

Abstract
Lizards, which are amniote vertebrates like humans, are able to lose and regenerate a functional tail. Understanding the molecular basis of this process would advance regenerative approaches in amniotes, including humans. We have carried out the first transcriptomic analysis of tail regeneration in a lizard, the green anole Anolis carolinensis, which revealed 326 differentially expressed genes activating multiple developmental and repair mechanisms. Specifically, genes involved in wound response, hormonal regulation, musculoskeletal development, and the Wnt and MAPK/FGF pathways were differentially expressed along the regenerating tail axis. Furthermore, we identified 2 microRNA precursor families, 22 unclassified non-coding RNAs, and 3 novel protein-coding genes significantly enriched in the regenerating tail. However, high levels of progenitor/stem cell markers were not observed in any region of the regenerating tail. Furthermore, we observed multiple tissue-type specific clusters of proliferating cells along the regenerating tail, not localized to the tail tip. These findings predict a different mechanism of regeneration in the lizard than the blastema model described in the salamander and the zebrafish, which are anamniote vertebrates. Thus, lizard tail regrowth involves the activation of conserved developmental and wound response pathways, which are potential targets for regenerative medical therapies.
Introduction
Regeneration of appendages in the adult is observed in a number of vertebrates, including in the lizard tail, the salamander limb and tail [1], and the zebrafish caudal fin [2]. Molecular and cellular analyses in these model organisms are beginning to reveal conserved versus divergent mechanisms for tissue regeneration [3]–[7], which impacts the translation of these findings to human therapies. Regeneration in newts is associated with proteins specific to urodele amphibians, casting doubt on the conservation of these regenerative pathways with other vertebrates [7]. In addition, muscle formation during limb regeneration differs between newts and the axolotl [8]. Mammals possess some neonatal regenerative capabilities, including mouse and human digit tip regeneration [9], [10] and heart regeneration in the mouse [11], but these processes are limited in the adult organism [12]. Lizards are capable of regrowing appendages, and as amniote vertebrates, are evolutionarily more closely related to humans than other models of regeneration, e.g., salamander and zebrafish. An examination of the genetic regulation of regeneration in an amniote model will advance our understanding of the conserved processes of regeneration in vertebrates, which is relevant to develop therapies in humans.
In response to threats, lizards have evolved the ability to autotomize, or self-amputate, their tails and regenerate a replacement (Figure 1A) [13], [14]. The patterning and final structure of the lizard tail is quite distinct between embryonic development and the process of regeneration [15], [16]. Whereas the original tail skeleton and muscular groups are segmentally organized, reflecting embryonic patterning, the regenerated tail consists of a single unsegmented cartilaginous tube surrounded by unsegmented muscular bundles [15], [16]. In addition, the segmental organization of the spinal cord and dorsal root ganglia in the original tail are absent in the replacement, with regenerated axons extending along the length of the endoskeleton [17], [18]. While the regenerative process in lizards has been described previously [14]–[16], [19], [20], both the source of regenerating tissue and the cellular and molecular mechanisms that are activated during the regenerative process remain unclear. Dedifferentiation has been proposed to be a major source of proliferating cells in the anamniote salamander blastema model [21]. However, no clear evidence of dedifferentiation has been identified in tail regeneration in the lizard, an amniote vertebrate [14], [15], [19], [20]. A temporal-spatial gradient of tissue patterning and differentiation along the regenerating tail axis has been described [14], [19], [20].
Figure 1. Overview of the stages of lizard tail regeneration.

(A) Anolis carolinensis lizard with a regenerating tail (distal to arrow). (B-E) Histology of the 10 dpa (B), 15 dpa (C), 20 dpa (D), and 25 dpa (E) regenerating tail by Gomori's trichrome stain, with which connective tissues and collagen stain green-blue, muscle, keratin, and cytoplasm stain red, and nuclei are black. (F) Immunohistochemistry of myosin heavy chain in a 25 dpa regenerating tail using the MY-32 antibody. e, wound epithelium; v, blood vessels; m, muscle; ct, cartilaginous tissue. Composites: B-F. Scale bars in black: 200 µm.
The green anole lizard, Anolis carolinensis, is an emerging model organism, and has provided insights in the fields of evolution and development [22], [23], population genetics [24], [25], reproductive physiology [26], behavior [27], and functional morphology [28]. Large-scale gene expression analyses of biological processes such as tail regeneration in the green anole have previously been limited by a lack of genomic resources. However, the A. carolinensis genome was recently made available [29]. In addition, our group has generated a robust genome annotation based on 14 deep transcriptomes using both directional and non-directional RNA-Seq data from a diverse number of tissues [30]. These genomic resources provide a platform for transcriptome-wide analysis of the genes involved in regeneration in the green anole. Here we describe, to our knowledge, the first transcriptomic analysis of lizard tail regeneration.
Materials and Methods
Animals and collection of regenerating tail samples
Animals were collected and maintained in strict accordance with Protocol Number 12-1247R approved by the Institutional Animal Care and Use Committee at Arizona State University. Adult A. carolinensis lizards were purchased from Marcus Cantos Reptiles (Fort Myers, FL) or Charles D. Sullivan Co., Inc. (Nashville, TN). Animals were housed as previously described [15], [16]. Autotomy was induced by applying pressure to the tail until it was released. Animal health was monitored following autotomy. We collected 5 biological replicates of regenerating tail sections at 25 days post autotomy (dpa). Regenerating tails (n = 5) at 25 dpa were divided into five sections (approximately 1 mm each) for RNA-Seq analysis.
RNA-Seq
RNA-Seq of the lizard embryos has been described previously [22]. Total RNA was isolated from tissue samples, including 25 dpa regenerating tail (n = 5) and satellite cells (n = 3; mirVana miRNA Isolation Kit total RNA protocol only, Ambion). The Ovation RNA-Seq kit (NuGEN) was used to synthesize double stranded cDNA. Paired-end sequencing libraries were then generated using manufacturer protocols and sequenced on an Illumina HiSeq 2000. For our analysis, 4 of the 5 regenerating tail replicates were multiplexed together and 2 of the 3 satellite cell replicates were multiplexed together.
Bioinformatic analysis
RNA-Seq reads were trimmed to eliminate nucleotide bias where necessary. Trimmed reads were then mapped to the A. carolinensis genome [29] using Bowtie2.1.0 and TopHat2.0.8 with the ASU_Acar_v2.2.1 annotation revised from Eckalbar et al., 2013 [30] (Table S1). For Cuffdiff analysis, TopHat aligned reads were assembled using Cufflinks2.1.1 and genes with differential expression were identified using Cuffdiff2.1.1 with the following options: —upper-quartile-norm —multi-read-correct. Cuffdiff data were then imported into CummeRbund [31], [32]. For DESeq2 analysis, raw counts were generated from TopHat aligned reads using HTSeq and normalized for library size in DESeq2 [33]–[35]. In order to identify variant genes using DESeq2, normalized data were fitted to a negative binomial general linear model and adjusted for multiple testing using the Benjamini-Hochberg method, and a likelihood ratio test was performed. CummeRbund and DESeq2 are part of the Bioconductor set of software packages [36], which use the R statistical programming environment (http://www.R-project.org). P-values for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of differentially expressed genes were generated using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) functional analysis tool [37], [38]. Significant GO terms (p<0.05) were mapped with the REViGO online tool (http://revigo.irb.hr), which removes redundant GO terms and visualizes the semantic similarity of remaining terms [39]. For all heatmaps, genes were clustered by Jensen-Shannon divergence of the log10(FPKM+1) value.
A. carolinensis genome annotation revision
An annotation of the A. carolinensis genome was reported using fourteen deep transcriptomes (ASU Acar v2.1) [30]. We further revised this annotation as follows: RNA-Seq data was assembled using the ABySS and Trans-ABySS pipeline [40]–[42]. Each of the 25 dpa regenerating tail sections was assembled individually in ABySS using every 5th kmer ranging from 26 bp to 96 bp. These assemblies were then combined using trans-ABySS to create a merged assembly with reduced redundancy. This merged assembly was then mapped to the genome using BLAT inside trans-ABySS. De novo assembled contigs were then filtered to require at least 90% coverage of the contig to the genome and to require at least one 25 bp gap. Seqclean was first used to remove Illumina adapters and any contaminants from the UniVec databases from the de novo assembled transcripts and the EST libraries. The cleaned de novo assembled transcripts from ABySS/Trans-ABySS were then assembled using the PASA reference genome guided assembly, and PASA alignment and assembly was executed using default parameters [43]–[46]. The PASA assemblies were then used to update the ASU Acar v2.1 annotations inside PASA to v2.2. The annotation was further updated to v2.2.1 with a subset of manual annotations.
Isolation of satellite cells from A. carolinensis
Lizard satellite cell isolation was adapted from mammalian [47]–[49] and avian [50], [51] methods. Following euthanasia, large limb muscle groups were dissected in PBS and minced. Cells were separated by protease treatment and suspensions were initially plated to remove adherent fibroblasts and other debris. Satellite cells remaining in suspension were then collected and plated onto Matrigel-coated tissue culture plates in growth medium (Ham's F-10, 20% FBS, 100 µg/mL penicillin, 100 µg/mL streptomycin, 40 µg/mL gentamicin, 20 ng/mL bFGF) at 30°C in a 5% CO2 humidified chamber. While a number of conditions were tested, 30°C was the optimal temperature identified.
Histological analysis
For paraffin sectioning, regenerated tails were fixed and embedded as described previously [15]. Embedded tails were sectioned into 20 µm sections using a CM1950UV Leica Cryostat and placed on HistoBond slides. Paraffin-embedded tissue sections were stained according to hematoxylin-eosin or Gomori's trichrome and mounted in Permount as described previously [15]. Hematoxylin stains nuclei and nucleoli blue and eosin stains cytoplasmic and extracellular matrix proteins pink/red, while hydrophobic cells such as adipocytes and myelin will remain clear. With Gomori's trichrome stain, connective tissues and collagen appear green-blue; muscle, keratin, and cytoplasm are red; and nuclei are black.
Immunohistochemistry
Paraffin-embedded tissue sections were deparaffinized, rehydrated, and bathed in sodium citrate buffer (pH 6.0). Cells were fixed in 100% methanol. Tissue sections and cells were stained using the Histostain-SP Broad Spectrum kit (Invitrogen) as follows: Tissue sections and cells were blocked in serum, incubated with primary antibody (MY-32, Sigma Aldrich, MFCD00145920; PCNA, Santa Cruz Biotechnology, sc-7907; MCM2, Abcam, ab4461) incubated with secondary antibody, and incubated with HRP-strepavidin complex, with blocking and antibody incubations at 37°C. Tissue sections and cells were counterstained with hematoxylin and mounted in Permount (Fisher Scientific).
Immunofluorescence
Cells were fixed in 100% methanol, blocked in serum, incubated with PAX7 antibody (Developmental Studies Hybridoma Bank), and incubated with secondary antibody, with blocking and antibody incubations at 37°C. Slides were then counterstained with DAPI.
Data Access
RNA-Seq data for the lizard embryo samples, which have been previously reported [22], are deposited in at the National Center for Biotechnology Information (NCBI), under BioProject PRJNA149661. RNA-Seq data for the lizard tail regeneration and satellite cell samples are deposited under BioProject PRJNA253971.
Results
Histology of early regenerative stages
Progressively increasing tissue patterning and differentiation are evident in the early regenerative stages of the lizard tail. The first 10 days are characterized by wound healing (0–10 days post autotomy (dpa); Figure 1B). By 10 dpa, a wound epithelium has formed over the autotomized stump and blood vessels have formed immediately below. There was no appreciable outgrowth at this stage. Outgrowth begins after the wound epithelium forms and is characterized by early growth of the ependyma from the spinal cord into the surrounding mesenchymal tissue (10–15 dpa). By 15 dpa, there was noticeable outgrowth of highly vascularized tissue and myofibers began to form (Figure 1C). With continued tail outgrowth, the central cartilage tube and surrounding skeletal muscle began to differentiate (15–20 dpa; Figure 1D). Note that the tip of the tail remains vascular (10–20 dpa, Figure 1B-D). By 25 dpa, further lengthening of the regenerating tail was observed, along with formation of muscle and cartilage surrounding the ependymal core (Figure 1E). Further outgrowth with continued tissue differentiation is evident post-25 dpa, and there is no significant outgrowth after 60 dpa [15]. In fact, by 25 dpa, myosin heavy chain (MHC) positive skeletal muscle was present along the length of the developing tail, except at the very distal tip (Figure 1F). Spatially, there is an increase in patterning and differentiation along the regenerating tail was observed at early outgrowth stages (e.g., 15–25 dpa, Figure 1C-E), with differences in tissue organization particularly evident along the proximal-distal axis.
Sequencing and differential expression testing of regenerating tail transcripts
To identify differentially expressed genes along the proximal-distal axis of regenerating tails, we carried out RNA-Seq analysis on five tails at 25 dpa (Table S2). Tails were sectioned into five segments of equal length (Figure 2A). RNA-Seq analysis identified 326 differentially expressed genes with p<0.05 after correcting for multiple testing using Cuffdiff2 [31], [52], 302 of which have mammalian orthologs (Figure 2B; Table S3). Data were also analyzed by DESeq2 [33], [34], which yielded 264 differentially expressed genes, 252 of which have mammalian orthologs (Figure 2C; Table S4). These Cuffdiff2 differentially expressed genes clustered into two major groups, representing genes elevated towards the proximal base (Cluster I, Figure 2B) or the distal tip (Cluster II, Figure 2B).
Figure 2. Transcriptomic analysis of gene expression in the 25 dpa regenerating lizard tail.

(A) 25 dpa regenerated tail tissue was divided into five equal sized segments (S1-S5) with S1 representing the most distal regenerating tip, and total RNA was extracted for RNA-Seq analysis. (B) A heatmap showing 326 genes that were differentially expressed, i.e., displayed significant differences between any two segments in the regenerating tail as determined by Cuffdiff (p<0.05). Genes were clustered by Jensen-Shannon divergence of the log10(FPKM+1) value into two major groups, as shown in the dendrogram on the left. 129 genes displayed increased expression distally towards the tail tip (Cluster II) while 197 displayed increased expression proximally (Cluster I). This clustering also demonstrated that the distal-most regenerating tail tip (S1) was the outlier among these samples. (C) Venn diagram of differentially expressed genes identified by DESeq2 and Cuffdiff2. (D-E) A treemap overview of differentially expressed genes in (D) Cluster I and (E) Cluster II based on representative Gene Ontology Biological Processes. The relative sizes of the treemap boxes are based on the |log10(p-value)| of the respective GO term. Related terms are visualized with the same color, with the representative category for each color group denoted in the legend.
Differential expression of genes involved in developmental and repair mechanisms in the regenerating tail
Our RNA-Seq analysis identified Gene Ontology (GO) groups associated with the differentiation of tissues present in the proximal regenerating tail, predominantly those that are specific to skeletal muscle (Figure 2D; Figure S1A; Table 1; Table S5). Sarcomeric proteins, including myosin heavy chains and actinins, were elevated in the proximal tail. This pattern of expression was validated by the presence of myosin heavy chain positive muscle fibers (Figure 1F). Myogenic regulatory factors associated with muscle growth and repair were also elevated in the proximal tail. These include the transcription factors pax7, mohawk (mkx), and tcf15, which are expressed in myogenic stem/progenitor cells [53]–[55], NFATc1, which regulates muscle hypertrophy [56], and the TGFβ family member myostatin (mstn), which modulates muscle mass [57] (Anolis Gene Nomenclature Committee standards used for gene symbols; [58]). Also, the MADS box factor mef2c, and the myogenic regulatory factor myod1, which synergize to activate muscle specific gene transcription, were elevated [59]. As growth and repair of skeletal muscle in vertebrates normally relies on the expansion and differentiation of muscle-specific progenitor cells, the enrichment for genes associated with the regulation of this population predicts a similar mechanism of muscle growth and repair occurring in a zone of active regeneration. Furthermore, the increase in mkx transcription raises the possibility of a coordinated growth between tendons and muscle in the regenerating tail, given that the orthologous gene is required for growth and repair in mammals [60].
Table 1. Selected Genes Ontology categories represented along the regenerating tail axis.
| Category | GO Term | Description | Count | P-value | Genes | |
| Cluster I | ||||||
| myogenesis | GO:0006936 | muscle contraction | 30 | 6.63E-29 | mybpc2, tnnc2, tnnc1, myl3, mybpc1, mybpc3, myl1, pgam2, myot, des, myom2, myl6b, myom1, chrna1, scn5a, dtna, kcnma1, actc1, acta1, actn2, myh6, tnni2, trdn, tnnt3, tnnt1, ryr1, stbd1, chrne, casq2, chrng | |
| GO:0007517 | muscle organ development | 28 | 3.44E-22 | mef2c, myod1, myl2, tnnc1, myl3, mybpc3, myl1, trim72, speg, myl6b, pax7, obsl1, mkx, mkl2, chrna1, actc1, acta1, mstn, mylpf, myh6, csrp3, flnb, murc, neb, xirp1, itga7, vgll2, tcf15 | ||
| GO:0007519 | skeletal muscle tissue development | 9 | 3.73E-07 | myod1, acta1, myl3, myl6b, pax7, mylpf, vgll2, chrna1, csrp3 | ||
| GO:0042692 | muscle cell differentiation | 11 | 4.86E-07 | myod1, actc1, acta1, xirp1, myl2, speg, lgals1, obsl1, myh6, mkl2, chrna1 | ||
| GO:0050881 | musculoskeletal movement | 6 | 1.14E-06 | tnnt3, tnnt1, tnnc2, tnnc1, chrna1, tnni2 | ||
| GO:0030029 | actin filament-based process | 14 | 1.28E-06 | actc1, tnxb, myl2, acta1, myl1, pdlim3, myh6, gas7, flnb, xirp1, xirp2, myl6b, limch1, obsl1 | ||
| GO:0007155 | cell adhesion | 21 | 3.41E-05 | hapln1, tnxb, mybpc2, clstn2, egfl6, lpp, mybpc1, col22a1, mybpc3, col28a1, mgp, actn2, col2a1, actn3, ecm2, col9a1, itga7, acan, susd5, col11a2, thbs4 | ||
| GO:0001501 | skeletal system development | 12 | 4.79E-04 | bmp3, col9a1, col9a2, tbx15, lect1, clec3a, pax7, acan, mgp, col2a1, col11a2, tcf15 | ||
| GO:0030198 | extracellular matrix organization | 7 | 7.29E-04 | csgalnact1, tnxb, adamts20, acan, col2a1, col11a2, ecm2 | ||
| GO:0030705 | cytoskeleton-dependent intracellular transport | 4 | 0.0166 | actc1, myl6b, myl1, myh6 | ||
| GO:0006873 | cellular ion homeostasis | 11 | 0.0055 | kcnma1, jph2, xirp1, pygm, atp2a1, ryr1, chrna1, chrne, csrp3, sypl2, chrng | ||
| chondrogenesis | GO:0051216 | cartilage development | 8 | 1.10E-05 | bmp3, col9a1, lect1, pax7, acan, mgp, col2a1, col11a2 | |
| GO:0002062 | chondrocyte differentiation | 4 | 7.90E-04 | col9a1, acan, col2a1, col11a2 | ||
| GO:0001502 | cartilage condensation | 3 | 0.0162 | acan, mgp, col2a1 | ||
| musculoskeletal activity | GO:0043462 | regulation of ATPase activity | 5 | 1.82E-05 | tnnt3, myl3, tnnc1, mybpc3, myh6 | |
| GO:0006029 | proteoglycan metabolic process | 4 | 0.0099 | csgalnact1, lect1, acan, col2a1 | ||
| biological adhesion | GO:0022610 | biological adhesion | 21 | 3.48E-05 | hapln1, tnxb, mybpc2, clstn2, egfl6, lpp, mybpc1, col22a1, mybpc3, col28a1, mgp, actn2, col2a1, actn3, ecm2, col9a1, itga7, acan, susd5, col11a2, thbs4 | |
| Cluster II | ||||||
| wound response | GO:0009611 | response to wounding | 10 | 0.0040 | pcsk1, scube1, pdgfra, pla2g7, entpd1, ptx3, mdk, igfbp4, f2r, spp1 | |
| GO:0009725 | response to hormone stimulus | 8 | 0.0059 | cga, pcsk1, krt19, tnfrsf11b, bsg, th, pdgfra, spp1 | ||
| GO:0007223 | Wnt receptor signaling pathway, calcium modulating pathway | 3 | 0.0067 | wnt5a, wnt16, ror2 | ||
| GO:0016055 | Wnt receptor signaling pathway | 5 | 0.0079 | dkk2, wnt5a, wnt16, ror2, wif1 | ||
| GO:0007166 | cell surface receptor signaling pathway | 20 | 0.0106 | wnt5a, cga, edn3, fgfr4, il1r1, wnt16, gpr158, bsg, maml2, ptpn22, thy1, dkk2, ednra, or5v1, pdgfra, ror2, wif1, pdgfc, entpd1, f2r | ||
| GO:0010033 | response to organic substance | 11 | 0.0098 | ednra, cga, pcsk1, krt19, il1r1, tnfrsf11b, bsg, th, pdgfra, f2r, spp1 | ||
| GO:0006954 | inflammatory response | 6 | 0.0433 | scube1, pla2g7, ptx3, igfbp4, f2r, spp1 | ||
| hormonal regulation | GO:0051050 | positive regulation of transport | 7 | 0.0020 | ednra, edn3, pcsk1, rab8b, ptx3, f2r, thy1 | |
| GO:0032844 | regulation of homeostatic process | 5 | 0.0046 | ednra, tnfrsf11b, f2r, spp1, thy1 | ||
| GO:0006590 | thyroid hormone generation | 2 | 0.0350 | cga, dio2 | ||
| embryonic morphogenesis | GO:0001501 | skeletal system development | 9 | 5.81E-04 | wnt5a, tnfrsf11b, pdgfra, ror2, mepe, cbfb, igfbp4, spp1, twist1 | |
| GO:0035295 | tube development | 7 | 0.0019 | wnt5a, ednra, fgfr4, sall1, pdgfra, ptk7, twist1 | ||
| GO:0048598 | embryonic morphogenesis | 7 | 0.0096 | wnt5a, sall4, th, ptk7, ror2, twist1, ptprq | ||
| immune response | GO:0006030 | chitin metabolic process | 2 | 0.0407 | chi3l1, chit1 |
Our transcriptome analysis identified multiple genetic pathways activated towards the tip of the regenerating tail. Genes differentially elevated at the tip were enriched for GO categories related to i.) wound response, ii.) hormonal regulation, and iii.) embryonic morphogenesis (Figure 2E; Figure S1B; Table 1; Table S6). Wound and inflammatory response genes elevated in the distal regenerating tail include igfbp4, mdk, ptx3, and pdgfra. Mouse Ptx3 is required for fungal resistance [61], and Mdk plays a role in angiogenesis [62]. Hormonal and homeostatic regulation genes included those involved in thyroid hormone generation, such as cga and dio2. Thyroid hormone plays a critical role in neuromuscular growth, both during normal development and in repair after injury. Dio2 has been shown to co-regulate myogenesis and muscle regeneration in the mouse [63]. In the rat model, triiodothyronine (T3) treatment after sciatic nerve injury has been shown to enhance reinnervation of muscles [64]. In the Xenopus laevis tadpole, thyroid hormone is critical for limb development during metamorphosis, where limb muscle growth, innervation of the limb, cartilage growth, and skin development are all thyroid hormone-dependent [65]. Genes involved in homeostatic regulation and vascular development include ednra and edn3, which are members of the endothelin family and regulate vasoconstriction and cell proliferation [66], the thrombin receptor f2r, which promotes vascular development by negatively regulating hematopoietic differentiation of mouse embryonic stem cells [67], and thy1, which is a marker of angiogenesis [68]. The wnt5a ligand and its receptor, ror2, were both significantly expressed at the tip, indicating non-canonical Wnt signaling, which can promote chondrogenesis [69], [70]. Skeletal system development genes elevated in the regenerating tail include the basic helix-loop-helix transcription factor twist1, which regulates a number of pathways, including FGF, by chromatin modification via histone acetyltransferases [71].
Differentially expressed genes analyzed for Kyoto Encyclopedia of Genes and Genomes (KEGG) categories (p<0.05) identified axon guidance and neural development genes, including slit homolog 2 (slit2), actin binding LIM protein family member 2 (ablim2), and netrin receptor unc-5 homolog C (unc5c) (Table 1; Table S7). KEGG groups enriched in the regenerating tail also include the Wnt and MAPK/FGF signaling pathways. FGF signaling plays a key role in developmental patterning, proliferation, and differentiation [72]. Differentially expressed MAPK/FGF pathway genes at the tail tip include pdgfra, il1r1, and cdc42 while mef2c, cacnb1, cacna2d1, flnb, flnc, and fgfr13 are elevated at the proximal region of the regenerating tail (Figure 3A). A number of recent reports from mouse digit tip and salamander limb regeneration identified Wnt pathway involvement [3], [4], [10]. Wnt signaling promotes the differentiation of embryonic stem cells as well as cells from skeletal muscle, osteogenic, and cardiogenic lineages [73]. The tip to the middle regions of the regenerating tail are enriched with Wnt inhibitors, including dkk2, igfbp4, wif1, and sgfrp2 (Figure 3B). The expression of soluble Wnt inhibitors from this region could create a proximal-distal gradient of Wnt signaling that is necessary to maintain the actively growing zone of the regenerating tail in a proliferative, undifferentiated state.
Figure 3. MAPK/FGF and Wnt pathway genes differentially expressed in the 25 dpa regenerating lizard tail.
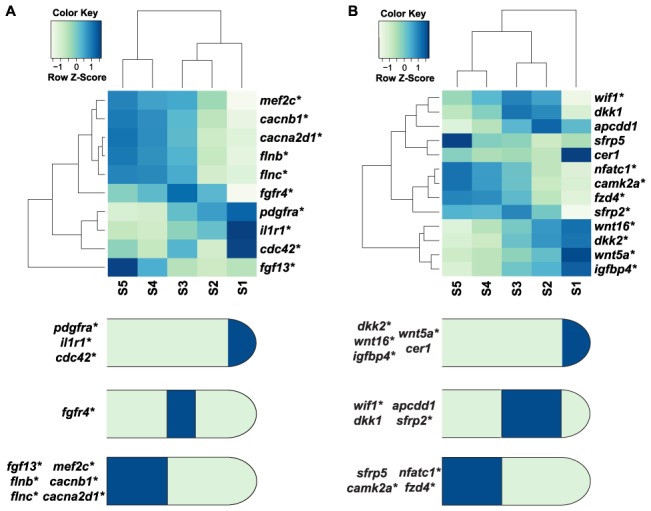
(A, B) Based on RNA-Seq analysis described in Figure 2, the heatmaps show the 10 MAPK/FGF pathway genes (A), or 9 Wnt pathway genes (B) defined by KEGG, that were differentially expressed, i.e., displayed significant differences between any two segments in the regenerating tail as determined by Cuffdiff2 (p<0.05), along with previously identified Wnt inhibitors. A diagram summarizing the tail segment(s) with highest expression level for each MAPK/FGF (A) or Wnt (B) pathway gene is also shown. Differentially expressed genes are denoted with an asterisk.
Novel and uncharacterized transcripts in the regenerating tail
We sought to characterize the 22 differentially expressed genes, representing 29 transcript isoforms, without clear orthology, i.e., BLAST alignment scores against the nonredundant protein database were either E≥1.0, identity was ≤50%, or no match was identified. These transcripts could potentially be protein-coding genes specific to squamate reptiles, either novel or highly divergent within the squamate lineage, or could represent noncoding RNA species. Transcripts were queried against the protein family (Pfam) [74] and RNA family (Rfam) [75] databases, and coding potential was evaluated using the Coding-Non-Coding Index (CNCI; [76]), which evaluates coding potential by profiling adjoining trinucleotide sequences (Table 2). Four transcripts were identified as retrotransposons, including the gag-pol polyprotein and RNA-directed DNA polymerase from mobile element jockey-like, which are enriched in the proximal regenerating tail. Of the remaining transcripts, 3 were predicted as protein-coding and 22 were characterized as non-coding by the CNCI. The protein-coding gene ASU_Acar_G.15880, which is differentially expressed in the proximal regenerating tail, has a DUF4585 (domain of unknown function) domain, and orthologous genes found in the king cobra (Ophiophagus hannah; GenBank: ETE69491.1) genome, the green sea turtle (Chelonia mydas; GenBank: EMP32806.1; NCBI: XP_007063098.1) genome, and the axolotl (Ambyostoma mexicanum) transcriptome. The 2 remaining protein-coding transcripts were not matched to any known domains in the Pfam database. Of the 22 non-coding transcripts, we identified 2 differentially expressed genes in the proximal tail categorized within the miRNA precursor families miR-133 and miR-324. miR-133 acts in a negative feedback loop with serum response factor (SRF) to promote myoblast differentiation in vitro, and suppresses BMP2-induced osteogenesis by targeting Runx2 [77], [78]. The remaining 20 non-coding transcripts represent potential modulators of genes down-regulated in regeneration. In summary, these unidentified transcripts represent novel protein-coding genes, long non-coding RNAs, and microRNAs that may regulate the regenerative process in concert with identified genes and signaling pathways.
Table 2. Novel and uncharacterized transcripts in the regenerating tail.
| Gene ID | Transcript ID | CNCI score | CNCI classification | Length (bp) | Longest ORF (bp) | Domain/Homology | Highest Section | |
| Predicted RNA only | ||||||||
| ASU_Acar_G.1063 | ASU_Acar_T.1063.1 | 0.0 | non-coding | 216 | 213 | lncRNA | S1 | |
| ASU_Acar_G.14483 | ASU_Acar_T.14483.1 | −0.0029 | non-coding | 698 | 153 | lncRNA | S4 | |
| ASU_Acar_G.14483 | ASU_Acar_T.14483.2 | −0.0784 | non-coding | 1256 | 195 | lncRNA | S4 | |
| ASU_Acar_G.14483 | ASU_Acar_T.14483.5 | −0.0029 | non-coding | 712 | 153 | lncRNA | S2 | |
| ASU_Acar_G.14483 | ASU_Acar_T.14483.7 | −0.0029 | non-coding | 1430 | 195 | lncRNA | - | |
| ASU_Acar_G.17546 | ASU_Acar_T.17546.1 | −0.0390 | non-coding | 225 | 222 | lncRNA | S1 | |
| ASU_Acar_G.17964 | ASU_Acar_T.17964.1 | −0.1550 | non-coding | 219 | 123 | lncRNA | S4 | |
| ASU_Acar_G.5235 | ASU_Acar_T.5235.1 | 0.0 | non-coding | 216 | 213 | lncRNA | S3 | |
| ASU_Acar_G.7180 | ASU_Acar_T.7180.1 | −0.0038 | non-coding | 243 | 240 | lncRNA | S5 | |
| ASU_Acar_G.8849 | ASU_Acar_T.8849.1 | −0.0532 | non-coding | 291 | 288 | lncRNA | S4 | |
| ASU_Acar_G.8944 | ASU_Acar_T.8944.1 | −0.2007 | non-coding | 279 | 276 | lncRNA | S1 | |
| ASU_Acar_G.20175 | ASU_Acar_T.20175.1 | −0.0204 | non-coding | 261 | 258 | lncRNA | S1 | |
| ASU_Acar_G.1922 | ASU_Acar_T.1922.1 | −0.0114 | non-coding | 2286 | 213 | mir-133 | S5 | |
| ASU_Acar_G.19355 | ASU_Acar_T.19355.1 | −0.0064 | non-coding | 2549 | 219 | mir-324 | S5 | |
| ASU_Acar_G.10886 | ASU_Acar_T.10886.1 | −0.1770 | non-coding | 637 | 384 | ncRNA | S1 | |
| ASU_Acar_G.13829 | ASU_Acar_T.13829.1 | −0.0563 | non-coding | 189 | 186 | ncRNA | S4 | |
| ASU_Acar_G.14483 | ASU_Acar_T.14483.4 | 0.0 | non-coding | 183 | 180 | ncRNA | S3 | |
| ASU_Acar_G.14483 | ASU_Acar_T.14483.6 | −0.0073 | non-coding | 459 | 456 | ncRNA | S4 | |
| ASU_Acar_G.14791 | ASU_Acar_T.14791.1 | 0.0 | non-coding | 199 | 114 | ncRNA | S1 | |
| ASU_Acar_G.1721 | ASU_Acar_T.1721.1 | −0.0170 | non-coding | 192 | 189 | ncRNA | S2 | |
| ASU_Acar_G.2935 | ASU_Acar_T.2935.1 | 0.0000 | non-coding | 195 | 192 | ncRNA | S1 | |
| ASU_Acar_G.3586 | ASU_Acar_T.3586.1 | 0.0000 | non-coding | 195 | 192 | ncRNA | S1 | |
| Protein Coding - Not Described | ||||||||
| ASU_Acar_G.15880 | ASU_Acar_T.15880.1 | 0.1481 | coding | 14705 | 4992 | DUF4585 | S2 | |
| ASU_Acar_G.14483 | ASU_Acar_T.14483.3 | 0.0510 | coding | 3395 | 2766 | unknown | S5 | |
| ASU_Acar_G.19198 | ASU_Acar_T.19198.1 | 0.0293 | coding | 264 | 261 | unknown | S1 | |
| Retrotransposons | ||||||||
| ASU_Acar_G.14133 | ASU_Acar_T.14133.1 | 0.2166 | coding | 3618 | 3615 | gag-pol polyprotein | S5 | |
| ASU_Acar_G.591 | ASU_Acar_T.591.1 | 0.1336 | coding | 762 | 759 | gag-pol polyprotein | S3 | |
| ASU_Acar_G.591 | ASU_Acar_T.591.2 | −0.0102 | non-coding | 198 | 195 | gag-pol polyprotein | S5 | |
| ASU_Acar_G.4168 | ASU_Acar_T.4168.1 | 0.0918 | coding | 2010 | 1863 | rna-directed dna polymerase from mobile element jockey-like | S5 |
Comparison of regenerating tail with stem/progenitor cells and developing embryo
Tissue regeneration in the lizard tail requires a source of cells; these could be tissue-specific oligopotent or progenitor stem cells, as in mammalian tissue repair, since there is no evidence of dedifferentiation in the lizard as observed in the salamander [14], [15], [19], [20]. We analyzed the regenerated tail in comparison with lizard embryos and satellite cells; both are highly enriched for stem cell populations (Figure S2). We profiled the transcriptome of lizard embryos at the 28–38 somite pair stages [79]. At this stage, the embryo contains paraxial mesoderm, a multipotent cell source for skeletal muscle, cartilage, bone, and tendon. Satellite cells capable of differentiating into skeletal muscle in response to injury serve as progenitor/stem cells for adult muscle repair in mammals [80]. We isolated a PAX7 positive cell population from adult lizard skeletal muscle that was morphologically comparable to mouse satellite cells (Figure S3A). These cells differentiated into multinucleated, MHC positive myotubes (Figure S3B), and express many of the same lineage-specific genes (Figure 4A, Figure S3C-F). The lizard embryos and satellite cells each possess distinct gene expression signatures based on gene markers for mouse and human embryonic, hematopoietic, and mesenchymal stem cells and satellite cells. In contrast, these genes are expressed at low levels without a distinct proximal-distal pattern in the regenerating tail (Figure 4A-D). These data predict a role for stem cells distributed throughout the regenerating tail, instead of being localized to the distal tip with a distal-to-proximal gradient of differentiation within the tail. While there are genes elevated in the regenerating tail relative to the embryo and satellite cells, genes elevated in the regenerating tail tip are primarily involved in the formation of tissues specific to the tail such as keratin-associated beta protein, and genes elevated in the proximal regenerating tail are primarily involved in tissue differentiation (Figure S2, Tables S8-S9). The lack of intensity in the signal compared to the embryo and satellite cells could be due to stem cells comprising only a minority population in the regenerating tail.
Figure 4. The 25 dpa regenerating tail has limited relative expression of stem cell markers.
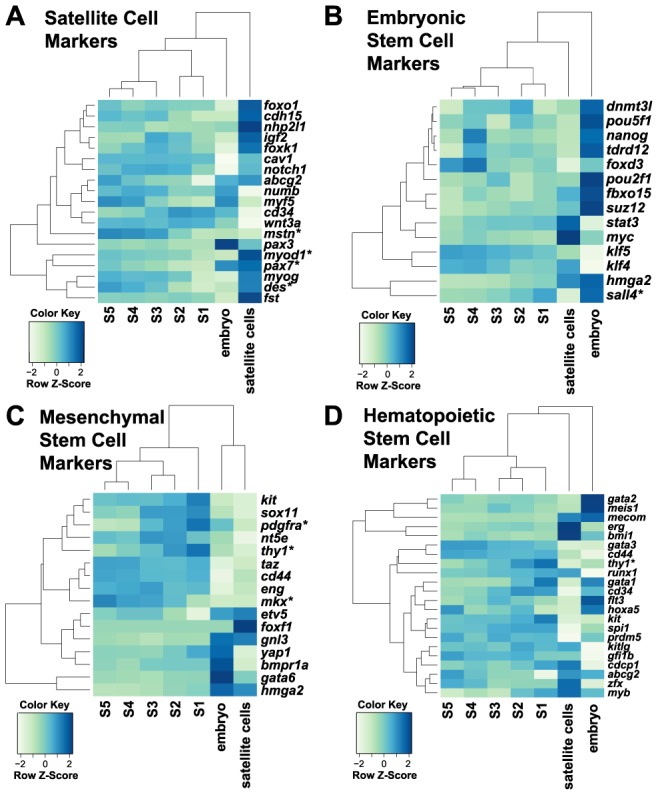
(A-D) Heatmap showing gene expression of satellite cell (A) and embryonic (B), mesenchymal (C), and hematopoietic stem cell markers in lizard embryos (n = 2), satellite cells (n = 3), and 25 dpa regenerating tail sections (n = 5). Differentially expressed genes along the regenerating tail axis are denoted with an asterisk.
Distributed pattern of cell proliferation in the regenerating tail
Proliferation and specification of progenitor cells is required for growth of the regenerating tail. While the regenerating tail did not express high levels of stem cell factors, selected progenitor/stem cell markers still displayed differential expression along the proximal-distal axis (Figure 4A-D). These genes included platelet-derived growth factor receptor pdgfra, which is expressed in subtypes of mesenchymal progenitor cells involved in muscle repair [81]. In addition, genes elevated in the tail tip include the kit ligand and sox11 transcription factor, and genes elevated towards the proximal tail included the previously discussed transcription factor mkx. To visualize the pattern of proliferating cells within the regenerating tail, we analyzed the distribution of minichromosome maintenance complex component 3 (MCM2) in the regenerating tail (Figure 5A-E). MCM2 positive cells are observed in distributed, discrete regions in the regenerating tail, including the condensing cartilage tube and ependymal core (Figure 5B-C) and in developing muscle (Figure 5D-E). A second marker of proliferation, proliferating cell nuclear antigen (PCNA), showed a similar pattern of expression, confirming that proliferating cells are distributed throughout the regenerating tail in comparison to low levels of proliferating cells in the original tail (Figure S4; Figure S5). This pattern of proliferation is corroborated by RNA-Seq analysis of proliferation markers along the regenerating tail (Figure 5F). No segment along the proximal-distal axis of the regenerating tail demonstrated elevated expression of these markers, indicating that there is no single growth zone.
Figure 5. Histological and RNA-Seq analysis of proliferation in the 25 dpa regenerating tail.

(A-E) MCM2 immunohistochemistry of the 25 dpa regenerating tail (brown nuclei), counterstained with hematoxylin (blue nuclei). (A) MCM2 is expressed throughout the regenerating tail, indicating a lack of a single proliferative zone. The condensing cartilage tube (B), ependymal core (C), developing muscles near the proximal base (D) and tip (E) of the regenerating tail are shown. (F) A heatmap showing gene expression of proliferative markers in the regenerating tail, the embryos, and satellite cells. DE genes along the regenerating tail axis are denoted with an asterisk. Composites: A. Scale bars in red: 200 µm (A) and 20 µm (B-E).
Discussion
While transcriptomic analysis has been carried out in anamniote regenerative models, including the zebrafish tail, the newt limb, and the axolotl limb [3], [4], [6], [7], the genetic profile of pathways activated in regeneration of amniote appendages has not been described. Through transcriptomic analysis of lizard tail regeneration, we have identified that genes in pathways involved in developmental processes, including myogenesis, chondrogenesis, and neurogenesis, as well as adult processes, such as wound and immune responses, and are differentially expressed along the regenerating tail axis. The Wnt pathway was significantly enriched along the regenerating lizard tail axis, and activation of this pathway has also been noted in salamander tail tip and mouse digit tip regeneration [3], [4], [10]. Specifically, the Wnt pathway members wnt5a and wif1 are differentially expressed in lizard as well as the salamander [3], [4]. The activation of Wnt signaling in two amniote lineages, mammals and squamate reptiles, as well as urodele amphibians supports a role for this pathway in regeneration that is conserved among tetrapod vertebrates. Transcriptomic analysis also revealed that genes involved in thyroid hormone generation (GO category GO:0006590; Table 1; Table S6) were differentially expressed, suggesting a regulatory connection between regeneration of the lizard tail and musculoskeletal transformations during amphibian metamorphosis. The lizard dio2 gene is the ortholog of deiodinase, iodothyronine, type I, which in mammals converts thyroxine prohormone (T4) to bioactive 3,3',5-triiodothyronine (T3) [82]. In Xenopus laevis, T3 is the key signal for the process of metamorphosis from tadpole to adult frog [83]. Many of the changes associated with metamorphosis are also observed in the remodeling of the tail stump and outgrowth of the lizard tail. The lizard cga gene is the ortholog of chorionic gonadotropin, alpha chain, which encodes the alpha chain of thyroid-stimulating hormone and other key hormones [84]. During tadpole metamorphosis, both thyroid hormone (TH) and thyroid-stimulating hormone (TSH) rise, despite the normal expectation that TH would down-regulate TSH [85]. Changes in TH regulation of TSH may also be altered in regeneration, which has not been studied in the lizard. It is possible that among the amniotes, the lizard retains genetic pathways associated with thyroid hormone regulation of metamorphosis in amphibian vertebrates. Similarly, we previously identified conserved features in Notch pathway regulation of lizard and amphibian development, specifically a gradient of hes6 expression in the presomitic mesoderm that was not observed in other amniote vertebrates and presumably lost [79]. Our transcriptomic analysis has highlighted the activation of multiple genetic pathways, sharing genes that have been identified as regulating development or wound response processes in other vertebrate model systems.
Developmental systems display different patterns of tissue outgrowth. For example, some tissues are formed from patterning from a localized region of a single multipotent cell type, such as the axial elongation of the trunk through production of somites from the presomitic mesoderm [86]. Other tissues are formed from the distributed growth of distinct cell types, such as the development of the eye from neural crest, mesenchymal, and placodal ectodermal tissue [87]. The regeneration of the amphibian limb involves a region of highly proliferative cells adjacent to the wound epithelium, the blastema, with tissues differentiating as they grow more distant from the blastema. However, regeneration of the lizard tail appears to follow a more distributed model. Stem cell markers and PCNA and MCM2 positive cells are not highly elevated in any particular region of the regenerating tail, suggesting multiple foci of regenerative growth. This contrasts with PNCA and MCM2 immunostaining of developmental and regenerative growth zone models such as skin appendage formation [88], liver development [89], neuronal regeneration in the newt [90], and the regenerative blastema [91], which all contain localized regions of proliferative growth. Skeletal muscle and cartilage differentiation occurs along the length of the regenerating tail during outgrowth; it is not limited to the most proximal regions. Furthermore, the distal tip region of the regenerating tail is highly vascular, unlike a blastema, which is avascular [92]. These data suggest that the blastema model of anamniote limb regeneration does not accurately reflect the regenerative process in tail regeneration of the lizard, an amniote vertebrate.
Regeneration requires a cellular source for tissue growth. Satellite cells, which reside along mature myofibers in adult skeletal muscle, have been studied extensively for their involvement in muscle growth and regeneration in mammals and other vertebrates [53], [55], [60], [80], [93]. For example, regeneration of skeletal muscle in the axolotl limb involves recruitment of satellite cells from muscle [8]. Satellite cells could contribute to the regeneration of skeletal muscle, and potentially other tissues, in the lizard tail. Mammalian satellite cells in vivo are limited to muscle, but in vitro with the addition of exogenous BMPs, they can be induced to differentiate into cartilage as well [80], [81]. High expression levels of BMP genes in lizard satellite cells could be associated with greater differentiation potential, and further studies will help to uncover the plasticity of this progenitor cell type.
In summary, we have identified a coordinated program of regeneration in the green anole lizard that involves both recapitulation of multiple developmental processes and activation of latent wound repair mechanisms conserved among vertebrates. However, the process of tail regeneration in the lizard does not match the dedifferentiation and blastema-based model as described in the salamander and zebrafish, and instead matches a model involving tissue-specific regeneration through stem/progenitor populations. The pattern of cell proliferation and tissue formation in the lizard identifies a uniquely amniote vertebrate combination of multiple developmental and repair mechanisms. We anticipate that the conserved genetic mechanisms observed in regeneration of the lizard tail may have particular relevance for development of regenerative medical approaches.
Supporting Information
Gene Ontology analysis of differentially expressed genes identified by both Cuffdiff2 and DESeq2. 130 genes were identified as differentially expressed by both methods (Figure 1B-C; Table S3; Table S4). (A-B) A treemap overview of differentially expressed genes in (A) Cluster I and (B) Cluster II based on representative Gene Ontology Biological Processes. The relative sizes of the treemap boxes are based on the |log10(p-value)| of the respective GO term. Related terms are visualized with the same color, with the representative category for each color group denoted in the legend.
(EPS)
Genes with high expression (>10-fold change) in the regenerating tail relative to the embryos and satellite cells. 44 differentially expressed genes had >10-fold change in S1 and S2 gene expression (FPKM) relative to the embryos and satellite cells (orange cluster) and 86 genes had >10-fold change in S4 and S5 (yellow cluster).
(EPS)
Satellite cells isolated from adult skeletal muscle express PAX7 and can differentiate into myotubes. (A-B) Detection of myosin heavy chain (MHC) in proliferating (A) and differentiated (B) A. carolinensis satellite cells. MHC was detected using MY-32 monoclonal antibody and HRP-conjugated anti-mouse secondary antibody with DAB stain. Immunofluorescence of lizard (C-F) and mouse (G-H) satellite cells. (C-D, G-H) PAX7 was detected using a monoclonal antibody and visualized by FITC-conjugated anti-mouse secondary antibody, and nuclei were stained with DAPI. (E-F) Cells with no primary antibody and FITC-conjugated anti-mouse secondary antibody only, and nuclei stained with DAPI.
(TIF)
Histological analysis of proliferation in the 25 dpa regenerating tail. (A-G) PCNA immunohistochemistry of the 25 dpa regenerating tail (brown nuclei), counterstained with hematoxylin (blue nuclei). (A) PCNA is expressed throughout the regenerating tail, indicating a lack of a single proliferative zone. The dermis near the proximal base (B) and tip (C) of the regenerating tail, condensing cartilage tube (D), ependymal core (E), and developing muscles near the proximal base (F) and tip (G) of the regenerating tail all show PCNA positive cells.
(TIF)
Immunohistochemistry shows few proliferating cells in the original tail. (A-D) Proliferating cell nuclear antigen (PCNA) immunohistochemistry of the original tail (brown nuclei), counterstained with hematoxylin (blue nuclei). (A) Transverse section of the original tail. (B-D) There are limited PCNA-positive cells in the centrum (B), skeletal muscle (C) and skin (D). There is some endogenous pigmentation due to chromatophores in the skin (D). (F) Original tail no primary antibody control, counterstained with hematoxylin. Composites: A & F. Scale bars: 200 µm (A, F), 20 µm (B-D).
(TIF)
A. carolinensis genome annotation version 2.2.1 in comparison with previous annotations.
(DOCX)
Summary of RNA-Seq reads.
(DOCX)
Differentially expressed genes in the lizard regenerating tail at 25 dpa analyzed by Cuffdiff2.
(DOCX)
Differentially expressed genes in the lizard regenerating tail at 25 dpa analyzed by DESeq2.
(DOCX)
GO Biological Process analysis (DAVID) on differentially expressed genes in the proximal regenerating tail (Cluster I).
(DOCX)
GO Biological Process analysis (DAVID) on differentially expressed genes in the regenerating tail tip (Cluster II).
(DOCX)
KEGG pathway analysis (DAVID) on differentially expressed genes in the 25 dpa regenerating tail.
(DOCX)
Differentially expressed genes elevated (10-fold) in the regenerating tip compared to embryo and satellite cells.
(DOCX)
Differentially genes elevated (10-fold) in the proximal regenerating tail compared to embryo and satellite cells.
(DOCX)
Acknowledgments
We thank Inbar Maayan, Joel Robertson, Allison Wooten, and John Cornelius for technical assistance; Stephen Pratt for statistical consultation; the Department of Animal Care and Technologies at Arizona State University for assistance in establishing and maintaining the lizard colony; Lorenzo Alibardi, Terry Ritzman, Eris Lasku, and Tonia Hsieh for discussions; and Fiona McCarthy and Sarah Stabenfeldt for comments. Support for GM, MT, and MA was provided by the School of Life Sciences Undergraduate Research (SOLUR) Program at Arizona State University. The PAX7 antibody developed by Kawakami, A. was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. RNA-Seq data for the lizard embryo samples, which have been previously reported [19], are deposited in at the National Center for Biotechnology Information (NCBI) BioProject (http://www.ncbi.nlm.nih.gov/bioproject/), under BioProject PRJNA149661. RNA-Seq data for the lizard tail regeneration and satellite cell samples are deposited under BioProject PRJNA253971.
Funding Statement
This work was supported by funding from the National Center for Research Resources and the Office of Research Infrastructure Programs (ORIP) grant R21 RR031305 (KK, JW-R); National Institute of Arthritis, Musculoskeletal, and Skin Diseases grant R21 AR064935 of the National Institutes of Health (KK); and funding from the Arizona Biomedical Research Commission grant 1113 (KK, REF). Computational analysis was supported by allocations from the Arizona State University Advanced Computing Center (A2C2). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. McCusker C, Gardiner DM (2011) The axolotl model for regeneration and aging research: a mini-review. Gerontology 57: 565–571 10.1159/000323761 [DOI] [PubMed] [Google Scholar]
- 2. Gemberling M, Bailey TJ, Hyde DR, Poss KD (2013) The zebrafish as a model for complex tissue regeneration. Trends Genet 29: 611–620 10.1016/j.tig.2013.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Knapp D, Schulz H, Rascon CA, Volkmer M, Scholz J, et al. (2013) Comparative transcriptional profiling of the axolotl limb identifies a tripartite regeneration-specific gene program. PLoS ONE 8: e61352 10.1371/journal.pone.0061352.s011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu C-H, Tsai M-H, Ho C-C, Chen C-Y, Lee H-S (2013) De novo transcriptome sequencing of axolotl blastema for identification of differentially expressed genes during limb regeneration. BMC Genomics 14: 1–1 10.1186/1471-2164-14-434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nacu E, Glausch M, Le HQ, Damanik FFR, Schuez M, et al. (2013) Connective tissue cells, but not muscle cells, are involved in establishing the proximo-distal outcome of limb regeneration in the axolotl. Development 140: 513–518 10.1242/dev.081752 [DOI] [PubMed] [Google Scholar]
- 6. Hui SP, Sengupta D, Lee SGP, Sen T, Kundu S, et al. (2014) Genome wide expression profiling during spinal cord regeneration identifies comprehensive cellular responses in zebrafish. PLoS ONE 9: e84212 10.1371/journal.pone.0084212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Looso M, Preussner J, Sousounis K, Bruckskotten M, Michel CS, et al. (2013) A de novo assembly of the newt transcriptome combined with proteomic validation identifies new protein families expressed during tissue regeneration. Genome Biol 14: R16 10.1186/gb-2013-14-2-r16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sandoval-Guzmán T, Wang H, Khattak S, Schuez M, Roensch K, et al. (2014) Fundamental differences in dedifferentiation and stem cell recruitment during skeletal muscle regeneration in two salamander species. Cell Stem Cell 14: 174–187 10.1016/j.stem.2013.11.007 [DOI] [PubMed] [Google Scholar]
- 9. Rinkevich Y, Lindau P, Ueno H, Longaker MT, Weissman IL (2011) Germ-layer and lineage-restricted stem/progenitors regenerate the mouse digit tip. Nature 476: 409–413 10.1038/nature10346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Takeo M, Chou WC, Sun Q, Lee W, Rabbani P, et al. (2013) Wnt activation in nail epithelium couples nail growth to digit regeneration. Nature 499: 228–232 10.1038/nature12214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, et al. (2011) Transient regenerative potential of the neonatal mouse heart. Science 331: 1078–1080 10.1126/science.1200708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fernando WA, Leininger E, Simkin J, Li N, Malcom CA, et al. (2011) Wound healing and blastema formation in regenerating digit tips of adult mice. Dev Biol 350: 301–310 10.1016/j.ydbio.2010.11.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alibardi L (2010) Morphological and cellular aspects of tail and limb regeneration in lizards. A model system with implications for tissue regeneration in mammals. Adv Anat Embryol Cell Biol 207: iii–v–x–1–109. [PubMed] [Google Scholar]
- 14. Cox PG (1969) Some aspects of tail regeneration in the lizard, Anolis carolinensis. I. A description based on histology and autoradiography. J Exp Zool 171: 127–149 10.1002/jez.1401710202 [DOI] [Google Scholar]
- 15. Fisher RE, Geiger LA, Stroik LK, Hutchins ED, George RM, et al. (2012) A histological comparison of the original and regenerated tail in the green anole, Anolis carolinensis . Anat Rec (Hoboken) 295: 1609–1619 10.1002/ar.22537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ritzman TB, Stroik LK, Julik E, Hutchins ED, Lasku E, et al. (2012) The gross anatomy of the original and regenerated tail in the green anole (Anolis carolinensis). Anat Rec (Hoboken) 295: 1596–1608 10.1002/ar.22524 [DOI] [PubMed] [Google Scholar]
- 17. Duffy MT, Simpson SB, Liebich DR, Davis BM (1990) Origin of spinal cord axons in the lizard regenerated tail: Supernormal projections from local spinal neurons. J Comp Neurol 293: 208–222 10.1002/cne.902930205 [DOI] [PubMed] [Google Scholar]
- 18. Simpson SB, Duffy MT (1994) The lizard spinal cord: a model system for the study of spinal cord injury and repair. Prog Brain Res 103: 229–241. [DOI] [PubMed] [Google Scholar]
- 19.Simpson SB (1965) Regeneration of the lizard tail. In: Kiortsis V, Trampusch HAL, editors. Regeneration in animals and related problems. Amsterdam: North-Holland Publishing Company. pp. 431–443. [Google Scholar]
- 20. Hughes A, New D (1959) Tail regeneration in the geckonid lizard, Sphaerodactylus . J Embryol Exp Morphol 7: 281–302. [PubMed] [Google Scholar]
- 21. Kintner CR, Brockes JP (1984) Monoclonal antibodies identify blastemal cells derived from dedifferentiating limb regeneration. Nature 308: 67–69 10.1038/308067a0 [DOI] [PubMed] [Google Scholar]
- 22. Eckalbar WL, Lasku E, Infante CR, Elsey RM, Markov GJ, et al. (2012) Somitogenesis in the anole lizard and alligator reveals evolutionary convergence and divergence in the amniote segmentation clock. Dev Biol 363: 308–319 10.1016/j.ydbio.2011.11.021 [DOI] [PubMed] [Google Scholar]
- 23. Koshiba-Takeuchi K, Mori AD, Kaynak BL, Cebra-Thomas J, Sukonnik T, et al. (2009) Reptilian heart development and the molecular basis of cardiac chamber evolution. Nature 461: 95–98 10.1038/nature08324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wordley C, Slate J, Stapley J (2011) Mining online genomic resources in Anolis carolinensis facilitates rapid and inexpensive development of cross-species microsatellite markers for the Anolis lizard genus. Mol Ecol Resour 11: 126–133 10.1111/j.1755-0998.2010.02863.x [DOI] [PubMed] [Google Scholar]
- 25. Tollis M, Boissinot S (2014) Genetic variation in the green anole lizard (Anolis carolinensis) reveals island refugia and a fragmented Florida during the quaternary. Genetica 142: 59–72 10.1007/s10709-013-9754-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lovern MB, Wade J (2003) Yolk testosterone varies with sex in eggs of the lizard, Anolis carolinensis . J Exp Zool A Comp Exp Biol 295: 206–210 10.1002/jez.a.10225 [DOI] [PubMed] [Google Scholar]
- 27. Wade J (2012) Sculpting reproductive circuits: relationships among hormones, morphology and behavior in anole lizards. Gen Comp Endocrinol 176: 456–460 10.1016/j.ygcen.2011.12.011 [DOI] [PubMed] [Google Scholar]
- 28. Montuelle SJ, Daghfous G, Bels VL (2008) Effect of locomotor approach on feeding kinematics in the green anole (Anolis carolinensis). J Exp Zool A Ecol Genet Physiol 309: 563–567 10.1002/jez.484 [DOI] [PubMed] [Google Scholar]
- 29. Alföldi J, Di Palma F, Grabherr M, Williams C, Kong L, et al. (2011) The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature 477: 587–591 10.1038/nature10390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eckalbar WL, Hutchins ED, Markov GJ, Allen AN, Corneveaux JJ, et al. (2013) Genome reannotation of the lizard Anolis carolinensis based on 14 adult and embryonic deep transcriptomes. BMC Genomics 14: 49 10.1186/1471-2164-14-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, et al. (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578 10.1038/nprot.2012.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, et al. (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotech 28: 516–520 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, et al. (2013) Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc 8: 1765–1786 10.1038/nprot.2013.099 [DOI] [PubMed] [Google Scholar]
- 35.Anders S, Pyl PT, Huber W (2014) HTSeq–A Python framework to work with high-throughput sequencing data. bioRxiv. doi:10.1101/002824 [DOI] [PMC free article] [PubMed]
- 36. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, et al. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5: R80 10.1186/gb-2004-5-10-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang DW, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 39. Supek F, Bošnjak M, Škunca N, Šmuc T (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6: e21800 10.1371/journal.pone.0021800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Birol I, Jackman SD, Nielsen CB, Qian JQ, Varhol R, et al. (2009) De novo transcriptome assembly with ABySS. Bioinformatics 25: 2872–2877 10.1093/bioinformatics/btp367 [DOI] [PubMed] [Google Scholar]
- 41. Robertson G, Schein J, Chiu R, Corbett R, Field M, et al. (2010) De novo assembly and analysis of RNA-seq data. Nat Methods 7: 909–912 10.1038/nmeth.1517 [DOI] [PubMed] [Google Scholar]
- 42. Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, et al. (2009) ABySS: a parallel assembler for short read sequence data. Genome Res 19: 1117–1123 10.1101/gr.089532.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Haas BJ (2003) Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 31: 5654–5666 10.1093/nar/gkg770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Loke JC, Stahlberg EA, Strenski DG, Haas BJ, Wood PC, et al. (2005) Compilation of mRNA polyadenylation signals in Arabidopsis revealed a new signal element and potential secondary structures. Plant Physiol 138: 1457–1468 10.1104/pp.105.060541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rhind N, Chen Z, Yassour M, Thompson DA, Haas BJ, et al. (2011) Comparative functional genomics of the fission yeasts. Science 332: 930–936 10.1126/science.1203357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shen Y, Ji G, Haas BJ, Wu X, Zheng J, et al. (2008) Genome level analysis of rice mRNA 3'-end processing signals and alternative polyadenylation. Nucleic Acids Res 36: 3150–3161 10.1093/nar/gkn158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Allen RE, Temm-Grove CJ, Sheehan SM, Rice G (1997) Chapter 8 Skeletal Muscle Satellite Cell Cultures. In: Emerson CP, Sweeney HL, editors. Methods in Cell Biology, Vol. 52 . Amsterdam: Elsevier. pp. 155–176. doi:10.1016/S0091-679X(08)60378-7. [DOI] [PubMed] [Google Scholar]
- 48. Lees SJ, Rathbone CR, Booth FW (2006) Age-associated decrease in muscle precursor cell differentiation. Am J Physiol Cell Physiol 290: C609–C615 10.1152/ajpcell.00408.2005 [DOI] [PubMed] [Google Scholar]
- 49. Tatsumi R, Yamada M, Katsuki Y, Okamoto S, Ishizaki J, et al. (2006) Low-pH preparation of skeletal muscle satellite cells can be used to study activation in vitro. Int J Biochem Cell Biol 38: 1678–1685 10.1016/j.biocel.2006.04.003 [DOI] [PubMed] [Google Scholar]
- 50. Yablonka-Reuveni Z, Quinn LS, Nameroff M (1987) Isolation and clonal analysis of satellite cells from chicken pectoralis muscle. Dev Biol 119: 252–259 10.1016/0012-1606(87)90226-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Feldman JL, Stockdale FE (1991) Skeletal muscle satellite cell diversity: Satellite cells form fibers of different types in cell culture. Dev Biol 143: 320–334 10.1016/0012-1606(91)90083-F [DOI] [PubMed] [Google Scholar]
- 52. Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, et al. (2013) Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotech 31: 46–53 10.1038/nbt.2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fan C-M, Li L, Rozo ME, Lepper C (2012) Making skeletal muscle from progenitor and stem cells: development versus regeneration. Wiley Interdisc Rev Dev Biol 1: 315–327 10.1002/wdev.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Anderson DM, Beres BJ, Wilson-Rawls J, Rawls A (2009) The homeobox gene Mohawk represses transcription by recruiting the sin3A/HDAC co-repressor complex. Dev Dyn 238: 572–580 10.1002/dvdy.21873 [DOI] [PubMed] [Google Scholar]
- 55. Takahashi Y, Takagi A, Hiraoka S, Koseki H, Kanno J, et al. (2007) Transcription factors Mesp2 and Paraxis have critical roles in axial musculoskeletal formation. Dev Dyn 236: 1484–1494 10.1002/dvdy.21178 [DOI] [PubMed] [Google Scholar]
- 56. Sakuma K, Nishikawa J, Nakao R, Watanabe K, Totsuka T, et al. (2003) Calcineurin is a potent regulator for skeletal muscle regeneration by association with NFATc1 and GATA-2. Acta Neuropathol 105: 271–280 10.1007/s00401-002-0647-0 [DOI] [PubMed] [Google Scholar]
- 57. Manceau M, Gros J, Savage K, Thomé V, McPherron A, et al. (2008) Myostatin promotes the terminal differentiation of embryonic muscle progenitors. Genes Dev 22: 668 10.1101/gad.454408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kusumi K, Kulathinal RJ, Abzhanov A, Boissinot S, Crawford NG, et al. (2011) Developing a community-based genetic nomenclature for anole lizards. BMC Genomics 12: 554 10.1186/1471-2164-12-554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wilson-Rawls J, Molkentin JD, Black BL, Olson EN (1999) Activated notch inhibits myogenic activity of the MADS-Box transcription factor myocyte enhancer factor 2C. Mol Cell Biol 19: 2853–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Anderson DM, George R, Noyes MB, Rowton M, Liu W, et al. (2012) Characterization of the DNA-binding properties of the Mohawk homeobox transcription factor. J Biol Chem 287: 35351–35359 10.1074/jbc.M112.399386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Garlanda C, Hirsch E, Bozza S, Salustri A, De Acetis M, et al. (2002) Non-redundant role of the long pentraxin PTX3 in anti-fungal innate immune response. Nature 420: 182–186 10.1038/nature01195 [DOI] [PubMed] [Google Scholar]
- 62. Reynolds AR, Reynolds LE, Nagel TE, Lively JC, Robinson SD, et al. (2004) Elevated Flk1 (vascular endothelial growth factor receptor 2) signaling mediates enhanced angiogenesis in beta3-integrin-deficient mice. Cancer Res 64: 8643–8650 10.1158/0008-5472.CAN-04-2760 [DOI] [PubMed] [Google Scholar]
- 63. Dentice M, Marsili A, Ambrosio R, Guardiola O, Sibilio A, et al. (2010) The FoxO3/type 2 deiodinase pathway is required for normal mouse myogenesis and muscle regeneration. J Clin Invest 120: 4021–4030 10.1172/JCI43670DS1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Panaite P-A, Barakat-Walter I (2010) Thyroid hormone enhances transected axonal regeneration and muscle reinnervation following rat sciatic nerve injury. J Neurosci Res 88: 1751–1763 10.1002/jnr.22344 [DOI] [PubMed] [Google Scholar]
- 65. Brown DD, Cai L, Das B, Marsh-Armstrong N, Schreiber AM, et al. (2005) Thyroid hormone controls multiple independent programs required for limb development in Xenopus laevis metamorphosis. Proc Natl Acad Sci USA 102: 12455–12458 10.1073/pnas.0505989102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Goldie RG (1999) Endothelins in health and disease: an overview. Clin Exp Pharmacol Physiol 26: 145–148. [DOI] [PubMed] [Google Scholar]
- 67. Yue R, Li H, Liu H, Li Y, Wei B, et al. (2012) Thrombin receptor regulates hematopoiesis and endothelial-to-hematopoietic transition. Dev Cell 22: 1092–1100 10.1016/j.devcel.2012.01.025 [DOI] [PubMed] [Google Scholar]
- 68. Lee WS, Jain MK, Arkonac BM, Zhang D, Shaw SY, et al. (1998) Thy-1, a novel marker for angiogenesis upregulated by inflammatory cytokines. Circ Res 82: 845–851. [DOI] [PubMed] [Google Scholar]
- 69. DeChiara TM, Kimble RB, Poueymirou WT, Rojas J, Masiakowski P, et al. (2000) Ror2, encoding a receptor-like tyrosine kinase, is required for cartilage and growth plate development. Nat Genet 24: 271–274 10.1038/73488 [DOI] [PubMed] [Google Scholar]
- 70. Day TF, Guo X, Garrett-Beal L, Yang Y (2005) Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell 8: 739–750 10.1016/j.devcel.2005.03.016 [DOI] [PubMed] [Google Scholar]
- 71. Hamamori Y, Sartorelli V, Ogryzko V, Puri PL, Wu HY, et al. (1999) Regulation of histone acetyltransferases p300 and PCAF by the bHLH protein twist and adenoviral oncoprotein E1A. Cell 96: 405–413 10.1016/S0092-8674(00)80553-X [DOI] [PubMed] [Google Scholar]
- 72.Pownall ME, Isaacs HV (2010) FGF Signalling in Vertebrate Development. Colloquium Series on Developmental Biology. San Rafael (CA):Morgan and Claypool Life Sciences. 75p. doi:10.4199/C00011ED1V01Y201004DEB002. [PubMed]
- 73. Cruciat C-M, Niehrs C (2013) Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb Perspect Biol 5: a015081 10.1101/cshperspect.a015081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, et al. (2012) The Pfam protein families database. Nucleic Acids Res 40: D290–D301 10.1093/nar/gkr1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, et al. (2013) Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 41: D226–D232 10.1093/nar/gks1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sun L, Luo H, Bu D, Zhao G, Yu K, et al. (2013) Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res 41: e166 10.1093/nar/gkt646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chen Y (2006) Control of muscle regeneration in the Xenopus tadpole tail by Pax7. Development 133: 2303–2313 10.1242/dev.02397 [DOI] [PubMed] [Google Scholar]
- 78. Li Z, Hassan MQ, Volinia S, van Wijnen AJ, Stein JL, et al. (2008) A microRNA signature for a BMP2-induced osteoblast lineage commitment program. Proc Natl Acad Sci USA 105: 13906–13911 10.1073/pnas.0804438105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Eckalbar WL, Infante C, Emmert N, Losos J, Rawls A, et al. (2010) Identification of a divergent notch pathway delta ligand in the segmentation clock of the reptile, Anolis carolinensis . Dev Biol 344: 528 10.1016/j.ydbio.2010.05.385 [DOI] [Google Scholar]
- 80. Asakura A, Komaki M, Rudnicki M (2001) Muscle satellite cells are multipotential stem cells that exhibit myogenic, osteogenic, and adipogenic differentiation. Differentiation 68: 245–253. [DOI] [PubMed] [Google Scholar]
- 81. Cairns DM, Liu R, Sen M, Canner JP, Schindeler A, et al. (2012) Interplay of Nkx3.2, Sox9 and Pax3 regulates chondrogenic differentiation of muscle progenitor cells. PLoS ONE 7: e39642 10.1371/journal.pone.0039642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Croteau W, Davey JC, Galton VA, St Germain DL (1996) Cloning of the mammalian type II iodothyronine deiodinase. A selenoprotein differentially expressed and regulated in human and rat brain and other tissues. J Clin Invest 98: 405–417 10.1172/JCI118806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Furlow JD, Neff ES (2006) A developmental switch induced by thyroid hormone: Xenopus laevis metamorphosis. Trends Endocrinol Metab 17: 40–47 10.1016/j.tem.2006.01.007 [DOI] [PubMed] [Google Scholar]
- 84. Boothby M, Ruddon RW, Anderson C, McWilliams D, Boime I (1981) A single gonadotropin alpha-subunit gene in normal tissue and tumor-derived cell lines. J Biol Chem 256: 5121–5127. [PubMed] [Google Scholar]
- 85. Buckbinder L, Brown DD (1993) Expression of the Xenopus laevis prolactin and thyrotropin genes during metamorphosis. Proc Natl Acad Sci USA 90: 3820–3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kusumi K, May CM, Eckalbar WL (2013) A large-scale view of the evolution of amniote development: insights from somitogenesis in reptiles. Curr Opin Genet Dev 23: 491–497 10.1016/j.gde.2013.02.011 [DOI] [PubMed] [Google Scholar]
- 87.Graw J (2010) Chapter Ten - Eye Development. In: Koopman P, editor. Organogenesis in Development. Current Topics in Developmental Biology, Volume 90 . San Diego: Academic Press. pp. 343–386. doi:10.1016/S0070-2153(10)90010-0. [DOI] [PubMed] [Google Scholar]
- 88. Chodankar R, Chang C-H, Yue Z, Jiang T-X, Suksaweang S, et al. (2003) Shift of localized growth zones contributes to skin appendage morphogenesis: role of the Wnt/beta-catenin pathway. J Invest Dermatol 120: 20–26 10.1046/j.1523-1747.2003.12008.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Suksaweang S, Lin C-M, Jiang T-X, Hughes MW, Widelitz RB, et al. (2004) Morphogenesis of chicken liver: identification of localized growth zones and the role of β-catenin/Wnt in size regulation. Dev Biol 266: 109–122 10.1016/j.ydbio.2003.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Berg DA, Kirkham M, Beljajeva A, Knapp D, Habermann B, et al. (2010) Efficient regeneration by activation of neurogenesis in homeostatically quiescent regions of the adult vertebrate brain. Development 137: 4127–4134 10.1242/dev.055541 [DOI] [PubMed] [Google Scholar]
- 91. Santos-Ruiz L, Santamaría JA, Ruiz-Sánchez J, Becerra J (2002) Cell proliferation during blastema formation in the regenerating teleost fin. Dev Dyn 223: 262–272 10.1002/dvdy.10055 [DOI] [PubMed] [Google Scholar]
- 92. Mescher AL (1996) The cellular basis of limb regeneration in urodeles. Int J Dev Biol 40: 785–795. [PubMed] [Google Scholar]
- 93. George RM, Biressi S, Beres BJ, Rogers E, Mulia AK, et al. (2013) Numb-deficient satellite cells have regeneration and proliferation defects. Proc Natl Acad Sci USA 110: 18549–18554 10.1073/pnas.1311628110 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gene Ontology analysis of differentially expressed genes identified by both Cuffdiff2 and DESeq2. 130 genes were identified as differentially expressed by both methods (Figure 1B-C; Table S3; Table S4). (A-B) A treemap overview of differentially expressed genes in (A) Cluster I and (B) Cluster II based on representative Gene Ontology Biological Processes. The relative sizes of the treemap boxes are based on the |log10(p-value)| of the respective GO term. Related terms are visualized with the same color, with the representative category for each color group denoted in the legend.
(EPS)
Genes with high expression (>10-fold change) in the regenerating tail relative to the embryos and satellite cells. 44 differentially expressed genes had >10-fold change in S1 and S2 gene expression (FPKM) relative to the embryos and satellite cells (orange cluster) and 86 genes had >10-fold change in S4 and S5 (yellow cluster).
(EPS)
Satellite cells isolated from adult skeletal muscle express PAX7 and can differentiate into myotubes. (A-B) Detection of myosin heavy chain (MHC) in proliferating (A) and differentiated (B) A. carolinensis satellite cells. MHC was detected using MY-32 monoclonal antibody and HRP-conjugated anti-mouse secondary antibody with DAB stain. Immunofluorescence of lizard (C-F) and mouse (G-H) satellite cells. (C-D, G-H) PAX7 was detected using a monoclonal antibody and visualized by FITC-conjugated anti-mouse secondary antibody, and nuclei were stained with DAPI. (E-F) Cells with no primary antibody and FITC-conjugated anti-mouse secondary antibody only, and nuclei stained with DAPI.
(TIF)
Histological analysis of proliferation in the 25 dpa regenerating tail. (A-G) PCNA immunohistochemistry of the 25 dpa regenerating tail (brown nuclei), counterstained with hematoxylin (blue nuclei). (A) PCNA is expressed throughout the regenerating tail, indicating a lack of a single proliferative zone. The dermis near the proximal base (B) and tip (C) of the regenerating tail, condensing cartilage tube (D), ependymal core (E), and developing muscles near the proximal base (F) and tip (G) of the regenerating tail all show PCNA positive cells.
(TIF)
Immunohistochemistry shows few proliferating cells in the original tail. (A-D) Proliferating cell nuclear antigen (PCNA) immunohistochemistry of the original tail (brown nuclei), counterstained with hematoxylin (blue nuclei). (A) Transverse section of the original tail. (B-D) There are limited PCNA-positive cells in the centrum (B), skeletal muscle (C) and skin (D). There is some endogenous pigmentation due to chromatophores in the skin (D). (F) Original tail no primary antibody control, counterstained with hematoxylin. Composites: A & F. Scale bars: 200 µm (A, F), 20 µm (B-D).
(TIF)
A. carolinensis genome annotation version 2.2.1 in comparison with previous annotations.
(DOCX)
Summary of RNA-Seq reads.
(DOCX)
Differentially expressed genes in the lizard regenerating tail at 25 dpa analyzed by Cuffdiff2.
(DOCX)
Differentially expressed genes in the lizard regenerating tail at 25 dpa analyzed by DESeq2.
(DOCX)
GO Biological Process analysis (DAVID) on differentially expressed genes in the proximal regenerating tail (Cluster I).
(DOCX)
GO Biological Process analysis (DAVID) on differentially expressed genes in the regenerating tail tip (Cluster II).
(DOCX)
KEGG pathway analysis (DAVID) on differentially expressed genes in the 25 dpa regenerating tail.
(DOCX)
Differentially expressed genes elevated (10-fold) in the regenerating tip compared to embryo and satellite cells.
(DOCX)
Differentially genes elevated (10-fold) in the proximal regenerating tail compared to embryo and satellite cells.
(DOCX)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. RNA-Seq data for the lizard embryo samples, which have been previously reported [19], are deposited in at the National Center for Biotechnology Information (NCBI) BioProject (http://www.ncbi.nlm.nih.gov/bioproject/), under BioProject PRJNA149661. RNA-Seq data for the lizard tail regeneration and satellite cell samples are deposited under BioProject PRJNA253971.