

α-Synuclein (aS), a small protein containing 140 amino acids, undergoes self-assembly into amyloid fibrils and plaques (Lewy bodies), which are pathological hallmarks of Parkinson’s disease (PD) as well as other neurodegenerative diseases.[1] While oligomeric species of aS are considered to exert the neurotoxic activity[2] that can be rescued by reducing those oligomers either by diversion to smaller oligomers[3] or acceleration of fibril formation, cell-to-cell transmission in nontransgenic mice points to a direct role of the fibrils in spreading the disease from peripheral to central neurons.[4] Understanding the molecular interactions that lead to misfolding strongly relies on the availability of suited biophysical methods that can access the structure of these states. For the monomeric form, magnetic resonance techniques gave evidence for a natively disordered yet partially folded protein[5a,b] that upon binding to lipid vesicles adopts an α-helical structure.[6–8] When aS is aggregated into fibrils, X-ray and electron diffraction studies[9a,b] revealed that it arranges in a classical cross-β conformation, where the individual β-sheets arrange perpendicularly to the fibril axis with spacing of 4.7–4.8 Å along the fibril axis and 10–11 Å perpendicular to the axis. In particular, aS stacks in a parallel, in-register arrangement as revealed by continuous-wave (CW) electron paramagnetic resonance (EPR)[10] and more recently supported by solid-state nuclear magnetic resonance (ss-NMR) spectroscopy.[11]
The structural features of monomeric aS consist of: 1) an amphipathic N-terminal region (residues 1-60), 2) a hydrophobic central domain, known as a non-Aβ component (NAC) region (residues 61–95), and 3) a highly negatively charged C-terminal region (residues 96–140).[12] Studies from ss-NMR and CW-EPR spectroscopy have shown that the β-sheet core region is located within the central NAC domain[10,13a–d] and extended toward the N-terminus[14a,b] whereas the C-terminal region appears less ordered. Figure 1 represents the amino acid sequence of human aS as well as β-strands within the NAC region assigned by different ss-NMR studies. All these studies identified at least five β-strands that could fold to generate the core unit of the fibril structure.
Figure 1.
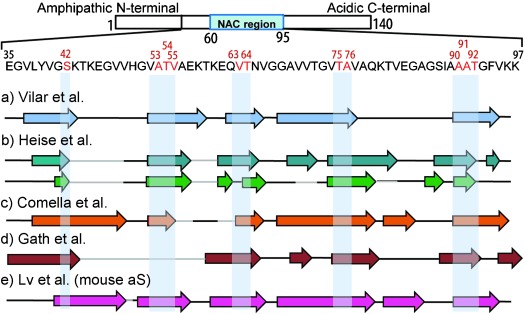
Top: Amino acid sequence of human αS in the NAC region. Mutation positions are marked in red. Bottom: Arrows indicate β-strand regions as identified by ss-NMR spectroscopy. a) Light blue, 13Cα–13Cβ chemical shift data from Ref. [13b]. b) β-Strand regions for two different types of fibril forms from Ref. [13a]. Cyan, form A and green, form B. c) Orange, from Ref. [13d]. d) Brown, from Ref. [14a]. e) Pink, mouse aS from Ref. [14b]. Gray lines indicate nonassigned amino acids. Bars denote conserved β-strands selected for labeling with methanethiosulfonate (NAC=non-Aβ component).
One model of the three-dimensional fold of the fibril consisting of five β-sheets per monomer has been proposed[13b] based on ss-NMR and cryo-electron microscopic data. However, long-range restraints at the molecular level in support of this model are still missing. Recently, we have reported pulsed EPR distance measurements to determine the intra-molecular distance between the extremal β-strands in aS fibrils.[15] A pair-labeling strategy was introduced to infer the orientation of the standard methanethiosulfonate (MTSL) label with respect to the plane of the β-strand, as the detected spin-bearing nitroxide of MTSL is located about 0.5 nm from the Cα of the amino acid the spin label is attached to. The resulting distance of (4.5±0.5) nm between the extremal strands was consistent with the size of the subfilament measured in AFM and cryo-electron microscopic studies.[12,13b] Nevertheless the dipolar oscillations observed in the pulsed EPR data, used to extract distances, were very weak and experiments at two different EPR frequencies were required to exclude artifacts. Similar observations were reported in other recent pulsed EPR distance measurements on amyloid fibrils of human islet polypeptide and tau protein.[16a,b] The challenge of these experiments is caused by the required labeling procedure combined with the structural instability of these proteins with respect to point mutations.[17] To date, despite the wealth of NMR and other spectroscopic data, no EPR distance measurements on aS fibrils have been reported by other groups. In the present study we illustrate how an optimized sample preparation has provided large signals in EPR distance measurements for several selected mutation sites. The result permitted to exploit a labeling strategy to measure distances between largely conserved β-strand regions and to obtain vectorial information on the spatial arrangements of the labels within the strands. The distances and two-dimensional coordinates reported here provide new long-range restraints for the structure of aS fibrils.
For distance measurements with EPR spectroscopy we employed pulsed electron–electron double resonance (PELDOR) also called double electron–electron resonance (DEER), a method frequently used to gain structural information in proteins and nucleic acids in a range of 2–8 nm.[18,19a–e] Experiments were performed at Q-band frequency (34 GHz) to optimize the spin sensitivity by maintaining reasonable modulation depths. Details on the PELDOR/DEER experimental setup are given in the Experimental Section (see the Supporting Information). In diamagnetic proteins, such as aS, the experiment requires the insertion of two spin-labels, most commonly the nitroxide derivative MTSL[20] that is reactive to a cysteine side-chain by formation of a disulfide bond. To detect intramolecular distances within the fibrils, cysteines were introduced pairwise in the monomeric protein by site-directed mutagenesis (Supporting Information). Moreover, intermolecular electron spin–spin interactions were suppressed by properly diluting the labeled protein with the wild type (wt) during fibril formation. In a first step, the double MTSL-labeled A90C/S42C mutant was selected to optimize the dilution conditions. The aggregation protocol was previously reported[15] and is described in the Supporting Information. In all samples, the aggregation kinetics was monitored by the Thioflavin-T (ThioT) fluorescence assay and the morphology of the fibrils was characterized by electron microscopy (EM). Room-temperature CW-EPR spectra were used to verify that the spin labels were magnetically diluted and immobilized in the fibrils. We performed a series of dilution experiments of labeled versus wt protein at molar ratios of 1:10, 1:20, and 1:40. Each sample at a given dilution was divided into three batches that were aggregated in parallel under identical conditions.
At increasing dilution we observed that differences in fibrillization kinetics and PELDOR/DEER modulation depths between parallel batches of samples were systematically attenuated (Figures S1 and S2). As expected, these measurements appeared to be most sensitive to both dilution and aggregation conditions. Data analysis revealed unambiguous intramolecular distances only for samples displaying clear dipolar oscillations (Figure S2 c). In these samples, the A90C/S42C mutant yielded a reproducible distance of 3.9 nm, which is consistent with our previous data[15] but in much higher quality. Considering that the optimal dilution ratio likely depends on the specific mutant, we first prepared all subsequent mutants at a ratio of 1:30 and in three parallel replicates. Samples that did not show modulation at this dilution were prepared at a second dilution of 1:50.
Double spin-labeled mutants were constructed to measure distances between conserved β-strands. In Figure 1 the selected labeling positions are displayed within the amino acid sequence and the reported β-strands. For each distance one label (reference) was inserted in either one of the extremal strands. A pair-labeling strategy was employed to infer the side-chain direction of the spin label within one strand and to report about the spatial arrangement of the labels and the strands. In the simplest case, this is accomplished by detecting two distances from one labeled residue to two adjacent ones, respectively. As the direction of the amino-acid side chains is alternating with respect to the plane of the β-strand, the two detected distances depend on the displacement of the third spin label with respect to the direction of the β-strand (Figure S8). From these two distances d1 and d2 as well as the distance between neighbor labels in the β-sheet Δz′, vectorial information about the location of spin label 3 can be defined according to Equations (1) and (2).
| (1) |
| (2) |
where (x, 0, z) are the coordinates of the third label with respect to labels 1 and 2. Here, a planar arrangement of the labels is assumed as a consequence of the characteristic cross-β sheet structure of the fibrils, in which β-sheets of the monomers stack in planes perpendicular to the fibril axis. (x′, 0, z′) are the coordinates of the third label in an intermediate frame with the z axis along Δz′. Employed vectors and angles are defined in Figure S8 in the Supporting Information. We note from Equation (2) that two symmetry-related solutions (coordinates) exist for each pair of measured distances.
Measured dipolar oscillations and extracted distance distributions are presented in Figure 2. Mutants of residues 42 versus 90/91/92, all located on the two extremal strands, lead to observable dipolar oscillations. The modulation depth was most pronounced (up to 8–9 %, this latter value also found with our Q-band setup in doubly spin-labeled model systems) for 90/42 but more shallow for the other two residues reflecting more heterogeneities in the latter cases. Three main distances could be determined at 3.9, 5.0, and 4.0 nm, respectively. The shorter distances for 42/90 and 42/92 with respect to 42/91 are consistent with an alternating direction of the MTSL side chain and indicate that the latter points toward residue 42 at position 90 and 92 and to the opposite direction at residue 91.
Figure 2.
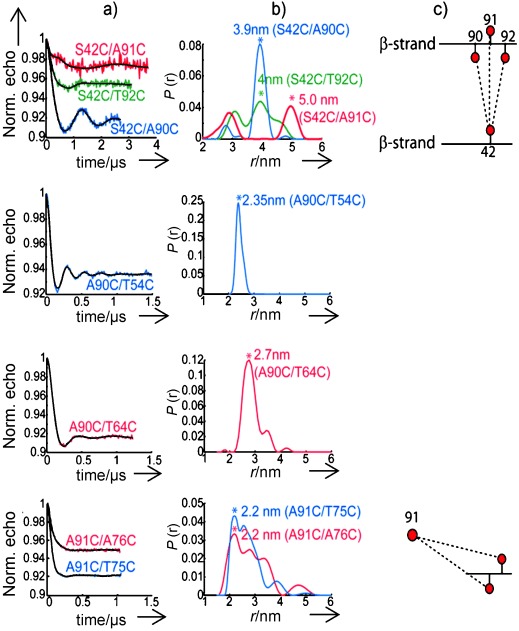
a) 34 GHz four-pulse PELDOR traces after background subtraction and the fit (black traces) of αS double mutants diluted in the wt protein at 1:30. b) Normalized distance distributions, P(r), obtained from a fit using DeerAnalysis.[21] The asterisks (*) indicate most probable distances. c) Scheme showing the observed distances between two β-strands containing the spin-labeled residues. The directions of the MTSL side chain within β-strands are also depicted, if known. Experimental parameters: Shot/point (SPP)=50, shot repetition time (SRT)=10 ms, acquisition time=24–36 h per trace.
The similar main distance for 42/90 and 42/92 (3.9–4.0 nm) strongly suggests an arrangement, in which the labels at residues 91 and 42 are almost on stack (Figure 2 c, first row). A more explicit model calculation of the spin label coordinates at residue 42 versus 90/91/92 is illustrated in Figure 3 (top, left). Knowledge of three distances allows us to locate residue 42 within a region centered at (x,z)=(−0.8±0.5 nm, −4.4±0.5 nm) with respect to the origin, which is set between Cα of residues 90 and 91. Symmetry-related solutions can be discarded because of the requirement of fulfilling all three distance constraints at once (Figure 3). If we additionally consider our previous distance measurement of 4.5±0.5 nm for residue 41 versus 90,[15] this distance is consistent with residue 42 pointing inside the β-sheets core and vice versa for residue 91 pointing towards the outside.
Figure 3.
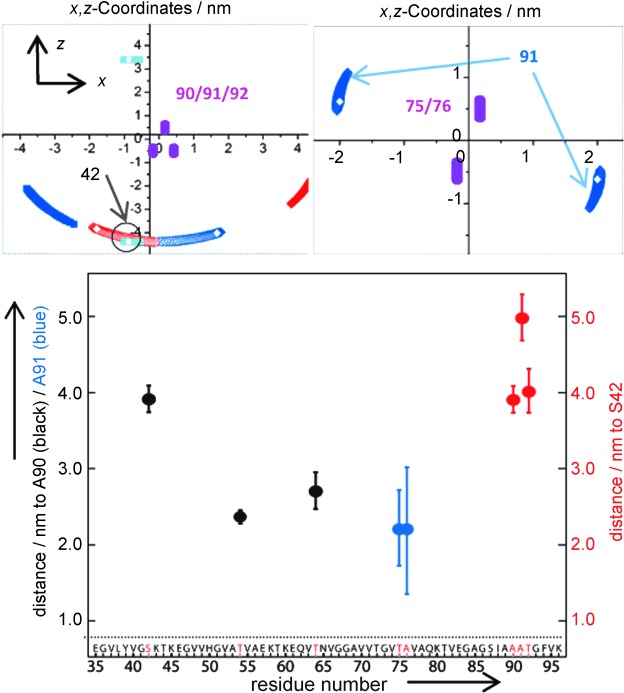
Top: Coordinates of MTSL at residue 42 versus 90/91/92 (left) and residue 91 versus 75/76 (right) using Equations (1) and (2), the coordinates of two adjacent labels, and main distances from Figure 2. Distances between the adjacent Cα of MTSL were set as 0.35±0.05 nm and MTSL chain extensions with 0.5±0.1 nm. For residue 42 versus 90/91/92, the three distances were analyzed pairwise and the corresponding solutions are plotted in different colors. The overlaying solutions are satisfying all three pairs of distances, as marked by a circle. Bottom: DEER distances as a function of labeling position within the protein sequence. Error bars correspond to twice the standard deviation from distance distributions.
From the three MTSL-labelled double mutants prepared between residues 90 and 53/54/55 in the next inner β-strand, only A90C/T54C revealed clear dipolar oscillations (Figure 2, second row). For the other two mutants the modulation depth was less than 1 % also after increasing the dilution ratio to 1:50. The weak signal of these mutants likely arose from intermolecular interactions, as shown by a comparative experiment with a single labeled sample (Figure S4). A similar situation was encountered for the adjacent mutants in the third inner strand, i.e. at residues 64 and 63. Only mutant A90C/T64C led to detectable dipolar oscillations (Figure 2, third row). Again, a higher dilution did not recover the signal of A90C/V63C. The results underline the difficulties in obtaining reliable signals for all mutants in fibrils, a fact which strongly depends on the site of mutation. Nevertheless, the signals for mutants A90C/T54C and A90C/T64C were of high quality and reproducible in almost all parallel batches (Figure S5). A sharp short distance of 2.3±0.1 nm was detected for pair 90/54 and a longer one of 2.7±0.3 nm for 90/64 (Figure 2, second and third row). Although the orientation of the MTSL chain remains unknown the result is striking as residue 54 is further remote in the sequence from 90 than residue 64. This information provides important constraints for a possible fold as discussed below.
The strand containing residues 75 and 76 is of considerable interest because of its large extension found in most fibril preparations (Figure 1). The mutual arrangement of the last two strands likely determines one dimension of the sub-filament. To detect a distance between these strands we constructed a pair of mutants containing residue 91, which points outside the core (see above), versus residues 75 and 76. We chose residue 91 instead of 90 as a reference to obtain longer distances (≥1.8 nm) that are possibly detectable by our method. For these two mutants, very similar dipolar oscillations and distance distributions were observed (Figure 2, fourth row). The same main distance to neighbor residues suggests a lateral displacement of residue 91 with respect to 75/76 along the direction of their β-strand (Figure 2 c, fourth row). An estimate of coordinates using Equations (1) and (2) and the main distances of Figure 2 c leads to two possible symmetry-related solutions, that is, (x1,z1)=(2±0.5 nm, −0.7±0.5 nm) and (x2,z2)=(−2±0.5 nm, 0.7±0.5 nm), as illustrated in Figure 3. The lateral displacement corresponds to the space of about six residues, the perpendicular one is on the order of or less of the X-ray diffraction distance between stacked β-sheets.
Finally, the measured distances are summarized in Figure 3 (bottom) as a function of the labeling position in the sequence. The distance of 4–5 nm between the extremal β-strands could in principle accommodate up to five β-sheets in parallel stack, as previously proposed by Vilar et al.[13b] However, our data provide more constraints. Starting from the strand containing residues 90/91/92, the short distance (2.2 nm) from 91 to 75/76 requires a turn between these strands. Further, the observed displacement between residues 75/76 and 91 appears on the order of one size of the subfilament (about 2 nm) observed in high-resolution cryo-EM.[13b] It could indicate that these residues are located towards the extremal of their respective strands. The next observed distance between residues 64/90 becomes longer (2.7±0.3) nm but the subsequent one 54/90 is shorter again (2.3±0.1) nm, Figure 3. Here the uncertainty about the direction of the MTSL side chain prevents detailed conclusions on the Cα–Cα distances. Nevertheless, the distance pattern strongly suggests that these distances are similar and that the two inner strands containing residues 54 and 64 might not stack in parallel and could arrange in a more complex fashion than proposed by the five β-sheet sandwich model.
In conclusion, the successful detection of multiple EPR distances combined with the pair-labeling strategy presented in this study provided a considerable number of long-range constrains at the molecular level on the fold of aS in fibrils. This represents a step towards a high-resolution structure of these fibrils, which will require input from multiple spectroscopic techniques combined with sophisticated molecular modeling. Moreover, detection of interstrand distances in fibrils will potentially allow for extending these measurements to oligomeric states of these protein families and obtain structural information on their role in protein misfolding.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1.Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 2.Karpinar DP, Balija MBG, Kügler S, Opazo F, Rezaei-Ghaleh N, Wender N, Kim H-Y, Taschenberger G, Falkenburger BH, Heise H, Kumar A, Riedel D, Fichtner L, Voigt A, Braus GH, Giller K, Becker S, Herzig A, Baldus M, Jäckle EH, Schulz JB, Griesinger C, Zweckstetter M. EMBO J. 2009;28:3256–3268. doi: 10.1038/emboj.2009.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wagner J, Ryazanov S, Leonov A, Levin J, Shi S, Schmidt F, Prix C, Pan-Montojo F, Bertsch U, Mitteregger-Kretzschmar G, Geissen M, Eiden M, Leidel F, Hirschberger T, Deeg AA, Krauth JJ, Zinth W, Tavan P, Pilger J, Zweckstetter M, Frank T, Bähr M, Weishaupt JH, Uhr M, Urlaub H, Teichmann U, Samwer M, Bötzel K, Groschup M, Kretzschmar H, Griesinger C, Giese A. Acta Neuropathol. 2013;125:795–813. doi: 10.1007/s00401-013-1114-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM-Y. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5a.Dedmon MM, Lindorff-Larsen K, Christodoulou J, Vendruscolo M, Dobson C. J. Am. Chem. Soc. 2005;127:476–477. doi: 10.1021/ja044834j. [DOI] [PubMed] [Google Scholar]
- 5b.Bertoncini CW, Jung YS, Fernandez CO, Hoyer W, Griesinger C, Jovin T, Zweckstetter M. Proc. Natl. Acad. Sci. USA. 2005;102:1430–1435. doi: 10.1073/pnas.0407146102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6a.Borbat P, Ramlall TF, Freed JH, Eliezer D. J. Am. Chem. Soc. 2006;128:10004–10005. doi: 10.1021/ja063122l. [DOI] [PubMed] [Google Scholar]
- 6b.Jao CC, Hegde BG, Chen J, Haworth IS, Langen R. Proc. Natl. Acad. Sci. USA. 2008;105:19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c.Drescher M, Rooijen BDv, Veldhuis G, Subramaniam V, Huber M. J. Am. Chem. Soc. 2010;132:4080–4082. doi: 10.1021/ja909247j. [DOI] [PubMed] [Google Scholar]
- 7a.Georgieva ER, Ramlall TF, Borbat PP, Freed JH, Eliezer D. J. Biol. Chem. 2010;285:28261–28274. doi: 10.1074/jbc.M110.157214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b.Shvadchak V, Yushchenko DA, Pievo R, Jovin T. FEBS Lett. 2011;585:3513–3519. doi: 10.1016/j.febslet.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 8.Mizuno N, Varkey J, Kegulian NC, Hegde BG, Cheng N, Langen R, Steven AC. J. Biol. Chem. 2012;287:29301–29311. doi: 10.1074/jbc.M112.365817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9a.Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Proc. Natl. Acad. Sci. USA. 2000;97:4897–4902. doi: 10.1073/pnas.97.9.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b.Conway KA, Harper JD, Peter J, Lansbury T. Biochemistry. 2000;39:2552–2563. doi: 10.1021/bi991447r. [DOI] [PubMed] [Google Scholar]
- 10.Chen M, Margittai M, Chen J, Langen R. J. Biol. Chem. 2007;282:24970–24979. doi: 10.1074/jbc.M700368200. [DOI] [PubMed] [Google Scholar]
- 11.Loquet A, Giller K, Becker S, Lange A. J. Am. Chem. Soc. 2010;132:15164–15166. doi: 10.1021/ja107460j. [DOI] [PubMed] [Google Scholar]
- 12.Fink AL. Acc. Chem. Res. 2006;39:628–634. doi: 10.1021/ar050073t. [DOI] [PubMed] [Google Scholar]
- 13a.Heise H, Hoyer W, Becker S, Andronesi OC, Riedel D, Baldus M. Proc. Natl. Acad. Sci. USA. 2005;102:15871–15876. doi: 10.1073/pnas.0506109102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b.Vilar M, Chou H-T, Lührs T, Maji SK, Riek-Loher D, Verel R, Manning G, Stahlberg H, Riek R. Proc. Natl. Acad. Sci. USA. 2008;105:8637–8642. doi: 10.1073/pnas.0712179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c.Cho M-K, Kim H-Z, Fernandez CO, Becker S, Zweckstetter M. Protein Sci. 2011;20:387–395. doi: 10.1002/pro.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13d.Comellas G, Lemkau LR, Nieuwkoop AJ, Kloepper KD, Ladror DT, Ebisu R, Woods WS, Lipton AS, George JM, Rienstra CM. J. Mol. Biol. 2011;411:881–895. doi: 10.1016/j.jmb.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14a.Gath J, Habenstein B, Bousset L, Melki R, Meier BH, Böckmann A. Biomol. NMR Assign. 2012;6:51–55. doi: 10.1007/s12104-011-9324-3. [DOI] [PubMed] [Google Scholar]
- 14b.Lv G, Kumar A, Giller K, Orcellet ML, Riedel D, Fernandez CO, Becker S, Lange A. J. Mol. Biol. 2012;420:99–111. doi: 10.1016/j.jmb.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 15.Karyagina I, Becker S, Giller K, Riedel D, Jovin TM, Griesinger C, Bennati M. Biophys. J. 2011;101:L1–L3. doi: 10.1016/j.bpj.2011.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16a.Bedrood S, Li Y, Isas JM, Hegde BG, Baxa U, Haworth IS, Langen R. J. Biol. Chem. 2012;287:5235–5241. doi: 10.1074/jbc.M111.327817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b.Siddiqua A, Luo Y, Meyer V, Swanson MA, Yu X, Wei G, Yheng J, Eaton GR, Ma B, Nussinov R, Eaton SS, Margittai M. J. Am. Chem. Soc. 2012;134:10271–10278. doi: 10.1021/ja303498q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertoncini CW, Fernandez CO, Griesinger C, Jovin T, Zweckstetter M. J. Biol. Chem. 2005;280:30649–30652. doi: 10.1074/jbc.C500288200. [DOI] [PubMed] [Google Scholar]
- 18.Milov AD, Ponomarev AB, Tsvetkov YD. Chem. Phys. Lett. 1984;110:67–72. [Google Scholar]
- 19a.Jeschke G. Macromol. Rapid Commun. 2002;23:227. [Google Scholar]
- 19b.Borbat PP, Freed JH. Methods Enzymol. 2007;423:52. doi: 10.1016/S0076-6879(07)23003-4. [DOI] [PubMed] [Google Scholar]
- 19c.Schiemann O, Prisner TF. Q. Rev. Biophys. 2007;40:1–53. doi: 10.1017/S003358350700460X. [DOI] [PubMed] [Google Scholar]
- 19d.Denysenkov VP, Biglino D, Lubitz W, Prisner TF, Bennati M. Angew. Chem. 2008;120:1244. doi: 10.1002/anie.200703753. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2008;47:1224. [Google Scholar]
- 19e.Tkach I, Pornsuwan S, Höbartner C, Wachowius F, Sigurdsson S, Baranova T, Diederichsen U, Sicoli G, Bennati M. Phys. Chem. Chem. Phys. 2013;15:3433–3437. doi: 10.1039/c3cp44415e. [DOI] [PubMed] [Google Scholar]
- 20.Altenbach C, Marti T, Khorana HG, Hubbell WL. Science. 1990;248:1088–1192. doi: 10.1126/science.2160734. [DOI] [PubMed] [Google Scholar]
- 21.Jeschke G, Chechik V, Ionita P, Godt A, Zimmermann H, Banham J, Timmel CR, Hilger D, Jung H. Appl. Magn. Reson. 2006;30:473–498. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information