

Abstract
To facilitate the ongoing research of Vibrio spp., a dedicated platform for the Vibrio research community is needed to host the fast-growing amount of genomic data and facilitate the analysis of these data. We present VibrioBase, a useful resource platform, providing all basic features of a sequence database with the addition of unique analysis tools which could be valuable for the Vibrio research community. VibrioBase currently houses a total of 252 Vibrio genomes developed in a user-friendly manner and useful to enable the analysis of these genomic data, particularly in the field of comparative genomics. Besides general data browsing features, VibrioBase offers analysis tools such as BLAST interfaces and JBrowse genome browser. Other important features of this platform include our newly developed in-house tools, the pairwise genome comparison (PGC) tool, and pathogenomics profiling tool (PathoProT). The PGC tool is useful in the identification and comparative analysis of two genomes, whereas PathoProT is designed for comparative pathogenomics analysis of Vibrio strains. Both of these tools will enable researchers with little experience in bioinformatics to get meaningful information from Vibrio genomes with ease. We have tested the validity and suitability of these tools and features for use in the next-generation database development.
1. Introduction
Vibrio is a genus of Gram-negative bacteria possessing a curved rod shape, of which several species are pathogenic. Vibrio cholerae [1] is one of the well-studied pathogenic species that causes foodborne infection and the cholera disease. Cholera is a diarrheal disease [2] that can kill within hours if it remains untreated. There are about 3 to 5 million cholera cases with 100,000 to 120,000 fatalities due to cholera infections every year [3]. Other notable members of pathogenic Vibrio spp. include the Vibrio parahaemolyticus [4], Vibrio vulnificus [5], and Vibrio harveyi [6]. These microorganisms are naturally found in saltwater and they are a leading cause of seafood-borne infections such as gastroenteritis and septicemia, carried by organisms such as crabs, prawns, or oysters. Infections with the noncholera Vibrio are commonly associated with eating undercooked seafood [7, 8] or open wounds infection [8]. Many other Vibrio spp. can also cause infections in sea-living organisms and are common causes of mortality in domestic marine life.
Many new Vibrio genomes have been sequenced and the availability of these genome sequences from different sources has made it possible to perform genome-wide comparative analyses [9–11]. Such comparative analysis may enhance our understanding of the biology, diversity, evolution, and virulence of the Vibrio bacteria, which may be useful towards combating the Vibrio pathogens. Recently, an online genome database, MabsBase, has been developed specifically for the Mycobacterium abscessus, a genus of Actinobacteria [12]. However, a similar genome database is not available for Vibrio spp. MBGD [13], IMG [14], PATRIC [15], and SEED database [16] do provide a wide array of microbial genomes including some of the Vibrio strains for comparative genomics, but without much emphasis on virulence factors for comparative pathogenomics and lacking user-friendly web interfaces. PATRIC [15] does provide virulence factors information but lack functionalities to cluster/compare and visualize user-selected Vibrio strains based on their virulence gene profiles, which are useful for comparative pathogenomics analysis.
To facilitate the ongoing research of Vibrio spp., we have developed VibrioBase, a dedicated platform for the Vibrio research community to host the fast-growing amount of genomic data and facilitate the analysis of these data. This online platform is empowered by advanced web technologies and in-house analysis tools for the Vibrio research community. The comprehensive set of genomic datasets in VibrioBase will facilitate analyses in comparative genomics and pathogenomics among different Vibrio strains or species. Here, we describe the overview and some key features of VibrioBase.
2. Database Content and Organization
VibrioBase currently houses a total of 252 genome sequences from 31 Vibrio spp. obtained from the NCBI (Supplementary Table 1 in Supplementary Material available online at http://dx.doi.org/10.1155/2014/569324). All the genome sequences in VibrioBase were annotated using the rapid annotation subsystem technology (RAST) pipeline [19]. The RAST pipeline is a fully automated annotation engine and helps in recognizing protein encoding genes, rRNAs, and tRNAs and subsequently ascribes functions to the genes. The protein assignments in the RAST pipeline are based on the functional properties within the subsystems (see the subsystems approach to genome annotation and its use in the project to annotate 1000 genomes) and FIGfams [20] maintained in the SEED system. The annotations include the functional prediction, protein translation of contigs, RNA and CDS classification, subsystem function prediction, and the start and stop positions of sequences. The PSORTb standalone version 3.0 [21] was used to predict the subcellular localization of each putative protein in the VibrioBase datasets. Protein subcellular localization describes the spatial arrangement of proteins within cells, thereby providing important functional information of proteins. The objective of this database is to provide resources for whole-genome annotations and state-of-the-art tools to support the expanding Vibrio research community and to collectively gather information on the strains of Vibrio spp. into a single database. With this annotation information, users are a step closer to the understanding of Vibrio spp. Information of all the strains in this database is linked to the NCBI database. For instance, by following the ORF ID, users will be taken to the NCBI database for further information on that specific region of each particular ORF. We present here some of the key features in our database which would be quintessential for the next-generation genome and annotation database development.
3. Real-Time Data Searching Feature
With the advances in next-generation sequencing technologies and bioinformatics tools, more Vibrio genomes and annotations will be progressively included in VibrioBase. Our goal is to enable users to browse rapidly and seamlessly along the huge genomic data. VibrioBase can be queried to obtain annotated features in different ways. We facilitated this by implementing a powerful real-time AJAX-based search function in this database. The AJAX is not a new technology but a combination of different existing technologies such as HTML, CSS, DOM, XML, and JavaScript. This allows our database to have a wider variety of controls and functions. It reduces the load on server which allows heavy analyses load to be processed simultaneously. Users can perform quick browsing of the database using keywords in a rapid, real-time manner which is a very important feature for browsing through database with massive datasets. The interactive JBrowse [22] genome browser incorporated in VibrioBase which uses the AJAX-based, server and client technology support fast and smooth animated genome navigation over the web. As we anticipate that many more Vibrio genomes are to be sequenced and annotated, the use of user friendly genome browser, the JBrowse, has become essential. Unlike other genome browsers such as the GBrowse, which is implemented using the Common Gateway Interface (CGI) protocol occupying much more resources and delaying in response for the out coming result, the server-client architecture of JBrowse minimizes server overhead problems as genomes are not rendered into images on the web server. This provides the users to have rapid genome browsing information without causing server overload when multiple users process simultaneously. JBrowse provides efficient panning and zooming of a genomic region in the genome via embedded navigation buttons, thus avoiding discontinuous transitions. Furthermore, JBrowse allows each track such as DNA, RNA, and CDS tracks to be turned on or off on clicking it. It allows users to hide unwanted information for better user experience.
4. Pairwise Genome Comparison (PGC) Tool: Information Aesthetic for Comparative Genomics
The pairwise genome comparison (PGC) tool is a newly designed in-house comparative analysis tool integrated to allow users to compare two different Vibrio genomes (Figure 1). Two main software components used in the automated pipeline were NUCmer in the MUMmer package [23] and Circos [24]. This visualization tool is useful in the identification and comparative analysis of two genomes which is reported in a circular ideogram layout, depicting the structural variations and positional relationships between two genomic intervals. The input form of PGC has three parameters which are the minimum percent identity, merge threshold, and link threshold. By default, the Circos in VibrioBase is set to 95% minimum percentage identity and 1,000 bp link threshold; users may change the parameter freely to get different comparative results.
Figure 1.
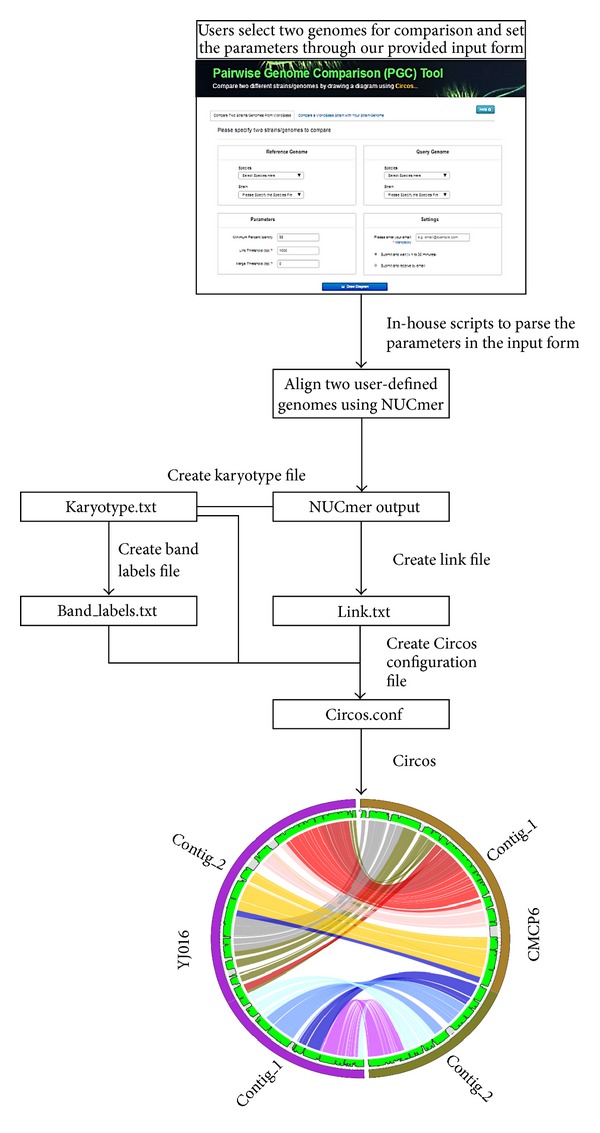
A workflow of the PGC tool. Through an input web interface on VibrioBase, users can choose two genomes of interest (reference versus query genomes) in VibrioBase and parameters for comparison. Three available useful parameters/thresholds which are the minimum percent identity, merge threshold, and link threshold. The minimum percent identity parameter will display aligned genomic regions (represented by colored links) once the sequence identity is higher than the user-defined cut-off. Similar to the merge threshold, this threshold will merge two links if the distance between the two regions is lower than the user-defined cut-off. The link threshold ignores links in the diagram if their widths are lower than the user-defined cut-off. Once the job is submitted to our server, PGC pipeline parses this information and starts the genome alignment with NUCmer. A series of in-house Perl and Python scripts are used to parse the NUCmer output and generate different text files: (1) Karyotype.txt stores information about the contigs and their colors used in Circos; (2) Links.txt stores information about the aligned genomic regions; (3) Band_labels.txt keeps the names of each contigs/chromosome. The Circos.conf configuration file will be created using the information in the above four files, which is needed for displaying the two aligned genomes with Circos.
As an example, we compared the genomes of two closely related Vibrio species: V. vulnificus CMCP6 and V. vulnificus YJ016. The two genomes are very similar or conserved in general. Interestingly, Circos plot has shown that there are two putative genomic translocations in the genome of V. vulnificus YJ016 and indels in the two genomes (Figure 2). Further analysis on the sequences of these regions revealed that the two regions in the genome of V. vulnificus CMCP6 are associated with putative prophages as predicted by PHAST [25], suggesting that these prophages might be inserted into V. vulnificus CMCP6. The insertion of prophages might be due to previous lysogenic bacteriophage infection and this probably confers pathogenicity to the bacterial host and changes its diversity. Here, we have demonstrated that PGC can be used to study the genetic differences between two genomes.
Figure 2.
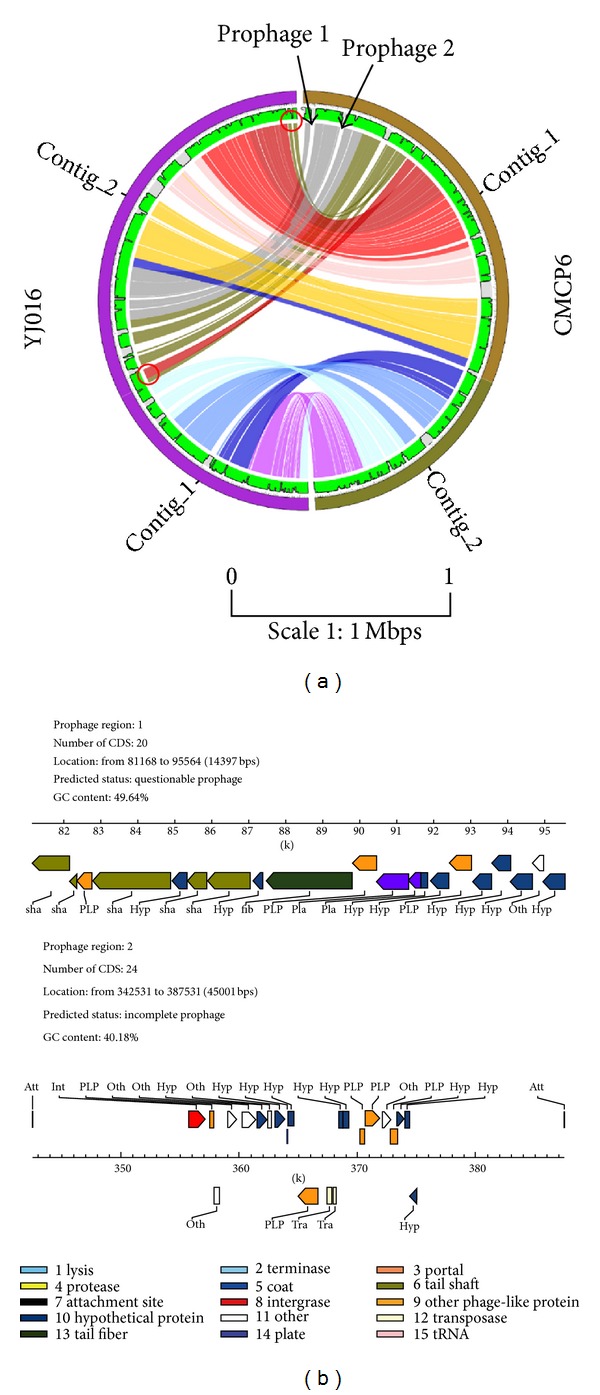
Example of a pairwise genome alignment between V. vulnificus YJ016 and V. vulnificus CMCP6. (a) Circos plot reveals differences between the two genomes. Red circle indicates the translocations possibly occur. Black arrowheads indicate phage insertions. (b) The structure of the two putative prophages. (Scale of the Circos 1 : 1 Mbps).
Although a similar tool named Circoletto [26] is available, there are many differences between this tool and our newly developed PGC. For instance, Circoletto aligns sequences using BLAST (local alignment), but PGC uses the NUCmer (global alignment) package in MUMmer 3.0 [27], which is advantageous for large-scale and rapid genome alignment. Moreover, PGC also allows users to adjust settings such as minimum percent identity, merge threshold, and link threshold through the provided web interface.
5. Pathogenomics Profiling Tool (PathoProT)
We developed a unique pathogenomics profiling tool (PathoProT) that could identify virulence genes and perform a comparative pathogenomics analysis. PathoProT predicts the virulence genes based on a sequence homology search of the putative protein sequences in each strain against the virulence factors database (VFDB) [28], depending on the user-defined cut-offs for protein identity and completeness. This tool clusters (agglomerative hierarchical cluster analysis) the predicted virulence genes across different strains using their virulence gene profiles. A user can visualize them in the form of heat map with dendrograms. Furthermore, it allows users to examine the similarities and differences of the virulence gene profiles between different groups of strains, for instance, nonpathogenic versus pathogenic strains. We compared the virulence gene profiles of all 134 strains of the highly pathogenic V. cholera in VibrioBase using PathoProT (Figure 3). Our analysis showed that all strains shared at least 58 conserved virulence genes, amongst which is the exopolysaccharide genes group (epsC-epsF-epsG-epsI-epsJ-epsK-epsL-epsM) [29–31] responsible for carrying out different virulence functions such as protection of bacteria from phagocytosis, phage invasion, and protection of cell against water absorption [32]. Besides showing the conserved virulence genes, the heat map is also able to show strain-specific or group-specific virulence genes.
Figure 3.

The profiling of virulence genes of 134 V. cholerae strains using PathoProT. The heat map was generated by using the thresholds of 50% completeness and 50% identity. It gave an overview of the clustered strains having closely related sets of virulence genes, sorted according to the level of similarities across the strains and genes. The flagella formation gene clusters (flgB-flgC-flgD-flgF-flgG-flgH-flgI-flgK-flgL) are the largest conserved virulence gene group among the V. cholera strains. The flg gene cluster works with other virulence gene clusters such as fla gene cluster (flaA-flaB-flaC-flaD-flaE) [17] and fli gene cluster (fliH-fliJ-fliL-fliM-fliN-flnO-fliP-fliQ-fliR-fliS) to form the flagella organelles on the bacteria [18], which is one of the common characteristics among all cholerae strains.
Interestingly, three strains (V. cholerae HENC 01, V. cholerae HENC 02, and V. cholera HENC 03) have very different virulence profiles as compared to other V. cholerae strains (Figure 4). One of the possible explanations is that these genes were not completely sequenced because the genomes of these strains are not complete. However, this may be unlikely because we observed that many virulence genes are also absent in the three genomes. Another possibility is that these strains were inappropriately classified as V. cholerae. To investigate the second possibility, we constructed a phylogenetic tree based on the 16S rRNA genes from different Vibrio spp. with MEGA4 using the maximum likelihood methods [27]. We found that these three strains are clustered into a group (Figure 4), but not with other V. cholerae strains, supporting our view that these three strains are not of V. cholera. It should be noted that, at the end of our analysis, these strains were recently renamed in the NCBI database to uncategorized Vibrio spp. Taken together, we have shown that PathoProT can be a useful tool for comparative pathogenomics analysis of the Vibrio strains or species, which may enhance our understanding of their virulence, evolution, and classification.
Figure 4.
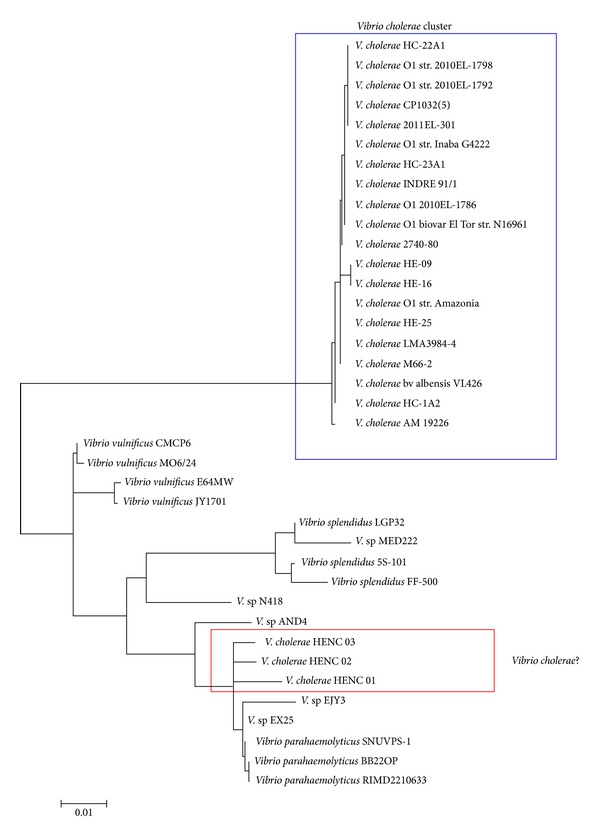
A 16S-based phylogenetic tree. V. cholera HENC 01, V. cholera HENC 02, and V. cholera HENC 03 are clustered into a group (red box), rather than with other V. cholera strains (blue box). The three strains are closely related to V. parahaemolyticus species. These results suggest that the three strains might be inappropriately classified into V. cholera.
6. Implementation and Future Development
VibrioBase is a HTML5 web application developed in PHP language using CodeIgniter and Twitter Bootstrap as back-end and front-end frameworks, respectively. MySQL database is used for storing information and the webserver is configured using Open Panel. To achieve higher level of performance, a job management and scheduler were developed to manage and monitor jobs, which include Circos plots and BLAST search. AJAX calls were emerged in different components of the application to provide a better user experience.
VibrioBase will be updated from time to time as more genome annotations and genomic sequences of Vibrio become available. To accelerate the development of this database for the use of the scientific community, we encourage other research groups to contact us if they would like to share annotations and related datasets with us. Suggestions on improving VibrioBase and requests for additional functions are also welcome.
7. Conclusion
Although there are many existing valuable genomic data and analysis tools from different sources, it is important to pool these resources together in order to allow rapid searching and browsing and also to facilitate the analysis of these data. We believe that a specialized resource such as the VibrioBase that is empowered with different analysis tools will prove to be invaluable to the Vibrio researchers both in basic science and clinical research areas.
Supplementary Material
Supplementary Table 1 shows the list of Vibrio species and strains available in VibrioBase. VibrioBase currently hosts a total of 252 genome sequences or strains of Vibrio species retrieved from the NCBI database. There are 24 complete genomes and 228 draft genomes.
Acknowledgment
This work was supported by research Grants UM.C/HIR/MOHE/08 and PG077-2012B from the University of Malaya, Kuala Lumpur, Malaysia.
Disclosure
Availability: http://vibrio.um.edu.my.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Thompson FL, Gevers D, Thompson CC, et al. Phylogeny and molecular identification of vibrios on the basis of multilocus sequence analysis. Applied and Environmental Microbiology. 2005;71(9):5107–5115. doi: 10.1128/AEM.71.9.5107-5115.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fasano A, Baudry B, Pumplin DW, et al. Vibrio cholerae produces a second enterotoxin, which affects intestinal tight junctions. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(12):5242–5246. doi: 10.1073/pnas.88.12.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cholera. (Fact Sheet No. 107). World Health Organization.
- 4.Shinoda S. Sixty years from the discovery of Vibrio parahaemolyticus and some recollections. Biocontrol Science. 2011;16(4):129–137. doi: 10.4265/bio.16.129. [DOI] [PubMed] [Google Scholar]
- 5.Linkous DA, Oliver JD. Pathogenesis of Vibrio vulnificus. FEMS Microbiology Letters. 1999;174(2):207–214. doi: 10.1111/j.1574-6968.1999.tb13570.x. [DOI] [PubMed] [Google Scholar]
- 6.Austin B, Zhang XH. Vibrio harveyi: a significant pathogen of marine vertebrates and invertebrates. Letters in Applied Microbiology. 2006;43(2):119–124. doi: 10.1111/j.1472-765X.2006.01989.x. [DOI] [PubMed] [Google Scholar]
- 7.Su YC, Liu C. Vibrio parahaemolyticus: a concern of seafood safety. Food Microbiology. 2007;24(6):549–558. doi: 10.1016/j.fm.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 8.Tacket CO, Brenner F, Blake PA. Clinical features and an epidemiological study of Vibrio vulnificus infections. The Journal of Infectious Diseases. 1984;149(4):558–561. doi: 10.1093/infdis/149.4.558. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez-Escalona N, Strain EA, de Jesús AJ, Jones JL, DePaola A. Genome sequence of the clinical O4:K12 serotype Vibrio parahaemolyticus strain 10329. Journal of Bacteriology. 2011;193(13):3405–3406. doi: 10.1128/JB.05044-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dias GM, Thompson CC, Fishman B, et al. Genome sequence of the marine bacterium Vibrio campbellii DS40M4, isolated from open ocean water. Journal of Bacteriology. 2012;194(4, article 904) doi: 10.1128/JB.06583-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park JH, Cho YJ, Chun J, et al. Complete genome sequence of Vibrio vulnificus MO6-24/O. Journal of Bacteriology. 2011;193(8):2062–2063. doi: 10.1128/JB.00110-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heydari H, Wee WY, Lokanathan N, et al. MabsBase: a Mycobacterium abscessus genome and annotation database. PLoS ONE. 2013;8(4) doi: 10.1371/journal.pone.0062443.e62443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uchiyama I, Mihara M, Nishide H, Chiba H. MBGD update 2013: the microbial genome database for exploring the diversity of microbial world. Nucleic Acids Research. 2013;41(1):D631–D635. doi: 10.1093/nar/gks1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Markowitz VM, Chen IM, Palaniappan K, et al. IMG: The integrated microbial genomes database and comparative analysis system. Nucleic Acids Research. 2012;40(1):D115–D122. doi: 10.1093/nar/gkr1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wattam AR, Abraham D, Dalay O, et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Research. 2014;42:D581–D591. doi: 10.1093/nar/gkt1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Overbeek R, Begley T, Butler RM, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Research. 2005;33(17):5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patenge N, Berendes A, Engelhardt H, Schuster SC, Oesterhelt D. The fla gene cluster is involved in the biogenesis of flagella in Halobacterium salinarum. Molecular Microbiology. 2001;41(3):653–663. doi: 10.1046/j.1365-2958.2001.02542.x. [DOI] [PubMed] [Google Scholar]
- 18.Barker CS, Meshcheryakova IV, Kostyukova AS, Samatey FA. FliO regulation of FliP in the formation of the Salmonella enterica flagellum. PLoS Genetics. 2010;6(9) doi: 10.1371/journal.pgen.1001143.e1001143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aziz RK, Bartels D, Best A, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9, article 75 doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyer F, Overbeek R, Rodriguez A. FIGfams: yet another set of protein families. Nucleic Acids Research. 2009;37(20):6643–6654. doi: 10.1093/nar/gkp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu NY, Wagner JR, Laird MR, et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26(13):1608–1615. doi: 10.1093/bioinformatics/btq249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Skinner ME, Uzilov AV, Stein LD, Mungall CJ, Holmes IH. JBrowse: a next-generation genome browser. Genome Research. 2009;19(9):1630–1638. doi: 10.1101/gr.094607.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kurtz S, Phillippy A, Delcher AL, et al. Versatile and open software for comparing large genomes. Genome Biology. 2004;5(2) article R12 doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krzywinski M, Schein J, Birol I, et al. Circos: An information aesthetic for comparative genomics. Genome Research. 2009;19(9):1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Research. 2011;39(2):W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Darzentas N. Circoletto: visualizing sequence similarity with Circos. Bioinformatics. 2010;26(20):2620–2621. doi: 10.1093/bioinformatics/btq484.btq484 [DOI] [PubMed] [Google Scholar]
- 27.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Molecular Biology and Evolution. 2007;24(8):1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 28.Chen L, Yang J, Yu J, et al. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Research. 2005;33:D325–D328. doi: 10.1093/nar/gki008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ali A, Rashid MH, Karaolis DKR. High-frequency rugose exopolysaccharide production by Vibrio cholerae. Applied and Environmental Microbiology. 2002;68(11):5773–5778. doi: 10.1128/AEM.68.11.5773-5778.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bramhachari PV, Dubey SK. Isolation and characterization of exopolysaccharide produced by Vibrio harveyi strain VB23. Letters in Applied Microbiology. 2006;43(5):571–577. doi: 10.1111/j.1472-765X.2006.01967.x. [DOI] [PubMed] [Google Scholar]
- 31.Rodrigues C, Bhosle NB. Exopolysaccharide production by Vibrio fischeri, a fouling marine bacterium. Biofouling. 1991;4(4):301–308. [Google Scholar]
- 32.Cerning J. Exocellular polysaccharides produced by lactic acid bacteria. FEMS Microbiology Reviews. 1990;87(1-2):113–130. doi: 10.1111/j.1574-6968.1990.tb04883.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 shows the list of Vibrio species and strains available in VibrioBase. VibrioBase currently hosts a total of 252 genome sequences or strains of Vibrio species retrieved from the NCBI database. There are 24 complete genomes and 228 draft genomes.