

Abstract
A series of mononuclear six-coordinate tungsten compounds spanning formal oxidation states from 0 to +VI, largely in a ligand environment of inert chloride and/or phosphine, was interrogated by tungsten L-edge X-ray absorption spectroscopy. The L-edge spectra of this compound set, comprised of [W0(PMe3)6], [WIICl2(PMePh2)4], [WIIICl2(dppe)2][PF6] (dppe = 1,2-bis(diphenylphosphino)ethane), [WIVCl4(PMePh2)2], [WV(NPh)Cl3(PMe3)2], and [WVICl6], correlate with formal oxidation state and have usefulness as references for the interpretation of the L-edge spectra of tungsten compounds with redox-active ligands and ambiguous electronic structure descriptions. The utility of these spectra arises from the combined correlation of the estimated branching ratio of the L3,2-edges and the L1 rising-edge energy with metal Zeff, thereby permitting an assessment of effective metal oxidation state. An application of these reference spectra is illustrated by their use as backdrop for the L-edge X-ray absorption spectra of [WIV(mdt)2(CO)2] and [WIV(mdt)2(CN)2]2– (mdt2– = 1,2-dimethylethene-1,2-dithiolate), which shows that both compounds are effectively WIV species even though the mdt ligands exist at different redox levels in the two compounds. Use of metal L-edge XAS to assess a compound of uncertain formulation requires: (1) Placement of that data within the context of spectra offered by unambiguous calibrant compounds, preferably with the same coordination number and similar metal ligand distances. Such spectra assist in defining upper and/or lower limits for metal Zeff in the species of interest. (2) Evaluation of that data in conjunction with information from other physical methods, especially ligand K-edge XAS. (3) Increased care in interpretation if strong π-acceptor ligands, particularly CO, or π-donor ligands are present. The electron-withdrawing/donating nature of these ligand types, combined with relatively short metal–ligand distances, exaggerate the difference between formal oxidation state and metal Zeff or, as in the case of [WIV(mdt)2(CO)2], exert the subtle effect of modulating the redox level of other ligands in the coordination sphere.
Short abstract
A series of six-coordinate tungsten compounds, primarily with phosphine and/or Cl− ligands and spanning formal oxidation states 0 → +VI, were examined by L-edge X-ray absorption spectroscopy. The estimated branching ratio derived from the L3- and L2-edge intensities and the L1 rising-edge energies correlate with formal oxidation state. The data reported were applied to compounds with ambiguous electronic structure descriptions due to the presence of π-accepting and redox-active ligands.
Introduction
The distinction that may be made between the formal and physical oxidation state of a metal atom in a transition metal complex was first articulated by Jørgensen.1 A formal oxidation state is a nonmeasurable integer commonly defined as the charge remaining on the metal after the ligands have been removed in their closed-shell form.2 In principle, a physical oxidation state is a value derived from a measurable quantity (e.g., from spectroscopy) that diagnoses a dn configuration.1 Where these two descriptions can appreciably diverge is in situations where the bonding between metal and ligand(s) is substantially covalent such that the charge associated with the metal atom, evaluated by accounting methods that assume integer charges or complete neutrality to specific ligand types,3 differs from a metal effective nuclear charge (Zeff) gauged by experimental methods. Redox-active ligands such as NO and heterodiene-type ligands, such as catecholates, o-diimines, and dithiolenes, are widely recognized as “noninnocent” ligands that can readily assume radical character and lead to significant departures from the electron-counting schemes that partition charge in a heterolytic fashion. Recent work reported from several research groups, in which computational insights are typically used in conjunction with information from a battery of physical methods, has demonstrated that ligand redox activity extends beyond NO and heterodiene ligands to include such other ligand types as phenolates,4 corroles,5,6 bis(imino)pyridines,7 and tris(amido)-8 and amido bis(phenolate)9 pincer-type ligands.
From the perspective of understanding the intrinsic nature of a metal complex and anticipating its reactivity and properties, the spectroscopic oxidation state may constitute a preferred basis for thought. Although no experimental method produces a metal “oxidation state” as immediate output, metal K-edge X-ray absorption spectroscopy (XAS), especially when taken in conjunction ligand K-edge XAS data,10,11 is a direct experimental method for evaluating a metal Zeff. The rising-edge is dominated by electric dipole-allowed 1s → np transitions and is effective for experimentally gauging Zeff because the core 1s orbital shifts to deeper binding energy upon oxidation of the metal. For first row metals, the core-hole lifetime is sufficiently long that the spectrum may be resolved into the pre-edge and near-edge constituents.12 The pre-edge arises from electric quadrupole-allowed excitations to vacancies in the 3d manifold, which gain intensity through d–p mixing via a departure from centrosymmetry and are diagnostic of the geometry of the complex.13 At this high resolution (1–1.5 eV), the pre-edge transition is the preferred metric for Zeff.14−17 The corresponding K-edge for third row metals occurs at prohibitively high energies (>60 keV). Consequently, attaining detail about the local electronic structure is confined to L-edge XAS. These edges comprise three components: L3- and L2-edges are dipole-allowed excitations of core 2p electrons to partially filled or empty 5d-based orbitals, while the L1-edge is dominated by 2s → 6p dipole-allowed transitions. The latter represents a viable alternative to K-edge XAS; however, tangible electronic structure details are washed out by lifetime broadening of the 2s core state.12 As such, data is almost exclusively restricted to oxides of 5d metals where well-resolved pre-edge peaks inherent to tetrahedral moieties are distinguished from the featureless rising-edge for octahedral centers.18,19
The application of L1-edge XAS to molecular systems with third row metals has yet to be undertaken. Its use hinges upon the availability of data for a well-defined set of reference compounds whose oxidation states are both relatively unambiguous and span a complete range of chemically accessible redox levels (Chart 1). Recent work20−22 in our laboratory with organometallic tungsten complexes bearing redox-active 1,2-dimethylethene-1,2-dithiolate, (mdt)2–, ligands has presented some challenge to finding a satisfactory description of their electronic structure and has motivated an effort to develop a small library of XAS reference compounds that would have some general utility for the assessment of Zeff and thereby assist a description of electronic structure in ambiguous situations (Chart 1). Accompanying these reference compounds are L-edge XAS data we have collected over a period of years on a broad set of mono-, bis-, and tris(dithiolene) complexes of tungsten. Here, we derive the estimated branching ratio (EBR), defined as the relative intensity of the L3-edge over the sum of the L3- and L2-edges, [I(L3)/I(L3 + L2)],23−26 and the L1 rising-edge energy for each reference compound. These two metrics are used to gauge whether the level of detail provided by L-edge XAS is sufficient to make an assessment of Zeff across a range of tungsten dithiolene compounds. As X-ray absorption spectroscopy continues to grow as a method to complement the insights obtained by other physical methods, we shall illustrate that appropriately calibrated L-edge XAS for third row metals is a useful and informative alternative to the inaccessible K-edge for these heavy elements.
Chart 1. Pictorial Representation of the Complexes Organized According to Tungsten Formal Oxidation Statea.

a For organizational purposes, all dithiolene-type ligands other than Me2pipdt are treated as fully reduced.
Experimental Section
Literature procedures were employed for the syntheses of [WIICl2(PMePh2)4],27 [WIICl2(dppe)2] (dppe =1,2-bis(diphenylphosphino)ethane),28 [WIVCl4(PMePh2)2],29 [WV(NPh)Cl3(PMe3)2],30 [WVI(xylidene)3],31 [W0(Me2pipdt)(CO4)] (Me2pipdt = 1,4-dimethylpiperazine-2,3-dithione),21 [WII(mdt)(CO)4],20,21 [WII(mdt)(CO)2(PMe3)2],20 [WIV(mdt)2(CO)2],20,32 [WIV(mdt)2(CO)(PMe3)],20 [WIV(mdt)2(PMe3)2],20 [WIV(mdt)2(CNtBu)2],22 [NEt4]2[WIV(mdt)2(CN)2],22 [Ph4P]2[WIV(mnt)3] (mnt2– = maleonitriledithiolate),33 [PPh4]2[WIV(bdt)3] (bdt2– = benzene-1,2-dithiolate),34 [NEt4]2[WIV(bdt)3],34 [Et4N]2[WIV(mdt)3],35 [PPh4][WV(bdt)3],34 [Et4N][WV(mdt)3],35 [WVI(bdt)3],36 [WVI(mdt)3],20 [WVI(pdt)3] (pdt2– = 1,2-diphenylethene-1,2-dithiolate).36 Commercial sources of [W0(CO)6] and [WVICl6] were purified by vacuum sublimation before use.
Syntheses
[WIIICl2(dppe)2][PF6]
A 100 mL Schlenk flask with a stir bar was charged with a sample of [WIICl2(dppe)2] (0.397 g, 0.378 mmol) and 25 mL of dry dichloromethane. In a separate 50 mL Schlenk flask, a crystalline sample of [FeCp2][PF6] (0.125 g, 0.378 mmol) was dissolved in 15 mL of dichloromethane. This solution was added to the yellow-orange solution of [WIICl2(dppe)2]. Over the first 15 min, a light green color developed in the solution; stirring was continued for another 2 h. The solvent was removed under reduced pressure, affording a light greenish solid, which then was washed with Et2O until the washings were colorless. After drying of the crude product overnight, orange crystals of [WIIICl2(dppe)2][PF6] were obtained by the diffusion of Et2O or pentane vapor into a concentrated 1,2-dichloroethane solution. Yield: 0.322 g (71%). Anal. Calcd for C52Cl2F6H48P5W: C, 52.20; H, 4.04; P, 12.94. Found: C, 51.95; H, 4.28; P, 12.72%.
X-ray Crystallographic Data Collection and Structure Refinement
Diffraction-quality, yellow-orange block-shaped crystals of [WIIICl2(dppe)2][PF6] were obtained by slow diffusion of Et2O vapor into a 1,2-dichloroethane solution, while orange block-shaped crystals of [WVI(xylidene)3] deposited from a hexanes solution upon standing. Crystals were coated with paratone oil and mounted on the end of a nylon loop attached to the end of a goniometer. Data were collected with a Bruker Smart APEX CCD diffractometer equipped with a Kryoflex attachment supplying a nitrogen stream at 100 K. Full spheres of data were obtained by the collection of three sets of 400 frames in ω (0.5°/scan), collected at φ = 0.00, 90.00, and 180.00° followed by two sets of 800 frames in φ (0.45°/scan) collected with ω constant at −30.00 and 210.00°. Frame times of 15 and 10 s were used for [WIIICl2(dppe)2][PF6] and [WVI(xylidene)3], respectively. Data were collected under control of the APEX2 software package.37 Raw data were reduced to F2 values using the SAINT(38) software, and a global refinement of the unit cell parameters was performed using ∼9900 selected reflections from the full data sets. Data were corrected for absorption on the basis of multiple measurements of symmetry-equivalent reflections with the use of SADABS.39 Structure solutions were obtained by Patterson methods using SHELXS,40 while refinements were accomplished by full-matrix least-squares procedures using SHELXL.41 Both the SHELXS and SHELXL programs are incorporated into the SHELXTL software suite.42
The asymmetric unit of the unit cell for [WIIICl2(dppe)2][PF6] was found to be composed of two independent [PF6]1– anions, one of which was disordered, and four independent half [WIIICl2(dppe)2]1+ cations residing on inversion centers. Because of the imposed centrosymmetry, three of the four independent half-cations showed disorder to varying extents in the diphosphine ligand. This disorder was modeled by treating the affected phenyl groups as rigid entities and restraining equivalent metrical parameters to be approximately equal. A similar treatment was accorded the disordered [PF6]1– anion. In the final stages of refinement, hydrogen atoms were added in calculated positions and included as riding contributions with isotropic displacement parameters 1.2–1.5 times those of the carbon atoms to which they were attached. The thermal ellipsoid plot in Figure 1 and in the supplementary data were created with the use of XP, which also is part of the SHELXTL package.42 Final unit cell data and refinement statistics are collected in Table 1.
Figure 1.

Thermal ellipsoid plot at the 50% probability level of one of the cations in the unit cell of crystals of [WIIICl2(dppe)2][PF6]. Hydrogen atoms are omitted for clarity.
Table 1. Crystallographic Data for [WIIICl2(dppe)2][PF6] and [WVI(xylidene)3].
| chem formula | C52H48Cl2F6P5W | C24H24W |
| fw | 1196.50 | 496.28 |
| crystal system | triclinic | triclinic |
| space group | P1̅ | P1̅ |
| color/shape | orange/slab | orange/block |
| a, Å | 13.2421(8) | 6.8375(4) |
| b, Å | 19.247(1) | 11.1829(6) |
| c, Å | 19.638(1) | 11.8409(6) |
| α, deg | 100.3790(1) | 105.100(1) |
| β, deg | 98.9950(1) | 92.924(1) |
| γ, deg | 90.9070(1) | 90.392(1) |
| V, Å3 | 4857.7(5) | 872.80(8) |
| Z | 4 | 2 |
| T, K | 100(2) | 100(2) |
| ρ calcd, g cm–3 | 1.636 | 1.888 |
| reflns collected/2Θmax | 85 478/56.00 | 15 457/56.50 |
| unique reflns/I > 2σ(I) | 23 262/16 625 | 4267/4102 |
| parameters/restraints | 1130/220 | 274/0 |
| λ, Å/μ (Kα), mm–1 | 0.710 73/2.713 | 0.710 73/6.620 |
| GoFa | 1.055 | 1.058 |
| R1b,c/wR2c,d | 0.0453/0.0986 | 0.147/0.0332 |
| residual density, e Å–3 | 3.58/–3.10 | 0.773/–0.579 |
GoF = {Σ[w(F02 – Fc2)2]/(n – p)}1/2, where n = number of reflections and p is the total number of parameters refined.
R1 = Σ∥F0| – |Fc∥/Σ|F0|.
R indices for data cut off at I > 2σ(I).
wR2 = {Σ[w(F02 – Fc2)2]/Σ[w(F02)2]}1/2, where w = 1/[σ2(F02) + (aP)2 + bP], P = (F02 + 2Fc2)/3.
X-ray Absorption Spectroscopy
XAS data were measured at the Stanford Synchrotron Radiation Lightsource (SSRL) with the SPEAR storage ring containing 200–300 mA at 3.0 GeV. Tungsten L-edge spectra were collected on beamline 7–3 operating with a wiggler field of 2 T. A Si(220) double-crystal monochromator was used. Beamline 7–3 is equipped with a rhodium-coated vertical collimating mirror upstream of the monochromator and a downstream bent-cylindrical focusing mirror (also rhodium-coated). Incident and transmitted X-ray intensities were monitored using nitrogen-filled ionization chambers. Data were measured in transmittance mode, and samples were maintained at 10 K using an Oxford Instruments CF1208 continuous flow liquid helium cryostat. Internal energy calibrations were performed by simultaneous measurement of the W reference foil placed between the second and third ionization chamber with inflection points assigned as 12 100, 11 544, and 10 207 eV for the L1-, L2-, and L3-edges, respectively. Data were processed by fitting a second-order polynomial to the pre-edge region and subtracting this background from the entire spectrum.43 A three-region cubic spline was used to model the smooth background above the edge. The data were normalized by subtracting the spline and normalizing the postedge to 1.0. Fits to the L2- and L3-edges modeled by pseudo-Voigt lines were carried out using the program EDG_FIT(43) with a fixed 1:1 ratio of Lorentzian to Gaussian contributions.
Other Physical Methods
Variable-temperature (2–290 K) magnetic susceptibility data were recorded in 1 T external field using an MPMS Quantum Design SQUID magnetometer. The experimental data were corrected for underlying diamagnetism using tabulated Pascal’s constants. S- and X-band fluid solution spectra were collected using a Bruker EMX Micro spectrometer, and frozen solution spectra were obtained using a Bruker E580 spectrometer. Simulations were performed using the Xsophe (Bruker Biospin GmbH) suite.44 The elemental analysis was performed by Midwest Microlab, LLC, of Indianapolis, IN.
Calculations
All calculations were performed using the Gaussian 09 package.45 Geometry optimizations and Kohn–Sham orbitals were calculated at Perdew–Burke–Ernzerhof (PBE)46 (for exchange and correlation) and B3LYP47 levels of density functional theory (DFT). The validity of all structures was confirmed by the absence of imaginary frequencies. The basis set chosen for all main group elements, except hydrogen, is the 6-31G(d,p). For tungsten a double-ζ (DZ) basis set with an effective electron core potential (LANL2DZ ECP) was implemented,48 and a Gaussian split valence (SV) basis set was used for the hydrogen atoms.49 The Mulliken population and atomic orbital composition analyses were calculated via a fragmentation approach using QMForge,50 and all molecular orbital images were rendered using Chemcraft51 and Jmol.52
Results and Discussion
Reference Compounds
Several criteria governed the choice of compounds for this tungsten L-edge XAS study: (1) Strict adherence to coordination number of six at tungsten. (2) Avoidance of any ligand with potential redox activity. (3) Preference for compounds with a minimal number of strongly perturbing π-acceptor or π-donor ligands. Compounds with varying numbers of chloride and phosphine ligands meet these conditions and are useful as reference compounds because they are readily accessible synthetically. To that end, we assembled a series comprising [W0(PMe3)6],53 [WIICl2(PMePh2)4],27 [WIIICl2(dppe)2][PF6], [WIVCl4(PMePh2)2],29 [WV(NPh)Cl3(PMe3)2],30 and [WVICl6], as they cover the full range of formal oxidation states at tungsten (Chart 1). The metal atom in [W0(PMe3)6] is as pure a zerovalent tungsten as might be conceptualized and thus is very useful as a spectroscopic benchmark compound. It is deemed superior to [W0(CO)6], which despite its classification as zerovalent tungsten, experiences significant metal-to-ligand charge transfer via π-backbonding.3 A switch of the number and positions of the chloride and phosphine ligands relates [WIICl2(PMePh2)4] and [WIVCl4(PMePh2)2]. The 1H and 31P NMR chemical shifts respond to the paramagnetism of the compounds.28 The WII complex possesses a (t2g)4 electron configuration, although a spin ground state cannot be applied given the large spin–orbit contribution from the 5d metal. A similar description is pertinent to [WIVCl4(PMePh2)2], which exhibits a nonzero magnetic moment due to mixing of paramagnetic excited states by spin–orbit coupling,54 a situation routinely encountered for octahedral d2 species.55,56 The WIII standard, [WIIICl2(dppe)2][PF6], with fewer π-donor ligands, was favored over [CoCp2][WIIICl4(PMePh2)2].57 Although previously generated in a reaction between [W0(N2)2(dppe)2] and CH2Cl2 in the presence of HFeCo3(CO)12,58 we describe a more direct and better-yielding synthesis via chemical oxidation of [WIICl2(dppe)2] by [FeCp2][PF6]. Its constitution was confirmed by X-ray crystallographic analysis (Figure 1); the structure is highly similar to that reported with a [BF4]1– counterion.58 Table 2 contrasts the salient metric parameters with the precursor, [WIICl2(dppe)2],59 and shows a noticeable shortening of the W–Cl distances commensurate with a one-electron depopulation of the degenerate dxz,yz highest occupied molecular orbital (HOMO), which are π-antibonding with the Cl– ligands (Figure 2). The breadth of the WP4 plane expands slightly upon oxidation of the metal ion.
Table 2. Comparison of Averaged Bond Distances (Å) and Angles (deg).
| a | [WIICl2(dppe)2]b | [WIIICl2(dppe)2][PF6] |
|---|---|---|
| W–Cl | 2.4228(8) | 2.3207(6) |
| W–P | 2.5008(7) | 2.5518(5)c |
| Cl–W–Cl | 173.65(4) | 180 |
| P–W–Pd | 79.14(3) | 78.27(2)c |
Values are averaged, where possible, with uncertainty propagations determined according to the general formula for uncertainty in a function of several variables detailed in ref (60).
Data from ref (59).
Values involving the disordered dppe ligand were excluded from the averages.
Ligand bite angle.
Figure 2.
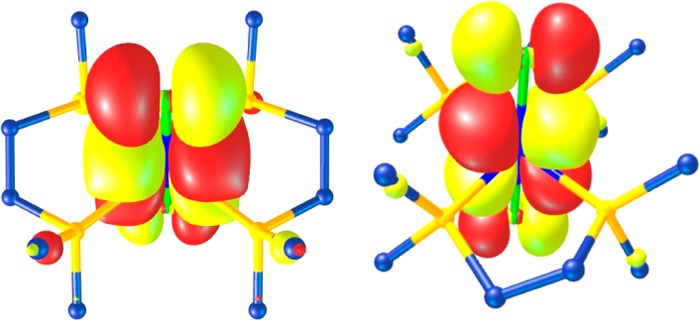
Degenerate SOMO comprising the ground state in [WIIICl2(dppe)2]1+ with ∼72% dxz,yz character.
The temperature dependence of the magnetic moment of this complex was examined by SQUID magnetometry (Figure S1). The room-temperature effective magnetic moment of 1.2 μB is consistent with a low-spin d3 ion (S = 1/2) affected by the sizable tungsten spin–orbit coupling constant that reduces the average g value to 1.35. Similar room-temperature magnetic moments have been reported for analogous complexes.56,58 The axial electron paramagnetic resonance (EPR) spectrum, measured at 20 K (Figure 3), was simulated using g = (1.359, 1.266, 1.006). The shift to high field leads to significant line broadening that obscures resolution of the 183W (I = 1/2, 14.3% abundant) and 31P (I = 1/2, 100% abundant) active nuclei. The substantial deviation away from 2.0023 is consistent with a (dxy)2(dxz,yz)1 electron configuration.61 The extent of the g shift, the fast electronic relaxation and low magnetic moment are hallmarks of a near degenerate ground state (2Eg) where orbital angular momentum is partially unquenched. To our knowledge, these are the lowest g values reported for a low-spin WIII complex.62 [WV(NPh)Cl3(PMe3)2] is a deliberate choice for WV representative rather than a more obvious [WVOCl3(PR3)2] compound, which could complicate the spectroscopy should it cocrystallize with the related chloro species [WIVCl4(PR3)2].63 The effect of substituting a terminal oxo for an imido ligand was explored by EPR spectroscopy. The simulation of chilled solution (190 K) spectra recorded at S- and X-band frequencies yielded giso = 1.903 (Figures S2 and S4), a value consistent with an effective magnetic moment lower than the spin-only value.30,64 In contrast, [WVOCl3(PPh3)2] exhibited g = 1.791,65 which identifies greater covalency within the {W=NPh}3+ unit. This conclusion is supported by a lower tungsten 4f7/2 binding energy in [WVI(NPh)Cl4]2 than in [WVIOCl4]x.66 Hyperfine coupling to two equivalent 31P (26 × 10–4 cm–1) nuclei and the 183W (60 × 10–4 cm–1) nucleus are observed with improved visibility at S-band (Figures S2 and S3).
Figure 3.
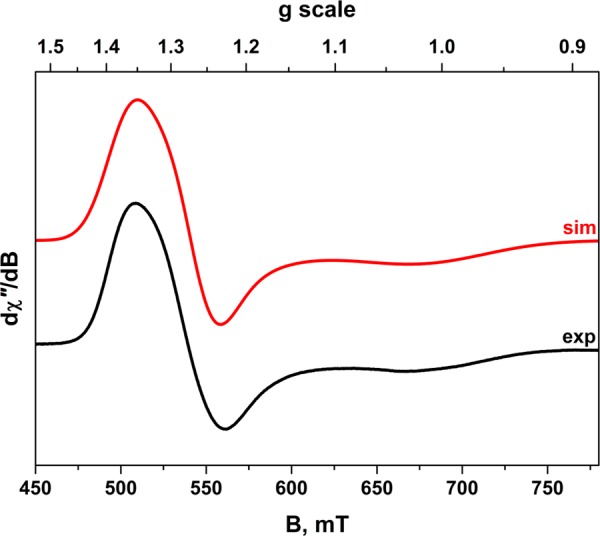
X-band EPR spectrum of [WIIICl2(dppe)2][PF6] recorded in CH2Cl2/THF solution at 20 K (experimental conditions: frequency, 9.6310 GHz; power 0.63 mW; modulation, 0.7 mT). Experimental data are depicted by the black line; the simulation is in red.
L3,2-Edges
The L3- and L2-edges exhibit prominent features referred to as white lines, identified as 2p → ns and 2p → nd electronic transitions, which are both dipole-allowed. These edges are separated by 2p spin–orbit coupling; the L2-edge for tungsten lies about 1337 eV above the L3-edge. Specifically, the L3-edge represents transitions from 2p3/2 to both 5d3/2 and 5d5/2 states, whereas the L2-edge pertains to excitations from 2p1/2 solely to the 5d3/2 state. Changes in the absorption are observed across the series and reflect variation in the number of vacancies in the 5d orbitals.67,68 The L3-edge is twice as intense as the L2, though there is deviation from this statistical ratio in the W L-edge study, as previously noted for 5d metals.69 Excitations to unoccupied s levels are considerably weaker and can be neglected.70 The L3- and L2-edge spectra recorded for each reference compound are presented in Figure 4. Each spectrum has been normalized to the step in the continuum across the absorption edge, and white-line energies are posted in Table 3.
Figure 4.
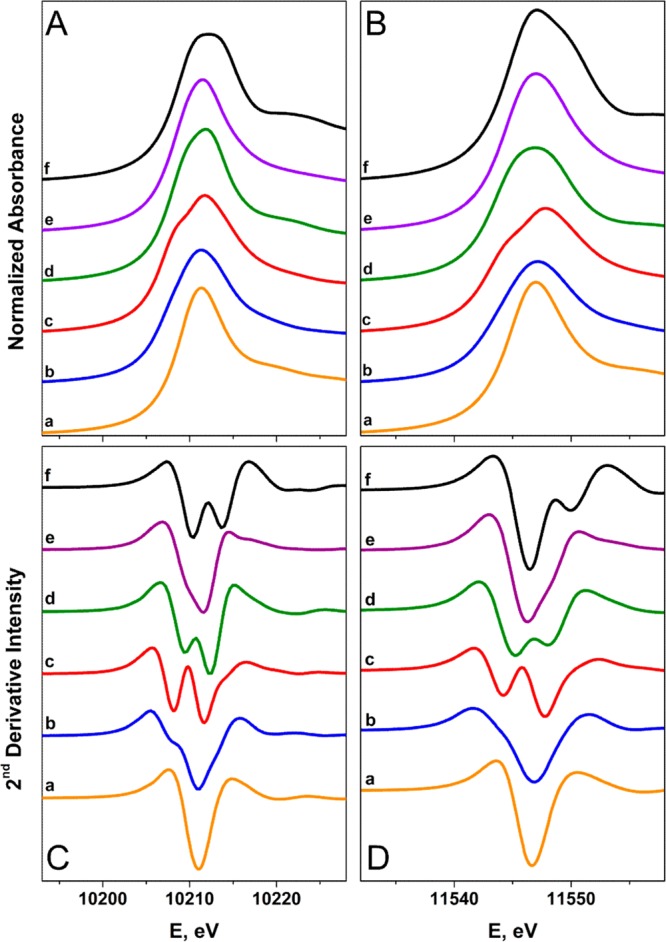
Comparison of the normalized W L3- (panel A) and L2-edge (panel B) and their FFT-smoothed second derivative spectra (panels C and D) of (a) [W0(PMe3)6], (b) [WIICl2(PMePh2)4], (c) [WIIICl2(dppe)2][PF6], (d) [WIVCl4(PMePh2)2], (e) [WV(NPh)Cl3(PMe3)2], and (f) [WVICl6].
Table 3. L-Edge Energies for Reference Compounds.
| L1 |
||||
|---|---|---|---|---|
| L3a | L2a | pre-edge | rising-edgeb | |
| Wc | 10 210.1 | 11 546.3 | 12 100.0 | |
| [W0(PMe3)6] | 10 211.3 | 11 547.0 | 12 091.6 | 12 095.2 |
| [WIICl2(PMePh2)4] | 10 211.3 | 11 547.1 | 12 092.3 | 12 096.0 |
| [WIIICl2(dppe)2][PF6] | 10 211.8 | 11 547.9 | 12 092.4 | 12 096.5 |
| [WIVCl4(PMePh2)2] | 10 211.8 | 11 547.0 | 12 090.4 | 12 097.6 |
| [WV(NPh)Cl3(PMe3)2] | 10 211.5 | 11 547.0 | 12 093.1 | 12 097.8 |
| [WVICl6] | 10 212.2 | 11 547.1 | 12 092.1 | 12 099.2 |
White-line peak maximum.
Energy of first inflection point determined from first derivative of the spectrum.
Tungsten reference foil.
The white-line energies are invariant at the L2-edge across the series, with only a subtle shift to higher values at the L3-edge for more oxidized W ions. With the exception of [W0(PMe3)6] and its (t2g)6 electron configuration, each spectrum displays splittings of the white-line peaks due to the ligand-field, which are the same at both edges. Ligand-field splittings, more readily observed in the second derivative plot, range from 1.1 to 3.6 eV (Figure 4) and compare well with those computed by DFT (Table S1). We note that the decreased shielding due to the creation of a core-hole will affect the acceptor orbitals differently, which is an effect not included in the calculations. Assuming near-octahedral geometry for the complexes, the lower energy peak is assigned as a transition to a vacancy in the t2g orbitals, while the higher energy peak is attributed to an excitation to the vacant eg set. The intensity of the first peak increases relative to the second peak for higher oxidation states of tungsten in a fashion commensurate with the greater number of electron vacancies in the t2g levels. The lower-valent compounds (0 to +IV) of this series are yellow or orange-yellow in color and devoid of low-energy ligand-field transitions consistent with large ΔOCT provided by phosphine ligands. The multiply bonded imido group in [WV(NPh)Cl3(PMe3)2] leads to a significant departure from an octahedral ligand-field, as reflected by the smaller splitting of the white-line peak. On the whole, this splitting makes precise determination of the peak energy problematic (Table 3).
The white-line intensity at both edges trends well with the assigned oxidation state of tungsten. The peak area was estimated by a simple curve-fitting method, where Gaussian broadened Lorentz functions were used to simulate the white-line peaks after removal of the edge continuum jump modeled by an arctan function (see Supporting Information).26,67,71 Because the optimum energy position for the arctan function is somewhat uncertain, they have been fixed to the intersection with the rising-edge of the white-line. The data plotted in Figure S6 show a monotonic increase in the intensity for the L3-edge with increasing oxidation state.71−73 Departure from linearity is ascribed to variation in the peak profile of the tail of the white line above the ionization threshold due to transitions to quasi-bound states leading to a deviation of the peak from a pseudo-Voigt line shape.69,72 Also 2p–5d multiplet effects will affect the spectral shape and peak area.74 A similar increase is noted for the L2-edge (Figure S6), though [W0(PMe3)6] departs significantly from expectation without an obvious explanation. This leads to a smaller EBR than anticipated based on its L1 rising-edge energy.
The branching ratio has been used as a metric of metal oxidation state in discrete molecules and extended solids by XAS6,23 and electron energy-loss spectroscopy.24,75 The EBR has been determined for each reference compound and plotted in Figure 5.23−26 Aside from the low value for [W0(PMe3)6], there is a decrease in the EBR as the series of reference compounds is traversed from low to high formal oxidation state. In an effort to assess the validity of assigning a physical oxidation state based on EBR, these values are compared to those obtained for an ensemble of W coordination compounds. The impact of potent π-accepting CO ligands is clearly observed for [W0(CO)6], where the computed EBR of 0.45 is indicative of a Zeff more akin to WV than to W0.
Figure 5.

Comparison of EBR (left) and L1 rising-edge energies (right).
At the other end of the scale, [WVI(xylidene)3] has a larger EBR than the corresponding standard, [WVICl6]. This suggests a somewhat more reduced central ion in [WVI(xylidene)3], which stems from its preference for a C3h symmetric paddle wheel structure as opposed to the octahedron in [WVICl6].31 Hexamethyltungsten, as an unambiguous WVI species, is the more ideal candidate for study inasmuch as the charge at tungsten cannot be alleviated by any π donation, there being no lone electron pairs. However, as the synthesis and handling of this compound are attended by some risk of explosion,76 [WVI(xylidene)3] is a safer alternative for measurement. A survey of the frontier MOs in an optimized structure of [WVI(Me)6] shows a lowest unoccupied molecular orbital (LUMO) that is essentially pure dz2 (Figure S42). A pair of deeper lying MOs (HOMO-1,2, e′; HOMO-3,4, e″) have moderate metal d character (32% and 34%, respectively), but the bonding is carbon p → tungsten d σ donating in character. The metal–ligand bonding description in [WVI(xylidene)3] is qualitatively similar to that in [WVI(Me)6] but is rendered more complicated largely by its lower symmetry (C3h).31 The symmetry is produced by a substantial fold of all three xylidene ligands along each intraligand CH2···CH2 vector, which orients the unit almost “side-on.” The methylene carbon atoms are positioned 2.2137(8) Å from the tungsten ion, while their corresponding aromatic carbons are 2.4817(8) Å away. Here the average dihedral angle between the W(CH2)2 and xylidene C8 planes is 83.8(3)° (Figure S62). This angle is considerably smaller than it is for xylidene complexes with other early transition metals77 and is a means by which the ligand further supplements the electron-deficient metal ion. The consequence of this structural distortion is mixing of the ligand-based HOMO with the metal-based LUMO, which transform to the same representation in C3h point symmetry.78,79 The mixing deposits metal character into the HOMO (now HOMO-3, Figure S41) and ligand character into the LUMO with overall stabilization of the molecule. Therefore, the EBR is closer to that of [WV(NPh)Cl3(PMe3)2] than that of [WVICl6] because the metal content of the LUMO is reduced. Tungsten’s preference for high oxidation states establishes [WVI(xylidene)3] as much closer to the limiting description of WVI with alkyl ligands than to a W0 species with olefinic ligands.
L3- and L2-edge spectra for a series of tungsten mono(dithiolene) and bis(dithiolene) compounds with various ancillary ligands (CO, PMe3, CNtBu, CN–) are collected and overlaid in Figure S8. The white-line energies listed in Table S2 show no discernible correlation with the formal oxidation state. As expected, the comparison of the white-line peak energy with the +II and +IV standards shows no correlation with the formal oxidation state at either the L3- or L2-edge (Figure S9), demonstrating the dominance of ligand-field over Zeff for excitation from p states. The second series, comprising a set of tris(dithiolene) complexes whose formal oxidation states span +IV to +VI, shows no variation in their white-line energies when plotted against their relevant standards (Figures S10 and S11). The L2-edge energies are the same within experimental error, whereas all L3-edge values are lower than the three standards. This reflects the similar spectroscopic oxidation state for all tris(dithiolene) complexes as revealed by EPR and S K-edge XAS,79,80 in that each member of the electron transfer series is related by predominantly ligand-centered redox steps.81 The electron-withdrawing effect of the cyano substituents in [PPh4]2[WIV(mnt)3] shifts the peak to an energy higher than for other dithiolene types, as has been observed repeatedly in S K-edge spectra.11,14,16,80
L1-Edge
An overlay of the L1-edge spectra for six tungsten compounds spanning the formal oxidation state range of 0 to +VI is presented in Figure 6. As further emphasized with the rising-edge energies listed in Table 3, the spectra are qualitatively similar and ordered according to increasing formal oxidation state but are progressively shifted to higher energy by irregular energy increments of 0.2–1.4 eV. Moreover, an inverse correlation between the L3,2-edge EBR and L1 rising-edge energy is achieved (Figure 5). The correspondence of the rising-edge energy order to the ordering by formal oxidation state is undoubtedly dependent upon juxtaposed members in the series being as alike as possible in the identity and nature of the ligand environment.
Figure 6.
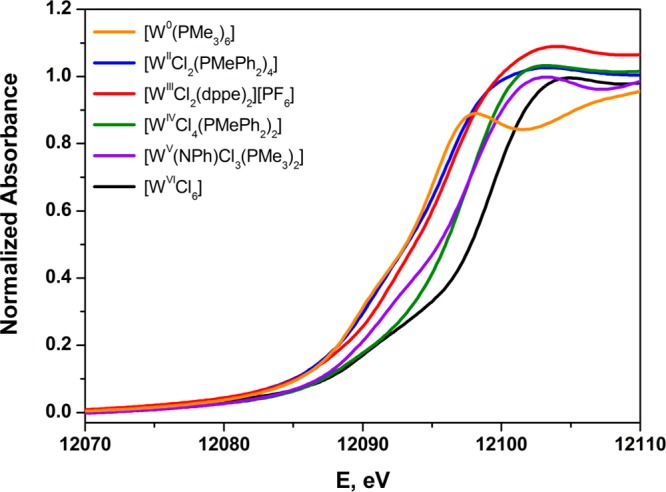
Overlay of the normalized W L1-edge X-ray absorption spectra of the reference compounds spanning formal oxidation states 0 → +VI.
The comparison between [WIICl2(PMePh2)4] and [WIIICl2(dppe)2]1+ is a noteworthy one, as the two compounds are essentially alike in coordination environment and differ only peripherally in the identity of the supporting phosphine ligand. The one-electron transfer that would oxidize [WIICl2(PMePh2)4] to [WIIICl2(PMePh2)4]1+ or reduce [WIIICl2(dppe)2]1+ to [WIICl2(dppe)2] involves a degenerate pair of MOs that is largely (∼72%) tungsten d orbital (dxz,yz) in character (Figure 2). The inference to which the foregoing leads is that a rising-edge energy change of ∼0.5 eV might be generally anticipated for a redox process that is metal-based, or largely so, for a third row transition element. For a first row transition metal, a change of ∼1.0 eV in the K-edge energy is generally associated with a metal-based redox process.11 A somewhat smaller value is plausible for the L1-edge energy for a transition metal inasmuch as the 2s core-hole, being further from the nucleus, is conceivably less sensitive to Zeff than a 1s core-hole.
The degree of the departure that can occur between formal and spectroscopic oxidation state is highlighted in spectacular fashion by the contrast between the L1-edge spectra for [W0(PMe3)6] and [W0(CO)6] (Figure 7). Gauged spectroscopically by L1-edge XAS, [W0(CO)6] bears more resemblance to WV than it does to W0. This strongly perturbing effect of CO has been noted previously in a K-edge XAS study of technetium compounds that included [Tc02(CO)10].82 The spectroscopic difference between [W0(CO)6] and [W0(PMe3)6] accords qualitatively with observation: [W0(CO)6] is a robust species and stable thermally in the open atmosphere for protracted times; [W0(PMe3)6] is extraordinarily reactive, capable of hard-to-effect C–C and C–H insertion reactions in heteroaromatic molecules,83 and prone to cyclometalation to form [WII(PMe3)4(η-CH2PMe2)H] following dissociation of PMe3.53
Figure 7.
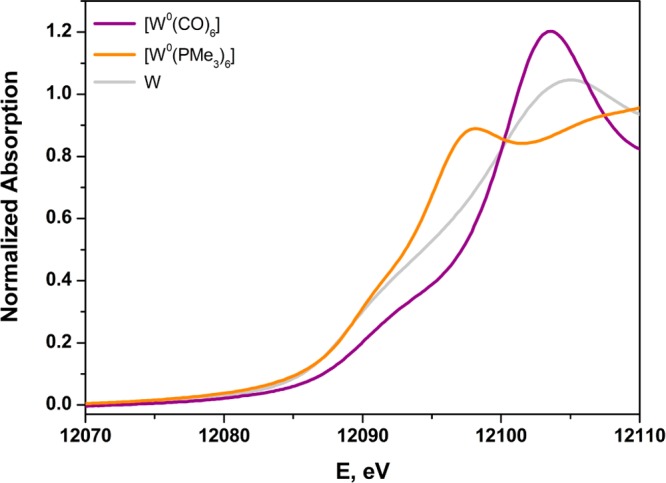
Overlay of the normalized W L1-edge X-ray absorption spectra of [W0(PMe3)6], [W0(CO)6], and W metal.
Although encompassing a broad energy window for formally zerovalent tungsten compounds, [W0(PMe3)6] and [W0(CO)6] are nevertheless useful for establishing limiting values for L1-edge energies. The former is particularly useful for the definition of a low-energy limit. All other common or conceivable zerovalent compounds would fill in a continuum between these two, in some cases with a plausible estimate of the L1-edge energy being possible. For example, [W0(CO)3(triphos)] (triphos = CH3C(CH2PPh2)3) would likely have an L1 rising-edge at the intermediate value of ∼12 098 eV. We note here that an increasing number of CO ligands in a series of related compounds correlates more strongly with a positive shift of the rising-edge than does the formal oxidation state of the metal ion. This point is exemplified in the series [W(dithiolene)n(CO)6–2n] (n = 1–3) whose L1-edge spectra are compared in Figure S48. The lowest energy corresponds to the compound with the highest formal oxidation state and an absence of CO ligands.
The usefulness of the L1-edge XAS data from reference compounds such as the set in Figure 6 is demonstrated by their use as a backdrop for comparison with the L1-edge spectra of metallodithiolene compounds [WIV(mdt)2(CO)2] and [WIV(mdt)2(CN)2]2– (Figure 8). The rising edges of these two metallodithiolene species are similar to one another, suggesting similar Zeff between the two, and roughly comparable to [WIVCl4(PMePh2)2] and [WVCl3(NPh)(PMe3)2] (Figure 5). The pre-edge shoulder is also comparable, given the same trigonal prismatic geometry.20,22 This observation affirms a description of [WIV(mdt)2(CN)2]2– as a WIV species, which is also the conclusion emerging from crystallographic data and a study of its frontier MO composition. The case of [WIV(mdt)2(CO)2], however, is considerably more subtle. Sulfur K-edge XAS data and the S–C and C–C bonds within the dithiolene ligands for both [WIV(mdt)2(CO)2] and [WIV(mdt)2(CN)2]2– indicate appreciably more oxidized sulfur in the former,22 which in the absence of W L-edge XAS data invite the conclusion that its tungsten ion is correspondingly more reduced. However, Figure 8 emphasizes that Zeff in [WIV(mdt)2(CO)2] is at least as high as it is in [WIV(mdt)2(CN)2]2–. Examination of the frontier MOs for [WIV(mdt)2(CO)2] shows not only effective W → CO π-backbonding, which renders tungsten Zeff relatively high, but also a key MO that is dithiolene π-donating and CO π-accepting via the same tungsten d orbital (Figure S49). This MO bears a strong analogy to cis-WIVO(CO) species in which oxo and carbonyl ligands interact in a synergistic way with the same tungsten d orbital as π donor and π acid (Figure S49).84 Thus, tungsten L1-edge XAS complements the S K-edge data, which only provides the dithiolene sulfur contribution to the frontier MOs.
Figure 8.
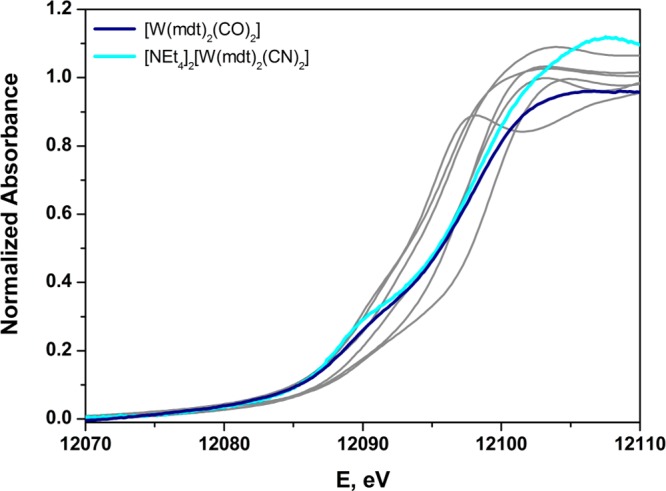
Normalized tungsten L1-edge X-ray absorption spectra of [WIV(mdt)2(CO)2] and [WIV(mdt)2(CN)2]2– overlaid upon the L1 spectra of the reference compounds in Figure 6.
The position of the rising-edge is also a function of the M–L distances in the first coordination sphere of the central ion.85 The distances of the non-dithiolene ancillary ligands from the tungsten ion in complexes of the type [WIV(mdt)2(X)(Y)] (X = Y = CO, PMe3, CNtBu, CN–; X = CO, Y = PMe3; Chart 1) trend as CO < CNtBu < CN– < PMe3,22 which correlates directly with the L1 rising-edge energy; the average W–S bond length remains unchanged across the series. Of course, π-backbonding is also enhanced by a short metal–ligand bond. The W–CO bond lengths in [WIV(mdt)2(CO)2] are shorter than the W–CN bond lengths in [WIV(mdt)2(CN)2]2– by 0.114(6) Å and contribute to its visibly higher energy tungsten L1 rising-edge (Figure 8). Substituting CO for PMe3 shifts the rising-edge to lower energy by the removal of an effective π acceptor and concomitant lengthening of the metal–ligand bond with the larger phosphorus atom. The shift in the L1 rising-edge corresponds directly to the energy of the first peak in the S K-edge spectrum.22
An overlay of two formally WVI compounds (Figure 9) similarly illustrates a difference of rising-edge energies, in this case 2.2 eV. While the separation between [WVICl6] and [WVI(xylidene)3] is less stark than that between [W0(PMe3)6] and [W0(CO)6], the order is opposite that given by the EBR. Although chloride is generally a modest π-donor ligand, the chloride lone pairs in [WVICl6] are situated to form multiple Cl p → W d π-type interactions with each of the metal t2g-type atomic orbitals. It is therefore unsurprising that that formal charge in [WVICl6] is attenuated to the extent that its L1 rising-edge is markedly lower than it is in [WVI(xylidene)3], where no ligand lone pairs are present on the atoms immediately coordinated to tungsten. The higher L1-edge energy for [WVI(xylidene)3] can also be ascribed to the shorter W–C distance of 2.2127(8) Å compared with an average W–Cl bond length of 2.276(2) Å in [WVICl6].86
Figure 9.
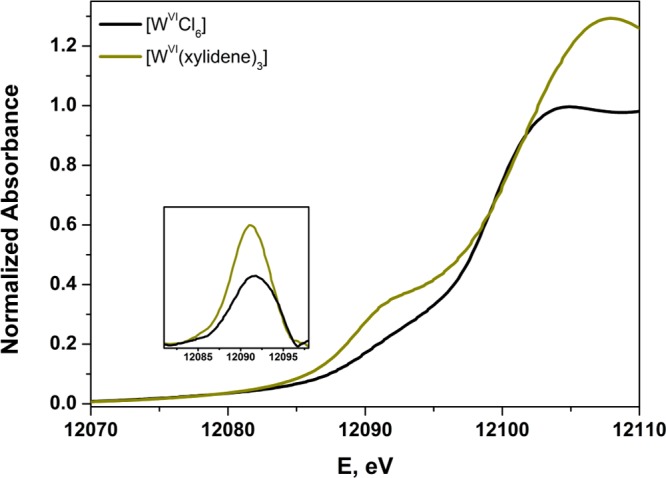
Overlay of the normalized W L1-edge X-ray absorption spectra of [WVICl6] and [WVI(xylidene)3]. Inset shows expansion of the pre-edge peak after subtraction of the rising-edge.
The energy gap between
the pre-edge and rising-edge diminishes
with increasing metal–ligand bond length because the energy
difference between empty metal d orbitals and filled ligand p orbitals
decreases. The pre-edge transition for [WVI(xylidene)3] occurs at slightly lower energy and with appreciably greater
intensity than that for [WVICl6] (Figure 9 inset). Its noncentrosymmetric geometry endows
the otherwise weak quadrupole-allowed 2s → 5d pre-edge excitation
with electric dipole character. For trigonal prismatic [WVI(xylidene)3], the pre-edge feature is defined as the 2s
→ 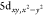 transition,
where DFT computed 2.1% W 6p
character is admixed into this e′ level (LUMO+1 and LUMO+2, Figure S41). The pre-edge peak energy, evaluated
after subtracting the rising-edge structure, shows the transition
in [WVI(xylidene)3] is 0.4 eV lower in energy
than it is for [WVICl6]. This difference is
in keeping with the EBR analysis, which showed the former is slightly
more reduced (vide supra).
transition,
where DFT computed 2.1% W 6p
character is admixed into this e′ level (LUMO+1 and LUMO+2, Figure S41). The pre-edge peak energy, evaluated
after subtracting the rising-edge structure, shows the transition
in [WVI(xylidene)3] is 0.4 eV lower in energy
than it is for [WVICl6]. This difference is
in keeping with the EBR analysis, which showed the former is slightly
more reduced (vide supra).
The impact of three redox-active dithiolene ligands is most apparent comparing the L1-edge position of the neutral tris(dithiolene) complexes — [WVI(bdt)3], [WVI(mdt)3], and [WVI(pdt)3] — with the +VI standard, [WVICl6] (Figure 5). The sulfur-donor species bring about a ∼2.5 eV shift to lower energy commensurate with the now generally accepted notion that tungsten is not in its highest oxidation state.79,87 Each compound forms a three-membered electron transfer series,79 where the rising-edge energy remains unresponsive to the successive addition of one electron to give monoanionic and dianionic species. For both the [W(mdt)3]0/1–/2– and [W(bdt)3]0/1–/2– series, the variation is ±0.1 eV (Table S4), which approaches the precision limit of the experiment. As noted for the L3,2-edges, the electronically unique [WIV(mnt)3]2– is 1 eV higher than the other dianions, as the degree of π donation is attenuated by the conjugated CN substituents. These data indicate that Zeff is the same for each member of the series. Recently, we detailed a spectroscopic and theoretical analysis of EPR-active monoanionic tris(dithiolene) complexes of Mo and W.79 For the Mo compounds there was an obvious difference between the aromatic and olefinic dithiolene ligand, with the former defined as a MoV central ion coordinated by three dianionic dithiolates, while the latter has a MoIV ion and an oxidized tris(dithiolene) ligand set. In the case of tungsten, the division is considerably more ambiguous, as the electronic structure of compounds with olefinic dithiolenes rests somewhere between the two extremes — WIV and WV — due to a second-order Jahn–Teller distortion as described for [WVI(xylidene)3] (vida supra).79 Similarly, all neutral complexes adopt the C3h symmetric paddle-wheel structure where the magnitude of the distortion defines the extent of mixing of metal and ligand orbitals, and therein the Zeff of W. Therefore, the ability to apply an integer oxidation state to any tungsten tris(dithiolene) complex is severely compromised in such strongly covalent systems, as reflected by the uniform rising-edge energies.
The conspicuous pre-edge shoulder in the L1-edge spectrum
of each tris(dithiolene) compound is a spectroscopic marker for complex
geometry. Neutral complexes are noted for their trigonal prismatic
WS6 polyhedron,20,32,79,88 as are the complex ions, [W(mdt)3]1–/2–, but with flat dithiolene
ligands.35,79 Each member of this series displays an equally
intense pre-edge peak (Figure
S50 inset), similar to other trigonal prismatic tris(dithiolene)
species.15,16 The calculated 6p content of the bound 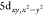 is uniform across
the series
(Table S4).
The analogous [W(bdt)3]0/1–/2– series shows marked differences in the pre-edge intensity (Figure S51 inset).
The degree of p–d mixing increases with decreasing Θ
(Table S4);
the distorted octahedral [WV(bdt)3]1– (Θ = 32.3°) has the weakest pre-edge peak.89 Perhaps the most satisfying demonstration rests
in the comparison of the two [WIV(bdt)3]2– complexes with different counterions. Crystallization
of this dianion with two tetraethylammonium cations yields a highly
trigonal prismatic structure (Θ = 1.9°),90 whose pre-edge intensity is noticeably reduced with tetraphenylarsonium
counterions, as the structure is shifted to distorted octahedral (Θ
= 23.0°).91 The effect of counterions
on the trigonal twist angle has been elegantly detailed in a crystallographic
study of [MIV(bdtCl2)3]2– (M = Mo, W; bdtCl22– = 3,6-dichlorobenzene-1,2-dithiolate),92 where those cocrystallized with tetraethylammonium
cations are more trigonal prismatic.
is uniform across
the series
(Table S4).
The analogous [W(bdt)3]0/1–/2– series shows marked differences in the pre-edge intensity (Figure S51 inset).
The degree of p–d mixing increases with decreasing Θ
(Table S4);
the distorted octahedral [WV(bdt)3]1– (Θ = 32.3°) has the weakest pre-edge peak.89 Perhaps the most satisfying demonstration rests
in the comparison of the two [WIV(bdt)3]2– complexes with different counterions. Crystallization
of this dianion with two tetraethylammonium cations yields a highly
trigonal prismatic structure (Θ = 1.9°),90 whose pre-edge intensity is noticeably reduced with tetraphenylarsonium
counterions, as the structure is shifted to distorted octahedral (Θ
= 23.0°).91 The effect of counterions
on the trigonal twist angle has been elegantly detailed in a crystallographic
study of [MIV(bdtCl2)3]2– (M = Mo, W; bdtCl22– = 3,6-dichlorobenzene-1,2-dithiolate),92 where those cocrystallized with tetraethylammonium
cations are more trigonal prismatic.
Conclusions
The complete L-edge XAS spectra of a broad set of six-coordinate tungsten compounds have been collected, some compounds being selected for use as references and others because they bear redox-active dithiolene ligands and varying degrees of ambiguity. We are unaware of any comparable study for a 5d metal. For third row metals, for which K-edge XAS spectra are inaccessible, it is clear that L-edge XAS has sufficient spectral resolution to probe metal Zeff, when appropriately calibrated. For the reference series employed here, which maintains a rough similarity in ligand environment from one series member to the next, the EBR derived from the relative intensity of the L3,2-edges is anticorrelated with the L1 rising-edge energy. As anticipated, the white-line peak energies at the L3- and L2-edges are independent of Zeff. Our observations regarding the EBR and L1 rising-edge energies demonstrate that these two metrics respond to changes in Zeff for a 5d metal and that the differences are significant as the reference series is traversed from 0 to +VI.
Important contributors to the EBR and rising-edge energy are the metal–ligand bond distances and the π-acid/π-donor character of the ligands, irrespective of the formal oxidation state classification. This point is underscored by [WII(mdt)(CO)4] and [WII(mdt)(CO)2(PMe3)2], where replacement of two strong π-accepting CO ligands with PMe3 shifts the L1 rising-edge 3 eV to lower energy. The position of the L1 rising-edge is strongly influenced by the number of π-acid ligands. Formally zerovalent [W0(CO)6] and [W0(Me2pipdt)(CO)4] have L1 rising-edge energies that are among the highest shown in Figure 5. As expected, the W0 atom in these compounds is depleted of charge by the CO ligands, and therefore the physical oxidation state, insofar as one can be gauged, is comparable to the WV and WVI reference compounds. Here, we suggest that the effort to assess a spectroscopic oxidation state has meaning as an attempt to rationalize or anticipate some property, such as reactivity, not as a general system for classifying molecules.
For the series of bis(dithiolene) compounds of the type [WIV(mdt)2XY] (X = Y = CO, PMe3, CNtBu, CN–; X = CO, Y = PMe3), the EBR and rising-edge energy are consistent with the interpretation of X-ray diffraction and S K-edge XAS studies.22 In the particular case of [WIV(mdt)2(CO)2] and [WIV(mdt)2(CN)2]2–, their comparable tungsten L1 rising-edge energies reveal that the strikingly greater degree of dithiolene ligand oxidation in the former compound, where the ligands appear to be at a radical monoanionic redox level, as compared to the latter compound, where the ligands appear to be fully reduced ene-1,2-dithiolates, is offset by transfer of charge density into the CO ligands with little net effect upon tungsten Zeff. For [WIV(mdt)2(PMe3)2], an increase in the bond length between metal and ancillary ligand and a decrease in π backdonation produce an L1 rising-edge that is ∼1.5 eV lower in energy than that of [WIV(mdt)2(CO)2]. This energy is similar to that found for [WIIICl2(dppe)2]1+, which has a similar array of σ- and π-donating ligands. The 1.743(1) Å S–C and 1.346(3) Å C–Cchelate dithiolene bond distances in [WIV(mdt)2(PMe3)2] are slightly shorter and longer, respectively, than the values typical of fully reduced ene-1,2-dithiolate.22 The covalency between metal and dithiolene implied by these bond lengths, taken with the absence of strongly perturbing π-acid ancillary ligand, makes it understandable that the tungsten ion in [WIV(mdt)2(PMe3)2] appears more reduced than in [WIV(mdt)2(CO)2] and [WIV(mdt)2(CN)2]2– (Figure 5).
The invariance of the EBR, L3-, and L2-edge white-line energy and the L1 rising-edge energy for the tris(dithiolene) electron-transfer series [W(mdt)3]z and [W(bdt)3]z (z = 0, 1–, 2−) align with crystallographic, EPR, and S K-edge XAS data, confirming that each member is related by a ligand-centered redox event. The L1 pre-edge peak intensity correlates with the magnitude of the trigonal twist angle in these compounds. This observation demonstrates that the L1-edge has sufficient resolution to identify molecular geometry both in the solid and solution state, which is analogous to, albeit different from, the diagnostic pre-edge peak for tetrahedral sites in tungsten oxide materials.19 The spectral detail will be improved by using high-energy resolution fluorescence detection XAS, which has enhanced L-edge spectral definition for several third row metals.93 The technique reduces the effect of lifetime broadening, thereby providing a target for time-dependent DFT simulation of the pre-edge and increasing the electronic structure detail obtained from these measurements. Furthermore, it would be advantageous to exploit solution-state studies to assess changes in geometry in the absence of lattice forces.16,81 Specifically for monoanionic tris(dithiolene) compounds, such measurements would provide a crucial link between the crystallographic structure and the putative solution-state geometry based on EPR and DFT results.79
A primary conclusion emerging from this effort is that the L-edge X-ray absorption spectra of molecular systems containing 5d metals offer useful and relatively direct insight into electronic structure from the perspective of what one may consider to be the compound’s most important part — the metal ion. The EBR and L1 rising-edge energy collectively provide information that is different from other physical methods about the state of the metal ion when it is strongly perturbed by π-acid or π-donor ligands and/or coordinated by redox-active ligands. Making the most of any third row metal L-edge XAS data is assisted when other data are available for comparison, and toward that end the EBR and L1 rising-edge values presented here are intended to be useful in establishing whether some tungsten compound of interest has a metal Zeff greater than, less than, or similar to one of the relatively simple calibrant compounds presented in Table 3. With due circumspection for the limitations and uncertainties in the methodology, we emphasize that metal L-edge X-ray absorption spectroscopy is most useful when complemented by data from other physical techniques, not when applied as a stand-alone method.
Acknowledgments
Prof. G. Parkin and Dr. A. Sattler of Columbia University are thanked for generously providing a sample of [W0(PMe3)6] and helpful discussions. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Science. The Louisiana Board of Regents is thanked for Enhancement Grant LEQSF-(2002-03)-ENH-TR-67 with which Tulane’s X-ray diffractometer was purchased, and Tulane University is acknowledged for its ongoing support with operational costs for the diffraction facility. Support from the National Science Foundation (Grant CHE-0845829 to J.P.D.) and the EPSRC National U.K. EPR Facility and Service at The University of Manchester (S.S.) is gratefully acknowledged.
Supporting Information Available
Complete crystallographic data for [WIIICl2(dppe)2][PF6] and [WVI(xylidene)3] in CIF format. Thermal ellipsoid plots of [WVI(xylidene)3] and of [WIIICl2(dppe)2][PF6] cation–anion pairs in the unit cell with complete atom labeling. Multifrequency EPR and magnetic data for [WIIICl2(dppe)2][PF6] and [WV(NPh)Cl3(PMe3)2]. Pseudo-Voigt deconvoluted and second derivative L3,2-edge spectra; comparative plots of L1-edge and first derivative spectra. Computational details and coordinates for optimized structures. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ Department of Chemistry, University of Illinois at Chicago, 845 West Taylor Street, MC 111, Chicago, Illinois, 60607.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Jørgensen C. K.Oxidation Numbers and Oxidation States; Springer: Heidelberg, Germany, 1969. [Google Scholar]
- Hegedus L. S.Transition Metals in the Synthesis of Complex Organic Molecules; University Science Books: Mill Valley, CA, 1994. [Google Scholar]
- Crabtree R. H.The Organometallic Chemistry of the Transition Metals, 3rd ed.; Wiley & Sons: New York, NY, 2001. [Google Scholar]
- Chaudhuri P.; Wieghardt K. Prog. Inorg. Chem. 2001, 50, 151. [DOI] [PubMed] [Google Scholar]
- Palmer J. H. Struct. Bonding Berlin, Ger. 2012, 142, 49. [Google Scholar]
- Palmer J. H.; Lancaster K. M. Inorg. Chem. 2012, 51, 12473. [DOI] [PubMed] [Google Scholar]
- Bart S. C.; Chłopek K.; Bill E.; Bouwkamp M. W.; Lobkovsky E.; Neese F.; Wieghardt K.; Chirik P. J. J. Am. Chem. Soc. 2006, 128, 13901. [DOI] [PubMed] [Google Scholar]
- Nguyen A. I.; Blackmore K. J.; Carter S. M.; Zarkesh R. A.; Heyduk A. F. J. Am. Chem. Soc. 2009, 131, 3307. [DOI] [PubMed] [Google Scholar]
- Heyduk A. F.; Zarkesh R. A.; Nguyen A. I. Inorg. Chem. 2011, 50, 9849. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Hedman B.; Hodgson K. O.; Dey A.; Szilagyi R. K. Coord. Chem. Rev. 2005, 249, 97. [Google Scholar]
- Sproules S.; Wieghardt K. Coord. Chem. Rev. 2011, 255, 837. [Google Scholar]
- Krause M. O.; Oliver J. H. J. Phys. Chem. Ref. Data 1979, 8, 329. [Google Scholar]
- Shulman R. G.; Yafet Y.; Eisenberger P.; Blumberg W. E. Proc. Natl. Acad. Sci. U.S.A. 1976, 73, 1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Banerjee P.; Sproules S.; Weyhermüller T.; DeBeer George S.; Wieghardt K. Inorg. Chem. 2009, 48, 5829. [DOI] [PubMed] [Google Scholar]; b Milsmann C.; Sproules S.; Bill E.; Weyhermüller T.; DeBeer George S.; Wieghardt K. Chem.—Eur. J. 2010, 16, 3628. [DOI] [PubMed] [Google Scholar]
- Sproules S.; Benedito F. L.; Bill E.; Weyhermüller T.; DeBeer George S.; Wieghardt K. Inorg. Chem. 2009, 48, 10926. [DOI] [PubMed] [Google Scholar]
- Sproules S.; Weyhermüller T.; DeBeer S.; Wieghardt K. Inorg. Chem. 2010, 49, 5241. [DOI] [PubMed] [Google Scholar]
- Szilagyi R. K.; Lim B. S.; Glaser T.; Holm R. H.; Hedman B.; Hodgson K. O.; Solomon E. I. J. Am. Chem. Soc. 2003, 125, 9158. [DOI] [PubMed] [Google Scholar]
- a Balerna A.; Bernieri E.; Burattini E.; Kuzmin A.; Lusis A.; Purans J.; Cikmach P. Nucl. Instrum. Methods Phys. Res., Sect. A 1991, 308, 240. [Google Scholar]; b Kuzmin A.; Purans J. J. Phys.: Condens. Matter 1993, 5, 9423. [DOI] [PubMed] [Google Scholar]; c Tougerti A.; Cristol S.; Berrier E.; Briois V.; La Fontaine C.; Villain F.; Joly Y. Phys. Rev. B 2012, 85, 125136. [Google Scholar]; d Uehara Y.; Kawase K.; Tsuchimoto J.; Shibano T. J. Electron Spectrosc. Relat. Phenom. 2005, 148, 75. [Google Scholar]
- a Barton D. G.; Soled S. L.; Meitzner G. D.; Fuentes G. A.; Iglesia E. J. Catal. 1999, 181, 57. [Google Scholar]; b Hilbrig F.; Göbel H. E.; Knözinger H.; Schmelz H.; Lengeler B. J. Phys. Chem. 1991, 95, 6973. [Google Scholar]; c Horsley J. A.; Wachs I. E.; Brown J. M.; Via G. H.; Hardcastle F. D. J. Phys. Chem. 1987, 91, 4014. [Google Scholar]; d Yamamoto T.; Orita A.; Tanaka T. X-Ray Spectrom. 2008, 37, 226. [Google Scholar]
- Chandrasekaran P.; Arumugam K.; Jayarathne U.; Pérez L. M.; Mague J. T.; Donahue J. P. Inorg. Chem. 2009, 48, 2103. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Chandrasekaran P.; Mague J. T.; DeBeer S.; Sproules S.; Donahue J. P. Inorg. Chem. 2012, 51, 346. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Keating C.; Chandrasekaran P.; Jayarathne U.; Mague J. T.; DeBeer S.; Lancaster K. M.; Sproules S.; Rubtsov I. V.; Donahue J. P. Inorg. Chem. 2013, 52, 6743. [DOI] [PubMed] [Google Scholar]
- Cho D.-Y.; Park J.; Yu J.; Park J.-G. J. Phys.: Condens. Matter 2012, 24, 055503. [DOI] [PubMed] [Google Scholar]
- a Daulton T. L.; Little B. J. Ultramicroscopy 2006, 106, 561. [Google Scholar]; b Zhang Z. Ultramicroscopy 2007, 107, 598. [DOI] [PubMed] [Google Scholar]
- a Koshino M.; Kurata H.; Isoda S.; Kobayashi T. Micron 2000, 31, 373. [DOI] [PubMed] [Google Scholar]; b Thole B. T.; van der Laan G. Phys. Rev. B 1988, 38, 3158. [DOI] [PubMed] [Google Scholar]
- Qi B.; Perez I.; Ansari P. H.; Lu F.; Croft M. Phys. Rev. B 1987, 36, 2972. [DOI] [PubMed] [Google Scholar]
- Sharp P. R. Organometallics 1984, 3, 1217. [Google Scholar]
- Over D. E.; Critchlow S. C.; Mayer J. M. Inorg. Chem. 1992, 31, 4643. [Google Scholar]
- Sharp P. R.; Bryan J. C.; Mayer J. M.; Templeton J. L.; Feng S. Inorg. Synth. 1990, 28, 326. [Google Scholar]
- Nielson A. J.; Waters J. M. Aust. J. Chem. 1983, 36, 243. [Google Scholar]
- Lappert M. F.; Raston C. L.; Rowbottom G. L.; Skelton B. W.; White A. H. J. Chem. Soc., Dalton Trans. 1984, 883. [Google Scholar]
- Goddard C. A.; Holm R. H. Inorg. Chem. 1999, 38, 5389. [Google Scholar]
- McCleverty J. A.; Locke J.; Wharton E. J.; Gerloch M. J. Chem. Soc. A 1968, 816. [Google Scholar]
- Sellmann D.; Kern W.; Moll M. J. Chem. Soc., Dalton Trans. 1991, 1733. [Google Scholar]
- Fomitchev D. V.; Lim B. S.; Holm R. H. Inorg. Chem. 2001, 40, 645. [DOI] [PubMed] [Google Scholar]
- Stiefel E. I.; Eisenberg R.; Rosenberg R. C.; Gray H. B. J. Am. Chem. Soc. 1966, 88, 2956. [Google Scholar]
- APEX2, Version 2009.11–0; Bruker-AXS, Inc.: Madison, WI, 2009. [Google Scholar]
- SAINT, Version 7.68A; Bruker-AXS, Inc.: Madison, WI, 2009. [Google Scholar]
- Sheldrick G. M.SADABS, Version 2008/2; Universität Göttingen: Göttingen, Germany, 2008. [Google Scholar]
- Sheldrick G. M.SHELXS-97, Universität Göttingen: Göttingen, Germany, 2008. [Google Scholar]
- Sheldrick G. M.SHELXL-97, Universität Göttingen: Göttingen, Germany, 2008. [Google Scholar]
- SHELXTL, Version 2008/4; Bruker-AXS, Inc.: Madison, WI, 2008. [Google Scholar]
- George G. N.EXAFSPAK & EDG_FIT; Stanford Synchrotron Radiation Laboratory, Stanford Linear Accelerator Center, Stanford University: Stanford, CA, 2000. [Google Scholar]
- Hanson G. R.; Gates K. E.; Noble C. J.; Griffin M.; Mitchell A.; Benson S. J. Inorg. Biochem. 2004, 98, 903. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam N. J.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09; Gaussian, Inc., Wallingford, CT, 2009. [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Phys. Rev. Lett. 1996, 77, 3865. [DOI] [PubMed] [Google Scholar]
- a Becke A. D. J. Chem. Phys. 1993, 98, 5648. [Google Scholar]; b Lee C. T.; Yang W. T.; Parr R. G. Phys. Rev. B 1988, 37, 785. [DOI] [PubMed] [Google Scholar]
- https://bse.pnl.gov/bse/portal (accessed November 15, 2013).
- Schäfer A.; Horn H.; Ahlrichs R. J. Chem. Phys. 1992, 97, 2571. [Google Scholar]
- Tenderholt A. L.QMForge: A Program to Analyze Quantum Chemistry Calculations, Version 2.1; http://qmforge.sourceforge.net (accessed November 15, 2013).
- Chemcraft, Version 1.6; http://chemcraftprog.com (accessed November 15, 2013).
- Jmol: an open source Java viewer for chemical structures in 3D. http://jmol.sourceforge.net (accessed November 15, 2013).
- a Rabinovich D.; Parkin G. J. Am. Chem. Soc. 1990, 112, 5381. [Google Scholar]; b Rabinovich D.; Zelman R.; Parkin G. J. Am. Chem. Soc. 1992, 114, 4611. [Google Scholar]
- Schaefer King M. A.; McCarley R. E. Inorg. Chem. 1973, 12, 1972. [Google Scholar]
- Blight D. G.; Kepert D. L. J. Chem. Soc. A 1968, 534. [Google Scholar]
- Boorman P. M.; Greenwood N. N.; Hildon M. A. J. Chem. Soc. A 1968, 2466. [Google Scholar]
- Jayarathne U.; Chandrasekaran P.; Jacobsen H.; Mague J. T.; Donahue J. P. Dalton Trans. 2010, 39, 9662. [DOI] [PubMed] [Google Scholar]
- Imaeda M.; Nishihara H.; Nakano K.; Ichida H.; Kobayashi A.; Saito T.; Sasaki Y. Inorg. Chem. 1985, 24, 1246. [Google Scholar]
- Filippou A. C.; Schnakenburg G.; Philippopoulos A. I. Acta. Crystallogr., Sect. E: Struct. Rep. Online 2003, 59, m602. [Google Scholar]
- Taylor J. R.An Introduction to Error Analysis; University Science Books: Sausalito, CA, 1997. [Google Scholar]
- Mabbs F. E.; Collison D.. Electron Paramagnetic Resonance of d Transition Metal Compounds; Elsevier: Amsterdam, 1992. [Google Scholar]
- a Adams C. J.; Anderson K. M.; Connelly N. G.; Harding D. J.; Hayward O. D.; Orpen A. G.; Patrón E.; Rieger P. H. Dalton Trans. 2009, 530. [DOI] [PubMed] [Google Scholar]; b Ceausescu E.; Cornilescu A.; Nicolescu E.; Popescu M.; Coca S.; Belloiu C.; Oprescu C.; Dimonie M.; Hubca G.; Dragutan V.; Chipara M. J. Mol. Catal. 1985, 28, 351. [Google Scholar]
- Yoon K.; Parkin G.; Rheingold A. L. J. Am. Chem. Soc. 1991, 113, 1437. [Google Scholar]
- Nielson A. J.; Boyd P. D. W.; Clark G. R.; Hunt P. A.; Hursthouse M. B.; Metson J. B.; Rickard C. E. F.; Schwerdtfeger P. A. J. Chem. Soc., Dalton Trans. 1995, 1153. [Google Scholar]
- Levason W.; McAuliffe C. A.; McCullough F. P. Jr. Inorg. Chem. 1977, 16, 2911. [Google Scholar]
- Nielson A. J.; Metson J. B. Polyhedron 2012, 31, 143. [Google Scholar]
- Horsley J. A. J. Chem. Phys. 1982, 76, 1451. [Google Scholar]
- Wang H.; Ge P.; Riordan C. G.; Brooker S.; Woomer C. G.; Collins T.; Melendres C. A.; Graudejus O.; Bartlett N.; Cramer S. P. J. Phys. Chem. B 1998, 102, 8343. [Google Scholar]
- Wei P. S. P.; Lytle F. W. Phys. Rev. B 1979, 19, 679. [Google Scholar]
- a Drube W.; Treusch R.; Sham T. K.; Bzowski A.; Soldatov A. V. Phys. Rev. B 1998, 58, 6871. [Google Scholar]; b Stöhr J. J. Electron Spectrosc. Relat. Phenom. 1995, 75, 253. [Google Scholar]
- Choi Y. G. Met. Mater. Int. 2009, 15, 993. [Google Scholar]
- Kosog B.; La Pierre H. S.; Denecke M. A.; Heinemann F. W.; Meyer K. Inorg. Chem. 2012, 51, 7940. [DOI] [PubMed] [Google Scholar]
- a Shimizu K.; Oda T.; Sakamoto Y.; Kamiya Y.; Yoshida H.; Satsuma A. Appl. Catal., B 2012, 111–112, 509. [Google Scholar]; b Yoshida H.; Nonoyama S.; Yazawa Y.; Hattori T. Phys. Scr. 2005, T115, 813. [Google Scholar]
- a de Groot F. Coord. Chem. Rev. 2005, 249, 31. [Google Scholar]; b de Groot F. M. F.; Hu Z. W.; Lopez M. F.; Kaindl G.; Guillot F.; Tronc M. J. Chem. Phys. 1994, 101, 6570. [Google Scholar]; c Sham T. K. J. Am. Chem. Soc. 1983, 105, 2269. [Google Scholar]
- a Keast V. J.; Scott A. J.; Brydson R.; Williams D. B.; Bruley J. J. Microscopy 2001, 203, 135. [DOI] [PubMed] [Google Scholar]; b Leapman R. D.; Grunes L. A. Phys. Rev. Lett. 1980, 45, 397. [Google Scholar]; c Pearson D. H.; Ahn C. C.; Fultz B. Phys. Rev. B 1993, 47, 8471. [DOI] [PubMed] [Google Scholar]; d Sparrow T. G.; Williams B. G.; Rao C. N. R.; Thomas J. M. Chem. Phys. Lett. 1984, 108, 547. [Google Scholar]
- a Galyer A. L.; Wilkinson G. J. Chem. Soc., Dalton Trans. 1976, 2235. [Google Scholar]; b Galyer L.; Mertis K.; Wilkinson G. J. Organomet. Chem. 1975, 85, C37. [Google Scholar]
- Bristow G. S.; Lappert M. F.; Martin T. R.; Atwood J. L.; Hunter W. F. J. Chem. Soc., Dalton Trans. 1984, 399. [Google Scholar]
- Campbell S.; Harris S. Inorg. Chem. 1996, 35, 3285. [DOI] [PubMed] [Google Scholar]
- Sproules S.; Banerjee P.; Weyhermüller T.; Yan Y.; Donahue J. P.; Wieghardt K. Inorg. Chem. 2011, 50, 7106. [DOI] [PubMed] [Google Scholar]
- Sproules S.; Weyhermüller T.; Goddard R.; Wieghardt K. Inorg. Chem. 2011, 50, 12623. [DOI] [PubMed] [Google Scholar]
- Sproules S. Prog. Inorg. Chem. 2014, 58, 1. [Google Scholar]
- Almahamid I.; Bryan J. C.; Bucher J. J.; Burrell A. K.; Edelstein N. M.; Hudson E. A.; Kaltsoyannis N.; Lukens W. W.; Shuh D. K.; Nitsche H.; Reich T. Inorg. Chem. 1995, 34, 193. [Google Scholar]
- Sattler A.; Parkin G. Nature 2010, 463, 523. [DOI] [PubMed] [Google Scholar]
- a Thomas S.; Tiekink E. R. T.; Young C. G. Organometallics 1996, 15, 2428. [Google Scholar]; b Thomas S.; Tiekink E. R. T.; Young C. G. Inorg. Chem. 2006, 45, 352. [DOI] [PubMed] [Google Scholar]
- Kutzler F. W.; Natoli C. R.; Misemer D. K.; Doniach S.; Hodgson K. O. J. Chem. Phys. 1980, 73, 3274. [Google Scholar]
- Cotton F. A.; Kibala P. A.; Sandor R. B. W. Acta Crystallogr., Sect. C 1989, 45, 1287. [Google Scholar]
- Kapre R. R.; Bothe E.; Weyhermüller T.; DeBeer George S.; Wieghardt K. Inorg. Chem. 2007, 46, 5642. [DOI] [PubMed] [Google Scholar]
- Huynh H.; Lügger T.; Hahn F. E. Eur. J. Inorg. Chem. 2002, 3007. [Google Scholar]
- Burrow T. E.; Morris R. H.; Hills A.; Hughes D. L.; Richards R. L. Acta Crystallogr., Sect C 1993, 49, 1591. [Google Scholar]
- Lorber C.; Donahue J. P.; Goddard C. A.; Nordlander E.; Holm R. H. J. Am. Chem. Soc. 1998, 120, 8102. [Google Scholar]
- Knoch F.; Sellmann D.; Kern W. Z. Kristallogr. 1993, 205, 300. [Google Scholar]
- Sugimoto H.; Furukawa Y.; Tarumizu M.; Miyake H.; Tanaka K.; Tsukube H. Eur. J. Inorg. Chem. 2005, 3088. [Google Scholar]
- a Glatzel P.; Singh J.; Kvashnina K. O.; van Bokhoven J. A. J. Am. Chem. Soc. 2010, 132, 2555. [DOI] [PubMed] [Google Scholar]; b Hämäläinen K.; Siddons D. P.; Hastings J. B.; Berman L. E. Phys. Rev. Lett. 1991, 67, 2850. [DOI] [PubMed] [Google Scholar]; c Makosch M.; Kartusch C.; Sá J.; Bessa Duarte R.; van Bokhoven J. A.; Kvashnina K.; Glatzel P.; Fernandes D. L. A.; Nachtegaal M.; Kleymenov E.; Szlachetko J.; Neuhold B.; Hungerbühler K. Phys. Chem. Chem. Phys. 2012, 14, 2164. [DOI] [PubMed] [Google Scholar]; d Safonova O. V.; Tromp M.; van Bokhoven J. A.; de Groot F. M. F.; Evans J.; Glatzel P. J. Phys. Chem. B 2006, 110, 16162. [DOI] [PubMed] [Google Scholar]; e Swarbrick J. C.; Skyllberg U.; Karlsson T.; Glatzel P. Inorg. Chem. 2009, 48, 10748. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.