5 and PTP1B D/A consistent with enzyme-substrate interaction occurring by the phosphatase active site. Collectively, these findings demonstrate an association between PTP1B and EphA5; however, the dynamics of this interaction and its direct contribution to GSIS, if any, require additional investigation. PTP1B-EphA5 interaction is modulated, at least in part, by their subcellular localization. PTP1B is anchored to the ER, limiting its access to substrates (8). PTP1B colocalizes with EphA5 at discrete intracellular sites where they can interact, but we cannot rule out interactions at other regions. Indeed, high-resolution imaging reveals interactions between PTP1B and EphA3 at endosomal vesicles as well as regions of cell-cell contact (33), in agreement with PTP1B's ability to engage targets at these sites (34). Although the potential contribution of PTP1B-EphA5 interaction to GSIS remains to be determined, one possibility is that high glucose leads to increased PTP1B activity, thereby decreasing EphA5 tyrosyl phosphorylation and insulin secretion. In support of this scenario, we observed increased PTP1B enzymatic activity after glucose stimulation (data not shown). Of note, PTP1B expression is increased in pancreas lysates of HFD-fed mice (21) and in islets of HFD-fed rats (44). However, in the context of the current findings, HFD-induced increased pancreatic PTP1B expression will decrease EphA5 tyrosyl phosphorylation and attenuate its signal to favor insulin secretion, which is counterintuitive to the effect of HFD on GSIS. It is worth noting that PTP1B can conceivably regulate pancreatic function through several effectors and signaling pathways. PTP1B is a key regulator of insulin (11, 12), leptin (13, 14), c-Src (47), and ER stress (21) signaling pathways, which play important roles in β-cell function (48–55). Indeed, preliminary studies indicate that panc-PTP1B islets exhibit increased expression of ER stress markers compared with controls (Supplemental Figure 5), suggesting that PTP1B deficiency may modulate islet function through an ER stress response. Finally, we recently identified Munc18c (26) and pyruvate kinase M2 (25) as PTP1B substrates in adipose tissue that can also contribute to islet function (40, 56).
In summary, our findings provide new insights into the role of pancreatic PTP1B in regulating glucose tolerance and identify EphA5 as a novel PTP1B substrate. These findings are directly relevant to type 2 diabetes and obesity, considering that PTP1B is already being harnessed as a potential therapeutic target.
Acknowledgments
We thank Dr Benjamin Neel (Ontario Cancer Institute) for PTP1Bfl/fl mice and Dr Doug Melton (Harvard University) for Pdx1-Cre transgenic mice.
This work was supported by research grant from the Juvenile Diabetes Research Foundation (1-2009-337) and National Institutes of Health Grants R56DK084317 and R01DK090492 to F.G.H. Research in the Kulkarni laboratory is funded by DK RO167536.
Disclosure Summary: S.L., Y.X., A.B., K.M., I.M., R.N.K., and F.G.H. have nothing to declare.
Footnotes
- D/A
- D181A
- ER
- endoplasmic reticulum
- GSIS
- glucose-stimulated insulin secretion
- GTT
- glucose tolerance test
- HFD
- high-fat diet
- hPTP1B
- human PTP1B
- IRS2
- insulin receptor substrate 2
- ITT
- insulin tolerance test
- KO
- knockout
- PTP
- protein-tyrosine phosphatase
- WT
- wild-type.
References
- 1. Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest. 2000;106:473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Biddinger SB, Kahn CR. From mice to men: insights into the insulin resistance syndromes. Annu Rev Physiol. 2006;68:123–158 [DOI] [PubMed] [Google Scholar]
- 3. Bell GI, Polonsky KS. Diabetes mellitus and genetically programmed defects in beta-cell function. Nature. 2001;414:788–791 [DOI] [PubMed] [Google Scholar]
- 4. Muoio DM, Newgard CB. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:193–205 [DOI] [PubMed] [Google Scholar]
- 5. Ravier MA, Güldenagel M, Charollais A, et al. Loss of connexin36 channels alters beta-cell coupling, islet synchronization of glucose-induced Ca2+ and insulin oscillations, and basal insulin release. Diabetes. 2005;54:1798–1807 [DOI] [PubMed] [Google Scholar]
- 6. Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell. 2008;133:38–52 [DOI] [PubMed] [Google Scholar]
- 7. Konstantinova I, Nikolova G, Ohara-Imaizumi M, et al. EphA-Ephrin-A-mediated beta cell communication regulates insulin secretion from pancreatic islets. Cell. 2007;129:359–370 [DOI] [PubMed] [Google Scholar]
- 8. Frangioni JV, Beahm PH, Shifrin V, Jost CA, Neel BG. The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid C-terminal sequence. Cell. 1992;68:545–560 [DOI] [PubMed] [Google Scholar]
- 9. Woodford-Thomas TA, Rhodes JD, Dixon JE. Expression of a protein tyrosine phosphatase in normal and v-src-transformed mouse 3T3 fibroblasts. J Cell Biol. 1992;117:401–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haj FG, Verveer PJ, Squire A, Neel BG, Bastiaens PI. Imaging sites of receptor dephosphorylation by PTP1B on the surface of the endoplasmic reticulum. Science. 2002;295:1708–1711 [DOI] [PubMed] [Google Scholar]
- 11. Elchebly M, Payette P, Michaliszyn E, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548 [DOI] [PubMed] [Google Scholar]
- 12. Klaman LD, Boss O, Peroni OD, et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol. 2000;20:5479–5489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheng A, Uetani N, Simoncic PD, et al. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev Cell. 2002;2:497–503 [DOI] [PubMed] [Google Scholar]
- 14. Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, et al. PTP1B regulates leptin signal transduction in vivo. Dev Cell. 2002;2:489–495 [DOI] [PubMed] [Google Scholar]
- 15. Delibegovic M, Zimmer D, Kauffman C, et al. Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes. 2009;58:590–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haj FG, Zabolotny JM, Kim YB, Kahn BB, Neel BG. Liver-specific protein-tyrosine phosphatase 1B (PTP1B) re-expression alters glucose homeostasis of PTP1B−/− mice. J Biol Chem. 2005;280:15038–15046 [DOI] [PubMed] [Google Scholar]
- 17. Delibegovic M, Bence KK, Mody N, et al. Improved glucose homeostasis in mice with muscle-specific deletion of protein-tyrosine phosphatase 1B. Mol Cell Biol. 2007;27:7727–7734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bence KK, Delibegovic M, Xue B, et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med. 2006;12:917–924 [DOI] [PubMed] [Google Scholar]
- 19. Owen C, Czopek A, Agouni A, et al. Adipocyte-specific protein tyrosine phosphatase 1B deletion increases lipogenesis, adipocyte cell size and is a minor regulator of glucose homeostasis. PloS One. 2012;7:e32700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kushner JA, Haj FG, Klaman LD, et al. Islet-sparing effects of protein tyrosine phosphatase-1b deficiency delays onset of diabetes in IRS2 knockout mice. Diabetes. 2004;53:61–66 [DOI] [PubMed] [Google Scholar]
- 21. Bettaieb A, Liu S, Xi Y, et al. Differential regulation of endoplasmic reticulum stress by protein tyrosine phosphatase 1B and T cell protein tyrosine phosphatase. J Biol Chem. 2011;286:9225–9235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447–2457 [DOI] [PubMed] [Google Scholar]
- 23. Kulkarni RN, Winnay JN, Daniels M, et al. Altered function of insulin receptor substrate-1-deficient mouse islets and cultured beta-cell lines. J Clin Invest. 1999;104:R69–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bettaieb A, Matsuo K, Matsuo I, et al. Protein tyrosine phosphatase 1B deficiency potentiates PERK/eIF2alpha signaling in brown adipocytes. PloS One. 2012;7:e34412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bettaieb A, Bakke J, Nagata N, et al. Protein tyrosine phosphatase 1B regulates pyruvate kinase M2 tyrosine phosphorylation. J Biol Chem. 2013;288:17360–17371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bakke J, Bettaieb A, Nagata N, Matsuo K, Haj FG. Regulation of the SNARE-interacting protein Munc18c tyrosine phosphorylation in adipocytes by protein-tyrosine phosphatase 1B. Cell Commun Signal. 2013;11:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tiganis T, Bennett AM. Protein tyrosine phosphatase function: the substrate perspective. Biochem J. 2007;402:1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang SS, Hao E, Yu J, et al. Coordinated regulation by Shp2 tyrosine phosphatase of signaling events controlling insulin biosynthesis in pancreatic beta-cells. Proc Natl Acad Sci U S A. 2009;106:7531–7536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bagdade JD, Bierman EL, Porte D., Jr The significance of basal insulin levels in the evaluation of the insulin response to glucose in diabetic and nondiabetic subjects. J Clin Invest. 1967;46:1549–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiao J, Gregersen S, Kruhøffer M, Pedersen SB, Ørntoft TF, Hermansen K. The effect of chronic exposure to fatty acids on gene expression in clonal insulin-producing cells: studies using high density oligonucleotide microarray. Endocrinology. 2001;142:4777–4784 [DOI] [PubMed] [Google Scholar]
- 31. Hennige AM, Ranta F, Heinzelmann I, et al. Overexpression of kinase-negative protein kinase Cdelta in pancreatic beta-cells protects mice from diet-induced glucose intolerance and beta-cell dysfunction. Diabetes. 2010;59:119–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jain R, Jain D, Liu Q, et al. Pharmacological inhibition of Eph receptors enhances glucose-stimulated insulin secretion from mouse and human pancreatic islets. Diabetologia. 2013;56:1350–1355 [DOI] [PubMed] [Google Scholar]
- 33. Nievergall E, Janes PW, Stegmayer C, et al. PTP1B regulates Eph receptor function and trafficking. J Cell Biol. 2010;191:1189–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Haj FG, Sabet O, Kinkhabwala A, et al. Regulation of signaling at regions of cell-cell contact by endoplasmic reticulum-bound protein-tyrosine phosphatase 1B. PloS One. 2012;7:e36633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang ZY, Maclean D, McNamara DJ, Sawyer TK, Dixon JE. Protein tyrosine phosphatase substrate specificity: size and phosphotyrosine positioning requirements in peptide substrates. Biochemistry. 1994;33:2285–2290 [DOI] [PubMed] [Google Scholar]
- 36. Flint AJ, Tiganis T, Barford D, Tonks NK. Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc Natl Acad Sci U S A. 1997;94:1680–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haj FG, Markova B, Klaman LD, Bohmer FD, Neel BG. Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatase-1B. J Biol Chem. 2003;278:739–744 [DOI] [PubMed] [Google Scholar]
- 38. Stuible M, Dubé N, Tremblay ML. PTP1B regulates cortactin tyrosine phosphorylation by targeting Tyr446. J Biol Chem. 2008;283:15740–15746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huyer G, Liu S, Kelly J, et al. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J Biol Chem. 1997;272:843–851 [DOI] [PubMed] [Google Scholar]
- 40. Merrins MJ, Van Dyke AR, Mapp AK, Rizzo MA, Satin LS. Direct measurements of oscillatory glycolysis in pancreatic islet β-cells using novel fluorescence resonance energy transfer (FRET) biosensors for pyruvate kinase M2 Activity. J Biol Chem. 2013;288:33312–33322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wicksteed B, Brissova M, Yan W, et al. Conditional gene targeting in mouse pancreatic β-cells: analysis of ectopic Cre transgene expression in the brain. Diabetes. 2010;59:3090–3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schwartz MW, Guyenet SJ, Cirulli V. The hypothalamus and β-cell connection in the gene-targeting era. Diabetes. 2010;59:2991–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Banno R, Zimmer D, De Jonghe BC, et al. PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice. J Clin Invest. 2010;120:720–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lu B, Wu H, Gu P, et al. Improved glucose-stimulated insulin secretion by intra-islet inhibition of protein-tyrosine phosphatase 1B expression in rats fed a high-fat diet. J Endocrinol Invest. 2012;35:63–70 [DOI] [PubMed] [Google Scholar]
- 45. Gunton JE, Kulkarni RN, Yim S, et al. Loss of ARNT/HIF1beta mediates altered gene expression and pancreatic-islet dysfunction in human type 2 diabetes. Cell. 2005;122:337–349 [DOI] [PubMed] [Google Scholar]
- 46. Holst B, Egerod KL, Jin C, et al. G protein-coupled receptor 39 deficiency is associated with pancreatic islet dysfunction. Endocrinology. 2009;150:2577–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bjorge JD, Pang A, Fujita DJ. Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J Biol Chem. 2000;275:41439–41446 [DOI] [PubMed] [Google Scholar]
- 48. Kulkarni RN, Brüning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell. 1999;96:329–339 [DOI] [PubMed] [Google Scholar]
- 49. Okada T, Liew CW, Hu J, et al. Insulin receptors in beta-cells are critical for islet compensatory growth response to insulin resistance. Proc Natl Acad Sci U S A. 2007;104:8977–8982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Covey SD, Wideman RD, McDonald C, et al. The pancreatic beta cell is a key site for mediating the effects of leptin on glucose homeostasis. Cell Metab. 2006;4:291–302 [DOI] [PubMed] [Google Scholar]
- 51. Morioka T, Asilmaz E, Hu J, et al. Disruption of leptin receptor expression in the pancreas directly affects beta cell growth and function in mice. J Clin Invest. 2007;117:2860–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tejedo JR, Cahuana GM, Ramírez R, et al. nitric oxide triggers the phosphatidylinositol 3-kinase/Akt survival pathway in insulin-producing RINm5F cells by arousing Src to activate insulin receptor substrate-1. Endocrinology. 2004;145:2319–2327 [DOI] [PubMed] [Google Scholar]
- 53. Kominato R, Fujimoto S, Mukai E, et al. Src activation generates reactive oxygen species and impairs metabolism-secretion coupling in diabetic Goto-Kakizaki and ouabain-treated rat pancreatic islets. Diabetologia. 2008;51:1226–1235 [DOI] [PubMed] [Google Scholar]
- 54. Harding HP, Zeng H, Zhang Y, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Molecular cell. 2001;7:1153–1163 [DOI] [PubMed] [Google Scholar]
- 55. Scheuner D, Song B, McEwen E, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176 [DOI] [PubMed] [Google Scholar]
- 56. Oh E, Thurmond DC. Munc18c depletion selectively impairs the sustained phase of insulin release. Diabetes. 2009;58:1165–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]