

Abstract
Osteogenesis imperfecta (OI) is an inherited brittle bone disorder characterized by bone fragility and low bone mass. Loss of function mutations in FK506-binding protein 10 (FKBP10), encoding the FKBP65 protein, result in recessive OI and Bruck syndrome, of which the latter is additionally characterized by joint contractures. FKBP65 is thought to act as a collagen chaperone, but it is unknown how loss of FKBP65 affects collagen synthesis and extracellular matrix formation. We evaluated the developmental and postnatal expression of Fkbp10 and analyzed the consequences of its generalized loss of function. Fkbp10 is expressed at low levels in E13.5 mouse embryos, particularly in skeletal tissues, and steadily increases through E17.5 with expression in not only skeletal tissues, but also in visceral tissues. Postnatally, expression is limited to developing bone and ligaments. In contrast to humans, with complete loss of function mutations, Fkbp10−/− mice do not survive birth, and embryos present with growth delay and tissue fragility. Type I calvarial collagen isolated from these mice showed reduced stable crosslink formation at telopeptide lysines. Furthermore, Fkbp10−/− mouse embryonic fibroblasts show retention of procollagen in the cell layer and associated dilated endoplasmic reticulum. These data suggest a requirement for FKBP65 function during embryonic connective tissue development in mice, but the restricted expression postnatally in bone, ligaments and tendons correlates with the bone fragility and contracture phenotype in humans.
INTRODUCTION
Osteogenesis imperfecta [OI (MIM 610915, 610682, 259420, 166210, 259420, 614856, 615066, 610967, 613848, 615220, 613849, 259440, 613982, 610968, 166220, 166210, 166200, 259420)] is the most common inherited bone dysplasia, characterized by bone fragility and low bone mass leading to increased fracture risk, as well as variable defects in other connective tissues. There is significant variable expressivity ranging from mild to neonatal lethality. It is most frequently caused by dominant mutations in one of the two type I collagen genes, COL1A1 and COL1A2 (1). However, other genes have now been implicated in both dominant and recessive forms of OI. While some function in type I collagen processing, others appear to regulate cell-cell and cell-matrix signaling (2,3). The first described were mutations in components of the prolyl-3-hydroxylase complex, which includes prolyl-3-hydroxylase (LEPRE1), cartilage associated protein (CRTAP) and cyclophilin B (PPIB) (4–7). This complex has two functions: (i) it serves to 3-hydroxylate proline 986 of the alpha 1 chain of type I collagen and other Clade A collagens and (ii) it acts as a collagen binding and assembly protein (8,9). Subsequently, mutations have been identified in both SERPINH1, encoding heat shock protein 47 (HSP47), as well as in FK506-binding protein 10 (FKBP10), encoding FKBP65, which both act as chaperones of type I collagen (10–15). The Serpinh1−/− mouse presents with embryonic lethality due to defects in collagen synthesis (16). Mouse embryonic fibroblasts (MEFs) isolated from these mice showed aggregates and accumulation of collagen within the endoplasmic reticulum (ER), possibly due to improper trafficking (17).
Mutations in FKBP10 can cause brittle bones in a recessive form of OI, as well as brittle bone variably associated with joint contractures, as seen classically in Bruck syndrome (MIM 259450, 609220) (11,12). Mutations in FKBP10 have also been associated with Kuskokwim syndrome (MIM 208200), which is a disorder of congenital contractures without skeletal manifestations (18). Prior to the association of FKBP10 mutations with Bruck syndrome, recessive mutations in PLOD2, encoding lysyl hydroxylase 2 (LH2), were identified in Bruck syndrome patients (19,20). The loss of LH2 results in a lack of hydroxylation of telopeptide lysines of type I collagen (20,21). Similarly, patients described with FKBP10 mutations also lack hydroxylation at these residues in bone (22). In vitro studies using patient fibroblasts have shown no evidence for over modification of type I collagen or alterations in prolyl-3 hydroxylation (11,22). These cells showed a minimal delay in collagen secretion and some patient fibroblasts showed retention of collagen within the ER (11,22).
The FKBP family of proteins is comprised of immunophilins that were identified through their ability to bind FK506, an immunosuppressive drug (23). They also have peptidyl–prolyl cis/trans–isomerase (PPIase) activity, which changes the peptidyl–prolyl bonds within a protein from the cis to trans conformation, which is essential for the proper folding of collagen (24,25). FKBP10, which encodes FKBP65, an ER resident protein, has been shown to chaperone both type I collagen and tropoelastin (15,26,27). The rate-limiting step of procollagen helix formation is the isomerization of proline residues from the cis to the trans conformation (28). The homolog of FKBP65 in Xenopus (xFKBP) was first characterized by expression studies using northern blotting and in situ hybridization. Expression was limited to the mesoderm of early gastrula stage embryos, with expression localizing to the notochord, somites and epidermis in later stages of development. Expression was retained only in the testes of adults (29). Subsequent to those studies, it was shown that E12 mouse embryos showed Fkbp10 expression in the aorta, lungs, brain and kidney by northern blotting. However, in adult tissues, this expression was limited, suggesting a role in organogenesis during development (30). Taken together, FKBP65 functions to chaperone type I collagen and elastin, and likely plays a role during development. However, FKBP65 has not been fully characterized, especially in the context of bone and other connective tissues.
Here, we utilize a murine model to temporally and spatially characterize Fkbp10 expression. Furthermore, we characterized the phenotypes of Fkbp10 null mice, as well as Fkbp10 null MEFs. We also analyzed the type I collagen biochemistry in bone to profile the telopeptide crosslinking pathway. We show that Fkbp10 is expressed at low levels at E13.5, with increasing levels in the bone and visceral tissues throughout embryonic development. In adult tissues, expression is limited to the developing bone and tendons/ligaments and correlates with the tissues affected in OI and Bruck syndrome in humans. Additionally, the Fkbp10 null mice display growth delay, tissue fragility and neonatal lethality. The collagen is retained within the cell layer of MEF cells, resulting in dilated ER with no major effects upon secretion, as determined by pulse chase experiments. However, the bone from these mice recapitulates the abnormal telopeptide crosslinking observed in bone isolated from patients with loss of function mutations in FKBP10 (22). Thus, this is a good model for the study of FKBP10 function that phenocopies in vitro human data in vivo (11,12).
RESULTS
Expression analysis of Fkbp10
We generated mice containing the European conditional mouse mutagenesis (EUCOMM) allele (Fig. 1A), which contains a splice acceptor with a β-galactosidase cassette (designated throughout this paper as Fkbp10−/−). The presence of this cassette generates a fusion transcript, allowing for the characterization of the temporal and spatial expression of Fkbp10 by β-galactosidase staining (Fig. 1A) (31). We stained for β-galactosidase activity in both whole-mount and sectioned samples at E13.5, E15.5, E17.5 and 2 months of age in heterozygous mice (Fkbp10+/−) and compared them with wild-type littermate controls (Fkbp10+/+) that lack the β-galactosidase cassette (n = 2–3 per group). Expression was first detected at relatively low levels at E13.5 (indicated by blue staining) and was increased by E15.5, which was especially evident in skeletal tissues (Fig. 1B, Supplementary Material, Fig. S1). Sections of E15.5 and E17.5 mice show a similar expression pattern, with more intense staining in skeletal tissues, including the long bones, vertebrae and ribs. Specifically, there was intense staining of developing cortical bone. Within the growth plate, there was strong staining of the resting zone, as well as expression in the condensing mesenchyme with low levels in the proliferative zone and no expression in the hypertrophic region (Fig. 1C and Supplementary Material, Fig. S1). In addition to these tissues, expression was also noted in the developing ligaments. Expression was also observed in the large vessels of the heart, large vessels of the lung and blood vessels of the glomeruli, tips of the villi, distal tubules and collecting ducts of the kidney (Fig. 1C). At 2 months of age, expression is limited to the bone, ligaments and tendon (Fig. 1D, Supplementary Material, Fig. S2). Staining was primarily evident in the developing bone and periosteum, although there is some level of nonspecific staining at the chondro-osseous junction, possibly due to osteoclast activity. No staining is observed in the growth plate or other visceral tissues (Fig. 1D and Supplementary Material, Fig. S2).
Figure 1.

In vivo expression analysis of Fkbp10. (A) Targeted ES cells from the European Conditional Mouse Mutagenesis Program were used for injections, which utilize a splice acceptor site with a β-gal cassette and polyA tail. This serves to replace the Fkbp10 transcript with β-galactosidase in a targeted manner. (B) Heterozygous mice at E13.5 and E15.5 (top panel) show LacZ staining of skeletal tissues, at a low level at E13.5 and higher by E15.5 as compared with wild-type control littermates (bottom panel). (C) Sagittal sections of E17.5 mice show staining of the skeletal tissues as well as visceral tissues. This includes the joint space and developing cortical bone in the long bones, growth plate, vertebral column, ribs, major vessels of the heart, vessels of the lung, villi of the intestine and vessels of the glomeruli in the kidney. No staining is present in control littermates. (D) At 2 months of age, staining is shown only in the developing bone, periosteum and tendons/ligaments. No staining is evident in the growth plate. Some nonspecific staining of the chondro-osseous junction is present in the control.
Phenotypic characterization of Fkbp10−/− mice
Although Fkbp10−/−mice were present at Mendelian ratios through E18.5, we were unable to recover null mice at weaning age (n = 99) nor at postnatal day 0 (n = 20–55) (Table 1). Mice display growth delay starting at approximately E13.5 and also exhibit generalized tissue fragility upon collection (not shown). Null embryos also have a downward facing orientation of the forelimbs and flattened craniofacial features (Fig. 2A). Histological sections of mice at E15.5 show no obvious defects in organs or tissues (n = 3, Fig. 2B). Furthermore, there is no patterning defect as indicated by skeletal preparations at E15.5 (n = 3, Fig. 2C).
Table 1.
Fkbp10−/− mice exhibit lethality at birth. A time course of isolated embryos and adult mice shows no null mice attainable after birth (P0, P21). Through E18.5 embryos were isolated at normal Mendelian ratios.
| Day | +/+ | +/− | −/− | Total |
|---|---|---|---|---|
| E11.5 | 7 (19%) | 17 (46%) | 13 (35%) | 37 |
| E12.5 | 10 (18%) | 34 (62%) | 11 (20%) | 55 |
| E13.5 | 2 (10%) | 15 (71%) | 4 (19%) | 21 |
| E15.5 | 3 (14%) | 12 (57%) | 6 (28%) | 21 |
| E18.5 | 13 (30%) | 19 (43%) | 12 (27%) | 44 |
| P0 | 5 (31%) | 11 (69%) | 0 | 16 |
| P21 | 41 (41%) | 58 (59%) | 0 | 99 |
Figure 2.
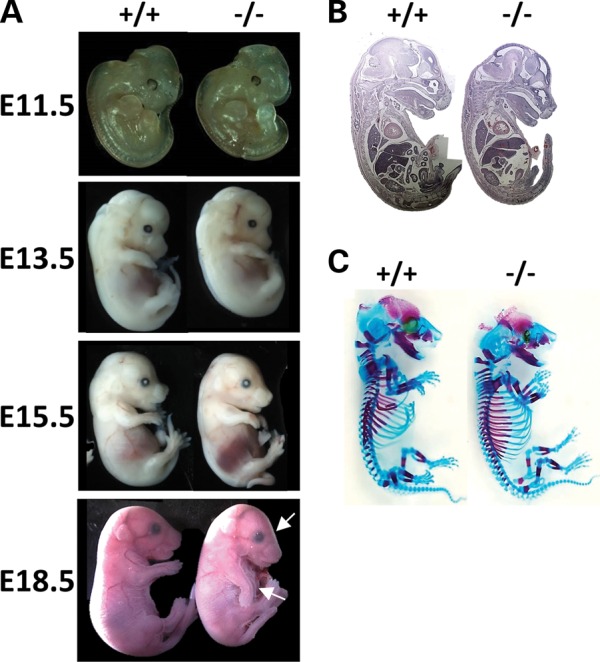
Phenotypic characterization of Fkbp10−/− embryos. (A) Fkbp10−/− embryos (right) show growth delay compared with their wild-type littermates (left) starting by E13.5. By E18.5, there is altered craniofacial shape and downward facing forelimbs (indicated with arrows). (B) Hematoxylin and eosin staining show no major defects in gross organ histology at E15.5. (C) Skeletal preparations at E17.5 show no skeletal patterning defects.
Histologic analysis of the aorta
To further identify potential causes of lethality, we performed histologic analysis of E18.5 mice on transverse sections of the heart and aorta by elastin staining (n = 6). Although there appears to be no obvious difference in the organization of elastin fibers, there is a reduction in the width of both the ascending and descending aortic walls (Fig. 3A and B). Additionally, trichrome staining for collagen in the descending aorta also fails to show any major differences in the organization of the collagen at the histologic level.
Figure 3.

Aortas of Fkbp10−/− mice have reduced wall thickness. (A) Elastin staining of E18.5 Fkbp10−/− embryos shows reduced wall thickness of the ascending aorta. No major defects in elastin fibers are noted. (B) Trichrome staining for collagen in the descending aorta shows no major defects in collagen and a reduced wall thickness of the descending aorta.
Cellular characterization of MEFs
To analyze the cellular phenotypes of Fkbp10−/− mice, we isolated MEFs from E13.5 embryos. Through quantitative real-time PCR, western blotting and immunofluorescence, we confirmed the ablation of FKBP65 in these cells (Fig. 4A–C). Furthermore, we also confirmed localization of FKBP65 to the ER by colocalization with concanavalin A, an ER marker, using immunofluorescence in wild-type cells (Fig. 4C). In order to examine the cellular morphology of these cells, we performed electron microscopy (n = 4 per group, Fig. 5). Notably, there is dilation of the ER, more so in mutant cells than wild-type cells. In addition to the presence of intracytoplasmic collagen fibers, the cytoplasm is also filled with dilated, irregular rough ER that are engorged with abnormal, poorly formed collagen matrix. All of these findings are consistent with previously reported phenotypes in fibroblasts isolated from human patients with FKBP10 mutations (11).
Figure 4.

FKBP65 localizes to the ER in MEF cells and is abolished in Fkbp10−/− cells. (A) Quantitative real-time PCR shows a reduced level of Fkbp10 transcript near to none. (B) Western blot analysis shows a complete ablation of FKBP65 protein in mutant cells. (C) Immunofluorescence shows FKBP65 localization (shown in green) to the ER as demonstrated by colocalization with Concanavalin A, an ER resident marker (shown in red) in wild-type mouse cells (top panel). There is no detectable FKBP65 in null cells (bottom panel).
Figure 5.

Electron microscopy shows abnormal cellular morphology and collagen retention. Fkbp10−/− cells show dilated ER with embedded matrix as well as intracytoplasmic collagen fibers (shown by arrows).
Alterations of collagen in vitro
It has been previously reported that FKBP10 loss of function patient fibroblasts show changes in collagen trafficking and that some contain aggregates of procollagen within the ER. Consistent with the human data, we observe an increase in procollagen levels in FKBP10 null MEF cells after ascorbic acid stimulation shown through both immunofluorescence and western blotting of the cell layer of independent MEF cell lines (Fig. 6A and B). Pulse chase analysis indicated no obvious changes in collagen secretion in null MEFs, consistent with observations in human fibroblasts, which show only a very mild delay in secretion (Fig. 6C) (11).
Figure 6.

Fkbp10−/− cells show retention of procollagen in the cell layer. (A) Immunofluorescence of MEF cells isolated from Fkbp10−/− embryos shows a low level of procollagen before ascorbic acid treatment. Upon stimulation of collagen synthesis with ascorbic acid, some cells show increased levels of procollagen compared with wild-type levels. The golgi is labeled with lectin HPA and the ER is labeled with an antibody to concanavalin A. (B) Western blotting of the cell layer isolated from MEF cells shows an increased level of procollagen in null cells from two independent MEF lines. (C) Pulse chase of MEF cells shows no dramatic effect on secretion of procollagen from these cells.
Mass spectrometry analysis of calvarial bone
In order to assess collagen modification, we isolated calvarial bone from E18.5 null mice and their wild-type littermates and performed mass spectrometry analysis. There were no differences in the migration pattern of collagen, suggesting no gross overmodification of the protein (Fig. 7A). However, the collagen was more soluble (2.5-fold) based on densitometry to quantify total collagen type I chains in the equal weights of tissue extracted and loaded on the gel. Both the amino- and carboxyl- telopeptide lysines were underhydroxylated (Fig. 7A–C), suggesting a reduction of telopeptide hydroxylysine–aldehyde crosslinking, which is consistent with that observed in patient bone (22).
Figure 7.

Telopeptide lysine underhydroxylation in type I collagen of Fkbp10−/− calvaria. (A) Heat denatured collagen extracted from E18.5 calvaria shows no changes in chain migration, suggesting no helical domain over or under modification. Fkbp10−/− collagen was more soluble, consistent with fewer stable crosslinks, and the prominence of beta dimers is more characteristic of skin than bone collagen. (B) The mass spectra of the bacterial collagenase-generated α1(I) carboxy-telopeptide and (C) the α1(I) amino-telopeptide from Fkbp10−/− and wild-type calvaria show a marked reduction in the ratio of hydroxylysine/lysine. In (C), lower case q and lower case m are used to identify, respectively, pyroglutamic acid and oxidized methionine residues.
DISCUSSION
OI has been shown to result from not only type I collagen mutations, but also from mutations in collagen chaperoning and modifying proteins. Many of the more recently identified causes have not been well studied and mouse models are needed. FKBP10 (which encodes FKBP65) was identified as a chaperone of tropoelastin and type I collagen prior to its implication as the disease gene in OI and Bruck syndrome (15,26,27). FKBP10 mutations are distinct from other causes of recessive OI, based on the loss of telopeptide hydroxylysine crosslinking in type I collagen of bone, and phenotypic overlap with mutations in PLOD2 (which encodes lysyl hydroxylase 2) (32). Loss of function of PLOD2 and FKBP10 results in OI and/or congenital joint contractures, highlighting the difference between this and classical OI cases (11,12,19,21,22). It also suggests that FKBP65 has other functions beyond that of chaperoning type I collagen in bone. Our studies highlight a comprehensive description of the in vivo expression pattern of Fkbp10 and a murine model of its global deletion and effects on connective tissues.
We show that Fkbp10 is limited in expression to bone and ligaments in postnatal animals, which is consistent with the bone and joint contracture phenotypes observed in patients. However, we also show that Fkbp10 is expressed in many other tissues throughout development, highlighting that it likely interacts with more proteins than type I collagen and tropoelastin. Based on its function as a chaperone and foldase, it may contribute to the proper folding of other proteins as well. Whether this is conserved in human development is unknown. Expression in the major vessels of the heart, lung and glomerulus highlights its potential role in the vascular system, particularly during embryogenesis where it likely chaperones elastin. Based on these expression data, it is clear that FKBP65 has multiple functions during development, but a more restricted function in mature tissues. There may be some low levels of endogenous Fkbp10 expression in other tissues of the adult that are undetectable by this method. Previously published data shows absent to very low expression levels by northern blotting in adult murine aorta, brain, kidney, liver and lung (33). It may be that FKBP65 is required during times of rapid tissue formation, including embryogenesis, but perhaps also in the context of healing. This is supported by data from Patterson et al, suggesting that Fkbp10 expression is increased in a lung injury model (30).
Fkbp10−/− mice could not be recovered at birth, but were viable until E18.5. By E13.5, these embryos show a growth delay phenotype and also present with generalized tissue fragility. They do not exhibit any gross patterning defects. The thickness of the aorta was reduced in Fkbp10−/− mice, compared with wild-type littermates. It is likely that the vessel defects and generalized tissue fragility result in the neonatal lethality that we observe, although it is also possible that there is compromised lung function in these mice. This degree of lethality is not seen in patients, and the reasons for this are unclear, but perhaps other genes in humans have functional redundancy for FKBP65 function. Similar differences in lethality are seen in OI patients with SERPINH1 mutations, where the mouse model exhibits very early lethality at E10; however, in this case, it is likely due to defects in collagen biosynthesis (10,16).
MEFs isolated from Fkbp10−/− mice and their wild-type littermates show a dilated ER with an improperly formed matrix in both the ER and cytoplasm. We also observe an increase in staining of procollagen within the ER of some of these cells and we further confirmed this by western blotting. We suspect that, owing to the loss of the chaperoning function of FKBP65, collagen fibrils cannot fold properly and become tangled to form aggregates. Over time, these aggregates accumulate at high levels in the cell, and it appears they have improper trafficking and also infiltrate the cytoplasm. Autophagosomes were observed in the electron microscopy of mutant cells and likely there is an increase in autophagy in response to these aggregates. Despite an increase in procollagen levels, the secretion of collagen was not obviously changed in these cells. This is consistent with patient fibroblasts that only have a very slight delay in secretion, and may be further confounded by the sensitivity of the assay and mixed cell types within the MEF cultures (11). It may be that the cells (both in human fibroblasts and MEFs) produce more procollagen as a compensatory mechanism to maintain a proper level of secretion similar to that of wild type.
Although electrophoresis revealed no changes in the migration pattern of type 1 collagen, suggesting no gross post-translational, over- or undermodification, we did observe a reduction in the hydroxylation status of telopeptide lysines present on type I collagen by mass spectroscopy analysis. Furthermore, the Fkbp10−/− collagen was more soluble than that isolated from wild-type bone. Taken together, these findings indicate a lack of telopeptide hydroxylysine–aldehyde crosslinking, with a pattern more similar to that observed in skin collagen than bone collagen. It is likely that the quality of the matrix is altered because of the change in crosslinking chemistry with subsequent effects on nanocrystal mineral structure within the collagen scaffold. These observations are consistent with bone collagen isolated from FKBP10 loss of function patients and phenocopies patients with PLOD2 loss of function mutations, who present with Bruck syndrome (22). It is possible that FKBP65 is required for LH2 to fold correctly. Thus, with loss of FKBP65 misfolded LH2 would not be able to fold properly and lacks enzymatic activity. However, this hypothesis has not been experimentally tested. It is also possible that FKBP65 functions in a complex with LH2 and they stabilize each other, similar to the situation with the P3H1 complex (4,5,9).
Our data highlight the importance of FKBP65 with respect to type I collagen assembly in skeletal tissues, but also suggest that it has other unidentified roles, in particular, during development. Loss of FKBP65 appears likely to cause other connective tissue defects, for example, in the vasculature. Despite some differences between the human and mouse phenotypes concerning early lethality, this mouse model recapitulates many of the basic molecular features associated with the disease and therefore is good model to further dissect the tissue-specific roles of FKBP65.
MATERIALS AND METHODS
Animal housing and genotyping
ES cells were obtained from EUCOMM and injected in-house. Mice were maintained on a C57/BL6J background. Mice were housed in the Baylor College of Medicine vivarium. These studies were approved by the Baylor College of medicine Institutional Animal Care and Use Committee and the Center for Comparative Medicine.
Genotyping was performed using primers flanking exon 2 for the wild-type allele (5′AATAGGTAGCACACAGTTCGG3′) and (5′GTGAACAGTATGACCTTGGCC3′). The mutant allele was identified by primers in the Bactin:neo cassette (5′AGCGTGCGCCGTTCCGAAAGT3′) and (5′GGCGGCGGTGGAATCGAAATC3′) as well as primers specific for the last loxP site (5′GGAGCGAAGGAGGATTACTGTGCC3′) and (5′TGAACTGATGGCGAGCTCAGACC3′).
Expression analysis
Heterozygous mice and their wild-type littermates were collected after timed matings at E13.5, E15.5, E17.5 and 2 months. Samples were then fixed for 2 h in 0.5% glutaraldehyde, 2.0% paraformaldehyde, 2 mm MgCl2 and 1.25 mm EGTA in 1× PBS, pH 7.3. Adult bone samples were decalcified in 10% EDTA for 3 days.
For whole-mount studies, embryos were then rinsed in PBS and stained in X-gal buffer for 1–24 h (2 mm MgCl2, 5 mm potassium ferrocyanide, 5 mm potassium ferricyanide, 1 mg/ml Xgal in 1× PBS). Samples were washed in 1× PBS, fixed in formalin, rinsed and then stored and imaged in 50% glycerol/PBS.
For sectioned samples, fixed embryos were rinsed in PBS, soaked in 30% sucrose overnight and then embedded in OCT media. Frozen sections were collected at 12 µm thickness and stored at −80°C. Slides were then fixed in 0.2% glutaraldehyde/PBS at room temperature for 10 min. Samples were washed three times for 5 min at room temperature in 2 mm MgCl2, 0.01% sodium deoxycholate, 0.02% NP40 and 1× PBS. Slides were stained overnight at 37°C in staining solution (above) in the dark. Slides were rinsed with PBS and then with water, and counterstained with Nuclear Fast Red for 2 min. They were washed three times in water, dehydrated in ethanol, cleared in xylenes and then mounted. Slides were imaged using a Zeiss Axioplan 2 microscope.
Histology
E15.5 and E18.5 embryos were collected and then fixed in 4% paraformaldehyde overnight at room temperature. They were stored in 70% ethanol and then embedded in paraffin. E15.5 whole embryos were sectioned sagittally at 7 µm. They were then stained with hematoxylin and eosin according to standard methods. E18.5 embryos were sectioned in the transverse plane at 7 µm thickness. They were stained with hematoxylin and eosin as well as elastin van Gieson stain (Sigma).
Cellular isolation
MEFs were isolated according to standard methods at E13.5 (34). Briefly, the head and internal organs were removed from embryos, which were then minced and trypsinized to single cells. Cells were grown and maintained in DMEM plus 10% fetal bovine serum. All cells used were between Passages 3 and 7.
Electron microscopy
Cells were pelleted and fixed in 4% paraformaldehyde, rinsed and then dehydrated in a graded ethanol series from 30–100%. The samples were washed in propylene oxide and embedded in Spurrs epoxy. Ultrathin sections were stained in uranyl acetate followed by Reynolds lead citrate and examined using a FEI Tecnai G2 TEM.
Pulse chase
Pulse chase studies were done as previously described (35). Briefly, MEF cells were plated at 2.5 × 105 cells on 35 mm dishes overnight. They were then treated with 50 mm ascorbic acid in DMEM plus 10% FBS for 24 h. Cells were washed with phosphate buffered saline and then treated with 140 µCi of l-[2,3,4,5-3H] proline under ascorbic acid stimulation in serum free DMEM. This was then chased using unlabeled proline in fresh DMEM for 0, 20, 40, 60 and 120 min. Medium was collected and inhibitor was added. Collagen was then concentrated from the samples with 25 ng of collagen carrier (Sigma) and absolute ethanol. Samples were electrophoresed on 5% acrylamide gels with urea under reducing conditions, dried and then exposed to film. This was repeated for four samples from each genotype to confirm consistent results.
Western blotting
Cells were either treated or not treated with 50 mm ascorbic acid for 24 h before collection. Cell lysates were collected by scraping cells into Laemmli buffer and boiling for 10 min. They were then loaded onto 4–15% gradient sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) gels (Biorad). Protein was transferred to PVDF membrane and blocked with 5% nonfat dry milk/PBS. Blots were probed with either polyclonal rabbit anti-FKBP65 antibody (Proteintech Group) or rabbit anti-Type I collagen antibody (kindly donated by Larry Fisher, LF-68) at 1 : 5000 and 1 : 10 000, respectively. They were coincubated with monoclonal mouse antitubulin at 1 : 10 000 in 1% milk overnight at 4°. Blots were washed with PBST. Secondary antibodies (antirabbit 488 and antimouse 594 Odyssey LiCor) were diluted 1 : 10 000 in 1% milk plus PBST for 1 h at room temperature. The Odyssey system was used to image the blots by fluorescence (LiCor).
For immunofluorescence, cells were plated in two-well chamber slides overnight and treated with ascorbic acid. Cells were fixed with 4% paraformaldehyde and then washed with PBS. They were then permeabilized with 0.1% Triton X100 in PBS for 5 min and washed with PBS. Blocking was done for 30 min in 10% goat serum plus 1% bovine serum albumin. Primary antibodies (FKBP65, proteintech group or LF-68, Larry Fisher) were incubated at 1 : 1000 in PBS plus 1% BSA for 1 h. Samples were washed with PBS and then incubated with secondary antibody in PBS (goat antirabbit 1 : 500, conjugated to fluorophore 488) plus anticoncanavalin antibody 594 (ER marker, 1 : 500, Invitrogen) or antilectin HPA antibody 488 (Golgi marker, 1 : 500, Invitrogen) for 1 h. Slides were then washed in PBS and mounted with antifade mounting solution plus DAPI stain.
Mass spectrometry analysis
Collagen was prepared from minced E18.5 calvaria. Type I α-chains were extracted by heat denaturation (90°C) in SDS–PAGE sample buffer, resolved on 6% SDS–PAGE gels (36), cut from gels and digested with trypsin in-gel (37).
Calvarial tissue was also digested with bacterial collagenase as described (38). Collagenase-generated peptides were separated by reversed-phase HPLC (C8, Brownlee Aquapore RP-300, 4.6 mm × 25 cm) with a linear gradient of acetonitrile : n-propanol (3 : 1v/v) in aqueous 0.1% (v/v) trifluoroacetic acid (39). Individual fractions were analyzed by LC–MS.
Peptides were analyzed by electrospray LC–MS using an LTQ XL ion-trap mass spectrometer (Thermo Scientific) equipped with in-line liquid chromatography using a C4 5 μm capillary column (300 μm × 150 mm; Higgins Analytical RS-15M3-W045) eluted at 4.5 μl/min. The LC mobile phase consisted of buffer A (0.1% formic acid in MilliQ water) and buffer B (0.1% formic acid in 3 : 1 acetonitrile : n-propanol, v/v). An electrospray ionization source introduced the LC sample stream into the mass spectrometer with a spray voltage of 3 kV. Proteome Discoverer search software (Thermo Scientific) was used for peptide identification using the NCBI protein database.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the National Institute of Health, P01 HD070394 (B.L., D.K., D.E.), F31DE022483 (C.L.), AR036794 and AR037318 (D.E.), the BCM Intellectual and Developmental Disabilities Research Center (HD024064) from the Eunice Kennedy Shriver National Institute Of Child Health & Human Development, the BCM Advanced Technology Cores with funding from the National Institute of Health (AI036211, CA125123 and RR024574), the Rolanette and Berdon Lawrence Bone Disease Program of Texas, and the BCM Center for Skeletal Medicine and Biology.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Larry Fisher (NIH) for his gift of type I collagen antibodies.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Rauch F., Glorieux F.H. Osteogenesis imperfecta. Lancet. 2004;363:1377–1385. doi: 10.1016/S0140-6736(04)16051-0. [DOI] [PubMed] [Google Scholar]
- 2.Wenstrup R.J., Willing M.C., Starman B.J., Byers P.H. Distinct biochemical phenotypes predict clinical severity in nonlethal variants of osteogenesis imperfecta. Am. J. Hum. Genet. 1990;46:975–982. [PMC free article] [PubMed] [Google Scholar]
- 3.Byers P.H., Pyott S.M. Recessively inherited forms of osteogenesis imperfecta. Annu. Rev. Genet. 2012;46:475–497. doi: 10.1146/annurev-genet-110711-155608. [DOI] [PubMed] [Google Scholar]
- 4.Marini J.C., Cabral W.A., Barnes A.M. Null mutations in LEPRE1 and CRTAP cause severe recessive osteogenesis imperfecta. Cell Tissue Res. 2010;339:59–70. doi: 10.1007/s00441-009-0872-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morello R., Bertin T.K., Chen Y., Hicks J., Tonachini L., Monticone M., Castagnola P., Rauch F., Glorieux F.H., Vranka J., et al. CRTAP is required for prolyl 3-hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. doi: 10.1016/j.cell.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 6.Cabral W.A., Chang W., Barnes A.M., Weis M., Scott M.A., Leikin S., Makareeva E., Kuznetsova N.V., Rosenbaum K.N., Tifft C.J., et al. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat. Genet. 2007;39:359–365. doi: 10.1038/ng1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pyott S.M., Schwarze U., Christiansen H.E., Pepin M.G., Leistritz D.F., Dineen R., Harris C., Burton B.K., Angle B., Kim K., et al. Mutations in PPIB (cyclophilin B) delay type I procollagen chain association and result in perinatal lethal to moderate osteogenesis imperfecta phenotypes. Hum. Mol. Genet. 2011;20:1595–1609. doi: 10.1093/hmg/ddr037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vranka J.A., Pokidysheva E., Hayashi L., Zientek K., Mizuno K., Ishikawa Y., Maddox K., Tufa S., Keene D.R., Klein R., et al. Prolyl 3-hydroxylase 1 null mice display abnormalities in fibrillar collagen-rich tissues such as tendons, skin, and bones. J. Biol. Chem. 2010;285:17253–17262. doi: 10.1074/jbc.M110.102228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baldridge D., Lennington J., Weis M., Homan E.P., Jiang M.M., Munivez E., Keene D.R., Hogue W.R., Pyott S., Byers P.H., et al. Generalized connective tissue disease in Crtap−/− mouse. PLoS One. 2010;5:e10560. doi: 10.1371/journal.pone.0010560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christiansen H.E., Schwarze U., Pyott S.M., AlSwaid A., Al Balwi M., Alrasheed S., Pepin M.G., Weis M.A., Eyre D.R., Byers P.H. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010;86:389–398. doi: 10.1016/j.ajhg.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alanay Y., Avaygan H., Camacho N., Utine G.E., Boduroglu K., Aktas D., Alikasifoglu M., Tuncbilek E., Orhan D., Bakar F.T., et al. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010;86:551–559. doi: 10.1016/j.ajhg.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelley B.P., Malfait F., Bonafe L., Baldridge D., Homan E., Symoens S., Willaert A., Elcioglu N., Van Maldergem L., Verellen-Dumoulin C., et al. Mutations in FKBP10 cause recessive osteogenesis imperfecta and Bruck syndrome. J. Bone Miner. Res. 2011;26:666–672. doi: 10.1002/jbmr.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koide T., Nishikawa Y., Asada S., Yamazaki C.M., Takahara Y., Homma D.L., Otaka A., Ohtani K., Wakamiya N., Nagata K., et al. Specific recognition of the collagen triple helix by chaperone HSP47. II. The HSP47-binding structural motif in collagens and related proteins. J. Biol. Chem. 2006;281:11177–11185. doi: 10.1074/jbc.M601369200. [DOI] [PubMed] [Google Scholar]
- 14.Ono T., Miyazaki T., Ishida Y., Uehata M., Nagata K. Direct in vitro and in vivo evidence for interaction between Hsp47 protein and collagen triple helix. J. Biol. Chem. 2012;287:6810–6818. doi: 10.1074/jbc.M111.280248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishikawa Y., Vranka J., Wirz J., Nagata K., Bachinger H.P. The rough endoplasmic reticulum-resident FK506-binding protein FKBP65 is a molecular chaperone that interacts with collagens. J. Biol. Chem. 2008;283:31584–31590. doi: 10.1074/jbc.M802535200. [DOI] [PubMed] [Google Scholar]
- 16.Nagai N., Hosokawa M., Itohara S., Adachi E., Matsushita T., Hosokawa N., Nagata K. Embryonic lethality of molecular chaperone hsp47 knockout mice is associated with defects in collagen biosynthesis. J. Cell Biol. 2000;150:1499–1506. doi: 10.1083/jcb.150.6.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishida Y., Kubota H., Yamamoto A., Kitamura A., Bachinger H.P., Nagata K. Type I collagen in Hsp47-null cells is aggregated in endoplasmic reticulum and deficient in N-propeptide processing and fibrillogenesis. Mol. Biol. Cell. 2006;17:2346–2355. doi: 10.1091/mbc.E05-11-1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barnes A.M., Duncan G., Weis M., Paton W., Cabral W.A., Mertz E.L., Makareeva E., Gambello M.J., Lacbawan F.L., Leikin S., et al. Kuskokwim syndrome, a recessive congenital contracture disorder, extends the phenotype of FKBP10 mutations. Hum. Mutat. 2013;34:1279–1288. doi: 10.1002/humu.22362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Puig-Hervas M.T., Temtamy S., Aglan M., Valencia M., Martinez-Glez V., Ballesta-Martinez M.J., Lopez-Gonzalez V., Ashour A.M., Amr K., Pulido V., et al. Mutations in PLOD2 cause autosomal-recessive connective tissue disorders within the Bruck syndrome—osteogenesis imperfecta phenotypic spectrum. Hum. Mutat. 2012;33:1444–1449. doi: 10.1002/humu.22133. [DOI] [PubMed] [Google Scholar]
- 20.van der Slot A.J., Zuurmond A.M., Bardoel A.F., Wijmenga C., Pruijs H.E., Sillence D.O., Brinckmann J., Abraham D.J., Black C.M., Verzijl N., et al. Identification of PLOD2 as telopeptide lysyl hydroxylase, an important enzyme in fibrosis. J. Biol. Chem. 2003;278:40967–40972. doi: 10.1074/jbc.M307380200. [DOI] [PubMed] [Google Scholar]
- 21.Hyry M., Lantto J., Myllyharju J. Missense mutations that cause Bruck syndrome affect enzymatic activity, folding, and oligomerization of lysyl hydroxylase 2. J. Biol. Chem. 2009;284:30917–30924. doi: 10.1074/jbc.M109.021238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwarze U., Cundy T., Pyott S.M., Christiansen H.E., Hegde M.R., Bank R.A., Pals G., Ankala A., Conneely K., Seaver L., et al. Mutations in FKBP10, which result in Bruck syndrome and recessive forms of osteogenesis imperfecta, inhibit the hydroxylation of telopeptide lysines in bone collagen. Hum. Mol. Genet. 2013;22:1–17. doi: 10.1093/hmg/dds371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schreiber S.L. Chemistry and biology of the immunophilins and their immunosuppressive ligands. Science. 1991;251:283–287. doi: 10.1126/science.1702904. [DOI] [PubMed] [Google Scholar]
- 24.Fischer G., Bang H., Mech C. [Determination of enzymatic catalysis for the cis-trans-isomerization of peptide binding in proline-containing peptides] Biomed. Biochim. Acta. 1984;43:1101–1111. [PubMed] [Google Scholar]
- 25.Fischer G., Bang H. The refolding of urea-denatured ribonuclease A is catalyzed by peptidyl-prolyl cis-trans isomerase. Biochim. Biophys. Acta. 1985;828:39–42. doi: 10.1016/0167-4838(85)90006-8. [DOI] [PubMed] [Google Scholar]
- 26.Cheung K.L., Bates M., Ananthanarayanan V.S. Effect of FKBP65, a putative elastin chaperone, on the coacervation of tropoelastin in vitro. Biochem. Cell Biol. 2010;88:917–925. doi: 10.1139/O10-137. [DOI] [PubMed] [Google Scholar]
- 27.Davis E.C., Broekelmann T.J., Ozawa Y., Mecham R.P. Identification of tropoelastin as a ligand for the 65-kD FK506-binding protein, FKBP65, in the secretory pathway. J. Cell Biol. 1998;140:295–303. doi: 10.1083/jcb.140.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bachinger H.P., Bruckner P., Timpl R., Prockop D.J., Engel J. Folding mechanism of the triple helix in type-III collagen and type-III pN-collagen. Role of disulfide bridges and peptide bond isomerization. Eur. J. Biochem. 1980;106:619–632. doi: 10.1111/j.1432-1033.1980.tb04610.x. [DOI] [PubMed] [Google Scholar]
- 29.Spokony R., Saint-Jeannet J.P. Xenopus FK 506-binding protein, a novel immunophilin expressed during early development. Mech. Dev. 2000;94:205–208. doi: 10.1016/s0925-4773(00)00315-4. [DOI] [PubMed] [Google Scholar]
- 30.Patterson C.E., Abrams W.R., Wolter N.E., Rosenbloom J., Davis E.C. Developmental regulation and coordinate reexpression of FKBP65 with extracellular matrix proteins after lung injury suggest a specialized function for this endoplasmic reticulum immunophilin. Cell Stress Chaperones. 2005;10:285–295. doi: 10.1379/CSC-118R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Friedel R.H., Seisenberger C., Kaloff C., Wurst W. EUCOMM—the European conditional mouse mutagenesis program. Brief Funct. Genomic Proteomic. 2007;6:180–185. doi: 10.1093/bfgp/elm022. [DOI] [PubMed] [Google Scholar]
- 32.Ha-Vinh R., Alanay Y., Bank R.A., Campos-Xavier A.B., Zankl A., Superti-Furga A., Bonafe L. Phenotypic and molecular characterization of Bruck syndrome (osteogenesis imperfecta with contractures of the large joints) caused by a recessive mutation in PLOD2. Am. J. Med. Genet. A. 2004;131:115–120. doi: 10.1002/ajmg.a.30231. [DOI] [PubMed] [Google Scholar]
- 33.Patterson C.E., Schaub T., Coleman E.J., Davis E.C. Developmental regulation of FKBP65. An ER-localized extracellular matrix binding-protein. Mol. Biol. Cell. 2000;11:3925–3935. doi: 10.1091/mbc.11.11.3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McElroy S.L., Reijo Pera R.A. Preparation of mouse embryonic fibroblast feeder cells for human embryonic stem cell culture. CSH Protoc. 2008 doi: 10.1101/pdb.prot5041. 2008, pdb prot5041. [DOI] [PubMed] [Google Scholar]
- 35.Bonadio J., Holbrook K.A., Gelinas R.E., Jacob J., Byers P.H. Altered triple helical structure of type I procollagen in lethal perinatal osteogenesis imperfecta. J. Biol. Chem. 1985;260:1734–1742. [PubMed] [Google Scholar]
- 36.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 37.Hanna S.L., Sherman N.E., Kinter M.T., Goldberg J.B. Comparison of proteins expressed by Pseudomonas aeruginosa strains representing initial and chronic isolates from a cystic fibrosis patient: an analysis by 2-D gel electrophoresis and capillary column liquid chromatography-tandem mass spectrometry. Microbiology. 2000;146(Pt 10):2495–2508. doi: 10.1099/00221287-146-10-2495. [DOI] [PubMed] [Google Scholar]
- 38.Hanson D.A., Eyre D.R. Molecular site specificity of pyridinoline and pyrrole cross-links in type I collagen of human bone. J. Biol. Chem. 1996;271:26508–26516. doi: 10.1074/jbc.271.43.26508. [DOI] [PubMed] [Google Scholar]
- 39.Wu J.J., Woods P.E., Eyre D.R. Identification of cross-linking sites in bovine cartilage type IX collagen reveals an antiparallel type II-type IX molecular relationship and type IX to type IX bonding. J. Biol. Chem. 1992;267:23007–23014. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.