

Background: It is crucial to identify caspase-3 substrates and their roles during neurodegeneration.
Results: Our approach identified 46 novel substrates. Furthermore, we found that anamorsin was cleaved by caspase-3 and mapped its cleavage site.
Conclusion: Anamorsin may play an antiapoptotic role in the central nervous system.
Significance: Our findings contribute to understanding the molecular mechanism underlying role for anamorsin in caspase-3-mediated cell death.
Keywords: Apoptosis, Caspase, Neurodegeneration, Neurotoxin, Proteomics
Abstract
Activated caspases play a central role in the execution of apoptosis by cleaving endogenous substrates. Here, we developed a high throughput screening method to identify novel substrates for caspase-3 in a neuronal cell line. Critical steps in our strategy consist of two-dimensional electrophoresis-based protein separation and in vitro caspase-3 incubation of immobilized proteins to sort out direct substrates. Among 46 putative substrates identified in MN9D neuronal cells, we further evaluated whether caspase-3-mediated cleavage of anamorsin, a recently recognized cell death-defying factor in hematopoiesis, is a general feature of apoptosis. In vitro and cell-based cleavage assays indicated that anamorsin was specifically cleaved by caspase-3 but not by other caspases, generating 25- and 10-kDa fragments. Thus, in apoptosis of neuronal and non-neuronal cells induced by various stimuli including staurosporine, etoposide, or 6-hydroxydopamine, the cleavage of anamorsin was found to be blocked in the presence of caspase inhibitor. Among four tetrapeptide consensus DXXD motifs existing in anamorsin, we mapped a specific cleavage site for caspase-3 at DSVD209↓L. Intriguingly, the 25-kDa cleaved fragment of anamorsin was also detected in post-mortem brains of Alzheimer and Parkinson disease patients. Although the RNA interference-mediated knockdown of anamorsin rendered neuronal cells more vulnerable to staurosporine treatment, reintroduction of full-length anamorsin into an anamorsin knock-out stromal cell line made cells resistant to staurosporine-induced caspase activation, indicating the antiapoptotic function of anamorsin. Taken together, our approach seems to be effective to identify novel substrates for caspases and has the potential to provide meaningful insights into newly identified substrates involved in neurodegenerative processes.
Introduction
Apoptosis is an active cell death process that occurs in development, homeostasis, and pathologic conditions of multicellular organisms (1). Apoptosis is controlled by a diverse range of internal and external signals encompassing hormone, growth factor, death ligands, reactive oxygen species, DNA damage, and hypoxia. Caspases, highly conserved cysteine-aspartate proteases, play an essential role in the transduction of apoptotic signaling pathways (2). There are two types of caspases: initiator caspases and effector caspases. Whereas initiator caspases specifically cleave proforms of inactive effector caspases, thereby activating them, effector caspases cleave cellular substrates, catalyzing diverse sets of biological phenomena (3). For example, changes such as DNA fragmentation (4) and membrane blebbing (5, 6) are mediated by caspase-3-induced cleavages of inhibitor of caspase-activated DNase and Rho-associated kinase (ROCK1), respectively.
To delineate the apoptotic pathway, the identification of caspase substrates is of great interest. To date, ∼1000 human protein substrates have been reported to be cleaved by caspases (7, 8). Various strategies are currently used to identify putative caspase substrates including the expression cloning strategy (9), yeast two-hybrid method (10), differential two-dimensional electrophoresis methods (11–13), diagonal gel proteomic approach (14, 15), and recent gel-free proteomic techniques (16, 17). Numerous studies have used gel-based high throughput technologies or gel-free proteomics in conjunction with mass spectrometry to identify putative caspase substrates in large scale (3). In these methods, analytical specimens are prepared in two different ways. In the first approach, cells are challenged to induce caspase activation (a forward approach), and in the second approach, cellular lysates from non-treated cells are incubated with a caspase of interest (a reverse approach). Considering the numbers of identified substrates (ranging from tens to over a few hundreds), both the forward and reverse approaches seem to be effective to identify putative caspase substrates in high throughput in diverse cell types. However, the possibility cannot be ruled out that the resulting cleavage products are not direct substrates of caspases but may be generated as a consequence of sequential or combinatorial action of caspases with other proteases that are activated downstream or upstream of caspases.
Although elevated caspase activity has been demonstrated in several neurodegenerative diseases including Alzheimer disease (AD),4 Parkinson disease (PD), and stroke (18–20), few attempts have been systematically made to identify potentially neuronal cell type-specific caspase substrates and/or disease progression-related caspase substrates. We therefore attempted to develop a novel high throughput substrate screening method to find caspase-3 substrates that are potentially related to neurodegenerative diseases. To identify substrates directly cleaved by caspase-3, we developed a gel-based protease proteomic approach in which proteins on immobilized isoelectric focusing (IEF) strips were incubated with exogenously added recombinant caspase-3. With this method, we identified 46 putative caspase-3 substrates in MN9D neuronal cells. Among the putative substrates identified, we further characterized anamorsin as a novel caspase-3 substrate and mapped its cleavage site sequence at DSVD209↓L. Intriguingly, caspase-3-mediated cleavage of anamorsin was detected in experimental models of neurodegeneration as well as in post-mortem brains of AD and PD patients. The study using anamorsin knock-out (KO) fetal liver stromal cells overexpressing the full-length anamorsin or either of two fragments indicated that full-length anamorsin may act as a novel antiapoptotic protein in the central nervous system, and its caspase-3-mediated cleavage may represent a loss-of-function phenotype. In sum, the present study suggests that our approach is effective to identify novel caspase-3 substrates and its associated cellular function in neurodegeneration.
EXPERIMENTAL PROCEDURES
Cell Culture, Drug Treatment, Transfection and Cell Viability
MN9D neuronal cells were cultivated as we described previously (21). U-2 OS cells were purchased from ATCC (Manassas, VA) and cultivated in DMEM (Sigma) supplemented with 10% heat-inactivated FBS (Invitrogen) in an atmosphere of 5% CO2 at 37 °C. For drug treatments, MN9D cells or U-2 OS cells were treated for the indicated time periods with 100 μm 6-hydroxydopamine (Sigma), 50 μm 1-methyl-4-phenylpyridinium (MPP+; Sigma), or 1 μm staurosporine in N2 serum-free medium or 50 μm etoposide (Sigma) in DMEM. In some cases, cells were co-treated with 100 μm N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (Z-VAD-fmk; MP Biomedicals, Eschwege, Germany). Fetal liver stromal cell lines were prepared as described previously (22, 23). Briefly, fetal liver stromal cells derived from wild-type or anamorsin-deficient E14.5 murine embryos were inoculated onto 0.1% gelatin-coated plastic dishes and allowed to proliferate for 2 days. Cell lines were established by transfection with an expression plasmid encoding simian virus 40 (SV40) large T antigen to the primary fetal liver stromal cells and then cloned by limiting dilution. Each cloned cell line was named as either FW (wild-type anamorsin) or FK (anamorsin-deficient). Cell lines were propagated over 30 times by splitting them 1:3. MN9D cells and mouse fetal liver cells were transiently transfected with the indicated vectors encoding FLAG- or V5-tagged wild-type or mutant anamorsin using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Briefly, MN9D cells or mouse fetal liver cells were seeded at a density of 1 × 106 cells or 5 × 105 cells on P-100 culture dishes, respectively. One day after plating, transfection was performed with 24 μg of vectors for MN9D cells or 10 μg of vectors for mouse fetal liver cells. Twenty-four hours after transfection, cells were used for experiments. The rate of cell survival was determined by the colorimetric measurement using the MTT reduction assay (24).
Primary Cultures of Cortical Neurons, Lentivirus-mediated RNA Interference, and Cell Viability
Primary cultures of cortical neurons were prepared from gestational day 14.5 mouse embryos. Briefly, dissociated cells were plated onto 6- or 24-well culture plates coated with 50 μg/ml poly-l-lysine (Sigma) and 1 μg/ml laminin (Invitrogen). Cultures were cultivated at 37 °C in a humidified 5% CO2 incubator in DMEM supplemented with 2 mm glutamine (Sigma), 5% fetal bovine serum (Hyclone), and 5% horse serum (Invitrogen). Three days after plating, 10 μm cytosine arabinoside was added to halt the growth of non-neuronal dividing cells. Cultures were used for experiments at 7 days in vitro. Under these culture conditions, more than 95% of cells were neurons. For transduction into primary cultures of cortical neurons, lentiviral vectors (in pLKO.1) containing anamorsin shRNA sequences (1, TRCN0000191007: CCGGCTTGAGTATTTATGCTGAGTACTCGAGTACTCAGCATAAATACTCAAGTTTTTTG; 2, TRCN0000189757: CCGGGACAAGTCATCTCCTGAGGAACTCGAGTTCCTCAGGAGATGACTTGTCTTTTTTG) and non-target shRNA control vector (shScrambled; SHC016) were purchased from Sigma. The lentivirus particles were generated by co-transfection of HEK293T cell with the above mentioned lentiviral vectors and three plasmids (pMDLg/pRRE, pMD2.G, and pRSV-Rev; all from Addgene). Lipofectamine 2000 was used for transfection as recommended by the manufacturer (Invitrogen). Two days after transfection, culture media were filtered using a 0.45-μm filter (Millipore). Primary cultures of cortical neurons at 7 days in vitro were exposed to lentiviral particles and incubated for an additional 4 days. Cultures were then treated with or without 200 nm staurosporine for 24 h. Anti-cell death effects of anamorsin were determined by immunoblot analysis for the cleaved caspase-3 and the MTT reduction assay. For the MTT reduction assay, cells cultured in 24-well culture plates and exposed to drug were subjected to a final concentration of 1 mg/ml MTT solution. Viability was expressed as a percentage over the untreated matched control cells (100%).
Caspase-3 Substrate Screening
Two-dimensional electrophoresis was carried out as described previously with some modifications (21). All chemicals used for two-dimensional electrophoresis were purchased from Sigma unless stated otherwise. Briefly, MN9D cells were solubilized in a 1× sample buffer containing 5 m urea, 2 m thiourea, 2% CHAPS, 0.25% Tween 20, 100 mm DTT, 10% isopropanol, 12.5% water-saturated butanol, 5% glycerol, and 1% immobilized pH gradient buffer (pH 4–7 linear Immobiline DryStrip, GE Healthcare). Protein concentrations of samples were measured by a 2D Quant kit (GE Healthcare) as recommended by the manufacturer. Prior to IEF, Immobiline DryStrips (24 cm, pI 4–7 linear; GE Healthcare) were rehydrated in a sample buffer containing 1.5 mg of cellular lysates. The gels were run for a total of 100 kV-h using progressively increasing voltage on an Ettan IPGphor (GE Healthcare). After IEF, the strips were washed three times with distilled water and then incubated with or without 25 μg of recombinant human caspase-3 in 5 ml of caspase-3 activation buffer (100 mm HEPES, 4 m NaCl, 0.5 m EDTA, 0.5 m EGTA, 2 m MgCl2). Recombinant human caspase-3 was prepared and purified as basically described by us (25–27). Prior to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), the strips were washed three times with distilled water followed by an equilibration step. SDS-PAGE was performed on an 8–18% gradient gel in an Ettan Dalt II System (GE Healthcare). Gels were then stained with 0.1% Coomassie Brilliant Blue G-250. The stained gels were scanned using a densitometer (Powerlook 2100XL, UMAX), and the gel images were analyzed by using the ProteomWeaver software system (Bio-Rad). For in-gel digestion, the protein spots of interest were manually excised from the two-dimensional electrophoresis gels. Gel slices were washed with a buffer containing 25 mm NH4HCO3 and 50% acetonitrile, dried completely using a SpeedVac evaporator (BioTron, Seoul, Korea), and digested with 10 μg/ml trypsin (Promega, Madison, WI) in 25 mm NH4HCO3 at 37 °C for 18 h. After peptides were solubilized with 0.1% trifluoroacetic acid (TFA), the peptide mixtures were desalted using a home-made column packed with C18 porus beads (Invitrogen). Subsequently, the bound peptides were eluted in 0.6 μl of elution buffer (1 mg/ml α-cyano-4-hydroxycinnamic acid solution in 60% acetonitrile, 0.1% TFA) and spotted onto a matrix-assisted laser desorption/ionization (MALDI) plate (Invitrogen). MALDI time-of-flight (TOF) mass spectra were acquired on a 4700 Proteomics Analyzer (Invitrogen). The peptide masses were matched with the theoretical peptide masses of all proteins from all species using the NCBI or Swiss-Prot database.
Construction of Vectors
Human anamorsin cDNA (BC002568) was purchased from ImaGenes (Berlin, Germany). Using this plasmid as a template, FLAG-tagged human anamorsin (wild type), N-terminal fragment (NT; amino acids 1–211), and C-terminal fragment (CT; amino acids 212–312) were generated by a standard PCR method with the primers 5′-CTCTCGAGATGGACTATAAGGACGATGATGACAAGATGGCAGATTTTGGG-3′ and 5′-CCTCTAGACTAGGCATCATGAAGATTGCTATC-3′ (wild type (WT)), 5′-CTCTCGAGATGGACTATAAGGACGATGATGACAAGATGGCAGATTTTGGG-3′ and 5′-CCTCTAGACTAATCCATGCTGTCGTCCTCC-3′ (NT), and 5′-CTCTCGAGATGGACTATAAGGACGATGATGACAAGCTCATTGACTCAGATG-3′ and 5′-CCTCTAGACTAGGCATCATGAAGATTGCTATC-3′ (CT) and subcloned into the pCI-Neo vector (Promega). V5-tagged mouse anamorsin was amplified by PCR with the primers 5′-CCGGTACCATGGAGGAGTTTGGGATC-3′ and 5′-CCTCTAGAGGCATCCTGGAGATTGCTATTG-3′ and subcloned into the pcDNA3.1/V5-His A type vector (Invitrogen). The pcDNA3.1/V5-His A vectors containing calponin-3, emerin, thymidylate synthase, or TDP43 were purchased from Cosmogenetech (Seoul, Korea). All constructs were verified by DNA sequencing. Based on search results for putative caspase-3 cleavage sites, pcDNA3.1/V5-His A vectors encoding D205A, D209A, D212A, or D221A anamorsin point mutant were made with the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer's instructions with the following primer sets: 5′-CTCAGCAAATGACATGGAGGCTGACAGTGTGGATCTCATTG-3′ and 5′-CAATGAGATCCACACTGTCAGCCTCCATGTCATTTGCTGAG-3′ (D205A), 5′-GGAGGATGACAGTGTGGCTCTCATTGACTCAGACG-3′ and 5′-CGTCTGAGTCAATGAGAGCCACACTGTCATCCTCC-3′ (D209A), 5′-CAGTGTGGATCTCATTGCCTCAGACGAGCTGCTTG-3′ and 5′-CAAGCAGCTCGTCTGAGGCAATGAGATCCACACTG-3′ (D212A), and 5′-GCTGCTTGATCCAGAGGCTTTGAAGAGGCCTGACC-3′ and 5′-GGTCAGGCCTCTTCAAAGCCTCTGGATCAAGCAGC-3′ (D221A). All constructs were confirmed by DNA sequencing. pRNAT-U6.1/Neo vector containing shRNA sequence against anamorsin was purchase from GenScript (Piscataway, NJ). The shRNA sequence against anamorsin is 5′-GGATCCCGGATGACAGTGTGGATCTCATTTGATATCCGATGAGATCCACACTGTCATCCTTTTTTCCAAAAGCTT-3′. pRNAT-U6.1/Neo vector containing shRNA sequence against firefly luciferase was used as control shRNA. The shRNA sequence against firefly luciferase is 5′-GGATCCCGCTTACGCTGAGTACTTCGATTGATATCCGTCGAAGTACTCAGCGTAAGTTTTTCCAAAAGCTT-3′.
In Vitro Caspase Cleavage Assay
For in vitro caspase-3 cleavage assays, vectors harboring the indicated sequences were transcribed and translated in the presence of [35S]methionine (PerkinElmer Life Sciences) by using the TNT coupled reticulocyte lysate system (Promega) according to the manufacturer's instructions. The resulting reactant (9 μl) was incubated with 50 ng of recombinant caspase-3 in the presence or absence of 50 μm Z-VAD-fmk (a pan-caspase inhibitor) in 20 μl of caspase reaction buffer (50 mm HEPES, 50 mm NaCl, 0.1% CHAPS, 10 mm EDTA, 5% glycerol, 10 mm dithiothreitol, pH 7.2) for 1.5 h at 37 °C (28). To examine the specificity of caspase-3-mediated cleavage, the metabolically labeled mouse anamorsin wild type was incubated with 50 ng of recombinant human caspase-2, -7, and -8 (R&D Systems, Minneapolis, MN) or recombinant human caspase-6 and -9 (Enzo Life Sciences, Plymouth Meeting, PA). Reactions were stopped by adding 5 μl of 5× sample buffer (250 mm Tris-HCl, 500 mm dithiothreitol, 10% SDS, 0.5% bromphenol blue, 50% glycerol, pH 6.8) followed by boiling for 5 min. Subsequently, reactants were separated by SDS-PAGE and subjected to autoradiography.
Antibodies and Immunoblot Analysis
Rat monoclonal anti-anamorsin antibody (KM3052; 1:30,000) recognizing the N-terminal region of anamorsin was used as reported previously by us (29). Primary antibodies used were HRP-conjugated anti-FLAG antibody (Sigma; 1:5000), anti-V5 antibody (Invitrogen; 1:5000), and anti-cleaved caspase-3 (Cell Signaling Technology, Boston, MA; 1:1000). Anti-GAPDH antibody (Millipore, Billerica, MA; 1:5000) and anti-actin antibody (Sigma; 1:5000) were used as loading controls. Following drug treatment, cells were lysed, and the resulting cell lysates were subjected to immunoblot analyses as described previously (30). Briefly, cells were washed with chilled PBS containing 2 mm EDTA and lysed on ice in radioimmune precipitation assay buffer (50 mm Tris, pH 7.0, 2 mm EDTA, 1.0% Triton X-100) with a protease inhibitor mixture (Roche Applied Science). Subsequently, cell lysates were homogenized using a 1-ml syringe with a 26-gauge needle attached. Protein contents were measured using a Bio-Rad protein assay kit. An equal amount of soluble proteins was separated by SDS-PAGE, blotted onto prewetted PVDF membranes, and processed for immunoblot analysis as we described previously (31). The HRP-conjugated secondary antibodies were goat anti-rabbit and anti-mouse IgG (Santa Cruz Biotechnology, Santa Cruz, CA; 1:5000). Enhanced chemiluminescence (ECL; PerkinElmer Life Sciences) was used to detect specific bands. The relative band intensity was measured using ImageJ imaging software (National Institutes of Health, Bethesda, MD).
Tissues
Autopsy brain tissue from the cortex of nine neuropathologically confirmed AD cases and four age-matched cases with no AD changes or any other neurodegenerative condition were provided by Boston University Alzheimer's Disease Center (Dr. Ann C. McKee). Tissue lysates of these post-mortem brains were obtained from Dr. Gwag at Ajou University (32). The substantia nigra of four PD cases and four age-matched controls were provided by the Department of Pathology, The Johns Hopkins University and processed for immunoblot analysis as described previously (33). Each brain underwent exhaustive neuropathological analysis. The substantia nigra of post-mortem human brain was homogenized in lysis buffer (10 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm EDTA, 0.5% Nonidet P-40, 10 mm sodium β-glycerophosphate, Phosphate Inhibitor Mixtures I and II (Sigma), Complete protease inhibitor mixture (Roche Applied Science)). After homogenization (Diax 900 homogenizer), samples were rotated at 4 °C for 30 min to complete lysis, then the homogenate was centrifuged at 14,400 rpm for 30 min, and the supernatant was collected. Protein levels were quantified using the BCA kit (Pierce) with BSA standards. Samples were subjected to immunoblot analyses using the indicated antibodies.
Statistics
Significant differences among groups were determined by Student's t tests. An unpaired parametric t test with Welch correction was used for comparison of data sets from human brain samples. A value of p <0.05 was considered statistically significant.
RESULTS
Identification of Putative Caspase-3 Substrates
Previously, the p75 subunit of complex I of the electron transport chain was identified as a substrate of caspase-3 by using a diagonal gel proteomic approach (15). Using the same approach, Shao et al. (14) successfully identified 41 putative substrates of caspase-1 including a series of proteins involved in the glycolysis pathway. This systematic approach to identify novel substrates is based on an assumption that the applied caspases can penetrate into the acrylamide gel and effectively cleave their potential substrates. In the present study, we further extended the diagonal gel proteomic approach to develop a modified gel-based protease proteomic analysis (see the schematic depiction in Fig. 1). After the IEF step, the resulting immobilized pH gradient strips were incubated with recombinant human caspase-3. By doing so, potential substrates on an immobilized pH gradient strip were cleaved directly by caspase-3 to generate its cryptic fragments. Because the subsequent SDS-PAGE step separates proteins by their molecular masses, the fragment(s) of caspase-3 substrates migrates faster than a full-length, uncleaved protein(s). Mass spectral analysis was carried out to identify any protein spots that decreased or newly appeared after caspase-3 treatment. Using MN9D neuronal cells, we performed this gel-based protease proteomic technique to identify novel caspase-3 substrates that may be involved in the regulation of neurodegenerative processes. As expected, many protein spots were degraded, and new protein spots with smaller molecular sizes appeared following incubation with recombinant human caspase-3. By densitometric analysis of gel images using ProteomWeaver, we selected 70 uncleaved, full-length protein spots (Fig. 1, colored in yellow) for mass spectrometry and identified 46 putative caspase-3 substrates with an expression level altered by at least 2-fold (n > 3). The putative caspase-3 substrates identified are listed in Table 1. Protein spots that were significantly altered are indicated by arrows and protein spot numbers (Fig. 2A). Fig. 3 shows comparative close-up views of the representative protein spots whose expression levels decreased in caspase-3-treated gels. These altered protein spots were subjected to MALDI-TOF mass spectrometry.
FIGURE 1.
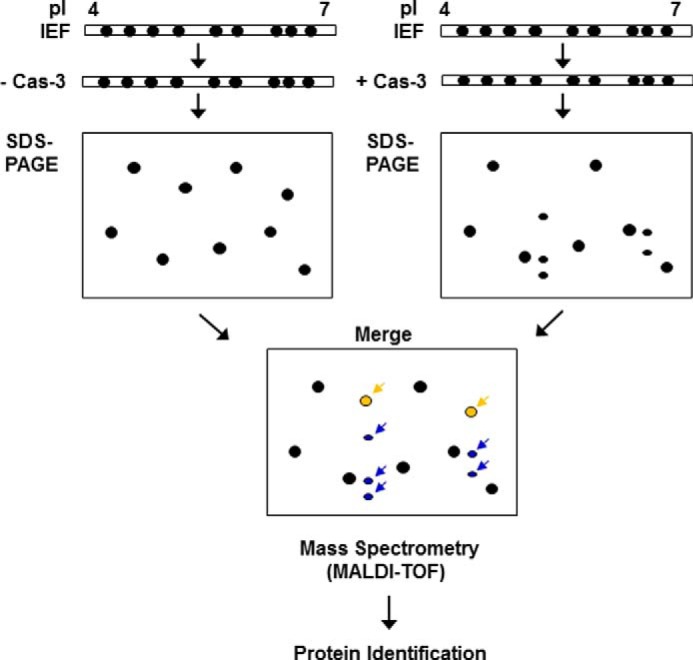
A schematic view of the novel caspase-3 substrate screening method. After the IEF step, the strips were incubated in a caspase-3 activation buffer with or without an empirically predetermined amount of the recombinant human caspase-3 (Cas-3). Subsequently, SDS-PAGE was performed, and the resulting gels were stained with Coomassie Brilliant Blue G-250. The separated protein spots were analyzed by using ProteomWeaver software system. Assuming that the cleaved forms of the putative caspase-3 substrates (indicated by blue arrows) appear below the uncleaved full-length forms (indicated by yellow arrows) on the merged gel, all of the spots indicated by arrows were subjected to MALDI-TOF mass spectrometry for identification.
TABLE 1.
Summary of the identified proteins
| Spot no. | Protein name | Accession no.a | Protein Mr/pIb | MOWSE scorec | Sequence coveraged |
|---|---|---|---|---|---|
| % | |||||
| 1 | 60-kDa heat shock protein, mitochondrial | P63038 | 60,956/5.9 | 3.17E + 13 | 58 |
| 2 | Inorganic pyrophosphatase | Q9D819 | 32,667/5.4 | 31,180,000 | 54 |
| 3 | Actin, cytoplasmic 1 | P60710 | 41,737/5.3 | 2.232E + 14 | 70 |
| 4 | Actin-like protein 6A | Q9Z2N8 | 47,448/5.4 | 1.087E + 10 | 46 |
| 5 | α-Enolase | P17182 | 49,797/7.7 | 1.527E + 18 | 80 |
| 6 | Glutaredoxin-3 | Q9CQM9 | 37,779/5.4 | 2.982E + 18 | 89 |
| 7 | Ubiquitin C-terminal hydrolase 14 | Q9JMA1 | 56,002/5.1 | 4.249E + 11 | 45 |
| 8 | Calponin-3 | Q9DAW9 | 36,429/5.5 | 4.411E + 15 | 53 |
| 9 | Microtubule-associated protein RP/EB family member 1 | Q61166 | 30,016/5.1 | 8.046E + 12 | 78 |
| 10 | TAR DNA-binding protein 43 | Q921F2 | 44,548/6.3 | 1.957E + 10 | 35 |
| 11 | Actin-related protein 3 | Q99JY9 | 47,358/5.6 | 7.59E + 20 | 69 |
| 12 | Thioredoxin domain-containing protein 5 | Q91W90 | 46,416/5.5 | 1.809E + 14 | 51 |
| 13 | 14-3-3 protein η | P68510 | 28,212/4.8 | 8.491E + 15 | 73 |
| 14 | Anamorsin | Q8WTY4 | 33,429/5.1 | 8.87E + 07 | 57 |
| 15 | Chloride intracellular channel protein 1 | Q9Z1Q5 | 27,013/5.1 | 41,380,000 | 73 |
| 16 | Adenine phosphoribosyltransferase | P08030 | 19,736/6.3 | 155,500,000 | 78 |
| 17 | Protein-disulfide isomerase A3 | P27773 | 56,622/6.0 | 7.671E + 17 | 64 |
| 18 | Dynactin subunit 2 | Q99KJ8 | 43,986/5.1 | 8.27E + 14 | 56 |
| 19 | Stress-70 protein, mitochondrial | P38647 | 73,529/5.9 | 6.763E + 30 | 64 |
| 20 | T-complex protein 1 subunit β | P80314 | 57,448/6.0 | 323,000,000 | 54 |
| 21 | NudC domain-containing protein 3 | Q8R1N4 | 40,891/5.2 | 3,826,000 | 41 |
| 22 | UPF0160 protein MYG1, mitochondrial | Q9JK81 | 42,723/6.5 | 2,007,000 | 24 |
| 23 | Nucleoporin Nup43 | P59235 | 42,015/5.1 | 19,080,000 | 31 |
| 24 | Ubiquitin C-terminal hydrolase isozyme L5 | Q9WUP7 | 37,617/5.2 | 9.203E + 20 | 64 |
| 25 | NG,NG-Dimethylarginine dimethylaminohydrolase 1 | Q9CWS0 | 31,381/5.6 | 636,000,000 | 63 |
| 26 | Optineurin | Q8K3K8 | 67,018/5.2 | 4,871,000,000 | 35 |
| 27 | Glutamate-rich WD repeat-containing protein 1 | Q810D6 | 45,972/4.7 | 3.658E + 10 | 50 |
| 28 | 26 S protease regulatory subunit 6A | O88685 | 49,493/5.1 | 1.046E + 24 | 55 |
| 29 | Protein SEC13 homolog | Q9D1M0 | 35,566/5.1 | 176,600,000 | 42 |
| 30 | Vimentin | P20152 | 53,557/5.1 | 2.372E + 14 | 60 |
| 31 | Splicing factor 3A subunit 3 | Q9D554 | 58,842/5.2 | 1.623E + 17 | 54 |
| 32 | Phosphomannomutase 2 | Q9Z2M7 | 27,657/6.0 | 4.216E + 10 | 60 |
| 33 | Pyruvate dehydrogenase E1 component subunit β, mitochondrial | Q9D051 | 38,937/6.4 | 5.189E + 17 | 58 |
| 34 | Protein KTI12 homolog | Q9D1R2 | 38,445/6.5 | 357,800,000 | 53 |
| 35 | Programmed cell death 6-interacting protein | Q9WU78 | 96,011/6.1 | 2.027E + 11 | 39 |
| 36 | Emerin | O08579 | 29,436/4.9 | 3,303,000 | 39 |
| 37 | Putative uncharacterized protein | Q3TIC8 | 52,753/5.7 | 6,250,000 | 28 |
| 38 | Putative uncharacterized protein | Q3THI5 | 45,236/5.4 | 1.612E + 11 | 46 |
| 39 | Sepiapterin reductase | Q64105 | 27,883/5.6 | 8.167E + 15 | 78 |
| 40 | Putative uncharacterized protein | Q3TVM1 | 88,577/5.1 | 1.17E + 11 | 34 |
| 41 | Nucleophosmin 1 | Q5SQB0 | 29,525/4.5 | 2,598,000,000 | 35 |
| 42 | Enolase | Q6PHC1 | 39,783/5.9 | 585,600,000 | 34 |
| 43 | Ras GTPase-activating protein-binding protein 1 | P97855 | 51,829/5.4 | 1.116E + 18 | 52 |
| 44 | Peripherin | P15331 | 54,268/5.4 | 1.319E + 22 | 69 |
| 45 | Heterogeneous nuclear ribonucleoprotein K | P61979 | 48,511/5.7 | 2.616E + 13 | 56 |
| 46 | Thymidylate synthase | P07607 | 34,958/6.0 | 14,430,000 | 39 |
a Accession numbers from Swiss-Prot database (December 3, 2012).
b Theoretical Mr and pI.
c From Swiss-Prot database. MOWSE scores were considered significant for identity (p < 0.05).
d From Swiss-Prot database.
FIGURE 2.
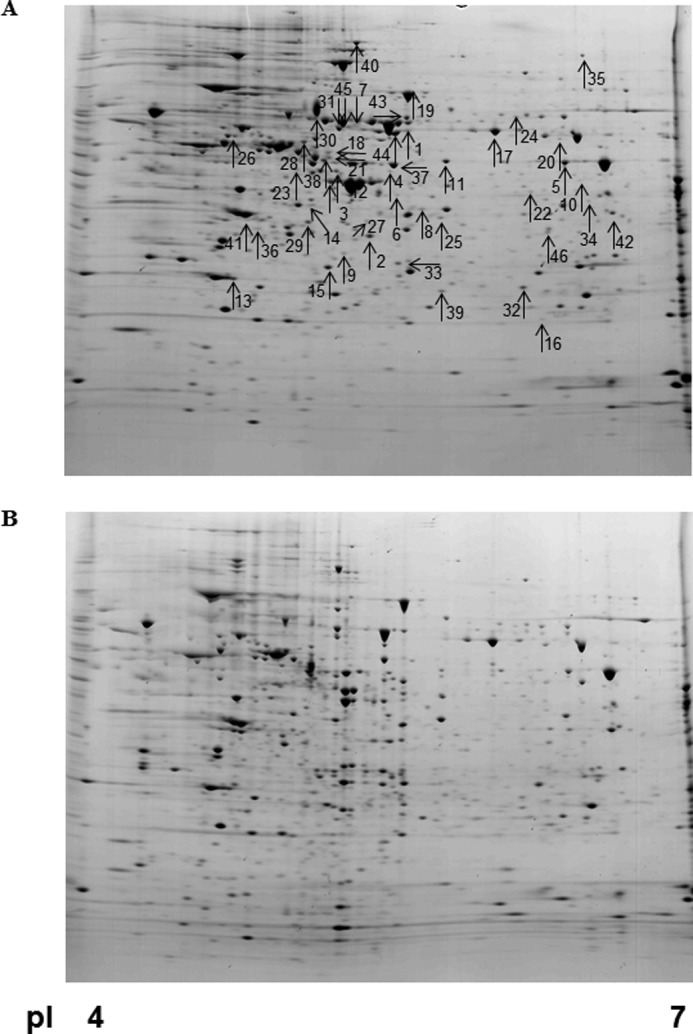
Representative gel images. Total cellular lysates (1.5 mg) were processed for protein separation by two-dimensional electrophoresis. Incubation with buffer only (A) and 25 μg of recombinant human caspase-3 in caspase-3 activation buffer (B) was done after the IEF step on Immobiline DryStrips (pI 4–7 linear). Representative gels demonstrate typical protein profiles of untreated and caspase-3-incubated samples. Arrows indicate protein spots that were altered significantly as summarized in Table 1.
FIGURE 3.
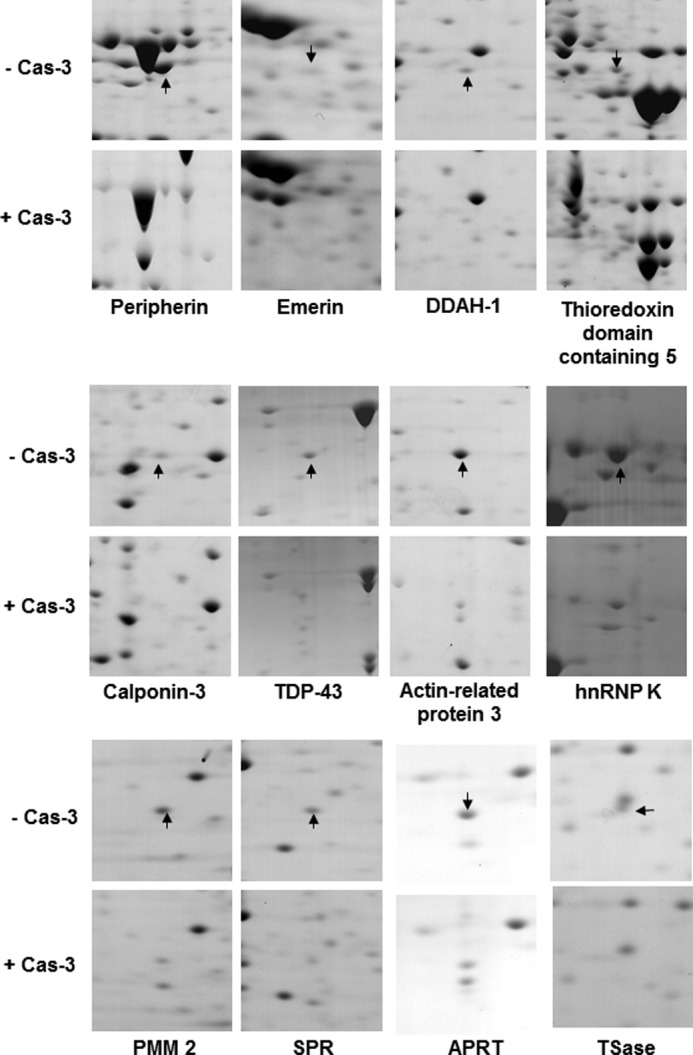
Comparative close-up view of the identified caspase-3 substrates. After MN9D cells were solubilized in 1× sample buffer, the resulting cellular lysates (1.5 mg) were absorbed into Immobiline DryStrips (pI 4–7 linear). Following the IEF step, the strips were incubated in caspase-3 activation buffer with or without 25 μg of recombinant human caspase-3 (Cas-3) followed by SDS-PAGE on an 8–18% gradient gel. The gels were stained with 0.1% Coomassie Brilliant Blue G-250 and analyzed by the ProteomWeaver system. Among several identified protein spots listed in Table 1, the arrows on the control gels indicate 12 putative caspase-3 substrates that were significantly decreased after incubation with caspase-3. hnRNP, heterogeneous nuclear ribonucleoprotein; SPR, sepiapterin reductase; APRT, adenine phosphoribosyltransferase; TSase, thymidylate synthase.
Validation of the Identified Protein as Putative Caspase-3 Substrates
To confirm the possibility that these identified proteins are bona fide substrates of caspase-3, we performed in vitro caspase-3 cleavage assays with four arbitrarily selected proteins: calponin-3, emerin, thymidylate synthase, and TDP43. In vitro transcribed and translated 35S-labeled proteins were incubated with recombinant human caspase-3 in the presence or absence of Z-VAD-fmk. The reactants were analyzed by SDS-PAGE followed by autoradiography. As shown in Fig. 4, these substrates were indeed cleaved by exogenously added caspase-3. As such, full-length proteins decreased, and simultaneously smaller fragments appeared in caspase-3-treated samples. This phenomenon was inhibited in the presence of Z-VAD-fmk. Although not extensively pursued as above, several other substrates listed in Table 1 have already been reported as caspase-3 substrates (30), supporting the validity of our gel-based protease proteomic screening method.
FIGURE 4.
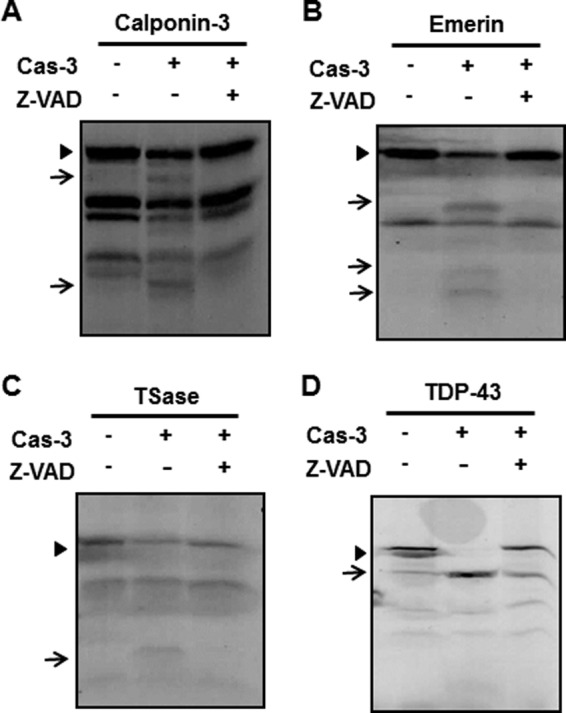
In vitro caspase-3 cleavage assay for the identified substrates. pcDNA3.1/V5-His vectors encoding calponin-3 (A), emerin (B), thymidylate synthase (TSase) (C), and TDP-43 (D) cDNAs were transcribed and translated in the presence of 35S-labeled methionine. To confirm whether these identified proteins are indeed cleaved by exogenously added caspase-3, each reactant was incubated for 1.5 h at 37 °C with or without 50 ng of recombinant human caspase-3 (Cas-3) in the presence or absence of 100 μm Z-VAD-fmk, a pan-caspase inhibitor. After incubation, the reactants were separated by SDS-PAGE followed by autoradiography. Arrowheads indicates the full-length proteins, whereas arrows indicate potentially caspase-3-cleaved fragments that do not appear or exist in low levels in the control and in lanes treated with caspase-3 plus Z-VAD-fmk.
Validation of Anamorsin as a Novel Caspase-3 Substrate
Anamorsin was first identified in a screen for proteins expressed in the variants of growth factor-dependent hematopoietic cells that did not undergo cell death upon growth factor withdrawal (29). Recent evidence indicates that anamorsin plays an important role in regulating cell death, and it is regarded as a cell death-defying factor that is indispensable for definitive hematopoiesis during mouse development as well as for conferring multidrug resistance (34, 35). Previously, we found that anamorsin is widely distributed in the central nervous system (36). As shown in Fig. 5A, we found that the intensity of the anamorsin spot in two-dimensional electrophoresis gel decreased in our caspase-3 protease proteomic screening method (also see Table 1). Interestingly, the anamorsin spot decreased in MN9D neuronal cells treated with 6-hydroxydopamine, which we previously demonstrated to be a caspase-3-inducing drug in these cells (31, 37) (Fig. 5B). We assumed that the time-dependent disappearance of the anamorsin spot correlated with the extent of neuronal cell death and accompanying caspase-3 activation. Therefore, we carried out an in vitro caspase-3 cleavage assay using 35S-labeled anamorsin. As shown in Fig. 5C, caspase-3 effectively cleaved both mouse and human full-length anamorsins, generating two fragments with molecular masses equivalent to 25 and 10 kDa. Co-treatment with Z-VAD-fmk blocked caspase-3-mediated cleavage of anamorsin. To further investigate whether anamorsin is cleaved selectively by caspase-3 or also non-selectively by other caspase members, 35S-labeled mouse anamorsin was incubated with various apoptosis-related caspases including caspase-2, -3, -6, -7, -8, and -9. As shown in Fig. 5D, mouse anamorsin was cleaved by caspase-3 but not by other caspases tested. Even amounts higher than 50 ng of caspase-2, -6, -7, -8, and -9 failed to cleave anamorsin (data not shown). Addition of calpain, the other cysteine protease, also did not generate these 25- and 10-kDa fragments. All results confirmed the caspase-3-mediated anamorsin cleavage and the consequent generation of caspase-3-dependent fragments.
FIGURE 5.
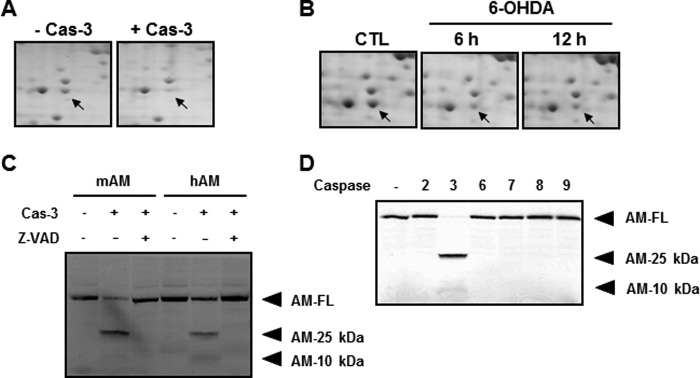
Confirmation of anamorsin as a novel caspase-3 substrate. A, MN9D cellular lysates (1.5 mg) were processed for two-dimensional electrophoresis following incubation with or without 25 μg of recombinant human caspase-3 (Cas-3). Arrows indicate the Coomassie Brilliant Blue G-250-stained protein spot, the level of which decreased in the presence of casapse-3 compared with untreated control. Mass spectrometry indicates that this protein spot is anamorsin. B, MN9D cells were treated with or without a prototypic apoptotic inducer, 100 μm 6-hydroxydopamine (6-OHDA) for the indicated time periods. Total cellular lysates were processed for two-dimensional electrophoresis, and the separated protein spots on the gel were stained with 0.1% Coomassie Brilliant Blue G-250. A close-up view of the same gel position as in A is demonstrated for each time period. Arrows indicate the expression level of anamorsin following 6-hydroxydopamine treatment or sham treatment (CTL). C, in vitro caspase-3 cleavage assay confirmed that the metabolically labeled mouse (mAM) and human anamorsin (hAM) were cleaved by 50 ng of caspase-3. Caspase-3-meidated cleavage of full-length anamorsin (AM-FL) resulted in the generation of two cryptic fragments with molecular sizes of 25 (AM-25 kDa) and 10 kDa (AM-10 kDa). This cleavage was blocked in the presence of 100 μm Z-VAD-fmk. D, specificity of caspase-3-mediated cleavage of anamorsin was confirmed by an in vitro caspase cleavage assay. Fifty nanograms of recombinant human caspase-2, -3, -6, -7, -8, and -9 were used for the reaction.
Because of the appearance of two fragments of anamorsin in the cleavage assay, anamorsin is likely to be cleaved at one site containing a conserved DXXD tetrapeptide motif that is recognized and cleaved by caspase-3 (38, 39). We identified four consensus sequences in mouse anamorsin (Fig. 6A). To determine the actual cleavage site of anamorsin by caspase-3, we performed cell-free caspase cleavage assays using the wild-type anamorsin and four anamorsin mutants in which aspartate in the P1 position was replaced by alanine. The mutations were D205A, D209A, D212A, and D221A. In vitro transcribed and translated 35S-labeled products were mock-treated or treated with caspase-3. As shown in Fig. 6B, the autoradiography showed that full-length anamorsin as well as anamorsin mutants except D209A were cleaved by caspase-3, and two fragments of anamorsin, an N-terminal 25-kDa fragment and a C-terminal 10-kDa fragment were generated. Because the D209A mutant showed no fragment upon caspase-3 exposure, the cleavage sequence of anamorsin was determined to be DSVD209↓L. Next, we tested whether the caspase-3-mediated anamorsin cleavage also occurs at DSVD209↓L in a cellular system. MN9D cells were transfected with either the wild type or each of four DXXD anamorsin mutants. Transiently transfected cells were treated with 1 μm staurosporine, which was previously demonstrated by us to be a caspase-3-inducing drug in MN9D cells (24, 37). Immunoblot analysis clearly showed that the wild-type anamorsin and three mutants except D209A were cleaved. Because V5 was C-terminally tagged to anamorsin, anti-V5 antibody recognized only the C-terminal fragment of anamorsin following staurosporine treatment (Fig. 6C). Taken together, our data indicated that anamorsin is indeed cleaved by caspase-3 at DSVD209↓L in neuronal cells.
FIGURE 6.

Determination of the anamorsin cleavage site by caspase-3. A, amino acid sequences of mouse anamorsin demonstrate four putative caspase-3 cleavage sites containing tetrapeptide motif DXXD (marked in bold and underlined). B, in vitro caspase-3 (Cas-3) cleavage assay was performed using 35S-labeled WT and each of four anamorsin DXXD mutants. Each reactant was incubated for 1.5 h at 37 °C with or without 50 ng of caspase-3. Note that the cleavage sequence of mouse anamorsin was mapped at DSVD209↓L. AM-NT and AM-CT represent 25- and 10-kDa anamorsin, respectively. C, MN9D cells overexpressing C-terminally V5-His-tagged WT anamorsin (AM-V5-His) or one of four anamorsin DXXD mutants were treated for 24 h with 1 μm staurosporine (STS). Following drug treatment, cellular lysates were processed for an immunoblot analysis using anti-V5 antibody that recognizes the C-terminally V5-tagged fragment of anamorsin and anti-cleaved caspase-3 antibody. Note that DSVD209↓L was also confirmed as a caspase-3-mediated cleavage site of anamorsin. AM-FL, full-length anamorsin.
Cleavage of Anamorsin in Various Forms of Neurodegeneration
We previously demonstrated that 6-hydroxydopamine or staurosporine causes caspase-3-dependent death over 12–24-h incubation periods, whereas MPP+ causes caspase-independent but calpain-dependent cell death in dopaminergic neuronal cells over 24–36-h incubation periods (37, 40). To determine whether anamorsin is cleaved in these paradigms, immunoblot analyses were performed using MN9D cells treated with 100 μm 6-hydroxydopamine or 1 μm staurosporine. We used anti-anamorsin antibody (KM3052) that recognizes the N-terminal region of anamorsin. Following drug treatment, N-terminal fragments accumulated in a time-dependent manner in parallel with the increase of cleaved caspase-3 (Fig. 7, A and B, middle panels). This was blocked by co-treatment with Z-VAD-fmk. In contrast, MPP+-treated MN9D cells did not show any discernible signs of anamorsin cleavage up to 36 h (Fig. 7C). To investigate whether the caspase-3-dependent cleavage of anamorsin is not restricted to neuronal cells but a general phenomenon associated with apoptosis, we then performed an immunoblot analysis using non-neuronal U-2 OS cells treated with 50 μm etoposide, another caspase-3-activating reagent. As shown in Fig. 7D, the N-terminal fragment of anamorsin appeared in only the lanes with caspase-3 activation, and this was blocked in the presence of Z-VAD-fmk. Taken together, the present data indicate that anamorsin is cleaved in a caspase-3-dependent manner in several apoptotic cell death paradigms.
FIGURE 7.

Cleavage of anamorsin in caspase-3-dependent apoptotic death. A and B, MN9D cells were treated for the indicated time periods with a prototypic apoptotic stimulus, 100 μm 6-hydroxydopamine (6-OHDA) (A) or 1 μm staurosporine (STS) (B), in the presence or absence of 100 μm Z-VAD-fmk. Following drug treatment, cellular lysates were processed for immunoblot analyses using anti-anamorsin that recognizes the N-terminal anamorsin fragment (KM3052) and anti-cleaved caspase-3 antibody. GAPDH and actin antibodies were used as loading controls. Note that the cleaved form of anamorsin (AM) was apparent only in activated caspase-3 (cas-3) lanes. C, MN9D cells were treated with 50 μm MPP+ for the indicated time periods. Lysates obtained from staurosporine-treated MN9D cells were used as a positive control for caspase-3-mediated anamorsin cleavage. An immunoblot analysis demonstrates no cleavage of anamorsin in MPP+-treated cells in which no sign of caspase-3 activation was found. D, U-2 OS cells were treated with 50 μm etoposide for 24 h in the presence or absence of 100 μm Z-VAD-fmk. An immunoblot blot analysis indicates that etoposide-induced cleavage of anamorsin was blocked in the presence of Z-VAD-fmk. CTL, control.
Our previous study demonstrated that anamorsin is widely expressed in various regions of the central nervous system including the cerebral cortex, spinal cord, and midbrain (36). Thus, we asked whether anamorsin is endogenously cleaved during apoptotic cell death in vivo. Based on previous findings demonstrating an elevated caspase-3 activity in post-mortem brains of patients with AD and PD (18, 20), we investigated whether anamorsin underwent caspase-3-dependent cleavage in these post-mortem brains. The information for AD and PD patients is provided in Tables 2 and 3, respectively. First, we found that cryptic fragments appeared in all tested cortical tissue lysates harvested from post-mortem AD brains (Fig. 8A). The molecular mass of fragments was ∼25 kDa, which corresponds to the caspase-3-cleaved N-terminal fragment of anamorsin. The overall intensity of this fragment in AD brains was significantly higher than that in age-matched control brains. The predicted 25-kDa fragment of anamorsin was also observed in the substantia nigra of PD brains, albeit it was only visible after longer exposure (Fig. 8B). Again, the intensity of the 25-kDa anamorsin fragment in PD brains was higher compared with that in age-matched control brains (Fig. 8B). However, the overall intensity of the 25-kDa fragment was much weaker in PD brains than in AD brains. In the middle cerebral artery occlusion model as well as the spinal cord injury model, we also observed the 25-kDa anamorsin fragment (data not shown). Taken together, our data suggest that caspase-mediated cleavage of anamorsin may be a general feature in various forms of neurodegeneration.
TABLE 2.
Post-mortem AD brains used
M, male; F, female; PMD, post-mortem delay.
| Sample | Age | Sex | PMD | Final diagnosis |
|---|---|---|---|---|
| h | ||||
| Control | ||||
| 1 | 66 | M | 8.5 | Control |
| 2 | 85 | M | 19 | Control |
| 3 | 73 | M | 17 | Control |
| 4 | 53 | M | 7 | Control |
| AD | ||||
| 1 | 71 | M | 15 | AD, Braak stage VI, 2 lacunes |
| 2 | 81 | M | 6 | AD, Braak stage I, multiple microinfarcts in distribution of basilar artery |
| 3 | 80 | M | 8.5 | AD, Braak stage VI |
| 4 | 77 | M | 10 | AD, Braak stage III, restricted to the hippocampus, amygdala, entorhinal cortex |
| 5 | 70 | M | 3 | AD, Braak stage VI |
| 6 | 79 | M | 7 | AD, Braak stage VI |
| 7 | 74 | M | 7 | AD, Braak stage VI |
| 8 | 83 | M | 12 | AD, Braak stage VI |
| 9 | 83 | M | 12 | AD, Braak stage VI |
TABLE 3.
Post-mortem PD brains used
M, male; F, female; PMD, post-mortem delay.
| Sample | Age | Sex | PMD | Final diagnosis |
|---|---|---|---|---|
| h | ||||
| Control | ||||
| 1 | 87 | F | 7 | Control |
| 2 | 89 | M | 8.5 | Control |
| 3 | 71 | M | 16 | Control |
| 4 | 79 | M | 16 | Control |
| PD | ||||
| 1 | 76 | M | 17 | PD with dementia |
| 2 | 83 | M | 5 | PD with dementia, neurodegeneration, occipital infarct |
| 3 | 71 | M | 8 | PD, neocortical |
| 4 | 73 | M | 6.5 | PD with dementia |
FIGURE 8.
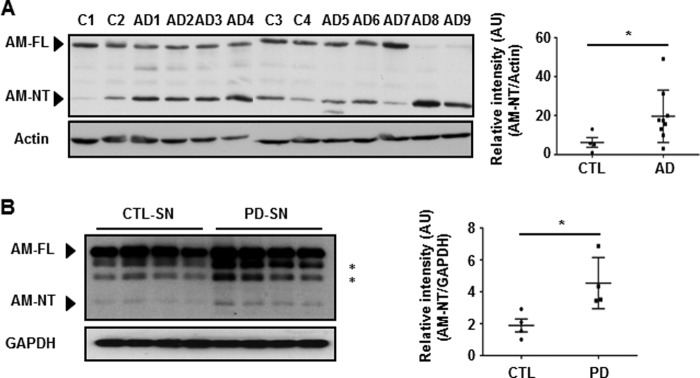
Appearance of the cleaved anamorsin fragment in the post-mortem brains of patients with AD or PD. Tissue lysates from the cortices of post-mortem brains from AD patients (A) or the substantia nigra region (SN) of post-mortem brains of patients with PD (B) were subjected to immunoblot analyses using antibodies against anamorsin (KM3052). The identity of two bands marked by asterisks was not determined. Equivalent tissues lysates from age-matched control brains were run in parallel. The intensity of the cleaved form of anamorsin (AM-NT) was plotted for both post-mortem brains (closed squares) and controls (CTL) brains (closed circles) for comparison. Error bars indicate ±S.D. on either side of the mean. *, p < 0.05. The information for AD and PD patients is provided in Tables 2 and 3, respectively. AU, arbitrary units.
Antiapoptotic Functions of Anamorsin
We then investigated whether caspase-3-mediated cleavage of anamorsin affects its proposed antiapoptotic role. As shown in Fig. 9A, the MTT reduction assay indicated that RNA interference-mediated knockdown of anamorsin left MN9D cells more vulnerable to staurosporine, suggesting its antiapoptotic function. To further confirm this possibility, mouse fetal liver stromal cell lines from anamorsin KO mice and wild-type littermates were established by transfection of SV40 large T antigen. The cell lines established were referred to as FK3-1 (anamorsin KO cells) and FW3-1 (anamorsin wild-type) cells. At 0.75 and 1 μm staurosporine, the intensity of cleaved caspase-3 increased in FK3-1 cells compared with that in FW3-1 cells (Fig. 9B, right panel). Higher concentrations of staurosporine (up to 4 μm for 6 h) also triggered significantly increased levels of cleaved caspase-3 in FK3-1 as compared with those in FW3-1 cells (data not shown). We then assessed whether the reintroduction of anamorsin into FK3-1 cells confers resistance to staurosporine-induced apoptosis. As shown in Fig. 9C, reintroduction of full-length anamorsin resulted in a reduced level of cleaved caspase-3 compared with control vector-transfected cells, indicating an antiapoptotic function of anamorsin. To compare the extent of antiapoptotic function of two anamorsin fragments, we reintroduced either N-terminal or C-terminal fragment of anamorsin into FK3-1 cells. Following staurosporine treatment, the intensity of cleaved caspase-3 was not altered in FK3-1 cells transfected with C-terminal or N-terminal fragment (data not shown), suggesting that caspase-3-mediated cleavage of anamorsin results in a loss of its proposed antiapoptotic function. To further examine anti-cell death functions of anamorsin in primary cultures of cortical neurons, lentivirus-mediated knockdown of mouse anamorsin was established using two anamorsin shRNAs and scrambled sequences. As shown in Fig. 10A, the anamorsin level decreased by approximately half in cells infected with lentiviral particles containing mouse anamorsin shRNA sequences. Immunoblot analysis showed that the intensity of cleaved caspase-3 significantly increased in anamorsin knockdown cultures (Fig. 10A). Similarly, the MTT reduction assay indicated that RNA interference-mediated anamorsin knockdown left primary cultures of cortical neurons more vulnerable to staurosporine treatment (Fig. 10B). In sum, our present data indicate that anamorsin leaves cells less vulnerable to any apoptotic stimuli. In this regard, it would be interesting to examine whether and how anamorsin may play any protective role in caspase-independent cell death paradigms.
FIGURE 9.
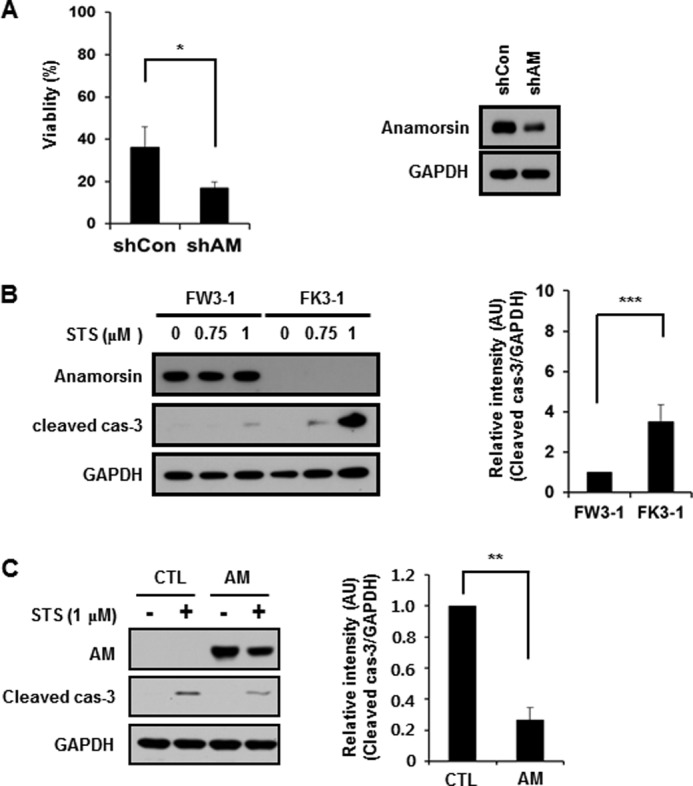
Evaluation of antiapoptotic function of anamorsin. A, MN9D control cells (shCon) and anamorsin-knockdown MN9D cells (shAM) were treated with 1 μm staurosporine (STS) for 18 h. The rate of viability was determined by MTT reduction assays. Values are expressed as a percentage of the untreated controls (100%). Bars represents the mean ± S.D. from three independent experiments done in triplicate. *, p < 0.05. Expression levels of anamorsin in cells transfected with anamorsin shRNA or scrambled sequences were determined by immunoblot analysis using anti-anamorsin antibody. B, FW3-1 (anamorsin WT) and FK3-1 (anamorsin KO) cells were treated for 12 h with the indicated concentrations of staurosporine. Cellular lysates were processed for immunoblot analyses using antibody recognizing anamorsin or cleaved caspase-3. The intensity of the cleaved caspase-3 in FK3-1 cells at 1 μm staurosporine was quantitated by densitometry and is expressed as a ratio over the staurosporine-treated FW3–1 cells after normalization against GAPDH. Bars represent the mean ± S.D. from three independent experiments. ***, p < 0.001. C, FK3-1 cells were transiently transfected with the vector alone (pCI-Neo; CTL) or vectors containing FLAG-tagged anamorsin wild type (AM). Twenty-four hours after transfection, cells were treated with 1 μm staurosporine for 12 h. Following drug treatment, cellular lysates were processed for immunoblot analyses using antibodies against anamorsin and cleaved caspase-3 (cas-3). Levels of the cleaved caspase-3 are expressed as a ratio over the staurosporine-treated control cells (1.0) after normalization against GAPDH. Bars represent the mean ± S.D. from three independent experiments. **, p < 0.01. All error bars represent S.D. AU, arbitrary units.
FIGURE 10.

Evaluation of anti-cell death function of anamorsin in primary cultures of cortical neurons. Primary cultures of cortical neurons were prepared using ED14.5 mouse cortices as described under “Experimental Procedures.” At 7 days in vitro, cultures were exposed to lentiviral particles containing one of the two shRNA anamorsin sequences (shAM 1 and shAM 2) or control shRNA sequences (shCon) for 4 days. Cultures were treated with 200 nm staurosporine (STS) for 24 h and subjected to immunoblot analyses using anti-anamorsin and anti-cleaved caspase-3 (A) or MTT reduction assay (B). A, relative intensity of cleaved caspase-3 (cas-3) is expressed as a ratio over the control cultures (1.0) after normalization against GAPDH. Bars represent the mean ± S.D. from three independent experiments. **, p < 0.01; ***, p < 0.001. B, viability was determined by MTT reduction assays. Values are expressed as a percentage of the untreated controls (100%). Bars represent the mean ± S.D. from three independent experiments. ***, p < 0.001. All error bars represent S.D. AU, arbitrary units.
DISCUSSION
Recent high throughput gel-based or gel-free proteomic approaches have detected hundreds of caspase substrates (16, 17, 41). However, the possibility cannot be ruled out that some of the identified substrates may be cleaved by other proteases that are activated either downstream or upstream of applied caspases. Indeed, the cross-talk between caspases and other proteases including calpains is well documented (42). In the present study, therefore, we developed a high throughput screening method to find endogenous substrates that are directly cleaved by caspase-3. The novelty of our strategy stems from the sequential steps that consist of protein separation by IEF followed by caspase-3 incubation of immobilized proteins on a strip. Because the incubation of proteins immobilized on a gel with caspase-3 ensures that only substrates directly cleaved by the applied proteases can be detected, the identified substrates are most likely caspase-3 substrates. Among 46 putative caspase-3 substrates identified, previously identified caspase-3 substrates such as TDP-43, dynactin subunit 2, vimentin, and nucleophosmin 1 are included, supporting the validity of our protease proteomic approach. A database search indicates that these altered protein spots belong to various biological categories including cytoskeleton (26%); chromatin structure and dynamics (17%); energy production and conservation (17%); replication, recombination, and repair (8%); defense mechanisms (8%); posttranslational modification and chaperones (8%); and translation, ribosomal structure, and biogenesis (8%). The cell death-regulating role of these identified proteins in neurodegenerative diseases and the consequence of its cleavage by activated caspase-3 remain to be determined. It would be also intriguing to examine the concerted roles of some of these proteins in regulating the cell death process of various neurodegenerative diseases. Although the IEF strips with a pI range covering from 4 to 7 were used in the present study, the strips with a wide range of pI (e.g. 3–10) or the strips whose pI range is highly zoomed (e.g. pI of 4–5) can also be used to find additional caspase-3 substrates. Moreover, more sensitive staining methods like silver staining or different separation techniques including difference gel electrophoresis can be applied to enhance the capability of detecting more caspase-3 substrates. In addition, our screening strategy consists of mass spectral analysis of any proteins that are cleaved to smaller fragments by the applied proteases (14, 15). Thus, our strategy can be extended to identify endogenous substrates of others caspases or calpains that are activated during neurodegeneration (43, 44).
Anamorsin was first identified as an antiapoptotic protein that makes BaF3 cells resistant to apoptotic stimuli and is indispensable in definitive hematopoiesis (29). Anamorsin has other important biological functions as well, contributing to cytosolic iron-sulfur cluster biogenesis (45) and multidrug resistance in several cancer cells (46). However, precise mechanisms for its antiapoptotic function are poorly understood. Most importantly, whether anamorsin exerts quite similar roles in the central nervous system has not been examined. Previously, we demonstrated that anamorsin mRNA and protein levels are higher in the brain and the spinal cord as compared with those in the peripheral organs, suggesting its critical role in the central nervous system (36). In the present study, we clearly showed that caspase-3-mediated cleavage of anamorsin is a general feature of neuronal apoptotic death as determined in various drug-induced death paradigms. Although we did not extensively explore whether other proteases may cleave anamorsin in pathological conditions, we clearly demonstrated the specific caspase-3-mediated cleavage of anamorsin at DSVD209↓L, generating 25- and 10-kDa fragments. Furthermore, we detected the 25-kDa anamorsin fragment in the post-mortem brains of patients with AD or PD, suggesting that caspase-3-mediated cleavage of anamorsin is a general feature of caspase-3-dependent cell death in the central nervous system. In the case of the post-mortem brains of patients with AD or PD, we found that a basal level of the 25-kDa fragment is present in all age-matched control brains. In general, the cleavage of anamorsin is significantly increased in AD and PD brains. Nevertheless, the reason why the intensity of 25-kDa anamorsin is weaker in the post-mortem brain with PD is not yet clear. Considering that dopaminergic neurons are typical of a diffused system, it may be that the signal of the cleaved anamorsin from a small population of dopaminergic neurons located in the substantia nigra pars compacta is very limited and therefore weak. In PD brain samples, two strong bands appear just below full-length anamorsin. Although the identity of these two bands has not yet been determined, they could be fragments generated by other unidentified proteases.
Previously, it has been indicated that the consequences of caspase-mediated cleavage of proteins can be gain-of-function, loss-of-function, or just bystander events (47). Anamorsin has two important functional domains: an N-terminal methyltransferase domain and a C-terminal DUF689 domain that contains two iron-sulfur redox centers (45, 48). The caspase-3-mediated cleavage of anamorsin at DSVD209↓L leads to separation of these two domains from each other. Our reconstitution assays showed that only full-length anamorsin but not its fragments confer resistance to anamorsin-deficient fetal liver stromal cells against staurosporine-induced apoptosis and activation of caspase-3 (see Fig. 9). However, our data do not exclude the possibility that each fragment can still play an unidentified, important role via caspase-3-independent routes during neurodegeneration. For example, Dre2, a yeast homolog of anamorsin, was identified as a cytosolic iron-sulfur cluster protein biogenerator (49, 50) and as a modulator of mitochondrial integrity and cell death after oxidative stress (51). Intriguingly, a previous report demonstrated that human anamorsin can complement the lethality of a dre2 deletion yeast strain and that the cysteine motifs at its C-terminal region are required to form a functional iron-sulfur cluster (49). By sequence alignment analysis of anamorsin orthologues, it has been shown that the C-terminal region of anamorsin is evolutionally conserved across species from yeast to human (49, 50), indicating a critical role of its C-terminal region for conferring potentially prosurvival activity to cells. Recently, a yeast two-hybrid assay indicated that anamorsin preferentially binds to PICOT, another iron-sulfur cluster protein (30, 52). In this case, however, it has been shown that the N-terminal regions of both anamorsin and PICOT are essential for their binding and their growth regulatory functions. Interestingly, the phenotype of anamorsin-deficient mice is very similar to that of PICOT, suggesting that anamorsin and PICOT may play essential roles in a cooperative manner during embryogenesis and perhaps in hematopoiesis (53). Considering that PICOT is also cleaved by caspase-3 (30, 52), it would be interesting to investigate whether and how combinatorial events of anamorsin-PICOT interactions may determine whether cells survive or die. Because both anamorsin KO and PICOT KO mice are embryonic lethal, conditional KO mice can be utilized to further analyze the function of anamorsin alone or in combination with PICOT during neurodegeneration.
This work was supported by National Research Foundation of Korea (NRF) Grant 2008-0061888 (to Y. J. O.) and NRF Bio and Medical Technology Development Program Grant 2011-0019440 (to W.-K. K.) funded by the Ministry of Science, Information/Communication Technology, and Future Planning.
- AD
- Alzheimer disease
- PD
- Parkinson disease
- IEF
- immobilized isoelectric focusing
- MPP+
- 1-methyl-4-phenylpyridinium
- Z
- N-benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NT
- N-terminal fragment
- CT
- C-terminal fragment
- PICOT
- protein kinase C-interacting cousin of thioredoxin.
REFERENCES
- 1. Thompson C. B. (1995) Apoptosis in the pathogenesis and treatment of disease. Science 267, 1456–1462 [DOI] [PubMed] [Google Scholar]
- 2. Degterev A., Boyce M., Yuan J. (2003) A decade of caspases. Oncogene 22, 8543–8567 [DOI] [PubMed] [Google Scholar]
- 3. Crawford E. D., Wells J. A. (2011) Caspase substrates and cellular remodeling. Annu. Rev. Biochem. 80, 1055–1087 [DOI] [PubMed] [Google Scholar]
- 4. Enari M., Sakahira H., Yokoyama H., Okawa K., Iwamatsu A., Nagata S. (1998) A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391, 43–50 [DOI] [PubMed] [Google Scholar]
- 5. Coleman M. L., Sahai E. A., Yeo M., Bosch M., Dewar A., Olson M. F. (2001) Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell Biol. 3, 339–345 [DOI] [PubMed] [Google Scholar]
- 6. Sebbagh M., Renvoizé C., Hamelin J., Riché N., Bertoglio J., Bréard J. (2001) Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat. Cell Biol. 3, 346–352 [DOI] [PubMed] [Google Scholar]
- 7. Lüthi A. U., Martin S. J. (2007) The CASBAH: a searchable database of caspase substrates. Cell Death Differ. 14, 641–650 [DOI] [PubMed] [Google Scholar]
- 8. Johnson C. E., Kornbluth S. (2008) Caspase cleavage is not for everyone. Cell 134, 720–721 [DOI] [PubMed] [Google Scholar]
- 9. Cryns V. L., Byun Y., Rana A., Mellor H., Lustig K. D., Ghanem L., Parker P. J., Kirschner M. W., Yuan J. (1997) Specific proteolysis of the kinase protein kinase C-related kinase 2 by caspase-3 during apoptosis. Identification by a novel, small pool expression cloning strategy. J. Biol. Chem. 272, 29449–29453 [DOI] [PubMed] [Google Scholar]
- 10. Kamada S., Kusano H., Fujita H., Ohtsu M., Koya R. C., Kuzumaki N., Tsujimoto Y. (1998) A cloning method for caspase substrates that uses the yeast two-hybrid system: cloning of the antiapoptotic gene gelsolin. Proc. Natl. Acad. Sci. U.S.A. 95, 8532–8537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gerner C., Frohwein U., Gotzmann J., Bayer E., Gelbmann D., Bursch W., Schulte-Hermann R. (2000) The Fas-induced apoptosis analyzed by high throughput proteome analysis. J. Biol. Chem. 275, 39018–39026 [DOI] [PubMed] [Google Scholar]
- 12. Gerner C., Gotzmann J., Fröhwein U., Schamberger C., Ellinger A., Sauermann G. (2002) Proteome analysis of nuclear matrix proteins during apoptotic chromatin condensation. Cell Death Differ. 9, 671–681 [DOI] [PubMed] [Google Scholar]
- 13. Lee A. Y., Park B. C., Jang M., Cho S., Lee D. H., Lee S. C., Myung P. K., Park S. G. (2004) Identification of caspase-3 degradome by two-dimensional gel electrophoresis and matrix-assisted laser desorption/ionization-time of flight analysis. Proteomics 4, 3429–3436 [DOI] [PubMed] [Google Scholar]
- 14. Shao W., Yeretssian G., Doiron K., Hussain S. N., Saleh M. (2007) The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J. Biol. Chem. 282, 36321–36329 [DOI] [PubMed] [Google Scholar]
- 15. Ricci J. E., Muñoz-Pinedo C., Fitzgerald P., Bailly-Maitre B., Perkins G. A., Yadava N., Scheffler I. E., Ellisman M. H., Green D. R. (2004) Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 117, 773–786 [DOI] [PubMed] [Google Scholar]
- 16. Van Damme P., Martens L., Van Damme J., Hugelier K., Staes A., Vandekerckhove J., Gevaert K. (2005) Caspase-specific and nonspecific in vivo protein processing during Fas-induced apoptosis. Nat. Methods 2, 771–777 [DOI] [PubMed] [Google Scholar]
- 17. Mahrus S., Trinidad J. C., Barkan D. T., Sali A., Burlingame A. L., Wells J. A. (2008) Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell 134, 866–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cribbs D. H., Poon W. W., Rissman R. A., Blurton-Jones M. (2004) Caspase-mediated degeneration in Alzheimer's disease. Am. J. Pathol. 165, 353–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Love S., Barber R., Wilcock G. K. (2000) Neuronal death in brain infarcts in man. Neuropathol. Appl. Neurobiol. 26, 55–66 [DOI] [PubMed] [Google Scholar]
- 20. Hartmann A., Hunot S., Michel P. P., Muriel M. P., Vyas S., Faucheux B. A., Mouatt-Prigent A., Turmel H., Srinivasan A., Ruberg M., Evan G. I., Agid Y., Hirsch E. C. (2000) Caspase-3: a vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 97, 2875–2880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee Y. M., Park S. H., Shin D. I., Hwang J. Y., Park B., Park Y. J., Lee T. H., Chae H. Z., Jin B. K., Oh T. H., Oh Y. J. (2008) Oxidative modification of peroxiredoxin is associated with drug-induced apoptotic signaling in experimental models of Parkinson disease. J. Biol. Chem. 283, 9986–9998 [DOI] [PubMed] [Google Scholar]
- 22. Maeda K., Yasumoto S., Tsuruda A., Andoh K., Kai K., Otoi T., Matsumura T. (2007) Establishment of a novel equine cell line for isolation and propagation of equine herpesviruses. J. Vet. Med. Sci. 69, 989–991 [DOI] [PubMed] [Google Scholar]
- 23. Murata K., Hanzawa K., Kasai F., Takeuchi M., Echigoya T., Yasumoto S. (2007) Robertsonian translocation as a result of telomere shortening during replicative senescence and immortalization of bovine oviduct epithelial cells. In Vitro Cell. Dev. Biol. Anim. 43, 235–244 [DOI] [PubMed] [Google Scholar]
- 24. Choi W. S., Lee E. H., Chung C. W., Jung Y. K., Jin B. K., Kim S. U., Oh T. H., Saido T. C., Oh Y. J. (2001) Cleavage of Bax is mediated by caspase-dependent or -independent calpain activation in dopaminergic neuronal cells: protective role of Bcl-2. J. Neurochem. 77, 1531–1541 [DOI] [PubMed] [Google Scholar]
- 25. Karki P., Seong C., Kim J. E., Hur K., Shin S. Y., Lee J. S., Cho B., Park I. S. (2007) Intracellular K+ inhibits apoptosis by suppressing the Apaf-1 apoptosome formation and subsequent downstream pathways but not cytochrome c release. Cell Death Differ. 14, 2068–2075 [DOI] [PubMed] [Google Scholar]
- 26. Karki P., Dahal G. R., Park I. S. (2007) Both dimerization and interdomain processing are essential for caspase-4 activation. Biochem. Biophys. Res. Commun. 356, 1056–1061 [DOI] [PubMed] [Google Scholar]
- 27. Park I. S., Moon H. R., Seok H., Lee M. (2004) Rearrangement of tryptophan residues in caspase-3 active site upon activation. Biochim. Biophys. Acta 1700, 5–9 [DOI] [PubMed] [Google Scholar]
- 28. Green K. A., Naylor M. J., Lowe E. T., Wang P., Marshman E., Streuli C. H. (2004) Caspase-mediated cleavage of insulin receptor substrate. J. Biol. Chem. 279, 25149–25156 [DOI] [PubMed] [Google Scholar]
- 29. Shibayama H., Takai E., Matsumura I., Kouno M., Morii E., Kitamura Y., Takeda J., Kanakura Y. (2004) Identification of a cytokine-induced antiapoptotic molecule anamorsin essential for definitive hematopoiesis. J. Exp. Med. 199, 581–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yun N., Kim C., Cha H., Park W. J., Shibayama H., Park I. S., Oh Y. J. (2013) Caspase-3-mediated cleavage of PICOT in apoptosis. Biochem. Biophys. Res. Commun. 432, 533–538 [DOI] [PubMed] [Google Scholar]
- 31. Choi W. S., Eom D. S., Han B. S., Kim W. K., Han B. H., Choi E. J., Oh T. H., Markelonis G. J., Cho J. W., Oh Y. J. (2004) Phosphorylation of p38 MAPK induced by oxidative stress is linked to activation of both caspase-8- and -9-mediated apoptotic pathways in dopaminergic neurons. J. Biol. Chem. 279, 20451–20460 [DOI] [PubMed] [Google Scholar]
- 32. Kang H. J., Yoon W. J., Moon G. J., Kim D. Y., Sohn S., Kwon H. J., Gwag B. J. (2005) Caspase-3-mediated cleavage of PHF-1 tau during apoptosis irrespective of excitotoxicity and oxidative stress: an implication to Alzheimer's disease. Neurobiol. Dis. 18, 450–458 [DOI] [PubMed] [Google Scholar]
- 33. Shin J. H., Ko H. S., Kang H., Lee Y., Lee Y. I., Pletinkova O., Troconso J. C., Dawson V. L., Dawson T. M. (2011) PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson's disease. Cell 144, 689–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lu D., Xiao Z., Wang W., Xu Y., Gao S., Deng L., He W., Yang Y., Guo X., Wang X. (2012) Down regulation of CIAPIN1 reverses multidrug resistance in human breast cancer cells by inhibiting MDR1. Molecules 17, 7595–7611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saito Y., Shibayama H., Tanaka H., Tanimura A., Kanakura Y. (2011) A cell-death-defying factor, anamorsin mediates cell growth through inactivation of PKC and p38MAPK. Biochem. Biophys. Res. Commun. 405, 303–307 [DOI] [PubMed] [Google Scholar]
- 36. Park K. A., Yun N., Shin D. I., Choi S. Y., Kim H., Kim W. K., Kanakura Y., Shibayama H., Oh Y. J. (2011) Nuclear translocation of anamorsin during drug-induced dopaminergic neurodegeneration in culture and in rat brain. J. Neural. Transm. 118, 433–444 [DOI] [PubMed] [Google Scholar]
- 37. Han B. S., Hong H. S., Choi W. S., Markelonis G. J., Oh T. H., Oh Y. J. (2003) Caspase-dependent and -independent cell death pathways in primary cultures of mesencephalic dopaminergic neurons after neurotoxin treatment. J. Neurosci. 23, 5069–5078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thornberry N. A., Rano T. A., Peterson E. P., Rasper D. M., Timkey T., Garcia-Calvo M., Houtzager V. M., Nordstrom P. A., Roy S., Vaillancourt J. P., Chapman K. T., Nicholson D. W. (1997) A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 272, 17907–17911 [DOI] [PubMed] [Google Scholar]
- 39. Talanian R. V., Quinlan C., Trautz S., Hackett M. C., Mankovich J. A., Banach D., Ghayur T., Brady K. D., Wong W. W. (1997) Substrate specificities of caspase family proteases. J. Biol. Chem. 272, 9677–9682 [DOI] [PubMed] [Google Scholar]
- 40. Choi W. S., Yoon S. Y., Oh T. H., Choi E. J., O'Malley K. L., Oh Y. J. (1999) Two distinct mechanisms are involved in 6-hydroxydopamine- and MPP+-induced dopaminergic neuronal cell death: role of caspases, ROS, and JNK. J. Neurosci. Res. 57, 86–94 [DOI] [PubMed] [Google Scholar]
- 41. Dix M. M., Simon G. M., Cravatt B. F. (2008) Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell 134, 679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang K. K. (2000) Calpain and caspase: can you tell the difference? Trends Neurosci. 23, 20–26 [DOI] [PubMed] [Google Scholar]
- 43. Camins A., Crespo-Biel N., Junyent F., Verdaguer E., Canudas A. M., Pallàs M. (2009) Calpains as a target for therapy of neurodegenerative diseases: putative role of lithium. Curr. Drug Metab. 10, 433–447 [DOI] [PubMed] [Google Scholar]
- 44. Venderova K., Park D. S. (2012) Programmed cell death in Parkinson's disease. Cold Spring Harb. Perspect. Med. 2, a009365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Banci L., Bertini I., Calderone V., Ciofi-Baffoni S., Giachetti A., Jaiswal D., Mikolajczyk M., Piccioli M., Winkelmann J. (2013) Molecular view of an electron transfer process essential for iron-sulfur protein biogenesis. Proc. Natl. Acad. Sci. U.S.A. 110, 7136–7141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hao Z., Li X., Qiao T., Du R., Hong L., Fan D. (2006) CIAPIN1 confers multidrug resistance by upregulating the expression of MDR-1 and MRP-1 in gastric cancer cells. Cancer Biol. Ther. 5, 261–266 [DOI] [PubMed] [Google Scholar]
- 47. Demon D., Van Damme P., Vanden Berghe T., Vandekerckhove J., Declercq W., Gevaert K., Vandenabeele P. (2009) Caspase substrates: easily caught in deep waters? Trends Biotechnol. 27, 680–688 [DOI] [PubMed] [Google Scholar]
- 48. Soler N., Craescu C. T., Gallay J., Frapart Y. M., Mansuy D., Raynal B., Baldacci G., Pastore A., Huang M. E., Vernis L. (2012) A S-adenosylmethionine methyltransferase-like domain within the essential, Fe-S-containing yeast protein Dre2. FEBS J. 279, 2108–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang Y., Lyver E. R., Nakamaru-Ogiso E., Yoon H., Amutha B., Lee D. W., Bi E., Ohnishi T., Daldal F., Pain D., Dancis A. (2008) Dre2, a conserved eukaryotic Fe/S cluster protein, functions in cytosolic Fe/S protein biogenesis. Mol. Cell. Biol. 28, 5569–5582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Netz D. J., Stümpfig M., Doré C., Mühlenhoff U., Pierik A. J., Lill R. (2010) Tah18 transfers electrons to Dre2 in cytosolic iron-sulfur protein biogenesis. Nat. Chem. Biol. 6, 758–765 [DOI] [PubMed] [Google Scholar]
- 51. Vernis L., Facca C., Delagoutte E., Soler N., Chanet R., Guiard B., Faye G., Baldacci G. (2009) A newly identified essential complex, Dre2-Tah18, controls mitochondria integrity and cell death after oxidative stress in yeast. PLoS One 4, e4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Saito Y., Shibayama H., Tanaka H., Tanimura A., Matsumura I., Kanakura Y. (2011) PICOT is a molecule which binds to anamorsin. Biochem. Biophys. Res. Commun. 408, 329–333 [DOI] [PubMed] [Google Scholar]
- 53. Cha H., Kim J. M., Oh J. G., Jeong M. H., Park C. S., Park J., Jeong H. J., Park B. K., Lee Y. H., Jeong D., Yang D. K., Bernecker O. Y., Kim do H., Hajjar R. J., Park W. J. (2008) PICOT is a critical regulator of cardiac hypertrophy and cardiomyocyte contractility. J. Mol. Cell. Cardiol. 45, 796–803 [DOI] [PMC free article] [PubMed] [Google Scholar]