

Abstract
Ventricular myosin (βMys) is the motor protein in cardiac muscle generating force using ATP hydrolysis free energy to translate actin. In the cardiac muscle sarcomere, myosin and actin filaments interact cyclically and undergo rapid relative translation facilitated by the low duty cycle motor. It contrasts with high duty cycle processive myosins for which persistent actin association is the priority. The only pharmaceutical βMys activator, omecamtive mecarbil (OM), upregulates cardiac contractility in vivo and is undergoing testing for heart failure therapy. In vitro βMys step-size, motility velocity, and actin-activated myosin ATPase were measured to determine duty cycle in the absence and presence of OM. A new parameter, the relative step-frequency, was introduced and measured to characterize βMys motility due to the involvement of its three unitary step-sizes. Step-size and relative step-frequency were measured using the Qdot assay. OM decreases motility velocity 10-fold without affecting step-size, indicating a large increase in duty cycle converting βMys to a near processive myosin. The OM conversion dramatically increases force and modestly increases power over the native βMys. Contrasting motility modification due to OM with that from the natural myosin activator, specific βMys phosphorylation, provides insight into their respective activation mechanisms and indicates the boilerplate screening characteristics desired for pharmaceutical βMys activators. New analytics introduced here for the fast and efficient Qdot motility assay create a promising method for high-throughput screening of motor proteins and their modulators.
Heart failure is a frequent cause of death, and those experiencing disease onset suffer significant loss in the quality of life. With systolic heart failure, modest physical exertion causes pain, weakness, or other symptoms indicative of inadequate cardiac performance. It can have a hereditary link focused principally on a malfunctioning myosin, the molecular motor powering heart contraction, but is most often associated with cardiac muscle damage caused by sudden or gradual arterial blockage. Pharmacological treatment frequently targets the β-adrenergic pathway to upregulate contractile function sometimes by enhancing calcium release into the cytoplasm. The β-adrenergic pathway is an upstream modulator of a multifunctional signaling pathway implying that unwanted effects associated with its modulation could be bypassed by treating myosin directly.
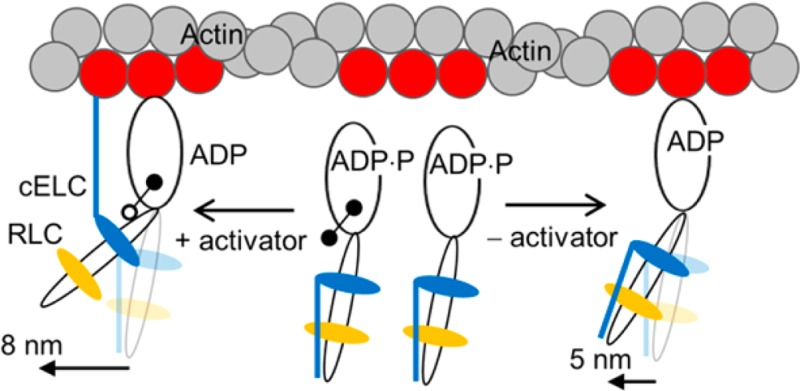
Myosin in cardiac muscle transduces ATP chemical energy into the mechanical work of moving blood volume under pressure. Myosin is the mover comprised of a catalytic motor domain containing ATP and actin binding sites and mechanical elements coupling motor-generated impulsive force to the myosin thick filament backbone. Myosin mechanical coupling elements consist of a lever-arm domain and two stabilizing light chains, essential (ELC) and regulatory (RLC), that undergo cyclical rotary movement to impel bound filamentous actin. Linear actin displacement due to lever-arm rotation is the myosin step-size. Post-translational modifications affect the myosin mover.1,2 Phosphorylation of S15 in RLC was specifically shown to enhance ventricle work productivity.3 We showed that tissue purified skeletal myosin and ventricular cardiac myosin (βMys and gene MYH7) have 1 and 3 unitary step-sizes in vitro, respectively, and suggested that this attribute contributes to tissue specific mechanisms affecting contractility performance.4,5
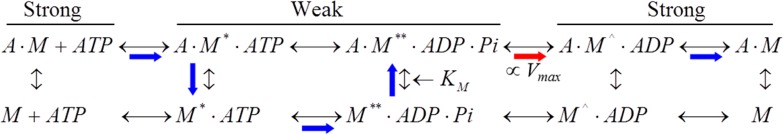
Scheme 1 shows the myosin (M) ATPase cycle in the presence of actin (A). Blue and red arrows indicate the predominant pathway, and the red arrow also indicates the weak to strong actin binding transition initiating force development. Vmax and KM are Michaelis–Menten constants measured from actin-activated myosin ATPase. Vmax scales with the phosphate release rate for muscle myosins as indicated but also depends on the weak actomyosin binding equilibrium.6,7 Actomyosin configurations are characterized as weak and strong binding states based in part on their ability to generate work with only the strongly bound state work-generating. In vitro motility has the myosin moving actin under unloaded conditions with a motility velocity vm such that
| 1 |
for myosin step-size d and duty cycle f.8 Duty cycle is the time fraction actomyosin is strongly bound during an ATPase cycle. Porcine cardiac myosin has f δ ≲ 0.05.9,10 Cardiac and skeletal muscles maintain myosin and actin filaments in a lattice favorable to their interaction. The filaments slide relatively during contraction shortening. The low duty cycle facilitates the rapid shortening in cardiac and skeletal muscle because a strongly actin-bound myosin will retard movement when it does not dissociate promptly after delivering its impulsive force.
Scheme 1.
Skeletal and cardiac myosin binding small molecule effectors are inhibitors, including blebbistatin11 and N-benzyl-p-toluene sulfonamide (BTS).12 Skeletal myosin inhibition by blebbistatin is attributed to its stabilization of an intermediate with a partially closed actin-binding cleft in the motor domain. Cleft closure accompanies strong actin binding at the end of the myosin power stroke. Blebbistatin inhibits strong binding and in vitro motility.13 BTS is structurally analogous to blebbistatin and likely to inhibit motility by a similar mechanism.14 A specific βMys activator in clinical trials for systolic heart failure, omecamtive mecarbil (OM), specifically binds the heavy chain near residue S148.15 It increases the myosin transitioning into the strongly actin-bound state probably by stabilizing its actin-bound conformation. In cardiomyocytes, the drug increases the cardiac myocyte contraction shortening length without affecting the Ca2+ transient.
We evaluated the OM mechanism for contractility enhancement by measuring the cardiac myosin step-size, motility velocity, relative step-frequency, and actin-activated myosin ATPase. Step-size and relative step-frequency were efficiently measured using the novel Qdot super-resolution in vitro motility assay.4,5 We find that OM has little impact on βMys actin-activated ATPase, in agreement with prior results,15 or its 3 unitary step-sizes but dramatically reduces motility velocity and affects the relative step-frequency. The results imply a large increase in duty cycle, while the quantitative change in relative step-frequency sharply contrasts with the natural phosphorylation-activated βMys. Preliminary results showing similar reduction in motility velocity were reported.16 Our data indicate that OM effectively converts the native βMys “muscle” motor characteristics near to that of a processive motor. Contrasting OM with natural βMys activation provides insight into activation mechanisms and defines criteria for pharmaceutical activators.
Materials and Methods
Chemicals
Omecamtiv mecarbil (OM) was purchased from Selleckchem (Houston, TX). Quantum dot 565 streptavidin conjugate (Qdot), phalloidin, rhodamine-phalloidin, and biotin-XX-phalloidin were obtained from Life Technologies (Grand Island, NY). Glucose oxidase was from MP Biomedicals (Santa Ana, CA). Other chemicals were purchased from Sigma-Aldrich (St. Louis, MO) or Affymetrix (Cleveland, OH). Protein concentrations were measured using the Bradford assay (Bio-Rad, Hercules, CA).
Protein Preparations
βMys was prepared from porcine heart ventriculum as described previously.4,17 Rabbit skeletal myosin was prepared from the leg and back muscles by the method of Tonomura et al.18 Rabbit skeletal heavy meromyosin (sHMM) was obtained by chymotryptic digestion of myosin.19 G-Actin was obtained from rabbit skeletal muscle acetone powder by using the method described by Pardee and Spudich20 and then stored immediately under argon gas in liquid nitrogen. The frozen G-actin was thawed and spun at 160000g for 90 min to remove denatured actin before use. Rhodamine-phalloidin or biotin-XX-phalloidin and rhodamine-phalloidin labeling of actin filaments was performed as described previously.4
Actin-Activated Myosin ATPase
Actin-activated myosin ATPase was measured as described previously21 with some modifications. βMys stored in 50% glycerol was precipitated with addition of 12 volumes of ice-cold water containing 2 mM DTT, collected by centrifugation, and then resuspended in 300 mM KCl, 25 mM imidazole (pH 7.4), 4 mM MgCl2, 1 mM EGTA, 10 mM DTT, and 10 μg/mL leupeptin. Myosin at a final concentration of 1 μM was titrated with 0.1, 2, 4, 8, 16, and 40 μM acin. The ATPase assay buffer contained 25 mM imidazole (pH 7.4), 4 mM MgCl2, 1 mM EGTA, 10 mM DTT, 10 μg/mL leupeptin, 1% DMSO, 0.1 or 100 μM OM, and a final KCl concentration of 25 mM. Control actin-activated ATPase had no DMSO or OM. ATPase reaction was initiated by the addition of 3 mM ATP, and the mixture was incubated at 21 °C for 5 min. OM was dissolved in DMSO before adding to the ATPase assay buffer. Inorganic phosphate production was assessed using the method of Fiske and Subbarow.22
Dose response of OM in actin-activated ATPase was measured as described above with 0.1–100 μM OM, 40 μM actin, and 1% DMSO.
In Vitro Motility
In vitro motility and Qdot assays of βMys and sHMM were performed as described previously,4 except for the presence of 1% DMSO and 0–1.5 μM OM. βMys motility buffer was 25 mM KCl, 25 mM imidazole (pH 7.4), 4 mM MgCl2, 1 mM EGTA, 20 mM DTT, 10 μg/mL leupeptin, 0.7% methylcellulose, 2 mM ATP, 3 mg/mL glucose, 0.018 mg/mL catalase, and 0.1 mg/mL glucose oxidase. All the motility assays were performed at 21 °C.
In vitro motility was observed with through-the-objective total internal reflection fluorescence (TIRF)23 on an Olympus IX71 inverted microscope using a 150×, 1.45 NA objective. Images were acquired with an Andor EMCCD camera (iXon3 897 with 16 μm × 16 μm pixels and 16 bit dynamic range) using the software supplied by the manufacturer (SOLIS). The actin sliding velocities were analyzed manually using ImageJ (National Institutes of Health, Bethesda, MD) plugin MtrackJ.24 The sliding velocity of actin filaments at each myosin or OM concentration was measured by averaging the speeds of 40–60 filaments in 2–3 slides. Each actin filament was tracked for 3–5 μm in the case of βMys or 20–40 μm in the case of sHMM. Control and OM-treated myosin experiments were performed on two independent preparations of myosin and actin on different days. More than 90% of the Qdot-labeled actin filaments translated in the assay under all conditions tested.
In experiments using the Qdot assay, images were collected at 5 or 2 frames per second and 30 or 50 ms exposures. The frame rates correspond to 200 or 500 ms intervals indicated by Δt. Intensity values were converted to photons using the conversion formula in SOLIS as appropriate for our camera and the images output in TIFF format for reading into ImageJ. A single myosin step is isolated in time and space and then characterized using super-resolution.
Super-resolution Measurements
Single-molecule dipole emitters are represented in the microscope image space by the point spread function (PSF). We estimate single molecule position with resolution below the diffraction limit by locating the center of the PSF with high precision.25,26 The process has been automated in the QuickPALM ImageJ plugin super-resolution fitting algorithm in 2 dimensions.27 QuickPALM identified and localized point objects that qualified for super-resolution fitting according to user specifications, including minimal SNR (>25 isolating Qdots) and maximal full width at half-maximum (fwhm) of 5 pixels (107 nm/pixel in object space for the 150× objective). The analysis produced a table (SRTable) listing each qualifying particle, particle position in pixels, position standard deviation, and frame identifier. Using the SRTable, QuickPALM rendered the super-resolved particle data as single pixels per particle in the frame sequence of the original data. Rendered frames were read into ImageJ and analyzed with MTrackJ.24 Single-pixel resolution (107 nm) of the rendered images is much less than super-resolution (<10 nm). Manual tracking was needed only to link the super-resolved particle positions into a Qdot-labeled actin track connecting time-ordered frames. A separate program, SRTrack written in Mathematica, linked the actual super-resolved particle coordinate to the track and then updated the SRTable with the frame-to-frame tracking linked list. SRTrack eliminated any incorrectly identified MTrackJ particles that did not have a super-resolved equivalent. The latter removed the effect of Qdot blinking. Representative Qdot displacement versus time data are included in Movie S1 of the Supporting Information.
In any motility assay a few Qdots did not visibly move due to apparent immobilization on the glass surface. These particles were tracked at super-resolution to quantitate thermal/mechanical fluctuations.
Simulation
We simulated motility assay velocity event density essentially as described previously for a 2.1 μm actin filament.4 Velocity data were separated into two data sets corresponding to the two independent protein preparations. These two data sets were analyzed by simulation separately and then together in a single pooled data set. The three results differ within the boundaries of uncertainties indicated in Results. It suggests quantitative results are readily reproducible with the standard protein preparations.
We input known Vmax and vm for the control and OM treated conditions. The unknown parameter set actively searched in the simulation consists of the actin binding probability for myosin, myosin step-size, and step-frequency. Simulation generates unitary myosin binding events during successive Δt’s that are converted to actin displacement by the myosin step-size and then to actin velocity by dividing by Δt. Simulated and measured velocity event density histograms are compared for fit to choose best fitting parameters. Unitary step events are counted and converted to relative step-frequency by dividing the unitary step count by the total number of unitary steps. The best fitting velocity event density histogram and the corresponding relative step-frequencies characterize the actomyosin interaction.
System Analytics
Unitary step-size is a fundamental structural feature of the myosin mover. We introduce the relative step-frequency, ωj, for unitary step j.4,5 It is a multiple unitary step motor characteristic proportional to the rate of unloaded cross-bridge cycling with the higher rate producing a more frequent jth step. Relative step-frequency is dimensionless and normalized such that ωS + ωI + ωL = 1, where subscripts S, I, and L are for the short (∼3 nm), intermediate (∼5 nm), and long (∼8 nm) nominal unitary steps, respectively. For native βMys, the intermediate step has highest cycling rate followed by the long step and then the short step with lowest cycling rate. Absolute cycling rate for step j, Vj, has Vj = Vmaxωj, and Vmax = ∑j=S,I,LVj.
In an ensemble of cross-bridges interacting with one actin filament, like the conditions in every muscle or motility assay, only one actin velocity is possible, and hence, motility velocity vm is the same for each unitary step-size, implying each step-size has a unique duty cycle. From eq 1, the step j duty cycle
| 2 |
Characteristics of the multistep motor derive from the step-frequency weighted averaged quantities indicated with broken brackets, such that average time cross-bridges spend strongly bound, ⟨ton⟩, computed from eq 2 is
| 3 |
Average force is proportional to the fraction of strongly actin bound cross-bridges28
| 4 |
for α the proportionality constant expressed in units of force (uf) where α = 1. Average power
| 5 |
is dependent on relative step-frequency, step-size, and Vmax but independent of vm. Dynamically averaged velocity, u, computed from average step-size, ⟨d⟩, and ⟨ton⟩
| 6 |
indicates trends in motility velocity accompanying step-frequency changes.
Step-sizes {dS,dI,dL} are constant due to immutable myosin structural constraints, and we observe that Vmax and ωS change only modestly under experimental conditions tested. Normalization implies {ωS,ωI,ωL} = {ωS,1 – ωS – ωL,ωL}; hence, ωL is the sole independent variable. ⟨P⟩ dependence on ωL contrasts natural and OM myosin activation mechanisms.
Results
βMys ATPase Activity Measurements
Michaelis–Menten Vmax and KM characterize actin-activated myosin ATPase with the maximal velocity for ATP hydrolysis and affinity for actin, respectively. Table 1 lists Vmax and KM values appropriate for the in vitro motility conditions. Figure 1 shows βMys ATPase rate versus actin concentration in the absence of OM and in the presence of 0.1 and 100 μM OM. All conditions indicate that the hydrolysis rate saturates at >10 μM actin. DMSO at ≤1% is introduced into the assay with the addition of the OM as indicated previously.15 ATPase is compared in assay buffers with or without 1% DMSO to indicate its negligible effect (filled triangles or empty inverted triangles, respectively).
Table 1. βMys Actin-Activated ATPase and in Vitro Motility.
| βMys | pβMysa | OM-treated βMysb | |
|---|---|---|---|
| Vmax (s–1) | 1.22 ± 0.05 | 1.26 ± 0.09 | 1.25 ± 0.08 |
| KM (μM) | 5.71 ± 0.81 | 3 ± 1 | 4.12 ± 0.91 |
| vm (μm/s) | 0.27 ± 0.02 | 0.32 ± 0.02 | 0.039 ± 0.002 |
| dS | 2.79 ± 0.6 | 2.8 ± 0.3 | 2.6 ± 0.6 |
| dI | 5.39 ± 0.4 | 4.8 ± 0.3 | 5.8 ± 0.5 |
| dL | 7.80 ± 0.06 | 7.3 ± 0.2 | 8.8 ± 0.6 |
| ωS | 0.181 ± 0.007 | 0.156 ± 0.007 | 0.154 ± 0.006 |
| ωI | 0.507 ± 0.008 | 0.118 ± 0.008 | 0.422 ± 0.009 |
| ωL | 0.312 ± 0.009 | 0.726 ± 0.008 | 0.424 ± 0.009 |
| fS (×103) | 2.29 ± 0.01 | 1.73 ± 0.01 | 12.6 ± 0.1 |
| fI (×103) | 12.34 ± 0.03 | 2.22 ± 0.02 | 78.6 ± 0.2 |
| fL (×103) | 10.99 ± 0.04 | 20.86 ± 0.04 | 119.8 ± 0.4 |
| ⟨d⟩ (nm) | 5.67 ± 0.04 | 6.30 ± 0.04 | 6.58 ± 0.05 |
| ⟨F⟩ (uf) | 10.5 ± 0.1 | 16.1 ± 0.3 | 89.5 ± 1.5 |
| ⟨P⟩ (μm/s) (uf) | 2.84 ± 0.04 | 5.1 ± 0.1 | 3.49 ± 0.06 |
Figure 1.

βMys actin-activated ATPase in the presence of 0.1 μM OM (▲) and 100 μM OM (●) and the control without OM (▽). Solid lines represent fitting curves with ATPase = (Vmax[actin])/(Km + [actin]). Parameter values for the curves are summarized in Table 1. Control βMys actin-activated ATPase had no OM or DMSO. Other conditions had OM and 1% DMSO.
Figure 2 shows the dose response of Vmax to OM for saturating actin concentration of 40 μM and 1% DMSO. Vmax is 1.16 ± 0.02 s–1 for <1 μM OM and increases to 1.33 ± 0.04 s–1 for >10 μM OM, indicating a 15% enhancement due to OM. The actin activation dose–response curve trends like that reported for bovine myosin subfragment 1 (S1).15Vmax amplitude differs from that reported previously due to species specificity (porcine vs bovine myosin) and different measurement conditions. Conditions for the actin-activated myosin ATPase and in vitro motility measurements described next overlap to allow duty cycle computation.
Figure 2.
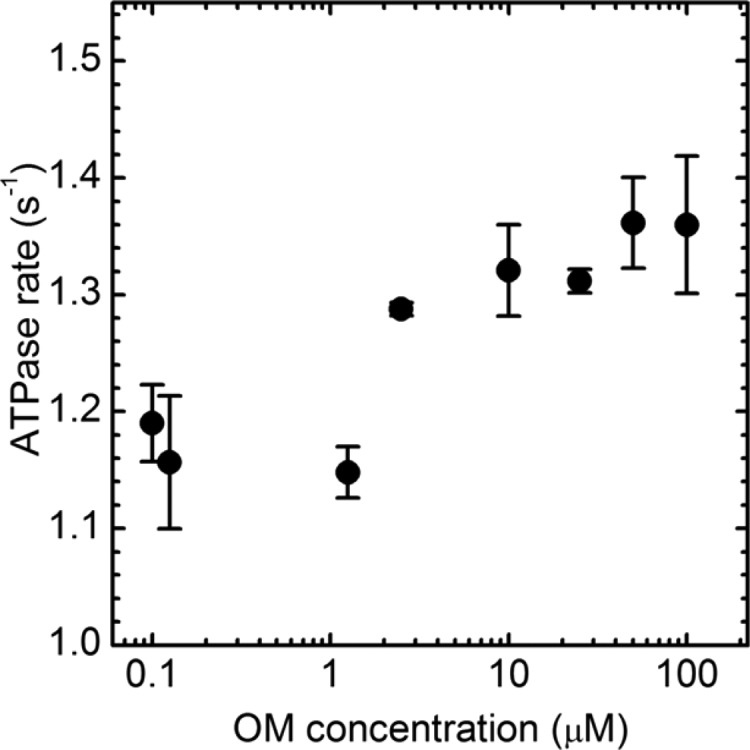
Dose response to OM of actin-activated βMys ATPase for 40 μM actin in 1% DMSO. The point at each OM concentration represents 2 replicates. Means ± SD are plotted.
βMys in Vitro Motility Velocity
Figure S2 of the Supporting Information shows βMys motility velocity, vm, at various βMys bulk concentrations, in 1.5 μM OM and 1% DMSO. vm increases with increasing [βMys] until reaching maximum (0.026 ± 0.001 μm/s) at 0.06 μM βMys and then slightly decreasing to a constant (0.021 ± 0.001 μm/s) at or beyond 0.08 μM. We showed previously that rabbit skeletal heavy meromyosin (sHMM) glides at its maximal velocity at or beyond the bulk concentration of 0.08 μM.4
The isoform specific dose response of vm to OM is shown in Figure 3. Rhodamine-phalloidin labeled actin moves over βMys or sHMM for 0.2 or 0.285 μM bulk protein concentration. DMSO at ≤1% is introduced into the assay with the addition of the OM. Motility is compared in assay buffers with or without 1% DMSO to indicate its negligible effect on motility velocity (filled or empty symbols, respectively). Actin sliding velocity for βMys decreases ∼10-fold with increasing OM and saturates for [OM] > 0.75 μM. Actin sliding velocity for sHMM decreases ∼20% under these conditions. Control experiments summarized by data in Figures 1 and 2 and the specificity of OM for βMys confirmed by Figure 3 indicate that porcine βMys recapitulates the previous bovine βMys characterization of OM.15
Figure 3.
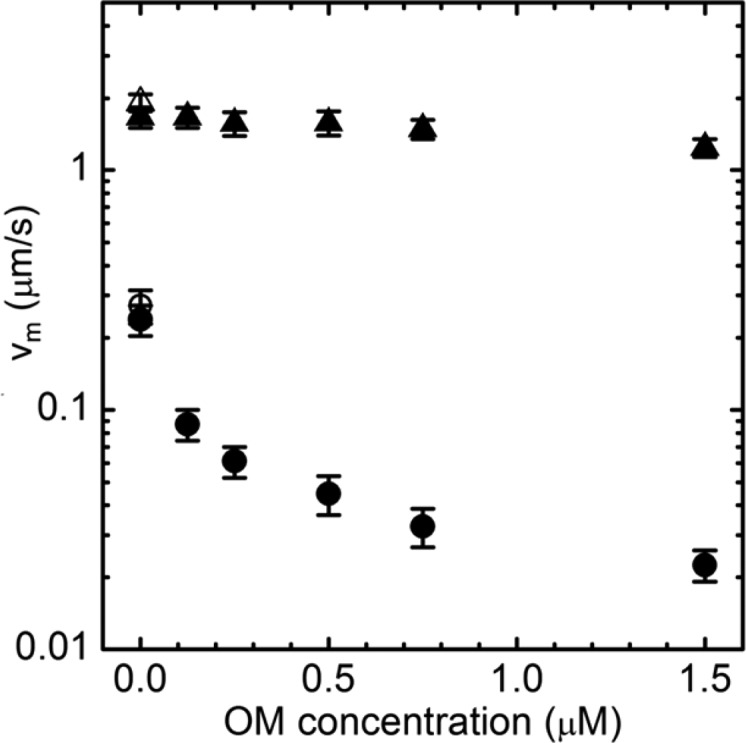
Dose response of βMys (circles) and sHMM (triangles) in vitro motility to OM and in motility assay buffers containing 1% DMSO (filled) or without DMSO (empty). Means ± SD are plotted. Bulk concentrations of βMys and sHMM in the motility were 0.2 and 0.285 μM, respectively.
Analysis of the Qdot assay data permits subtraction of the baselines due to thermal/mechanical fluctuations that tend to remove the slowest velocities in the velocity histogram and increasing the mean velocity. These slowest velocities are randomly directed and contribute negligibly to the standard in vitro motility velocity except when directed movement is very slow. OM treated βMys requires a 10–20 s Δt to allow actin the time to move ≥1 pixel. This condition underestimates velocity as the particle does not move along a line due to the thermal/mechanical fluctuations. Subpixel movement is quantifiable with the Qdot assay, and the Δt needed is 0.5 s. Average velocity computed from super-resolution data suggests vm = 0.039 ± 0.002 (standard error) μm/s (Table 1). This value for vm is used in all subsequent calculations as the best estimate for vm in the presence of OM.
Figure S3 of the Supporting Information indicates the percentage of moving actin filaments in βMys motility for various concentrations of OM. The high percentage of moving filaments across all drug concentrations indicates assay quality.
βMys Step-Size and Relative Step-Frequency
The Qdot assay has labeled actin translating over surface bound βMys at 0.16 μM bulk protein concentration in the absence and presence of 1.5 μM OM (with 1% DMSO). Inspection of actin filaments labeled with Qdot/rhodamine shows that the Qdot sparsely labels intact filaments with average length ∼2.1 μm long. A single myosin step is isolated in time and space and then characterized using super-resolution. Panels A and C of Figure 4 show actin sliding velocity event density in the low-velocity domain of 0–15 nm/(Δt = 1) for Δt the 200 or 500 ms frame capture interval in the absence or presence of OM, respectively. Baselines due to thermal/mechanical fluctuations were subtracted as described previously.4 Measured (■ and dashed line) and simulated (red line) data are shown for the velocity curve. Peaks in Figure 4 correspond to short (red), intermediate (green), and long (blue) step-sizes. Some step combinations are indicated with the appropriate symbol combinations and their lengths for the best fitting simulation shown. Step-size estimates and their standard error are computed by combining results from the 80 best fitting simulations and are summarized in Table 1 in the rows labeled dS, dI, and dL. All values are equivalent within error to our previous results for βMys and pβMys.4,5 Previously measured average step-size for βMys is 5–9 nm.4,29−31
Figure 4.
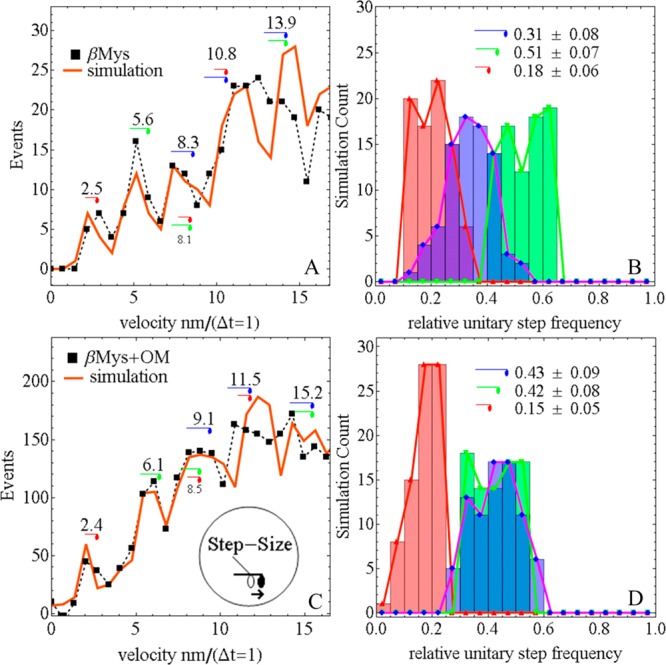
Qdot assay velocity event density histogram (left) and step-frequency histogram (right) for βMys in the absence (top row) or presence (bottom row) of OM and 1% DMSO. Measured (■ and dashed line) and simulated (red line) data are shown for the velocity curve. The inset in panel C indicates translation of the motor domain associated with a unitary step-size and a lever-arm rotation. Unitary steps of ∼3 nm (red), ∼5 nm (green), and ∼8 nm (blue) are indicated symbolically near their event distribution peak. Several unitary step combinations are also indicated. The simulated velocity curve corresponds to the best fitting single simulation, while the histograms summarize findings from the 80 best simulations. Each simulated curve provided the number of unitary events for 3, 5, and 8 nm steps that is converted to relative unitary step-frequency by dividing the unitary step count by the total number of 3, 5, and 8 nm unitary steps, respectively. Relative unitary step-frequency summed over the 3 unitary steps is 1 for each simulation.
Panels B and D of Figure 4 show relative step-frequency histograms and numerical averages ± SD for the ∼3, ∼5, and ∼8 nm unitary steps. Numerical average relative step-frequencies for control βMys are identical within error to previously reported values.4 The 5 nm step-size is predominant in control βMys. In the presence of 1.5 μM OM, the frequency for the 5 nm step is diminished in favor of the 8 nm unitary step. The 5 and 8 nm steps are equally frequent. The short step-frequency is nearly unchanged. Relative step-frequency estimates and their standard error are computed by combining results from the 80 best fitting simulations and are summarized in Table 1 in the rows labeled ωS, ωI, and ωL.
Low probability event combinations falling into the range occupied by unitary steps are indicated under the velocity curve in panels A and C of Figure 4 in the smaller font. The short-step probability is too small to contribute significantly as a doublet. The ∼8 nm step is similar in length to the short and medium steps in combination. We adapted the simulation to investigate the relative contributions of the combined steps and unitary 8 nm step to the probability peak at 8 nm as described previously.4 We obtained results identical to those reported previously, where with the 8 nm unitary step included in the simulation, the best fits fully account for observation (red line in Figure 4A,C). Without the 8 nm unitary step, best fitting causes the simulation to substantially overshoot the 3 and 5 nm peaks in the event histogram indicating the event deficit at 8 nm. The peak assigned to the combined 5 and 8 nm steps is substantially underoccupied in the simulation, again demonstrating the need for the unitary 8 nm step. Representative curves were already shown for this case (blue line in Figure 5A,B of ref (4)).
Comparison of Natural and Pharmaceutical Activated βMys
Natural activation of βMys was accomplished by its phosphorylation at S15 in RLC as described previously.32 All phosphorylated βMys (pβMys) data summarized here were taken from ref (5). Quantities {ωS,ωI,ωL}, {dS,dI,dL}, vm, and Vmax were measured for native (control), OM treated (Figure 4), and pβMys. They are summarized in Table 1 along with computed quantities {fS,fI,fL}, ⟨F⟩, and ⟨P⟩ (eqs 2–5) and their standard errors.
Systemic Performance
Average power ⟨P⟩ tests system performance under conditions of varying relative step-frequencies, {ωS,ωI,ωL}, where ωS is a constant taken from the data in Table 1 for βMys, pβMys, and OM treated βMys and ωI = 1 – ωS – ωL; hence, ωL is the sole dependent variable. This simplifies the subsequent discussion without altering the generality of the results. Figure 5 indicates ⟨F⟩, u, and ⟨P⟩ for actual step-sizes {dS,dI,dL} versus ωL. Panels A and B indicate parabolic ⟨F⟩ and u with a minimum and maximum, respectively, at ωL ≈ 0.31, i.e., the value used by the native βMys where average motility velocity vm = 270 nm/s. Clearly, the native βMys is optimized for peak speed. Panel C shows that for any motor (βMys, pβMys, OM+βMys), ⟨P⟩ increases parabolically (like ⟨F⟩) in ωL indicating the significance of the relative step-frequency in system optimization. Curves in panels A and B assume constant vm for a particular species (Table 1) over a changing ωL. This is unlikely to happen in the real system because vm will probably vary but the curves agree with the observation at the beginning and ending of the arrows where vm is known. Curves in panel C are independent of vm; hence, they model the real system at all points. Curves differ for βMys, pβMys, and OM+βMys species also due to their slightly different step-sizes, ωS, and Vmax. The rising power at the low and high ends of the ordinate in panel C reflects higher duty cycles.
Figure 5.
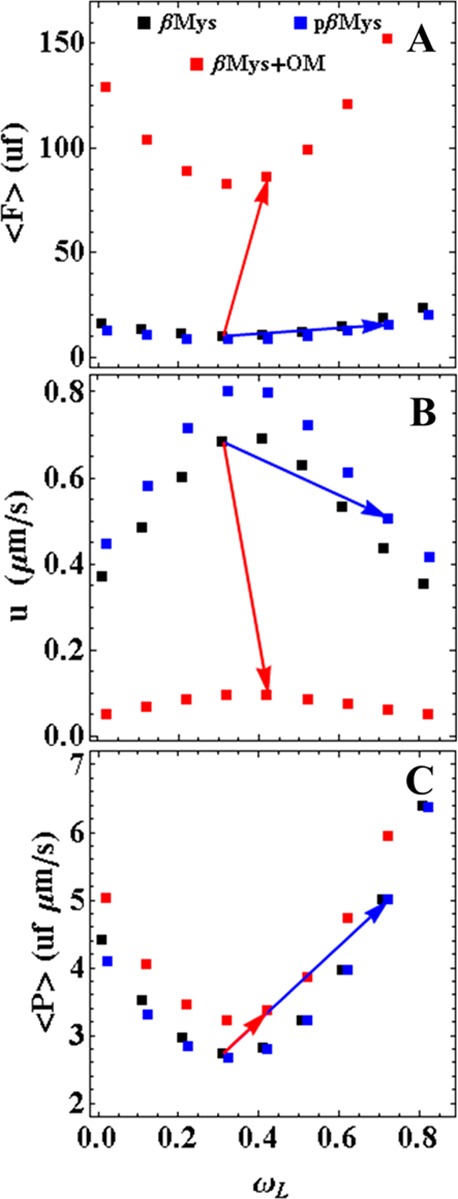
Qdot assay analytics comparing native (black), phosphorylated (blue), and OM treated (red) βMys. The average force, ⟨F⟩, in units of force [uf and eq 5 (A)], dynamic velocity, u [eq 6 (B)], and average power, ⟨P⟩ [eq 7 (C)], were computed as a function of the relative step-frequency for the long step, ωL, assuming relative step-frequency for the short step, ωS, was constant and taken from Table 1. System optimization calls for maximizing ⟨P⟩ with βMys activation. Panel C shows that maximal ⟨P⟩ occurs for maximal ωL. Activated ⟨P⟩ due to phosphorylation (blue arrow) is larger than activated ⟨P⟩ by OM treatment (red arrow). Blue and red arrows follow the changes in ⟨F⟩ and u due to phosphorylation and OM treatment, respectively. Arrows begin and end on values listed in Table 1.
Activation by phosphorylation or treatment with OM in Figure 5 involves crossing from black to blue or black to red square symbols, respectively. The blue or red arrow indicates the effect of activation from native to pβMys or OM+βMys, respectively. Transition in ⟨F⟩ and u has steeper slope for OM versus natural activation. These characteristics indicate the underlying mechanism for activation, i.e., increased duty cycle due to higher actomyosin affinity for the OM treatment and higher relative step-frequency for phosphorylation. We propose that natural activation with phosphorylation is the gold standard to which pharmaceutical myosin activators be compared.
Discussion
OM is the unique drug selectively targeting cardiac myosin in vivo to activate contraction. Earlier work with OM showed it causes greater shortening of contracting cardiac myocytes without affecting the calcium transient and that the activator stabilized the strongly actin bound myosin conformation to increase force.15 A consequence of the strong actin binding stabilization is that OM dramatically increases the βMys duty cycle. The duty cycle is widely recognized as a key myosin characteristic naturally adapted to fulfill the varied roles myosin has in living organisms. Cellular myosins working in isolation have large duty cycle (τ ≳ 0.5) to maintain nearly uninterrupted actin contact to avoid losing their way in the cytosol.33 At the opposite extreme are the muscle myosins like βMys. These molecules function in densely packed thick filaments interdigitated with actin thin filaments in the muscle fiber lattice. They are adapted for high velocity movement by their low duty cycle (δ ≲ 0.05).33 Naturally, intermediate duty cycle myosins fulfill functions requiring fewer myosins working in concert to produce work outside of muscle. The average duty cycle of βMys increased from ∼0.01 to ∼0.09 due to treatment with OM implying OM treated βMys is intermediate between muscle and processive myosins.
The resting human heart completes a cycle in ∼1 s. It is shortening for about half of that time, so we expect individual sarcomeres to shorten at a rate of 0.3 μm/0.5 s or ∼0.6 μm/s. The half-sarcomere, where we measure actin/myosin filament sliding velocity, then translates at ∼0.3 μm/s. Unloaded shortening of βMys in the in vitro motility assay is ∼0.27 μm/s in good agreement. A tripled heart rate in a stressed human would require additional translation velocity that is very likely matched by the in vitro assay at in vivo conditions of ionic strength and temperature.34 It is reasonable to surmise that the sliding velocity of the low duty cycle native βMys is fully utilized under the normal range of physiological conditions. OM treatment of βMys should place drug-induced limitations on normal human physiology. For example, a stressed human may not be able to triple heart rate because the ton needed is too small to achieve in the presence of OM. Human clinical trials indicated a small decrease in resting heart rate due to OM treatment.35 A 15% heart rate decrease was registered in dogs with systolic heart failure.36 These data are consistent with an inhibited sliding velocity for the OM treated βMys even at resting heart rate.
With porcine βMys, we detected 3 unitary step-sizes using the Qdot assay4 versus the apparent single 5 nm unitary step for rabbit sHMM.37 In βMys we observed most frequently a 5 nm step like that in sHMM, a less frequent 8 nm step, and a rare 3 nm step. Relative frequencies of the 3 and 5 nm steps are very different making it unlikely that they are only substeps of the longer 8 nm unitary step. We explore implications of the dramatic rise in duty cycle for the OM treated βMys in the context of its three unitary step-sizes with comparison of native, phosphorylated, and OM treated myosin movers in Table 1 and using the new system analytics described in Materials and Methods. The pβMys is 81–89% specifically modified at S15 of its regulatory light chain (RLC),32 and we use the motility data from ref (5). We compare phosphorylated and OM treated myosin because they are natural and pharmaceutical myosin activators potentially useful for compensating heart failure.38 In both cases, myosin performance divergence from native βMys occurs with minimal or no change in Vmax suggesting (for the case of the natural activator) that modification of the fundamental catalytic activity is impossible or counterproductive. The relative step-frequencies and duty cycles,{ωS,ωI,ωL} and {fS,fI,fL}, indicate a shift in force production significance away from the intermediate step in native βMys to the long step in phosphorylated and OM treated βMys. The shift is accompanied by rising average duty cycle, ⟨f⟩, and average force, ⟨F⟩, that is incremental or dramatic for pβMys or OM treated βMys, respectively. The average power, ⟨P⟩, follows a different trend with pβMys the best power generator. Although the OM treatment dramatically increases ⟨F⟩, it does so at the cost of lowering motility velocity. In average power, OM treated is modestly higher than native βMys while phosphorylation almost doubles native βMys power. The effect for either activator is to cause healthy cross-bridges to develop more force in a failing heart.
System analytics in the form of quantities [average force (⟨F⟩), dynamic velocity (u), and average power (⟨P⟩) for the actual step-sizes vs ωL in Figure 5] strikingly indicate that the native βMys is optimized for speed and that the natural activator holds speed nearly constant while increasing ⟨F⟩ and ⟨P⟩ by increasing the relative step-frequency of the long step. In contrast, OM treatment has a mixed impact that sacrifices speed for ⟨F⟩ thereby limiting its potential for increasing power. The natural activator enhances force and power by adjusting relative step-frequency in the system rather than adjusting the fundamental catalytic activity of the motor. The OM activator enhances force by increasing ton; however, this apparently limits relative frequency for the long step to δ ≲ 0.4. The mechanistic insights just described explain the role of phosphorylation in heart regulation and how a synthetic enhancer differs from a natural one. βMys phosphorylation is a nonlinear mechanism to adjust force-velocity in the cardiac muscle. It is a target for heart failure therapy that has already been recognized.39 OM follows an entirely different mechanism. Contrasting motility modification due to OM with that from the pβMys as indicated in Table 1 identifies the boilerplate screening characteristics for the next-generation βMys activators.
The Qdot assay routinely estimates step-size from low duty cycle muscle myosins using a standard research microscope setup.4 Typical experimental preparations are identical to those for the in vitro motility assay except for the Qdot labeling of actin. The latter is one additional incubation step following preparation of the in vitro motility slide. Optimizing conditions for estimating step-size follows the previously established guidelines4 usually requiring 2 or 3 measurement–analysis cycles to pinpoint optimal parameters for exciting light intensity, Δt, camera exposure time, and bulk concentration of motor protein. Once the conditions have been optimized, measurement consists of properly imaging the movement of 40–60 Qdot labeled filaments in 2–3 slides for each condition or motor protein investigated. At present, analysis is partially automated with the manual tracking of the Qdot required just for linking individual Qdots over the sequential images that undergo super-resolution analysis. One measurement–analysis cycle requires 2–4 days of effort not including protein preparation. Full automation of the analysis, with emphasis on making the assay amenable to high-throughput screening, is in progress. Overall, the Qdot assay is efficient, accurate, and inexpensive when compared to the modern laser-trap assay.40
Conclusions
Omecamtive mecarbil (OM) is known to specifically target βMys and upregulate cardiomyocyte contractile displacement and cardiac contractile force. Actin-activated myosin ATPase, in vitro step-size, relative step-frequency, and motility were measured to critically compare the characteristics of βMys in control native, OM treated, and phosphorylated βMys. Relative step-frequency is a newly defined and fundamental characteristic of the multiple unitary step-size βMys motor. Phosphorylation is the natural activation mechanism for this motor. Principal analytical characteristics are the average force (⟨F⟩), dynamic velocity (u), and average power (⟨P⟩). They strikingly demonstrate that the native βMys is optimized for speed and that the natural activator holds speed near constant while maximizing ⟨F⟩ and ⟨P⟩ by increasing the relative step-frequency for the longest unitary step-size of βMys. OM treatment sacrifices speed for ⟨F⟩ by increasing actomyosin affinity and duty cycle thereby lowering potential for increasing power. The natural activator enhances force and power by adjusting relative step-frequency in the system rather than adjusting the fundamental catalytic activity of the motor. The former mechanism is a template for βMys activator screening. The Qdot assay is an efficient, accurate, and inexpensive platform technology for characterizing step-size and step-frequency in low duty cycle myosins. The Qdot assay and associated analytics are promising new techniques amenable to high-throughput screening of motor protein modulators.
Glossary
Abbreviations
- βMys
porcine ventricular cardiac myosin (heavy chain gene MYH7)
- d
myosin step-size
- ELC
ventricular myosin essential light chain (gene MYL3)
- ⟨F⟩
average force
- f
myosin duty cycle
- KM
actin binding constant in the presence of ATP
- OM
omecamtive mecarbil
- ⟨P⟩
average power
- pβMys
RLC S15-phosphorylated βMys
- RLC
ventricular myosin regulatory light chain (gene MYL2)
- SD
standard deviation
- u
dynamically averaged velocity
- vm
myosin in vitro motility velocity
- Vmax
maximal actin-activated myosin ATPase
- ω
relative step-frequency.
Supporting Information Available
Movie S1 of the Qdot-labeled actin moving over βMys (Movie S1), motility velocity versus bulk βMys concentration (Figure S2), and the percentage of moving actin filaments versus OM concentration (Figure S3). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was supported by National Institutes of Health Grants R01AR049277 and R01HL095572 and by the Mayo Foundation.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Scruggs S. B.; Solaro R. J. (2011) The significance of regulatory light chain phosphorylation in cardiac physiology. Arch. Biochem. Biophys. 510, 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh F.; Ouyang K.; Campbell S. G.; Lyon R. C.; Chuang J.; Fitzsimons D.; Tangney J.; Hidalgo C. G.; Chung C. S.; Cheng H.; Dalton N. D.; Gu Y.; Kasahara H.; Ghassemian M.; Omens J. H.; Peterson K. L.; Granzier H. L.; Moss R. L.; McCulloch A. D.; Chen J. (2012) Mouse and computational models link Mlc2v dephosphorylation to altered myosin kinetics in early cardiac disease. J. Clin. Invest. 122, 1209–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toepfer C.; Caorsi V.; Kampourakis T.; Sikkel M. B.; West T. G.; Leung M.-C.; Al-Saud S. A.; MacLeod K. T.; Lyon A. R.; Marston S. B.; Sellers J. R.; Ferenczi M. A. (2013) Myosin Regulatory Light Chain (RLC) Phosphorylation Change as a Modulator of Cardiac Muscle Contraction in Disease. J. Biol. Chem. 288, 13446–13454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Ajtai K.; Burghardt T. P. (2013) Qdot Labeled Actin Super-Resolution Motility Assay Measures Low Duty Cycle Muscle Myosin Step-Size. Biochemistry 52, 1611–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Ajtai K.; Burghardt T. P. (2014) Ventricular Myosin Modifies In Vitro Step-Size When Phosphorylated. J. Mol. Cell. Cardiol. 72, 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White H. D.; Taylor E. W. (1976) Energetics and mechanism of actomyosin adenosine triphosphatase. Biochemistry 15, 5818–5826. [DOI] [PubMed] [Google Scholar]
- Marston S. B.; Taylor E. W. (1980) Comparison of the myosin and actomyosin ATPase mechanisms of the four types of vertebrate muscles. J. Mol. Biol. 139, 573–600. [DOI] [PubMed] [Google Scholar]
- Kron S. J.; Spudich J. A. (1986) Fluorescent actin filaments move on myosin fixed to a glass surface. Proc. Natl. Acad. Sci. U.S.A. 83, 6272–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyeda T. Q. P.; Kron S. J.; Spudich J. A. (1990) Myosin step size: Estimation from slow sliding movement of actin over low densities of heavy meromyosin. J. Mol. Biol. 214, 699–710. [DOI] [PubMed] [Google Scholar]
- Harris D. E.; Warshaw D. M. (1993) Smooth and skeletal muscle myosin both exhibit low duty cycles at zero load in vitro. J. Biol. Chem. 268, 14764–14768. [PubMed] [Google Scholar]
- Limouze J.; Straight A. F.; Mitchison T.; Sellers J. R. (2005) Specifity of blebbistatin, an inhibitor of myosin II. J. Muscle Res. Cell Motil. 25, 337–341. [DOI] [PubMed] [Google Scholar]
- Cheung A.; Dantzig J. A.; Hollingworth S.; Baylor S. M.; Goldman Y. E.; Mitchison T. J.; Straight A. F. (2001) A small-molecule inhibitor of skeletal muscle myosin II. Nat. Cell Biol. 4, 83–88. [DOI] [PubMed] [Google Scholar]
- Sakamoto T.; Limouze J.; Combs C.; Straight A. F.; Sellers J. R. (2005) Blebbistatin, a myosin II inhibitor, is photoinactivated by blue lights. Biochemistry 44, 584–588. [DOI] [PubMed] [Google Scholar]
- Allingham J. S.; Smith R.; Rayment I. (2005) The structural basis of blebbistatin inhibition and specificity for myosin II. Nat. Struct. Mol. Biol. 12, 378–379. [DOI] [PubMed] [Google Scholar]
- Malik F. I.; Hartman J. J.; Elias K. A.; Morgan B. P.; Rodriguez H.; Brejc K.; Anderson R. L.; Sueoka S. H.; Lee K. H.; Finer J. T.; Sakowicz R.; Baliga R.; Cox D. R.; Garard M.; Godinez G.; Kawas R.; Kraynack E.; Lenzi D.; Lu P. P.; Muci A.; Niu C.; Qian X.; Pierce D. W.; Pokrovskii M.; Ion S.; Sylvester S.; Tochimoto T.; Valdez C.; Wang W.; Katori T.; Kass D. A.; Shen Y.-T.; Vatner S. F.; Morgans D. J. (2011) Cardiac Myosin Activation: A Potential Therapeutic Approach for Systolic Heart Failure. Science 331, 1439–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; White H. D.; Winkelmann D. A.; Forgacs E. (2013) The affect of omecamtive mecarbil on the phosphate dissociation and motile properties of the recombinant human β-cardica heavymeromyosin. Biophys. J. 104, 153a. [Google Scholar]
- Ajtai K.; Garamszegi S. P.; Park S.; Velazquez Dones A. L.; Burghardt T. P. (2001) Structural characterization of b-cardiac myosin subfragment 1 in solution. Biochemistry 40, 12078–12093. [DOI] [PubMed] [Google Scholar]
- Tonomura Y.; Appel P.; Morales M. (1966) On the molecular weight of myosin II. Biochemistry 5, 515–521. [DOI] [PubMed] [Google Scholar]
- Weeds A. G.; Taylor R. S. (1975) Separation of subfragment-1 isoenzymes from rabbit skeletal muscle myosin. Nature 257, 54–56. [DOI] [PubMed] [Google Scholar]
- Pardee J. D.; Spudich J. A. (1982) Purification of muscle actin. Methods Enzymol. 85, 164–179. [DOI] [PubMed] [Google Scholar]
- Pant K.; Watt J.; Greenberg M.; Jones M.; Szczesna-Cordary D.; Moore J. R. (2009) Removal of the cardiac myosin regulatory light chain increases isometric force production. FASEB J. 23, 3571–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiske C. H.; Subbarow Y. (1925) The Colorimetric Determination of Phosphorus. J. Biol. Chem. 66, 375–400. [Google Scholar]
- Stout A. L.; Axelrod D. (1989) Evanescent field excitation of fluorescence by epi-illumination microscopy. Appl. Opt. 28, 5237–5242. [DOI] [PubMed] [Google Scholar]
- Meijering E., Dzyubachyk O., and Smal I. (2012) Chapter nine: Methods for Cell and Particle Tracking. In Methods in Enzymology (Conn P. M., Ed.) pp 183–200, Academic Press, New York. [DOI] [PubMed] [Google Scholar]
- Bobroff N. (1986) Position measurement with a resolution and noise-limited instrument. Rev. Sci. Instrum. 57, 1152–1157. [Google Scholar]
- Thompson R. E.; Larson D. R.; Webb W. W. (2002) Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 82, 2775–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques R.; Lelek M.; Fornasiero E. F.; Valtorta F.; Zimmer C.; Mhlanga M. M. (2010) QuickPALM: 3D real-time photoactivation nanoscopy image processing in ImageJ. Nat. Methods 7, 339–340. [DOI] [PubMed] [Google Scholar]
- Gordon A. M.; Huxley A. F.; Julian F. J. (1966) The variation in isometric tension with sarcomere length in vertebrate muscle fibres. J. Physiol. (Oxford, U.K.) 184, 170–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura S.; Kobayakawa N.; Fujita H.; Yamashita H.; Momomura S.-i.; Chaen S.; Omata M.; Sugi H. (1998) Comparison of Unitary Displacements and Forces Between 2 Cardiac Myosin Isoforms by the Optical Trap Technique: Molecular Basis for Cardiac Adaptation. Circ. Res. 82, 1029–1034. [DOI] [PubMed] [Google Scholar]
- Palmiter K. A.; Tyska M. J.; Dupuis D. E.; Alpert N. R.; Warshaw D. M. (1999) Kinetic differences at the single molecule level account for the functional diversity of rabbit cardiac myosin isoforms. J. Physiol. (Oxford, U.K.) 519, 669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debold E. P.; Saber W.; Cheema Y.; Bookwalter C. S.; Trybus K. M.; Warshaw D. M.; VanBuren P. (2010) Human actin mutations associated with hypertrophic and dilated cardiomyopathies demonstrate distinct thin filament regulatory properties in vitro. J. Mol. Cell. Cardiol. 48, 286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephson M. P.; Sikkink L. A.; Penheiter A. R.; Burghardt T. P.; Ajtai K. (2011) Smooth muscle myosin light chain kinase efficiently phosphorylates serine 15 of cardiac myosin regulatory light chain. Biochem. Biophys. Res. Commun. 416, 367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell C. B.; Tyska M. J.; Mooseker M. S. (2007) Myosin at work: Motor adaptations for a variety of cellular functions. Biochim. Biophys. Acta 1773, 615–630. [DOI] [PubMed] [Google Scholar]
- Homsher E.; Wang F.; Sellers J. R. (1992) Factors affecting movement of F-actin filaments propelled by skeletal muscle heavy meromyosin. Am. J. Physiol. 31, C714–C723. [DOI] [PubMed] [Google Scholar]
- Cleland J. G. F.; Teerlink J. R.; Senior R.; Nifontov E. M.; Mc Murray J. J. V.; Lang C. C.; Tsyrlin V. A.; Greenberg B. H.; Mayet J.; Francis D. P.; Shaburishvili T.; Monaghan M.; Saltzberg M.; Neyses L.; Wasserman S. M.; Lee J. H.; Saikali K. G.; Clarke C. P.; Goldman J. H.; Wolff A. A.; Malik F. I. (2011) The effects of the cardiac myosin activator, omecamtiv mecarbil, on cardiac function in systolic heart failure: A double-blind, placebo-controlled, crossover, dose-ranging phase 2 trial. Lancet 378, 676–683. [DOI] [PubMed] [Google Scholar]
- Shen Y. T.; Malik F.; Zhao X.; Depre C.; Dhar S. K.; Abarzua P.; Morgans D. J.; Vatner S. F. (2010) Improvement of cardiac function by a cardiac myosin activator in conscious dogs with systolic heart failure. Circ.: Heart Failure 3, 522–527. [DOI] [PubMed] [Google Scholar]
- Steffen W.; Smith D.; Simmons R.; Sleep J. (2001) Mapping the actin filament with myosin. Proc. Natl. Acad. Sci. U.S.A. 98, 14949–14954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthu P.; Kazmierczak K.; Jones M.; Szczesna-Cordary D. (2012) The effect of myosin RLC phosphorylation in normal and cardiomyopathic mouse hearts. J. Cell. Mol. Med. 16, 911–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes-Ferreira P.; De Keulenaer G. W.; Leite-Moreira A. F.; Brás-Silva C. (2013) Therapeutic potential of neuregulin-1 in cardiovascular disease. Drug Discovery Today 18, 836–842. [DOI] [PubMed] [Google Scholar]
- Capitanio M.; Canepari M.; Maffei M.; Beneventi D.; Monico C.; Vanzi F.; Bottinelli R.; Pavone F. S. (2012) Ultrafast force-clamp spectroscopy of single molecules reveals load dependence of myosin working stroke. Nat. Methods 9, 1013–1019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.