

Abstract
C-type cytochromes are distinguished by the covalent attachment of a heme cofactor, a modification that is typically required for its subsequent folding, stability, and function. Heme attachment takes place in the mitochondrial intermembrane space and, in most eukaryotes, is mediated by holocytochrome c synthase (HCCS). HCCS is the primary component of the eukaryotic cytochrome c biogenesis pathway, known as System III. The catalytic function of HCCS depends on its ability to coordinate interactions between its substrates: heme and cytochrome c. Recent advancements in the recombinant expression and purification of HCCS have facilitated comprehensive analyses of the roles of conserved residues in HCCS, as demonstrated in this study. Previously, we proposed a four-step model describing HCCS-mediated cytochrome c assembly, identifying a conserved histidine residue (His154) as an axial ligand to the heme iron. In this study, we performed a systematic mutational analysis of 17 conserved residues in HCCS, and we provide evidence that the enzyme contains two heme-binding domains. Our data indicate that heme contacts mediated by residues within these domains modulate the dynamics of heme binding and contribute to the stability of the HCCS–heme–cytochrome c steady state ternary complex. While some residues are essential for initial heme binding (step 1), others impact the subsequent release of the holocytochrome c product (step 4). Certain HCCS mutants that were defective in heme binding were corrected for function by exogenous aminolevulinic acid (ALA, the precursor to heme). This chemical “correction” supports the proposed role of heme binding for the corresponding residues.
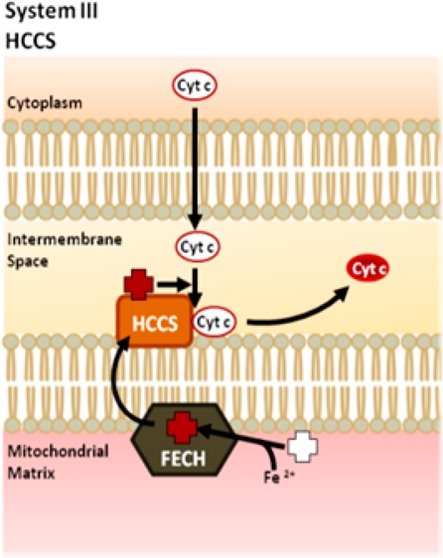
In eukaryotes, c-type cytochromes are important metalloproteins that function in diverse cellular processes such as mitochondrial respiration and programmed cell death.1,2 Cytochromes c are covalently attached to heme via thioether bonds formed between the two vinyl groups of the heme and two cysteinyl thiol groups in the conserved heme-binding motif of the apocytochrome protein at CysXxxXxxCysHis (CXXCH). The covalent attachment of the heme cofactor occurs post-translationally within the mitochondrial intermembrane space.3,4 The enzyme that catalyzes heme attachment in mitochondria is known as holocytochrome c synthase (HCCS). It has been demonstrated that thioether formation is spontaneous as long as the cysteinyl thiols of cytochrome c and the heme iron are both in the reduced state.5,6 Therefore, the role of HCCS is likely to facilitate proper stereospecific positioning of these two substrates at its active site. Fungi possess two related HCCS proteins; one matures cytochrome c7 and another matures cytochrome c1.8−10 However, humans and other animals express a single HCCS that recognizes both c-type cytochromes. HCCS is an essential protein in animals,11 with mutations in the X-linked human HCCS gene resulting in lethality in males and in a rare disease called microphthalmia with linear skin defects (MLS) in females.12
HCCS-mediated cytochrome c maturation is referred to as System III in eukaryotes and performs a function similar to the well-characterized Systems I and II found in prokaryotes and plants.13−15 However, these pathways are unrelated to System III since each protein in Systems I and II appears to be divergent from HCCS at the sequence level.4,13 Unlike the elaborate multiprotein, integral membrane complexes of Systems I and II, System III is rather simple, with HCCS peripherally associated with the inner mitochondrial membrane, though predicted to be a soluble protein.7,12,16 Despite the apparent simplicity of HCCS, and its discovery over 25 years ago, only recently has a greater understanding of its structure and function emerged. In 1987, Dumont and colleagues complemented a respiratory-deficient Saccharomyces cerevisiae strain with the yeast HCCS gene, designating it the cytochrome c heme lyase.7 Many of the earlier studies on HCCS relied on isolated mitochondrial fractions to indirectly assess the functional capabilities of this enzyme, in which it was also shown that HCCS is necessary for import of apocytochrome c into the mitochondrial intermembrane space.17−19 A recent breakthrough in the purification of a functional recombinant HCCS has facilitated the present study on the mechanistic and structure/function properties of HCCS.20
We recently purified the human HCCS from Escherichia coli membranes (as a GST-fusion) showing that it possesses endogenously synthesized heme.20 The HCCS protein is purified from isolated E. coli membranes using detergent, whereas its product, holocytochrome c, is released into the E. coli cytoplasm. It was discovered that a conserved histidine in human HCCS, His154, provides an essential axial ligand to the heme substrate. By copurifying the human HCCS and its cognate human cytochrome c, it was shown that an HCCS: heme-scaffold is required for the recruitment of the cytochrome c acceptor substrate.20 These data provided the basis for the following proposed four-step cytochrome c assembly mediated by HCCS (Figure 1A): (1) HCCS binds to the heme-cofactor with His154 serving as an axial ligand. (2) Heme-bound HCCS recognizes and binds to cytochrome c (referred to as apocytochrome c in its nascent state), with the histidine (of CXXCH) from apocytochrome c (His19) supplying the second axial ligand. (3) HCCS catalyzes (by orchestrating substrate proximity and orientation) formation of the covalent thioether bonds between cytochrome c and heme, forming holocytochrome c (i.e., the heme-bound state of cytochrome c). And (4) HCCS releases holocytochrome c to fold into its native conformation. (Note that only when the two thioether bonds are formed (step 3) is the holocytochrome c released (step 4) from the complex.) The recombinant functional assays and purification of the human HCCS now make it possible to perform a comprehensive structure/function study on this previously uncharacterized protein. The analysis of key residues in HCCS that participate in the above steps is presented in the current study.
Figure 1.

HCCS model for cytochrome c assembly and primary sequence. (A) Model of HCCS-mediated cytochrome c assembly adapted from ref (20). (B) ClustalW-generated alignment of HCCS homologues from various eukaryotic organisms. Sequences with extensive gaps in conservation were condensed, with the number of intervening residues enclosed by brackets. Residues shaded in gray are conserved across diverse phyla. Residues shaded in yellow are conserved in HCCS homologues from animals. Putative functional and/or structural domains are indicated (DI, DII, DIII, and DIV) and are enclosed by black boxes. Domains that mediate mitochondrial import are enclosed by red-dashed boxes. Black dots indicate residues that were substituted either previously (H154 and H211 in ref (20)) or for the present study. The His154 ligand to heme iron is shaded blue. Red asterisks indicate residues that undergo mutation in patients with MLS (E159 and R217). Hs, Homo sapien (UniProt P53701); Ec, Equus caballus (UniProt F6YF82); Xl, Xenopus laevis (UniProt Q6DOG6); Dm, Drosophila melanogaster (UniProt Q9VD55); Ts, Trichinella spiralis (UniProt E5SBT9); Vc, Volvox carteri (UniProt D8U027); Nc, Neurosporo crassa (UniProt P14187).
The sequence alignment of HCCS homologues from diverse eukaryotic organisms, including fungi, protozoa, and metazoans, reveals that HCCS maintains less than 20% sequence identity, with 46 residues completely conserved (Figure 1B). Results presented here suggest that the conserved amino acid residues in HCCS group together in what we describe as four putative domains (Figure 1B, solid lined boxes). Previous characterization of the mitochondrial targeting signal of HCCS identified two internal regions that were both necessary and sufficient to mediate its translocation across the outer membrane of the mitochondria and into the intermembrane space21 (Figure 1B, dashed red-lined boxes). The sequence limits of one of these sorting domains overlaps with one of the functional assignments described here (Figure 1B, domain II), signifying a potential dual role for the conserved residues contained therein. In the present study, we performed a systematic mutational analysis to uncover the roles of highly conserved amino acids and establish whether “domains” with distinguishable functional roles might indeed be present.
We present evidence that conserved residues in two of the putative domains (I and II) are involved in heme binding (Figure 1A, step 1), which in some instances also affected the release of the holocytochrome c product (Figure 1A, step 4). In addition, our data suggest that the conserved residues in the C-terminal domain (IV) contribute to the overall folding and/or stability of HCCS (see Discussion). The functional impacts of the HCCS substitutions associated with the human disease MLS (E159K and R217C, red asterisks in Figure 1B) were investigated, ultimately revealing defects in heme-binding (E159K) and potentially protein folding (R217C). Finally, results using exogenously added δ-aminolevulinic acid (ALA), a heme precursor that increases the levels of endogenous heme available for heme proteins, supported the functional assignments of certain mutations involved in heme binding. Notably, some of these HCCS defects could be corrected for function by exogenous ALA. These analyses not only provide insight into the mechanistic roles of the conserved residues involved in HCCS-mediated cytochrome c assembly but also support our model identifying heme as the core component driving the synthetase activity of HCCS.
Materials and Methods
Construction of Strains and Plasmids
Plasmids used in this study, pRGK403 (N-terminal GST-tagged human HCCS) and pRGK405 (human cytochrome c (CYCS)), have been described previously.20 All oligonucleotide primer sequences and derived plasmids are reported in Supplemental Table 1, Supporting Information (Table S1). Nucleotide substitutions were engineered using the QuikChange II Site-Directed Mutagenesis kit (Agilent Technologies) according to the manufacturer’s specifications. All cloning steps were confirmed by sequencing. Verified clones were transformed into the E. coli Δccm strain RK103.22
BPER Functional Assay
E. coli strains were grown overnight and used to inoculate 5 mL of LB broth supplemented with appropriate antibiotics. These cultures were grown at 37 °C with shaking at 200 rpm for 3 h, followed by induction with 0.1 mM IPTG and 0.8% arabinose (wt/vol) for an additional 3 h. For ALA-supplemented cultures, ALA (Sigma) was added at 50 μg/mL during induction. Cells were harvested by centrifugation at 4500g, and the cell pellet was lysed in 200 μL of B-PER reagent (Thermo Scientific). Total protein was quantified using the Nanodrop 1000 spectrophotometer (Thermo Scientific), and 100 μg of extracted protein was resolved by SDS-PAGE, transferred to nitrocellulose, and analyzed by heme stain.
Protein Expression and Purification
GST-HCCS proteins were expressed (with or without cytochrome c) and purified from the E. coli Δccm strain RK103 as described previously.20 Briefly, 100 mL starter cultures were grown overnight at 37 °C with shaking at 200 rpm and used to inoculate 1 L of LB broth supplemented with the appropriate antibiotics. Following 1 h growth of the 1 L cultures at 37 °C with shaking at 120 rpm, the cultures were induced with 0.1 mM IPTG for expression of pGEX-HCCS plasmids for 5 h. For coexpression of pBAD-cycS (cytochrome c), the cultures were induced with 0.2% arabinose (wt/vol) 2 h after the induction of HCCS expression. Cells were harvested by centrifugation at 4500g, resuspended in PBS with 1 mM PMSF, and sonicated. The crude sonicate was cleared by centrifugation at 24000g for 20 min, and the membrane fraction was isolated by ultracentrifugation at 100000g for 45 min. Membrane pellets were solubilized in 50 mM Tris (pH 8), 150 mM NaCl, 1% Triton X-100 on ice for 1 h. Solubilized membranes were loaded onto glutathione agarose (Pierce) for an overnight batch pull-down of GST-HCCS protein (with or without the copurified cytochrome c). Bound GST-HCCS protein or cocomplexes were eluted with 20 mM reduced glutathione in 50 mM Tris (pH 8), 150 mM NaCl, 0.02% Triton X-100, concentrated in an Amicon Ultra Centrifugal Filter (Millipore), and the total protein concentration was determined using the Bradford reagent (Sigma).
Heme Stain
Heme stains were performed as described previously.23 Briefly, to preserve the heme signal, protein samples were prepared for SDS-PAGE with loading dye at 1:1 (v/v) that did not contain reducing agents, and the samples were not boiled. Following electrophoresis, proteins were transferred to nitrocellulose, and the chemiluminescent signal for the heme stain was developed using the SuperSignal Femto kit (Thermo Scientific) and detected with the ImageQuant LAS4000 mini detection system (Fujifilm-GE Healthcare). For the BPER functional assay, the heme stained cytochrome c signal was quantified by densitometry using ImageJ analysis software (NIH).
Antibodies and Immunoblotting
A GST tagged version of the WT human HCCS was expressed, purified, and injected into rabbits for the production of antisera against GST-HCCS by Cocalico Biologicals. To purify the antibodies, sodium dextran sulfate was added to the serum at 0.25% (wt/vol) followed by the addition of CaCl2 at 11% (wt/vol) and centrifugation at 11000g. A saturated 76% ammonium sulfate-PBS solution (wt/vol) was added to the supernatant and incubated with gentle agitation at 4 °C overnight. The antibodies were pelleted by centrifugation at 11000g, washed in the ammonium sulfate-PBS solution, resuspended in PBS at 1/10 the starting serum volume, and dialyzed with PBS overnight. A working solution of the prepared antisera against GST-HCCS was made and used at a dilution of 1:5000. Protein A peroxidase (Sigma) was used as a secondary label for detection. The chemiluminescent signal for anti-GST-HCCS immunoblots was developed using the Immobilon Western kit (Millipore) and detected by the ImageQuant LAS 4000 mini detection system (Fujifilm-GE Healthcare).
Reduced Pyridine Hemochrome
Heme type and concentration in purified protein preparations was determined by pyridine extraction as described previously.24 Briefly, 0.5 M NaOH and pyridine were added to 100 μg of purified protein to yield final concentrations of 100 mM NaOH and 20% pyridine (v/v). Samples were chemically reduced with the addition of solid sodium dithionite (sodium hydrosulfite), and UV–vis spectra were recorded from 500 to 600 nm. Heme concentration was determined using an extinction coefficient of 23.9 mM–1 cm–1.24
UV–vis Absorption Spectroscopy
UV–vis absorption spectra were recorded with a Shimadzu UV-2101 PC UV–vis scanning spectrophotometer at room temperature as described previously.25 All spectra were obtained in the same buffer in which the proteins were purified: 50 mM Tris (pH 8), 150 mM NaCl, and 0.02% Triton X-100. Chemically reduced spectra were generated upon the addition of solid sodium dithionite (sodium hydrosulfite) to the purified sample. Where specified, imidazole (1 M, pH 7) was added to purified protein samples at 100 mM prior to spectra acquisition.
Results
We engineered 29 HCCS mutants, changing the conserved residues to (1) an alanine, (2) an amino acid of opposite or like charge, and/or (3) an amino acid with disease relevance (i.e., MLS mutations E159K and R217C). In most cases, we decided not to substitute conserved residues that lacked reactive side chains (e.g., alanines, glycines, prolines, and leucines). In short, we discovered that 17 of the mutants were nonfunctional, and, of those, 12 were poorly expressed or unstable (see below). To determine the functional consequences of each mutation, we coexpressed each HCCS variant along with an arabinose-inducible gene for the human cytochrome c in an E. coli strain lacking its own endogenous cytochrome c maturation system (Δccm). HCCS mutants that maintained synthetase activity (i.e., attached heme and released the 12 kDa holocytochrome c product) yielded holocytochrome c at levels comparable to or better than wild type (WT) HCCS; mutants with functional defects released significantly less holocytochrome c (Figures 2 and S1). We define “function” or “activity” as the ability to both attach heme to and subsequently release the cytochrome c acceptor as detected either by heme stain (Figure 2) or spectrally (Figure S1). For clarity, the functional data for each mutant will be discussed according to the respective domains. Note that a priori we evaluated the “domains” initially based on their natural clustering in the HCCS alignment (Figure 1B). We suggest that the data in general also support the presence of functional “domains.” However, regardless of the validity of domain assignment, results concerning the structure/function contributions of individual residues are important to the understanding of this enzyme.
Figure 2.

Substitution of conserved residues in HCCS putative domains modulate synthetase function. Recombinant GST-HCCS variants with substitutions in (A) Domain I, (B) Domain II, (C) Domain III, and (D) Domain IV were coexpressed with cytochrome c in Δccm E. coli. Cells were lysed with BPER reagent, and protein extracts were resolved by SDS-PAGE and transferred to nitrocellulose. Released cytochrome c was detected by heme stain and signal intensity was quantified by densitometry and plotted in GraphPad Prism. Data shown represent the average amount of cytochrome c released by each mutant relative to the level released by WT ± SEM, n = 4.
Synthetase Function of HCCS Variants
Domain I
We tested four of the nine amino acid residues in domain I (human HCCS amino acid positions 115–133) that are completely conserved across diverse phyla, along with a Met residue at position 130 in human HCCS, which is conserved in animals (Figure 1B). Initial assays were carried out in small scale cultures with BPER-extracted protein assayed for holocytochrome c by heme staining post-SDS-PAGE. We observed that HCCS variants with mutations in domain I retained activity, with some mutants releasing >1.5 fold more holocytochrome c than WT (Figure 2A and Table S2; e.g., see W118A and N128A). These results suggest that certain mutations can enhance HCCS activity, perhaps through altered heme/product-binding properties (e.g., increasing step 4, Release, see below and Discussion). Even double mutants Y120A/P121A and N128A/M130A maintained activity similar to WT (Figure 2A and Table S2), indicating that these residues do not significantly contribute to HCCS synthetase activity.
For a more quantitative assessment of mutant activity, and as a complement to the small-scale assay, we used UV–vis spectroscopy to analyze the holocytochrome c released by the HCCS mutants and the WT HCCS control. Covalently attached heme in cytochrome c absorbs at 550 nm, producing a sharp alpha peak when reduced, which was measured to determine relative amounts of cytochrome c present. Consistent with our densitometric analysis of heme stains (i.e., the small-scale BPER assay in Figure 2A), the UV–vis absorption spectra of the respective soluble fractions confirmed that the W118A and N128A HCCS variants yielded up to 2-fold more matured cytochrome c than WT (Figure S1A and Table S2). Below we show data that is consistent with the proposal that residues in this domain contribute to HCCS: heme interactions leading to increases in released holocytochrome c.
Domain II
Of the nine conserved residues in domain II (human HCCS amino acid residues 154–169) (Figure 1B), we made substitutions at six positions. Residues in domain II were previously designated as part of the mitochondrial import signal of HCCS;21 however, we have already shown that the His154 residue serves as an essential ligand required for heme binding and subsequent cytochrome c maturation.20 The synthetase activity of the H154A HCCS mutant was completely abrogated. Similar to the H154A HCCS mutant, substitution of several other conserved residues in domain II resulted in significantly reduced activity (Figure 2B and Table S2). For instance, alanine substitutions at residues Trp162, Trp168, and Glu169 largely abolished HCCS activity, yielding low to undetectable levels of holocytochrome c, as determined by heme stain and UV–vis spectroscopy (Figure 2B, Figure S1B, and Table S2). An exception was observed in the substitution of Asn155, the conserved residue adjacent to His154. Mutation of Asn155 resulted in a 2-fold increase in HCCS activity (Figure 2B). Spectral quantitation of the released holocytochrome c from the N155A HCCS mutant further confirmed this increase (Figure S1B and Table S2).
Domain II contains the conserved residue Glu159, which is mutated to a lysine in some patients with MLS disease.26 A previous report has shown that the equivalent substitution in yeast HCCS at the corresponding glutamate residue renders the protein nonfunctional in recombinant E. coli.27 We observed that the production of holocytochrome c by human E159K HCCS was significantly reduced in small scale cultures, yielding less than 20% of the WT levels (Figure 2B and Table S2). However, when we substituted an alanine residue for Glu159 in human HCCS (E159A), holocytochrome c yields increased 2-fold above that of WT (Figure 2B, Figure S1B, and Table S2). Moreover, when we replaced Glu159 with an aspartate residue, thus maintaining the charge, WT activity was restored (Figure 2B, Figure S1B, and Table S2). These results suggest that the positive charge of the MLS-associated lysine substitution at Glu159 in HCCS causes functional perturbations that ultimately lead to the MLS disease phenotype (see below).
Domain III
The seven conserved residues in domain III (human HCCS amino acids 207–213) are part of the bipartite mitochondrial targeting signal (human HCCS amino acids 189–216) (Figure 1B) identified near the C-terminal portion of the yeast HCCS, previously shown to be necessary and sufficient for mitochondrial import.21 We made substitutions at two of these residues, His211 and Trp213. We mutated the conserved histidine at position 211 in our earlier study and determined that this residue did not make a major contribution to the synthetase activity of HCCS, in that cytochrome c production remained within 2-fold of WT levels when His211 was substituted with other amino acids.20 In the current study, we expressed a W213A HCCS mutant and also observed synthetase activity similar to WT (Figure 2C, Figure S1C, and Table S2). We thus focused on residues in the other domains for this study, with the likelihood that domain III represents the mitochondrial targeting domain.
Domain IV
Of the 13 conserved residues in domain IV (human HCCS amino acids 217–258), we made substitutions at 5 positions, along with an aspartate at position 257 (Figure 1B). While designating the boundaries of the assigned domains, we observed that the C-terminal portion of HCCS (composed of domains III and IV) contained several alternating charged residues, particularly arginines and aspartates, most of which were evolutionarily invariant (Figure 1B). We hypothesized that the frequency of the highly conserved DR/DXR motif may have functional significance; therefore, we made single and double substitutions at many of these Asp and Arg residues in domain IV and measured the cytochrome c levels produced by these mutants. We found that, in nearly every case, substitution of the C-terminal aspartate and arginine residues (e.g., Asp227 and Arg246) resulted in inactive HCCS proteins (Figure 1D, Figure S1D, and Table S2). For instance, when we changed Asp257 and Arg258 to alanines in tandem, generating the HCCS double mutant D257A/R258A, we observed that cytochrome c yields were approximately 10-fold less than that of WT (Figure 2D, Figure S1D, and Table S1), indicating that at least one of these residues is important for HCCS activity. Only the single mutation of Asp257 to alanine (D257A) yielded WT levels of function (Figure 2D, Figure S1D, and Table S2). Interestingly, Asp257 is only conserved among HCCS enzymes in animals (Figure 1B). When we tested the R258A single mutant, HCCS activity was reduced to a level similar to the D257A/R258A double mutant (Figure 2D). The large scale assays were also consistent with the synthetase activity observed in the small-scale preparations for these substitutions (Figure S1D and Table S2).
Amino acid Arg217 in domain IV is the second of two residues implicated in MLS disease, where it is mutated to a cysteine residue. When the corresponding mutation was generated in yeast, HCCS activity was severely attenuated.28 Likewise in the current study with human HCCS, the R217C mutant exhibited a clear defect in cytochrome c maturation, yielding no detectable product by heme stain or spectral analysis (Figure 2D, Figure S1D, and Table S2). To test whether the positive charge of Arg217 was important, we substituted either a glutamate (negatively charged), a lysine residue (positively charged), or an alanine for Arg217. We observed that no Arg217 variants retained activity.
Expression and Purification of HCCS Mutants
Of the 29 HCCS variants generated, 17 exhibited defects in the release of matured cytochrome c, with substitutions in domain IV comprising the majority of these nonfunctional mutants. We considered that the lack of activity may be due to HCCS expression defects and/or protein instability caused by certain mutations. Accordingly, subsequent purification of some of the nonfunctional mutants produced poor protein yields when expressed either alone or with the cytochrome c substrate (Table S3), suggesting that the corresponding mutations perturbed the structural integrity of HCCS. For example, the MLS HCCS mutant R217C fit into this category, as well as almost all of the domain IV substitutions, in which less than 25% WT protein yields were obtained. Therefore, HCCS mutants that yielded insufficient protein (namely, W162A, W168A/E169A, W168A, E169A, E169K, R127C, R217K, R217D, R217A, R246A, D257K, and R258E HCCS mutants) were not pursued further.
Mutations in Domains I and II of HCCS Affect Heme Interactions
Domain I: Trp118, Asn128, and Met130
Since several mutants in domain I appeared to display enhanced activity, we hypothesized that the corresponding residues mediate interactions with heme, thus leading to the increased release of the cytochrome c product (see Figure 1A, step 4) when mutated. Our previous study established that while HCCS by itself purifies with a b-type heme partially in the ferrous (Fe2+) state, recombinant WT HCCS largely copurifies in complex with holocytochrome c, when coexpressed.20 To investigate the substrate interactions of the domain I mutants, we expressed each HCCS mutant with (or without) the cytochrome c substrate. These proteins were then purified from detergent-solubilized membranes and assessed for their respective spectral properties.
As previously reported,20 the Soret peak of the WT HCCS–heme–cytochrome c ternary complex corresponded to 409 nm (characteristic of ferric (Fe3+) heme), while the longer wavelength alpha and beta peaks appeared broader and less defined between 500–600 nm (Figure 3A, black). Chemical reduction of the heme-containing WT complex with sodium dithionite resulted in a characteristic red-shift of the Soret peak to 420 nm and increased absorption of the alpha and beta peaks at 555 and 524 nm, respectively (Figure 3A, red). A pyridine hemochrome of the reduced WT complex showed a peak absorption at 551 nm (inset, Figure 3A), consistent with the conclusion that the majority of the heme in the complex is c-type (i.e., covalently attached to the bound cytochrome c).24 By contrast, the HCCS mutants W118A and N128A/M130A displayed spectral features that differed significantly from the WT HCCS–cytochrome c cocomplex (Figures 3B–C). Notably, the wavelength maxima of the Soret peaks for both of these mutants indicated the presence of heme iron in the characteristically ferrous (Fe2+) state (418 nm for W118A and 419 nm for N128A/M130A) and remained unchanged upon the addition of reducing agent (Figure 3B,C). The alpha peaks of both mutants sharpened at 552 nm under reducing conditions (Figure 3B,C), and the pyridine spectra for both complexes yielded alpha peak maxima at 553–554 nm (Figure 3B,C, inset). These results indicate the presence of b-type heme (i.e., a pyridine spectrum alpha maximum in between that expected for predominate c-type (550 nm) or b-type heme (556 nm)24). Therefore, we propose that these mutant complexes contain a mixture of b- and c-type heme species.
Figure 3.

Mutation of HCCS Domain I residues alter heme interactions. Recombinant GST-HCCS protein (alone) and GST-HCCS: cytochrome c cocomplexes were purified from Δccm E. coli and prepared for UV/vis absorption spectroscopy and SDS-PAGE. Shown are spectra for (A) WT HCCS/cyt c, (B) W118A HCCS/cyt c, and (C) N128A/M130A HCCS/cyt c following purification (black line), chemical reduction with sodium dithionite (red), and extraction with pyridine (inset). (D) Soret peak spectra were obtained from cocomplexes representing WT HCCS/cyt c (left) and W118A HCCS/cyt c (right) following purification (black) and treatment with 100 mM imidazole (purple). UV/vis spectra between 500–580 nm (alpha/beta region) of cocomplexes treated with 100 mM imidazole following chemical reduction with sodium dithionite are shown for (E) WT HCCS/cyt c, (F) W118A HCCS/cyt c, (G) N128A/M130A HCCS/cyt c, (H) N128A HCCS/cyt c, and (I) M130A HCCS/cyt c. (J) Heme stain of the indicated purified cocomplexes following SDS-PAGE and transfer to nitrocellulose. (K) Heme stain (top) and GST-HCCS immunoblot (bottom) of the indicated purified cocomplexes following SDS-PAGE and transfer to nitrocellulose. (L) UV–vis spectra of sodium dithionite reduced purified cocomplexes from WT HCCS (black), Y120A HCCS (red), and P121A HCCS (blue). (M) UV–vis spectra of HCCS proteins (alone) purified from WT HCCS (black) and Y120A/P121A HCCS (green). (N) UV–vis spectra of purified cocomplexes from WT HCCS/cyt c (black) and Y120A/P121A HCCS/cyt c (orange). (O) Heme stain (top) and GST-HCCS immunoblot (bottom) of the indicated purified cocomplexes following SDS-PAGE and transfer to nitrocellulose. (P) UV–vis spectra of HCCS proteins (alone) purified from WT HCCS (black), Y120A HCCS (red), and P121A HCCS (blue). Arrows indicate wavelength (nm) of peak absorption maxima. All spectra were performed with equal amounts (50–100 μg) of total purified protein. All SDS-PAGE samples were equally loaded (2–5 μg of total purified protein each). For all proteins, Bradford quantitation was confirmed by Coomassie staining, which also indicated that GST-HCCS proteins were obtained at >90% purity.
To further examine the qualities of the heme interactions of these mutants, we added exogenous imidazole (a structural mimic of histidine that can displace transient heme ligands25,29,30 to each purified HCCS cocomplex and obtained UV–vis absorption spectra. In the presence of imidazole, the Soret peak of the WT cocomplex does not shift from its initial position at 410 nm (Figure 3D, left), consistent with our prior conclusion that the heme ligands of the HCCS–cytochrome c complex (i.e., His154 and His19, respectively) stably occupy the axial coordination positions of the heme iron (see Figure 1A, step 2). However, the Soret peak of the W118A HCCS cocomplex is blue-shifted 3 nm (from 418 to 415 nm) in the presence of imidazole, suggesting that a labile heme ligand was indeed replaced upon addition of the compound (Figure 3D, right). This spectral shift in the Soret peak is characteristic of HCCS protein purified alone (i.e., in the absence of the cytochrome c acceptor substrate) with the b-type heme stabilized by the His154 ligand of HCCS and an unidentified ligand occupying the other axial coordination site.20 Chemical reduction of imidazole-treated W118A HCCS cocomplexes resulted in the resolution of a broad split-alpha peak with maxima at 553 and 559 nm, unlike the WT complex (Figure 3E,F). We propose that the appearance of an alpha peak/shoulder at 553 and 559 nm indicates the presence of both c- and b-type heme, respectively. These results support the proposition that the mutant complex contains a mixture of heme types, a feature perhaps associated with the enhanced release function. That is, less holocytochrome c is trapped in complex (see steps 2 and 3 of Figure 1A) with HCCS mutants that exhibit higher release activity (step 4).
Similar to the W118A HCCS variant, reduction of imidazole-treated cocomplexes from the N128A/M130A HCCS double mutant resulted in the formation of a broad split alpha peak-shoulder at 552 and 560 nm (Figure 3G), suggesting that this mutant also contained a mixture of heme species. When we assessed the single HCCS mutants N128A and M130A, we observed that their individual spectral contributions could be distinguished in the composite features of the N128A/M130A double mutant. For instance, the Soret peak of cocomplexes from the N128A HCCS mutant appeared at 419 nm and did not red-shift upon the addition of sodium dithionite to further reduce the heme iron (Figure S2A), similar to the N128A/M130A double mutant (Figure 3C). However, chemical reduction of N128A HCCS cocomplexes did not resolve an alpha peak (Figure S2A), suggesting that any copurified cytochrome c was significantly below spectral limits of detection. Consistent with this data, heme staining of the N128A HCCS cocomplex revealed that the heme signal for cytochrome c was significantly lower than the levels copurified with WT HCCS (Figure 3J, Holo-cyt c). The lower levels of cytochrome c copurified with the N128A HCCS mutant are also consistent with the observed functional enhancement in the release of the holocytochrome c product (Figure 2A, Figure S1A, and Table S2). Furthermore, chemical reduction of imidazole-treated N128A HCCS cocomplexes yielded an alpha peak at 560 nm (Figure 3H), indicating b-type heme as the predominate species present. Similar to the role of Trp118, these data suggest that the Asn128 residue in HCCS also contributes to the formation and stability of the cocomplex and potentially participates in the controlled release of the matured cytochrome c product.
Analysis of the M130A HCCS cocomplex revealed that this mutant possessed heme characteristics similar to WT (Figure S2B and Table 1). Addition of imidazole to purified M130A HCCS cocomplexes did not shift the Soret peak (data not shown), and subsequent chemical reduction of the imidazole-treated cocomplexes resulted in a comparable alpha peak maximum at 552 nm (Figure 3I, compare to Figure 3E), suggesting that both axial coordination sites of the bound heme were occupied by stable ligands (presumably His154 from HCCS and His19 from cytochrome c). Consistent with these data, heme stain analysis of M130A HCCS cocomplexes indicated that this mutant copurified with holocytochrome c at levels similar to WT HCCS (Figure 3J, Holo-cyt c). In Figure 3J, it is shown that some b-type heme remains associated with HCCS, while in the WT cocomplex, most heme is associated with the holocytochrome c, as noted previously.20 Though conserved in animal HCCS enzymes, Met130 is variant in the yeast HCCS homologue (Figure 1B). Our results suggest that Met130 is not required for the HCCS functions evaluated here.
Table 1. Quantitation of Heme Bound to Purified HCCSa.
| |
total heme |
||
|---|---|---|---|
| putative domain | HCCS variant | expressed alone | coexpressed with cytochrome c acceptor |
| WT | 1 | 1 | |
| Domain I | W118A | 1.2 ± 0.06 | 0.7 ± 0.02 |
| Y120A/P121A | <0.10 | <0.10 | |
| Y120A | <0.10 | 1.0 ± 0.06 | |
| P121A | 0.5 ± 0.14 | 0.7 ± 0.13 | |
| N128A/M130A | 0.9 ± 0.03 | 0.6 ± 0.07 | |
| N128A | 1.3 ± 0.51 | 0.8 ± 0.17 | |
| M130A | 1.1 ± 0.18 | 1.2 ± 0.07 | |
| Domain II | N155A | 1.0 ± 0.09 | 0.5 ± 0.12 |
| E159K | 0.5 ± 0.10 | 0.3 ± 0.08 | |
| E159A | 0.4 ± 0.07 | 0.3 ± 0.05 | |
| E159D | 1.2 ± 0.23 | 0.9 ± 0.09 | |
Bound heme was determined from reduced pyridine hemochrome spectra using 100 μg of total purified protein. Each value is relative to the amount of total heme purified with WT HCCS under each respective condition, which has been set to 1. Data shown represent the average of at least two separate experiments ± SEM.
Domain I: Tyr120 and Pro121
Functional analysis of the Y120A and P121A HCCS single variants established that these mutants displayed WT-like and enhanced release function, respectively (Figure 2A, Figure S1A, and Table S2). On the basis of these data, we predicted that the heme profile of the Y120A HCCS mutant would be similar to WT, while that of the P121A HCCS mutant would be considerably different. Accordingly, heme staining and spectral analysis of the purified Y120A HCCS cocomplexes were similar to the features displayed by WT HCCS, particularly in the resolution of the reduced alpha peak at 554 nm (Figure 3K,L and Table 1). Co-complexes from the P121A HCCS mutant exhibited a heme profile characteristic of HCCS purified in the absence of the cytochrome c acceptor (Figure 3K,L and Table 1), consistent with the observed enhanced release function (Figure 2A, Figure S1A, and Table S2).
While evaluating the function of the Y120A/P121A HCCS double mutant, we discovered that the UV–vis absorption-based quantitative measurement of the released cytochrome c in the soluble fraction largely conflicted with the near-WT levels detected by heme stain from our small scale functional assay. Following coexpression and purification, the UV–vis absorption spectra of the soluble fraction from the Y120A/P121A HCCS double mutant indicated that the levels of matured cytochrome c present were less than half that obtained from the WT (Figure S1A, and Table S2), representing a significant decrease in function. To uncover the basis for this, we investigated the spectral properties of the Y120A/P121A HCCS double mutant, both in the presence and absence of the cytochrome c acceptor substrate. Interestingly, in both cases (i.e., with and without cytochrome c) we found that the Y120A/P121A HCCS mutant exhibited very little spectral evidence of heme binding (Figure 3M,N and Table 1), which was also consistent with the heme stain analysis of the purified protein (Figure 3K,O). Despite the lack of copurified heme, Y120A/P121A HCCS retains at least 50% WT function (Table S2), which distinguishes this mutant from the completely nonfunctional H154A HCCS variant described in our previous study.20 Although the Y120A/P121A HCCS double mutant was significantly deficient in heme binding, the single Y120A HCCS mutant purified with WT-levels of heme when coexpressed with the cytochrome c substrate (Figure 3K,L and Table 1). However, when we expressed the single Y120A and P121A HCCS mutants alone (i.e., in the absence of the cytochrome c acceptor), we discovered that both mutants exhibited heme-binding defects, with the former displaying a defect nearly as severe as the Y120A/P121A HCCS double mutant (Figure 3O–P and Table 1). We speculate in the Discussion that heme-binding “defects” can impact both steps 1 and 4 (Figure 1A) of HCCS-mediated cytochrome c assembly, so that levels of the final released holocytochrome c product ultimately depend on the strength of the interaction between heme and HCCS.
Domain II: Asn155
The functional data for the N155A HCCS mutant indicated that this substitution resulted in a 2-fold increase in cytochrome c synthesis, suggesting an enhanced release function. When we examined the spectral features of purified N155A HCCS cocomplexes, we observed characteristics shared by the domain I mutants that exhibited a similar increase in function. For instance, the Soret peak of the N155A HCCS mutant cocomplexes (422 nm) indicated the presence of heme in the ferrous (Fe2+) state (Figure 4A, black). Chemical reduction with sodium dithionite did not cause a shift in the Soret (Figure 4A, red), similar to WT HCCS when purified alone. These data were confirmed by heme stain analysis showing that the N155A HCCS cocomplex purified with predominantly b-type heme bound to the HCCS protein, while the majority of the heme in the WT cocomplex was covalently attached to the cytochrome c substrate (Figure 4B), a result consistent with the respective reduced pyridine hemochrome spectra (Figure 4A, inset).
Figure 4.
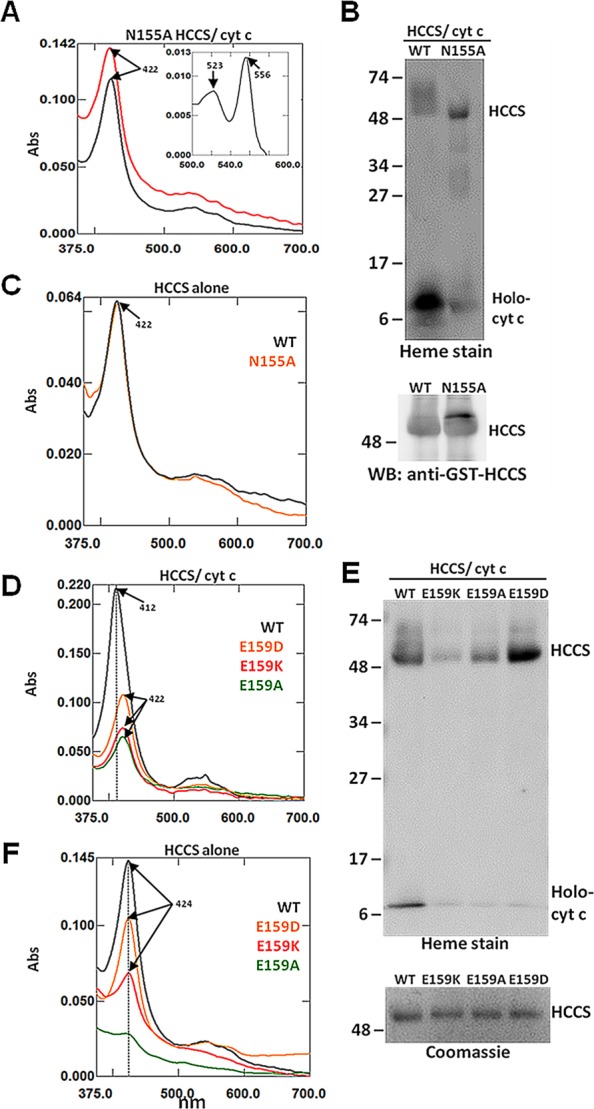
Mutation of HCCS Domain II residues alter heme interactions. Recombinant GST-HCCS protein (alone) and GST-HCCS: cytochrome c cocomplexes were purified from E. coli and prepared for UV/vis absorption spectroscopy and SDS-PAGE. (A) UV–vis spectra of purified (black), sodium dithionite reduced (red), and pyridine extracted (inset) N155A HCCS/cyt cocomplexes. (B) Heme stain (top) and GST-HCCS immunoblot (bottom) of the indicated purified cocomplexes following SDS-PAGE and transfer to nitrocellulose. (C) UV–vis spectra of purified WT HCCS protein (black) and N155A HCCS protein (orange). (D) UV–vis spectra of purified cocomplexes from WT HCCS/cyt c (black), E159D HCCS/cyt c (orange), E159K HCCS/cyt c (red), and E159A (green). (E) Heme stain (top) of the indicated purified cocomplexes following SDS-PAGE and transfer to nitrocellulose, and corresponding Coomassie stain (bottom). (F) UV–vis spectra of purified WT HCCS protein (black), E159D HCCS protein (orange), E159K HCCS protein (red), and E159A HCCS protein (green). Arrows indicate wavelength (nm) of absorption maxima. All spectra were performed with equal amounts (50–100 μg) of total purified protein. All SDS-PAGE samples were equally loaded (2–5 μgof total purified protein each). For all proteins, Bradford quantitation was confirmed by Coomassie staining, which also indicated that GST-HCCS proteins were obtained at >90% purity.
Considering the proximity of Asn155 to the essential heme iron ligand of HCCS (His154), the heme-binding properties of this mutant were of particular interest. When we expressed the N155A HCCS mutant in the absence of the cytochrome c acceptor, we observed that the mutant purified with heme at levels comparable to WT HCCS (Figure 4C and Table 1), indicating that Asn155 is not required for the initial heme binding step that facilitates the recruitment of the cytochrome c substrate (Figure 1A, step 1). Collectively, these data suggest that Asn155 may play a role in stabilizing the heme-dependent interactions between cytochrome c and HCCS, thus impacting the step 4 release (see Discussion).
Domain II: Glu159
We showed that substitution of the MLS disease-relevant HCCS residue Glu159 with different amino acids had various functional impacts ranging from significantly reduced activity (MLS E159K) to 2-fold enhancement (E159A) (Figure 2B, Figure S1B, and Table S2). Accordingly, the heme profiles of the Glu159 HCCS mutants also exhibited features suggestive of altered substrate interactions. For instance, heme stains and UV–vis absorption spectra indicated that the MLS-associated HCCS variant, E159K, copurified with up to 3-fold less heme relative to WT when expressed in both the presence (Figure 4D,E and Table 1) and absence (Figure 4F and Table 1) of the cytochrome c acceptor, suggesting a heme-binding defect (see Figure 1A, step 1) consistent with its decreased function. Interestingly, the E159A HCCS mutant, which exhibited enhanced function, also copurified with substantially less heme compared to WT HCCS (Figure 4D–F and Table 1). These data are consistent with the proposal that Glu159 contributes to heme binding, with side-chains that effect largely step 1 (E159K) or steps 1 and 4 (E159A). In line with this hypothesis, we observed that the E159D HCCS mutant, which exhibited WT-like function (Figure 2B), bound to heme at near WT-levels (Figure 4F and Table 1), but did not copurify with holocytochrome c (Figure 4E). These data indicate that both the charge and size of the Glu159 side-chain play a role in HCCS function and identify an inherent heme-binding defect caused by the MLS-associated lysine mutation that likely contributes to the disease-causing phenotype. Moreover, the results with this individual residue reflect the idea that a single side chain that interacts with the heme in HCCS can impact step 1 or step 4, resulting in quite different levels of released holocytochrome c.
Addition of ALA Rescues and Enhances Function of HCCS Heme-Binding Domain Mutants
Aminolevulinic acid (ALA) synthesis is the rate-limiting step of heme biosynthesis.31 Because ALA is the first committed precursor, exogenously added ALA often increases the level of heme in recombinant heme proteins.32 We have demonstrated that several HCCS residues play a role in heme binding, resulting in either enhanced or impaired synthetase function when mutated. Therefore, we investigated whether modulating the level of heme in the presence of the cytochrome c substrate would elicit a functional response in HCCS-mediated holocytochrome c production.
When we added ALA to bacterial cultures coexpressing cytochrome c and WT HCCS, we observed a 1.5-fold increase in the level of released holocytochrome c (Figures 5 and S3), suggesting that the ALA-mediated increase in heme had a slight effect on the activity of the WT protein. As a control, we treated the H154A HCCS variant, which does not bind heme,20 with ALA and observed that this mutant remained completely defective (Figures 5 and S3). When we examined the effects of ALA on the domain I HCCS mutants W118A and Y120A/P121A, the Y120A/P121A HCCS mutant showed nearly a 5-fold increase in holocytochrome c production while the W118A HCCS mutant showed a 2-fold increase (Figures 5 and S3). The functionally impaired domain II HCCS mutant W162A showed nearly a 10-fold increase in cytochrome c production in the presence of ALA (Figures 5 and S3), indicating that the increased cellular availability of heme effectively rescued the function of this mutant. Finally, we compared the effects of ALA on the MLS mutants E159K HCCS (domain II) and R217C HCCS (domain IV), which exhibited defects in heme binding and protein folding/stability, respectively. We observed that the holocytochrome c product of the E159K HCCS mutant increased by 6-fold, while that of the R217C HCCS mutant appeared unaffected (Figures 5 and S3). These data suggest that the increased levels of heme generated from the addition of ALA drives the substrate interaction dynamics of the HCCS mutants with heme-binding defects (i.e., defects that are independent of the heme iron coordination chemistry).
Figure 5.

ALA treatment rescues synthetase function of HCCS mutants with heme binding defects. Recombinant GST-HCCS variants were coexpressed with cytochrome c in Δccm E. coli either in the presence (gray bars) or absence of ALA (black bars). Cells were lysed with BPER reagent and protein extracts were resolved by SDS-PAGE and transferred to nitrocellulose. Released cytochrome c was detected by heme stain and signal intensity was quantified by densitometry and plotted in GraphPad Prism. Data shown represent the average amount of cytochrome c released by each mutant relative to the level released by WT in the absence of ALA ± SEM, n = 3.
Discussion
Our data indicate that the conserved residues of HCCS contribute to its function by modulating the interaction dynamics of the enzyme with its substrates: heme and cytochrome c. Our conclusions regarding the roles of these conserved residues are discussed below in the context of their domain assignments.
Domains I and II: HCCS–Substrate Interaction
In our proposed model of HCCS-mediated cytochrome c assembly (Figure 1A), the interaction of HCCS with heme, facilitated by domain II residue His154 (step 1), is required for the docking of the apocytochrome c substrate (step 2), which is followed by the covalent attachment of heme to apocytochrome c (step 3), and subsequent release of the heme-bound cytochrome c product (step 4). The current study identified several residues (Domain I: Trp118, Pro121, Asn128, and Domain II: Asn155) wherein substitutions resulted in a steady-state decrease in complex formation (step 3) and enhanced release of holocytochrome c (step 4). The majority of the heme in the purified WT HCCS cocomplex is c-type (i.e., covalently attached to the cytochrome c). However, pyridine hemochrome absorption spectra of W118A and N128A HCCS cocomplexes revealed that these mutants purify with a mixture of b- and c-type heme, which presumably stems from the rapid release of the holocytochrome c product from the HCCS active site and subsequent binding of another heme molecule, in preparation for the next maturation cycle. Spectral analysis indicates that when cytochrome c is in complex with HCCS, the axial coordination positions of the heme iron (Fe) are occupied by His154 from HCCS and His19 from cytochrome c.20 Release of the holocytochrome c product from HCCS would therefore require the displacement of His154 from its axial position, followed by the discharge of the heme cofactor from HCCS as holocytochrome c. Therefore, the observed enhancement in holocytochrome c release by these substitutions might be due to a more facilitated displacement of His154 or through weaker associations directly with the heme molecule.
Although complex formation with cytochrome c is observed with WT HCCS in our bacterial model system, the remarkable stability of this interaction is not well understood. It is possible that such interactions occur in the native mitochondrial environment and are related to the import-associated binding interactions of HCCS with the apocytochrome c.19 It is likely that a balance between heme binding (step 1) and release (step 4) has evolved to optimize both the import of apocytochrome c and the synthesis of holocytochrome c, with conserved residues impacting either or both steps. In this respect, heme appears to be the central hub to direct HCCS function.
Heme binding domains of proteins typically contain residues that can coordinate the heme iron (Fe) (e.g., histidine, methionine, and cysteine) as well as make contacts with the hydrophobic porphyrin ring structure (e.g., tryptophan, phenylalanine, and tyrosine).33 While a confirmed heme axial ligand of HCCS resides in domain II (His154), we conclude that the heme binding property of HCCS spans at least two domains of conserved residues (Domain I, e.g., Tyr120 and Domain II, e.g., Glu159, Trp162) in the protein. On the basis of our previous study characterizing the H154A HCCS mutant,20 we proposed that binding of the heme substrate to HCCS (step 1) primes the enzyme for cytochrome c interaction. Although His154 (in domain II) has been established as a true heme ligand of HCCS that is absolutely required for heme binding and subsequent function, our data indicate that the domain I residue Tyr120 also mediates direct interactions with the heme substrate and potentially contributes to its stabilization with the enzyme. Heme binding was severely abrogated in both the Y120A/P121A double and Y120A single HCCS mutants; however, unlike H154A HCCS, these mutants retained synthetase function. This indicates that these mutants could mediate sufficient heme interactions in vivo to drive cytochrome c assembly, but these interactions were likely inadequate to withstand our protein purification methods. The fact that the addition of exogenous ALA dramatically enhanced (5-fold) the activity of the Y120A/P121A HCCS mutant further supports this conclusion (Figure 5). The Y120A HCCS/cytochrome c cocomplex purified with WT-levels of heme; therefore, we conclude that the weak heme interactions at the active site of the Y120A HCCS mutant are stabilized by the binding of the cytochrome c substrate, as proposed for WT HCCS.20 It is possible that Tyr120 represents the “unknown” weak, second ligand (with His154) for binding the heme iron. This ligand is replaced by the incoming His19 of apocytochrome c.
Defects in heme-binding were also observed with the MLS-associated E159K HCCS mutant; however, the synthetase activity of this mutant varied widely from 10% to 70% WT levels, depending on the culture volumes used to measure the HCCS-mediated release of matured cytochrome c (Table S2). Another study examining the functional effects of human E159K HCCS in a respiratory-deficient yeast model system reported that this mutant remains competent in mediating apocytochrome c translocation across the mitochondrial membrane; however, yeast viability via respiration was compromised, indicating a synthetase defect.34 Any impairment in HCCS function is not well-tolerated by animal cells, which depend on optimal cytochrome c levels to satisfy energy requirements and regulate developmental progression.35 Although Glu159 is located in the previously reported targeting signal of HCCS (ref (21) and Figure 1B), its mitochondrial localization is not disrupted by the lysine substitution.26 Therefore, the heme-binding defect of E159K HCCS identified in our study likely accounts for the functional perturbations of the enzyme. Consistent with this conclusion, we observed that the addition of ALA restored the activity of E159K HCCS from 15% to WT-levels in our small culture volume functional assay (Figure 5). Further studies are warranted to determine if the E159K defect is corrected by ALA in eukaryotic mitochondria.
C-Terminal Domain: HCCS Localization, Folding, and Stability
The conserved residues of domains III and IV, including the second MLS-associated residue Arg217, reside in the C-terminal portion of the HCCS protein (Figure 1B). Although Arg217 borders one of the internal targeting motifs (outside of domain III), the MLS-associated cysteine mutation does not disrupt the mitochondrial targeting of HCCS;28 therefore, we designated it as being part of domain IV since the phenotype of the R217C mutation resulted in both function- and folding/stability-related defects, similar to those observed with the other variants in this domain. The synthetase defect of the Arg217Cys mutation in human HCCS has been established previously;34 however, we determined that HCCS function is abolished with virtually any substitution of Arg217, suggesting that this residue is essential to HCCS activity/structure.
The collective data for the domain IV mutants suggest that the C-terminal portion of HCCS is required for the proper folding and/or stability of the protein. We previously demonstrated that in recombinant E. coli, the human HCCS required an N-terminal GST-fusion for optimal expression. Furthermore, we discovered that recombinant GST-HCCS tightly associates with the membrane and that nearly all soluble (i.e., cytoplasmic) forms were degraded.20 We thus cannot rule out that domain IV is required for membrane localization, without which, proteolysis occurs.
Acknowledgments
We thank Vivien Goh and the Washington University Bio437 class (2012) for assistance in constructing HCCS mutants and Dr. Molly Sutherland for reading our manuscript and providing helpful comments and suggestions.
Glossary
Abbreviations
- HCCS, holocytochrome c synthase
- ALA, aminolevulinic acid
- MLS, microphthalmia with linear skin defects
- Ccm, cytochrome c maturation
- BPER, bacterial protein extraction reagent
- WT, wild-type
- GST, glutathione-S-transferase
- UV/vis, ultraviolet/visible
- cyt c, cytochrome c
- nm, nanometer
- IPTG, isopropyl β-d-1-thiogalactopyranoside
- PBS, phosphate buffered saline
- PMSF, phenylmethanesulfonyl fluoride
Supporting Information Available
Figures S1 (UV/vis absorption spectra (alpha peak region) of cytochrome c released by HCCS (WT and variants)), S2 (UV/vis absorption spectra of N128A and M130A HCCS mutants), and S3 (heme stain of cytochrome c released by HCCS (WT and variants) in the presence and absence of ALA). Tables S1 (description of oligonucleotide primer sequences used for the site-directed mutagenesis of HCCS and the derived plasmids), S2 (quantitation of the cytochrome c released by HCCS (WT and variants)), and S3 (quantitation of total GST HCCS protein purified in the presence and absence of cytochrome c). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
‡ (B.S.) The Institute for Genomic Biology, University of Illinois at Urbana–Champaign, IL 61801.
The authors declare no competing financial interest.
This work was funded by National Institutes of Health Grants GM47909.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Liu X.; Kim C. N.; Yang J.; Jemmerson R.; Wang X. (1996) Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147–157. [DOI] [PubMed] [Google Scholar]
- Nicholls D. G., and Ferguson S. (2013) Bioenergetics, 4th ed., Academic Press, New York. [Google Scholar]
- Hamel P.; Corvest V.; Giege P.; Bonnard G. (2009) Biochemical requirements for the maturation of mitochondrial c-type cytochromes. Biochim. Biophys. Acta 1793, 125–138. [DOI] [PubMed] [Google Scholar]
- Allen J. W. (2011) Cytochrome c biogenesis in mitochondria--Systems III and V. FEBS J. 278, 4198–4216. [DOI] [PubMed] [Google Scholar]
- Barker P. D.; Ferrer J. C.; Mylrajan M.; Loehr T. M.; Feng R.; Konishi Y.; Funk W. D.; MacGillivray R. T.; Mauk A. G. (1993) Transmutation of a heme protein. Proc. Natl. Acad. Sci. U. S. A. 90, 6542–6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson D. W.; Neupert W. (1989) Import of cytochrome c into mitochondria: reduction of heme, mediated by NADH and flavin nucleotides, is obligatory for its covalent linkage to apocytochrome c. Proc. Natl. Acad. Sci. U. S. A. 86, 4340–4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont M. E.; Ernst J. F.; Hampsey D. M.; Sherman F. (1987) Identification and sequence of the gene encoding cytochrome c heme lyase in the yeast Saccharomyces cerevisiae. EMBO J. 6, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visco C.; Taniuchi H.; Berlett B. S. (1985) On the specificity of cytochrome c synthetase in recognition of the amino acid sequence of apocytochrome c. J. Biol. Chem. 260, 6133–6138. [PubMed] [Google Scholar]
- Zollner A.; Rodel G.; Haid A. (1992) Molecular cloning and characterization of the Saccharomyces cerevisiae CYT2 gene encoding cytochrome-c1-heme lyase. Eur. J. Biochem. 207, 1093–1100. [DOI] [PubMed] [Google Scholar]
- Bernard D. G.; Gabilly S. T.; Dujardin G.; Merchant S.; Hamel P. P. (2003) Overlapping specificities of the mitochondrial cytochrome c and c1 heme lyases. J. Biol. Chem. 278, 49732–49742. [DOI] [PubMed] [Google Scholar]
- Prakash S. K.; Cormier T. A.; McCall A. E.; Garcia J. J.; Sierra R.; Haupt B.; Zoghbi H. Y.; Van Den Veyver I. B. (2002) Loss of holocytochrome c-type synthetase causes the male lethality of X-linked dominant microphthalmia with linear skin defects (MLS) syndrome. Hum. Mol. Genet. 11, 3237–3248. [DOI] [PubMed] [Google Scholar]
- Schaefer L.; Ballabio A.; Zoghbi H. Y. (1996) Cloning and characterization of a putative human holocytochrome c-type synthetase gene (HCCS) isolated from the critical region for microphthalmia with linear skin defects (MLS). Genomics 34, 166–172. [DOI] [PubMed] [Google Scholar]
- Kranz R. G.; Richard-Fogal C.; Taylor J. S.; Frawley E. R. (2009) Cytochrome c biogenesis: mechanisms for covalent modifications and trafficking of heme and for heme-iron redox control. Microbiol. Mol. Biol. Rev. 73, 510–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders C.; Turkarslan S.; Lee D. W.; Daldal F. (2010) Cytochrome c biogenesis: the Ccm system. Trends Microbiol. 18, 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens J. M.; Mavridou D. A.; Hamer R.; Kritsiligkou P.; Goddard A. D.; Ferguson S. J. (2011) Cytochrome c biogenesis System I. FEBS J. 278, 4170–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervera A. M.; Gozalbo D.; McCreath K. J.; Gow N. A.; Martinez J. P.; Casanova M. (1998) Molecular cloning and characterization of a Candida albicans gene coding for cytochrome c haem lyase and a cell wall-related protein. Mol. Microbiol. 30, 67–81. [DOI] [PubMed] [Google Scholar]
- Dumont M. E.; Ernst J. F.; Sherman F. (1988) Coupling of heme attachment to import of cytochrome c into yeast mitochondria. Studies with heme lyase-deficient mitochondria and altered apocytochromes c. J. Biol. Chem. 263, 15928–15937. [PubMed] [Google Scholar]
- Dumont M. E.; Cardillo T. S.; Hayes M. K.; Sherman F. (1991) Role of cytochrome c heme lyase in mitochondrial import and accumulation of cytochrome c in Saccharomyces cerevisiae. Mol. Cell. Biol. 11, 5487–5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson D. W.; Hergersberg C.; Neupert W. (1988) Role of cytochrome c heme lyase in the import of cytochrome c into mitochondria. J. Biol. Chem. 263, 19034–19042. [PubMed] [Google Scholar]
- San Francisco B.; Bretsnyder E. C.; Kranz R. G. (2013) Human mitochondrial holocytochrome c synthase’s heme binding, maturation determinants, and complex formation with cytochrome c. Proc. Natl. Acad. Sci. U. S. A. 110, E788–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diekert K.; Kispal G.; Guiard B.; Lill R. (1999) An internal targeting signal directing proteins into the mitochondrial intermembrane space. Proc. Natl. Acad. Sci. U. S. A. 96, 11752–11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feissner R. E.; Richard-Fogal C. L.; Frawley E. R.; Loughman J. A.; Earley K. W.; Kranz R. G. (2006) Recombinant cytochromes c biogenesis systems I and II and analysis of haem delivery pathways in Escherichia coli. Mol. Microbiol. 60, 563–577. [DOI] [PubMed] [Google Scholar]
- Feissner R.; Xiang Y.; Kranz R. G. (2003) Chemiluminescent-based methods to detect subpicomole levels of c-type cytochromes. Anal. Biochem. 315, 90–94. [DOI] [PubMed] [Google Scholar]
- Berry E. A.; Trumpower B. L. (1987) Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra. Anal. Biochem. 161, 1–15. [DOI] [PubMed] [Google Scholar]
- Frawley E. R.; Kranz R. G. (2009) CcsBA is a cytochrome c synthetase that also functions in heme transport. Proc. Natl. Acad. Sci. U. S. A. 106, 10201–10206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimplinger I.; Shaw G. M.; Kutsche K. (2007) HCCS loss-of-function missense mutation in a female with bilateral microphthalmia and sclerocornea: a novel gene for severe ocular malformations?. Mol. Vis. 13, 1475–1482. [PubMed] [Google Scholar]
- Moore R. L.; Stevens J. M.; Ferguson S. J. (2011) Mitochondrial cytochrome c synthase: CP motifs are not necessary for heme attachment to apocytochrome c. FEBS Lett. 585, 3415–3419. [DOI] [PubMed] [Google Scholar]
- Wimplinger I.; Morleo M.; Rosenberger G.; Iaconis D.; Orth U.; Meinecke P.; Lerer I.; Ballabio A.; Gal A.; Franco B.; Kutsche K. (2006) Mutations of the mitochondrial holocytochrome c-type synthase in X-linked dominant microphthalmia with linear skin defects syndrome. Am. J. Hum. Genet. 79, 878–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick D. (1994) Replacement of the proximal ligand of sperm whale myoglobin with free imidazole in the mutant His-93→ Gly. Biochemistry 33, 6546–6554. [DOI] [PubMed] [Google Scholar]
- San Francisco B.; Bretsnyder E. C.; Rodgers K. R.; Kranz R. G. (2011) Heme ligand identification and redox properties of the cytochrome c synthetase, CcmF. Biochemistry 50, 10974–10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajioka R. S.; Phillips J. D.; Kushner J. P. (2006) Biosynthesis of heme in mammals. Biochim. Biophys. Acta 1763, 723–736. [DOI] [PubMed] [Google Scholar]
- Kery V.; Elleder D.; Kraus J. P. (1995) Delta-aminolevulinate increases heme saturation and yield of human cystathionine beta-synthase expressed in Escherichia coli. Arch. Biochem. Biophys. 316, 24–29. [DOI] [PubMed] [Google Scholar]
- Li T.; Bonkovsky H. L.; Guo J. T. (2011) Structural analysis of heme proteins: implications for design and prediction. BMC Struct. Biol. 11, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indrieri A.; Conte I.; Chesi G.; Romano A.; Quartararo J.; Tate R.; Ghezzi D.; Zeviani M.; Goffrini P.; Ferrero I.; Bovolenta P.; Franco B. (2013) The impairment of HCCS leads to MLS syndrome by activating a non-canonical cell death pathway in the brain and eyes. EMBO Mol. Med. 5, 280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttemann M.; Helling S.; Sanderson T. H.; Sinkler C.; Samavati L.; Mahapatra G.; Varughese A.; Lu G.; Liu J.; Ramzan R.; Vogt S.; Grossman L. I.; Doan J. W.; Marcus K.; Lee I. (2012) Regulation of mitochondrial respiration and apoptosis through cell signaling: cytochrome c oxidase and cytochrome c in ischemia/reperfusion injury and inflammation. Biochim. Biophys. Acta 1817, 598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.