

Abstract
Particulate methane monooxygenase (pMMO) catalyzes the oxidation of methane to methanol in methanotrophic bacteria. As a copper-containing enzyme, pMMO has been investigated extensively by electron paramagnetic resonance (EPR) spectroscopy, but the presence of multiple copper centers has precluded correlation of EPR signals with the crystallographically identified monocopper and dicopper centers. A soluble recombinant fragment of the pmoB subunit of pMMO, spmoB, like pMMO itself, contains two distinct copper centers and exhibits methane oxidation activity. The spmoB protein, spmoB variants designed to disrupt one or the other or both copper centers, as well as native pMMO have been investigated by EPR, ENDOR, and ESEEM spectroscopies in combination with metal content analysis. The data are remarkably similar for spmoB and pMMO, validating the use of spmoB as a model system. The results indicate that one EPR-active Cu(II) ion is present per pMMO and that it is associated with the active-site dicopper center in the form of a valence localized Cu(I)Cu(II) pair; the Cu(II), however, is scrambled between the two locations within the dicopper site. The monocopper site observed in the crystal structures of pMMO can be assigned as Cu(I). 14N ENDOR and ESEEM data are most consistent with one of these dicopper-site signals involving coordination of the Cu(II) ion by residues His137 and His139, the other with Cu(II) coordinated by His33 and the N-terminal amino group. 1H ENDOR measurements indicate there is no aqua (HxO) ligand bound to the Cu(II), either terminally or as a bridge to Cu(I).
Introduction
The conversion of methane to methanol by methanotrophic bacteria is catalyzed by methane monooxygenases (MMOs).1 MMOs activate the inert methane C–H bond (104 kcal/mol) at ambient temperature and pressure, in contrast to costly and inefficient industrial catalysts.2 There are two distinct types of MMO. Almost all methanotrophs produce a membrane-bound MMO, particulate MMO (pMMO), and some strains produce a soluble MMO (sMMO), primarily under conditions of copper starvation.3 sMMO is well characterized and contains a diiron active site.4 pMMO is less well understood,5 but has attracted intense attention as a target for the development of gas-to-liquid bioconversion processes. Such efforts require a detailed understanding of the pMMO catalytic site and mechanism.
Several crystal structures of pMMO have been determined and reveal an α3β3γ3 trimer comprising three copies each of the pmoB (α), pmoA (β), and pmoC (γ) subunits.6−8 The structure of pMMO from Methylococcus capsulatus (Bath) includes three metal binding sites. Two distinct copper centers are located in the soluble domains of the pmoB subunit (Figure 1). The first site is modeled as a single copper ion coordinated by residues His48 and His72 and is not present in other pMMOs. The second site, located near the membrane interface, is coordinated by residues His33, His137, and His139 and is highly conserved among pMMOs and related enzymes, with the exception of the Verrucomicrobia family.5,9 This site has been modeled as dinuclear, consistent with a Cu–Cu distance of 2.5–2.6 Å observed in extended X-ray absorption fine structure (EXAFS) data for multiple pMMOs.7,8,10,11 A third metal center resides in the pmoC transmembrane subunit and is occupied by either zinc or copper in the crystal structures.6−8
Figure 1.
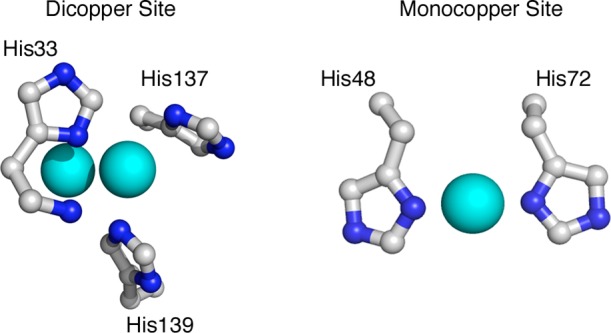
Copper centers modeled in the pmoB subunit of M. capsulatus (Bath) pMMO (pdb 3RGB). The dicopper site (left) is located at the N-terminus of the pmoB subunit, and conserved residues His33, His137, and His139 coordinate the copper ions. The monocopper site (right), located ∼21 Å distant, is not conserved in other pMMO structures, and residues His48 and His72 coordinate the copper ion.
The identity of the pMMO active site has been a longstanding controversy with several distinct models proposed in the literature,5,12 including a trinuclear copper site not observed in any of the crystal structures13 and a diiron center similar to that in sMMO, modeled at the pmoC zinc/copper site,14 and supported by recent mutagenesis studies on a hydrocarbon monooxygenase homologous to pMMO.15 However, our work indicates that pMMO activity is dependent on copper, not iron, and that the active site is located at the dicopper center modeled in the soluble, periplasmic region of the pmoB subunit.16 Key evidence in support of this model was obtained by the construction of a soluble monomeric pmoB protein fragment in which the two cupredoxin-like domains are connected by an artificial linker rather than the two transmembrane helices present in native pmoB. This protein, denoted soluble pmoB (spmoB), contains the ligands for both the mono- and dicopper sites and exhibits methane oxidation activity. Most important, analysis of site-specific spmoB variants implicates the dicopper center as the pMMO active site.16 Consistent with this hypothesis is the recent detection of a 345 nm optical feature upon reaction of reduced pMMO with H2O2 or reduced spmoB with either H2O2 or O2.17 This feature is suggestive of oxygen reacting at a dicopper center and disappears in the presence of methane.17
One unresolved issue surrounding the pMMO copper centers, including the proposed dicopper active site, is their oxidation states. Native M. capsulatus (Bath) pMMO has been examined with electron paramagnetic resonance (EPR) spectroscopic methods (Table S1).10,18−22 Both membrane-bound and purified pMMO exhibit a typical type 2 Cu(II) signal that corresponds to 40%–60% of total copper, consistent with the mixture of Cu(I) and Cu(II) observed in X-ray absorption near edge spectral (XANES) data.11,23 It is of central importance to determine the oxidation states of the copper ions of the proposed dicopper active site, which requires identification of the type 2 signal with copper sites present in the crystal structures, but this has been impossible without a recombinant expression system for pMMO.24 The development of spmoB provides a new toolkit for systematically investigating the EPR properties of pMMO.
In this work, we have employed advanced EPR techniques to study the previously designed spmoB_wt protein,16 several new metal-binding variants, and native pMMO itself, all in the as-isolated state. Using a combination of EPR and inductively coupled plasma atomic emission spectroscopies (ICP-AES), we have determined the oxidation states of the copper ions in each site of these enzymes. All the spmoB variants and pMMO contain Cu(II), and in each case, we have quantified this ion and structurally characterized its coordination sphere by electron spin echo envelope modulation ESEEM and/or electron nuclear double resonance (ENDOR) spectroscopies. These measurements allow us to assign the oxidation states of both the dicopper and monocopper centers of spmoB and pMMO.
Experimental Section
Preparation of spmoB, spmoB Variants, and pMMO
Site-specific variants spmoB_H48N,H72A, spmoB_H33,137,139A, and spmoB_penta (H33,72,137,139A,H48N) were generated using the QuikChange site-directed mutagenesis kit (Stratagene). Previously reported variants (spmoB_H48N, spmoB_H137,139A, spmoB_H137,139A,H48N)16 were used as the starting templates for mutagenesis. Expression and purification were performed as described previously.16,25 Briefly, recombinant spmoBs were expressed in E. coli strain BL21(DE3) (Invitrogen) in liquid LB medium incubated at 37 °C to an OD600 of 0.4–0.6. Protein expression was induced by the addition of 1 mM isopropyl-β-d-thiogalactopyranoside. After induction, the cultures were incubated at 37 °C for 3 h and the cells were harvested by centrifugation. Cells were washed with 50 mM Tris pH 8.0, 150 mM NaCl, and frozen at −20 °C until thawed and lysed by sonication. The spmoB proteins express mostly in inclusion bodies, which were isolated by centrifuging the lysate at 3 000g for 30 min at 4 °C. The pellet containing spmoB was washed repeatedly with 50 mM Tris pH 8.0, 150 mM NaCl containing 1.0% Triton X-100. A final wash with buffer lacking Triton X-100 was then performed. The solubilized inclusion bodies were denatured by incubation with 8 M urea. After a 1 h incubation, the sample was centrifuged at 15 000g for 30 min at 20 °C. The supernatant containing the spmoB protein was used for refolding and copper incorporation. Protein concentration was determined by A280 using ε280 = 46 410 M–1 cm–1 as determined by amino acid analysis (Texas A&M University, Protein Chemistry Lab). To obtain native pMMO, M. capsulatus (Bath) cells were cultivated in a fermenter, and pMMO was solubilized and purified with n-dodecyl-β-d-maltopyranoside (DDM) as described previously.10
Metal Incorporation and Quantitation
The copper content of pMMO was determined by ICP-AES (Varian Vista MPX) at the Integrated Molecular Structure Education and Research Center (IMSERC) of Northwestern University. All samples of pMMO used for EPR, ENDOR, and ESEEM experiments contained three copper ions per αβγ protomer. Copper incorporation into spmoB was performed as described previously by a stepwise reduction of 8 M urea to buffer containing no urea.16,25 Copper (1 mM CuSO4) was added to the 6 M urea refolding solution. Copper-loaded, active spmoB is obtained by this method. The spmoB copper concentrations were determined by ICP-AES.
Enzyme Activity Assays
Methane oxidation activity was measured by gas chromatography (GC) (Hewlett-Packard 7890A) with an HP-Plot Q capillary column (Agilent). Copper loaded spmoB samples (∼100 μM) were reacted with CH4 (2 mL headspace) and O2 (1 mL headspace) in the presence of duroquinol. Samples were incubated at 45 °C with shaking at 200 rpm for 1 h. Reactions were then quenched at 95 °C for 5 min, cooled on ice, centrifuged to remove the solid duroquinol, and the clear supernatant (5 μL) was injected onto the GC. GC analysis was performed using a gradient program of 85 to 125 °C with increases of 25 °C per min, followed by ramping to 225 °C at 50 °C per min, and an additional 5 min at 225 °C (total run time 8.6 min). Peak intensities were compared to a calibration curve generated from methanol standards (Sigma-Aldrich, spectrophotometric grade, >99%).
Quantitative EPR Spectroscopy
Quantitative X-band continuous wave (CW) EPR spectra of spmoB, spmoB variants, and pMMO were collected on a modified Varian E-4 at 77 K with 100 kHz field modulation (4.0 G modulation amplitude) under nonsaturating conditions by comparison to a CuSO4 standard calibration of 40 to 360 μM in 50 mM Tris pH 8.0, 150 mM NaCl, 1 mM EDTA, 10% v/v glycerol. Double integration of background-corrected spectra was performed digitally using Spincount software.26 All spectra were collected under the same conditions and integrated over the same magnetic field range. Multiple measurements indicated the combined EPR measurement, and the integration error is approximately ±7%. Simulations of EPR spectra were performed using the MATLAB EasySpin v4.5 toolbox (easyspin.org).27
ENDOR Spectroscopy
All ENDOR samples were concentrated to approximately 100 μM in 50 mM Tris pH 8.0, 150 mM NaCl, 10% v/v glycerol for spmoB and 200 μM in 50 mM HEPES pH 7.5, 400 mM NaCl, 0.05% DDM for pMMO and frozen in custom Q-band tubes. Deuterated samples were prepared in the same buffer using D2O and d8-glycerol (Sigma-Aldrich). Pulsed Q-band ENDOR spectra were collected at 2 K on previously described instrumentation.28,29 ENDOR collection employed a Davies microwave pulse sequence (π – T – π/2 – τ – π – τ – echo) in which the RF pulse is applied during time T.30 Pulse data acquisition was performed with the SpecMan software package31 (http://specman4epr.com) in conjunction with a Spin-Core PulseBlaster ESR_PRO 400 MHz word generator and Agilent Technologies Acquiris DP235 500MS/sec digitizer and employed random hopping of the RF frequency.32 CW Q-band ENDOR spectra were collected on a previously described modified Varian E-110 by the digitization of the RC-smooth output signal under “rapid adiabatic” conditions at 2 K with 100 kHz field modulation.33,34 RF noise broadening was employed to improve ENDOR response intensity.35
The ENDOR spectrum from a nucleus with a spin of I = 1/2 (1H) and from the ms = ±1/2 electron-spin manifold exhibits a doublet at frequencies,
| 1 |
where vn is the nuclear Larmor frequency and A is the hyperfine coupling. When I ≥ 1 (14N), a nuclear quadrupole interaction (P) introduces further splitting of the ν± manifolds. Absolute hyperfine coupling signs were obtained through the PESTRE protocols (see Supporting Information).36−38
ESEEM Spectroscopy
ESEEM experiments were carried out at 9.72 GHz using a Bruker EleXsys E580 spectrometer with an EN4118X-MD4 resonator and an Oxford Instruments CF935 helium flow cryostat/ITC503S temperature controller at 6 K. A three-pulse sequence, π/2 – τ – π/2 – ΔT – π/2 – τ – echo, was employed with four-step phase cycling to suppress unwanted echoes. ESEEM was collected with τ = 348 ns to suppress the strong water protons’ response.
Simulations of the ESEEM results were performed using the MATLAB based OPTESIM software package.39 The spin Hamiltonian for interaction of the Cu(II) electron spin (S = 1/2) with the remote 14N nuclear spin (I = 1) of a histidyl imidazole ligand is formulated with a nuclear Zeeman, a hyperfine (hf, A), and a nuclear quadrupole (nqi) term, as follows:
| 2 |
In this expression, gn is the nuclear g-value, βn is the nuclear magneton, S is the electron spin operator, I is the nuclear spin operator, A is the hf coupling tensor, and Q is the nqi tensor. The hf tensor has the principal components, A = [AxxAyyAzz], and is composed of an isotropic part, aiso = (1/3) ΣiAii, and a dipolar part, Tdip= A – Aiso. The nqi tensor has the principal components, Q = [QxxQyyQzz] and is defined by the nuclear quadrupole coupling constant, e2qQ/h, and the electric field gradient asymmetry parameter, η. In its principal axis system (PAS), Q is related to e2qQ/h and η by the following expressions:
| 3 |
| 4 |
The orientation between the nqi tensor PAS and the hf tensor PAS is defined by the Euler angles, Q[α, β, γ]. The single field spectra with simulations of two coupled nuclei include relative A-tensor orientation of the second nuclei to the first as described by a set of Euler angles and keeping the first nuclei A[α, β, γ] = [0, 0, 0]°.
We considered the two distinct coordination environments of a Cu(II) in the dicopper site, one with two histidine imidazoles bound to Cu(II) and one with a single histidine and the terminal amine; in this latter case, the amine 14N would not give an ESEEM response, and the ESEEM would be described as arising from a single remote histidyl nitrogen. As explained in the Results, for spmoB, the Cu(II) is localized in a single location, whereas, for pMMO, simulations sum equal weights of the simulations for the di- and mono-His locations. Final simulations parameters are presented in Table 1. The OPTESIM optimization uses the Nelder–Mead simplex method.
Table 1. ESEEM Simulation Parameters.
| spmoB_H48N,H72A | pMMO |
||
|---|---|---|---|
| Cu(II)His2 | Cu(II)His2 | Cu(II)His | |
| A (MHz) | [1.57, 1.07, 2.03] | [1.68, 1.18, 2.13] | [1.68, 1.18, 2.13] |
| e2Qq (MHz) | 1.51 | 1.53 | 1.53 |
| η | 0.91 | 0.87 | 0.87 |
| A[α, β, γ]° | [240, 70, 0] | [240, 65, 0] | – |
| Q[α, β, γ]° | [0, 55, 0] | [0, 60, 0] | – |
Results
New spmoB Variants
The original spmoB variant that was designed to disrupt both copper sites, spmoB_H48N,H137,139A, still binds approximately one copper ion per protein16 (Figure 2), suggesting that even a single histidine in either site (Figure 1) can bind copper. If this is the case, the other original variants, spmoB_H48N and spmoB_H137,139A, might also contain some copper in both sites, rendering correlation of EPR signals with specific sites difficult. To address this issue, we prepared three new variants in which all the metal coordinating histidines are replaced with alanine or asparagine (Figure 1). These include spmoB_H48N,H72A, which completely eliminates the monocopper site, spmoB_H33,137,139A, which completely eliminates the dicopper site, and spmoB_penta, in which all five histidines are replaced with either alanine or asparagine. All three variant proteins express similarly to wildtype spmoB (spmoB_wt) (Figure S1) as inclusion bodies and can be refolded in the presence of copper.
Figure 2.

Copper content (equivalents per protein monomer) of refolded spmoB_wt and variants determined by ICP-AES. Error bars represent the average of at least three independent protein preparations.
Residual copper binding is indeed diminished in these variants. The spmoB_wt protein binds 2.8 ± 0.4 copper ions per protein monomer, spmoB_H48N,H72A binds 2.0 ± 0.3 copper ions, spmoB_H33,137,139A binds 0.9 ± 0.1 copper ions, and spmoB_penta binds 0.2 ± 0.1 copper ions (Figure 2). These values are consistent with the M. capsulatus (Bath) pMMO crystal structure.6 The small amount of copper present in spmoB_penta suggests that some adventitious binding can still occur outside of the two crystallographic copper sites. Only spmoB_wt and the spmoB_H48N,H72A variant exhibit methane oxidation activity (Figure S2), in agreement with our previous results and assignment of the active site location.16 The activity of spmoB_H48N,H72A is ∼10% that of spmoB_wt, similar to what we observed for the spmoB_H48N variant.16 These new variants could thus be used to isolate EPR signals deriving from the mononuclear (spmoB_H33,137,139A) and dinuclear (spmoB_H48N,H72A) copper sites.
EPR Characterization
The copper ions in the M. capsulatus (Bath) pMMO crystal structure were modeled as occupying one monocopper center and one dicopper center.6 Phylogenetic and sequence alignments indicate that the pMMOs from Methylomicrobium album BG8 and likely Methylococcus capsulatus (strain M) contain ligands that could also support both mono- and dicopper sites. In contrast, the pMMOs from Methylocystis species (strain M)8 and Methylosinus trichosporium OB3b7 lack the monocopper site ligands required for copper binding and retain only the ligands present in the dicopper site. Previous copper quantifications of pMMOs from various methanotrophic species show that there is variability in the amounts of total copper and EPR-active Cu(II) between species and/or type of sample (Table S1).
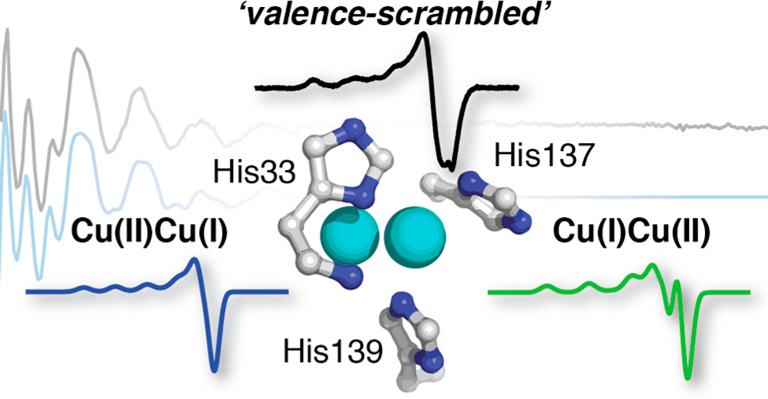
As previously reported,10,11 we find that purified pMMO from M. capsulatus (Bath) binds three total copper ions per protomer, one of which is an EPR-active Cu(II). The EPR spectrum for pMMO (Figure 3) cannot be attributed to the signal of a valence-delocalized Cu(1.5)–Cu(1.5) dicopper center, which would exhibit a 7-line g⊥ pattern extending to higher fields than observed. The presence of one Cu(II) ion would permit a heterogeneous dicopper site, with ∼0.5[Cu(II)–Cu(II)] EPR-active dicopper and ∼0.5[Cu(I)–Cu(I)] EPR-silent copper, but this also is excluded since no strong Cu–Cu dipolar interactions are observed.
Figure 3.
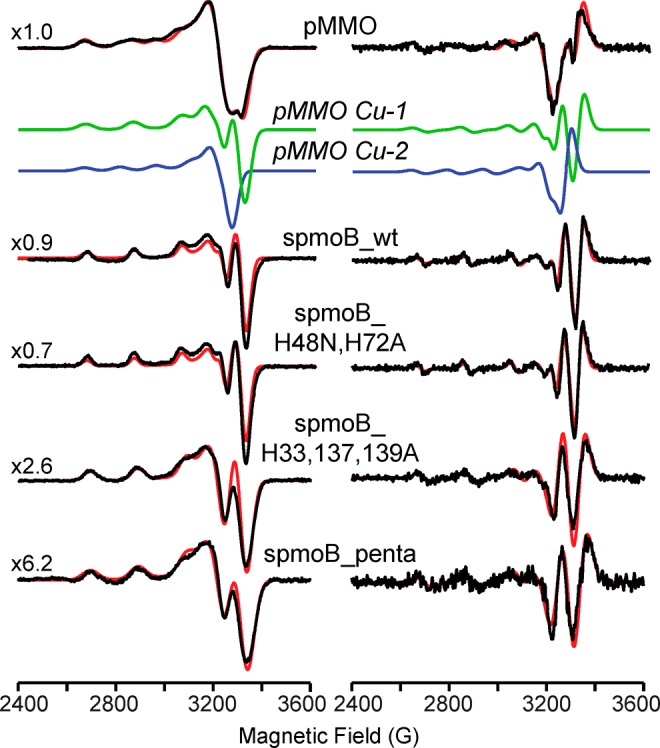
X-band (∼9.23 GHz) EPR spectra (left, black) and smoothed numerical derivatives (right, black) with simulations in red of pMMO (equal equivalents of Cu-1: g⊥ = 2.047, g∥ = 2.285, and A⊥ = 65; A∥ = 585 MHz, 50 G fwhm line width with additional 150 MHz fwhm unresolved hyperfine broadening along g∥, green; Cu-2: g⊥ = 2.047, g∥ = 2.225, and A⊥ = 40; A∥ = 450 MHz, blue; 50 G fwhm line width with additional 150 MHz fwhm unresolved hyperfine broadening along g∥ for each Cu-1 and Cu-2), spmoB_wt (g⊥ = 2.047, g∥ = 2.285, and A⊥ = 65; A∥ = 585 MHz, 40 G fwhm line width), spmoB_H48N,H72A (g⊥ = 2.047, g∥ = 2.285, and A⊥ = 65; A∥ = 585 MHz, 32.5 G fwhm line width), spmoB_H33,137,139A (g⊥ = 2.047, g∥ = 2.210, and A⊥ = 50; A∥ = 595 MHz, 60 G fwhm line width), and spmoB_penta (g⊥ = 2.047, g∥ = 2.210, and A⊥ = 50; A∥ = 595 MHz, 72.5 G fwhm line width). Signal intensities were normalized for receiver gain and protein concentration and expanded to identical heights for presentation with expansion factors given in the figure. Spectra were collected at 77 K with 100 kHz field modulation, 4.0 G modulation amplitude, 300 ms time constant, 2 min scans minimum of 5 scans. Spectra are baseline corrected by subtraction of a cavity background signal obtained from HEPES buffer solution under identical conditions.
The EPR signal observed for pMMO instead is characteristic of type 2 Cu(II) centers (Figure 3), but with significant line broadening and poorly resolved copper hyperfine features compared to spectra of well-defined type 2 Cu(II) centers. The EPR spectrum for the single Cu(II) ion per pMMO protomer can be well simulated with equal contributions from two distinct type 2 Cu(II) species: Cu-1 (g⊥ = 2.047, g∥ = 2.285, A⊥ = 65; A∥ = 585 MHz) and Cu-2 (g⊥ = 2.047, g∥ = 2.225, A⊥ = 40; A∥ = 450 MHz) (Figure 3). This situation can be attributed to three possible scenarios: (i) a fully occupied [Cu(II) Cu(I)] dicopper site with “valence scrambling” between the two locations that make up this site, (ii) the presence of 50% of a well-defined [Cu(II) Cu(I)] site plus 50% Cu(II) occupancy of the monocopper site, or (iii) equal Cu(II) occupancy of a well-defined [Cu(II) Cu(I)] site and a well-defined Cu(II) adventitious site (Figure 1).
EPR analysis and quantification on recombinant spmoB and its variants were carried out to identify the location of the EPR active Cu(II). The spmoB_wt enzyme exhibits an EPR spectrum of a well-defined type 2 Cu(II) species, with the same parameters as the pMMO Cu-1 species: g⊥ = 2.047, g∥ = 2.225, and A⊥ = 65, A∥ = 585 MHz (Figure 3). Elimination of the ligands at the monocopper site in the spmoB_H48N,H72A variant does not significantly alter the EPR parameters from those observed for spmoB_wt, indicating that the Cu(II) ions of spmoB_wt and spmoB_H48N,H72A do not bind at the monocopper site, but rather that the Cu-1 species of pMMO and spmoB is bound at one particular location within the dicopper site. Additionally, ICP-AES measurements show that the spmoB_wt sample contains nearly three total copper ions, consistent with the M. capsulatus (Bath) pMMO crystal structure,6 and that one copper is lost when the monocopper site is disrupted in spmoB_H48N,H72A (Figures 2, 4). However, spmoB_wt and spmoB_H48N,H72A exhibit the same amount of Cu(II) by EPR spectroscopy (Figure 4 and Table S2). As the monocopper site has been disrupted in the spmoB_H48N,H72A variant, this site must therefore contain an EPR-silent Cu(I), eliminating scenario (ii) in which the enzyme contains 50% of a well-defined [Cu(II) Cu(I)] site plus 50% Cu(II) occupancy of the monocopper site.
Figure 4.
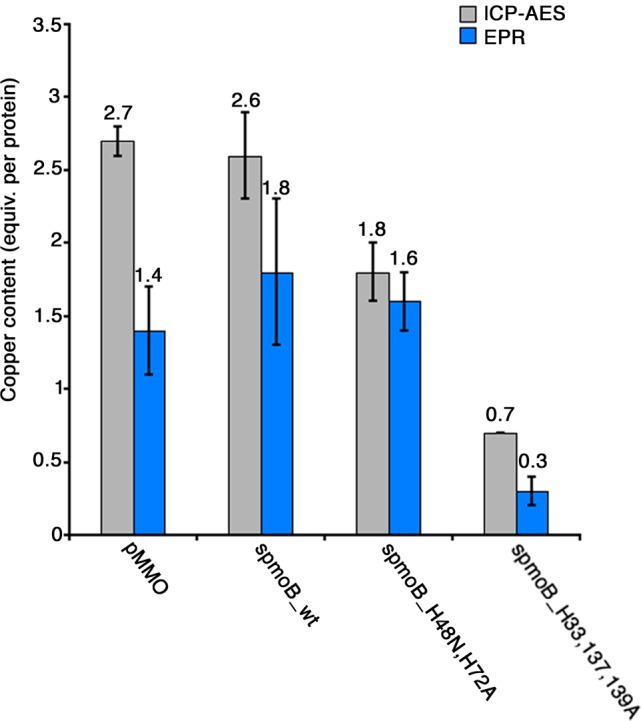
Copper content of M. capsulatus (Bath) pMMO (equivalents per αβγ protomer), spmoB_wt, and the new spmoB variants (equivalents per monomer) determined by ICP-AES (gray) and EPR (blue). The amounts of EPR-active copper in spmoB_wt and spmoB_H48N,H72A are comparable, but spmoB_wt binds an additional equivalent of copper. Error bars represent an average of three experiments. These values are normalized against values from the spmoB_penta variant.
Disruption of the dicopper site in spmoB_H33,137,139A and of both sites in spmoB_penta causes the loss of most of the EPR-active Cu(II) (Figure 4 and Table S2). This observation supports the assignment of the type 2 Cu(II) of pMMO as being coordinated within the dicopper center. The small residual signal in spmoB_penta has different characteristics from that of spmoB_wt, with g⊥ = 2.047, g∥ = 2.210, A⊥ = 50, A∥ = 595 MHz, as expected if it is bound elsewhere on the protein. The spmoB_H33,137,139A variant has lost a significant portion of the Cu(II) EPR intensity compared to spmoB_wt and spmoB_H48N,H72A, but still contains approximately one copper ion by ICP-AES. The Cu(II) retained in spmoB_H33,137,139A has different EPR parameters from spmoB_wt and spmoB_H48N,H72A, consistent with its assignment as adventitious binding. Taken together, the measurements on both pMMO and the suite of spmoB variants indicate that the monocopper site contains one EPR-silent Cu(I)40 and that the dicopper site contains one EPR-active Cu(II) species and one Cu(I), present as a valence-localized but scrambled pair. We favor the scrambling scenario (i) over scenario (iii), equal Cu(II) occupancy of a well-defined [Cu(II) Cu(I)] site and a well-defined Cu(II) adventitious site, because the crystal structure would reveal an additional copper site with 50% occupancy, but this is not observed.6 For completeness, we emphasize that 50% of Cu distributed among numerous sites would not be detected crystallographically, but also could not give the well-defined EPR spectrum observed.
14N ENDOR Characterization
14N ENDOR spectroscopy was employed to characterize and compare the coordination environments of the Cu(II) of pMMO, spmoB, and its variants. The 14N (I = 1) Davies ENDOR responses at g∥ for the EPR-active Cu(II) of the dicopper sites of pMMO, spmoB_wt, and spmoB_H48N,H72A each show a quadrupole-split doublet-of-doublets from 14N, denoted 14N1, with a hyperfine coupling of A∥ = +35.5 MHz and resolved quadrupole splitting, 3P∥ = 2.8 MHz (Figure 5); the absolute signs of A were determined through a Pulsed ENDOR Saturation and Recovery (PESTRE) experiment (Figure S3). The Cu(II) is shown above to be associated with the proposed valence-scrambled dicopper site of pMMO and the valence-localized sites of spmoB_wt and spmoB_H48N,H72A. In the case of spmoB, where Cu(II) is associated with one of the two copper locations in the dicopper site, the crystal structure shows that the candidates for the 14N ligands are either His137 and His139 or His33 and the N-terminal amino group. The 14N1 response does not distinguish between these options because the hyperfine/quadrupole couplings for bound 14N of histidine and an amino group are so similar.34,41−44 Correspondingly, it is expected that the 14N ENDOR spectra of the valence-localized and valence-scrambled sites are not distinguishable, as observed.
Figure 5.
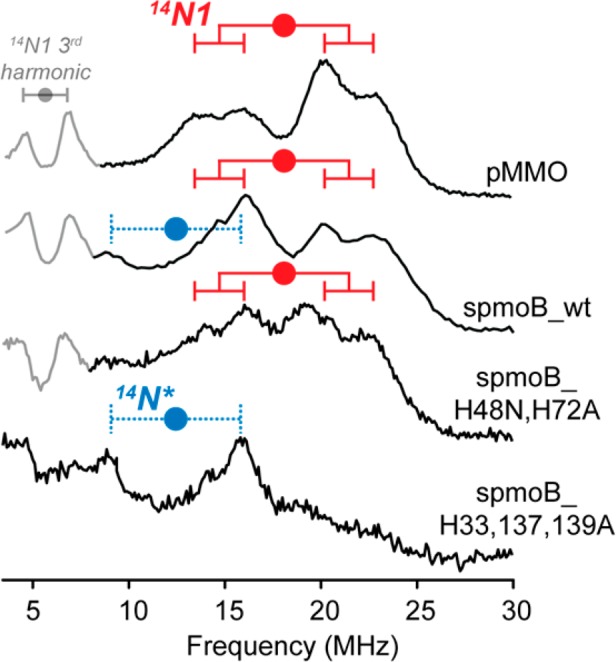
∼34.8 GHz ENDOR Davies traces (π = 80 ns, τ = 600 ns, T = 20 μs, repetition rate = 20 ms) of pMMO, spmoB_wt, spmoB_H48N,H72A, and spmoB_H33,137,139A collected at 2 K and g = 2.22. The nitrogen couplings are A/2 centered with circle goalposts: A∥(14N1) A = +35.5 (red); A∥(14N*) = +25 MHz (blue). Only 14N1 has clear quadrupole splitting, P∥(14N1) = 1.83 MHz. The third harmonic centered at 6 MHz of 14N1 is observed in spmoB_wt, spmoB_H48N,H72A, and pMMO and is shaded out in gray.
CW and pulsed X-band ENDOR signals from 14N1 of membrane-isolated pMMO from M capsulatus (strains M and Bath) were reported previously,45,46 but this signal was assigned differently,45 as discussed below. In contrast, spmoB_H33,137,139A shows no ENDOR intensity from 14N1, confirming the disruption of the histidine coordination of the Cu(II) within the dicopper site.
Figure 5 also shows that the Cu(II) of spmoB_H33,137,139A, which is not associated with the dicopper site, has a weak ν+/ν–14N doublet without quadrupole splitting that corresponds to a different type of 14N, denoted 14N*, with smaller hyperfine coupling, A = +25 MHz. Careful examination of the spmoB_wt spectrum also suggests the presence of a weak ν– feature from 14N*.
14N ESEEM Characterization
The three-pulse ESEEM time-domain waveforms and FT frequency-domain spectra collected near g⊥ for spmoB_H48N,H72A and for M. capsulatus (Bath) pMMO are quite similar, but with clear, albeit subtle differences (Figure 6). The waveforms and corresponding FT spectra are characteristic of the coupling of an electron spin with the remote 14N of imidazole.42,47−51 It was previously suggested by Yuan et al.200 that membrane fractions of pMMO from M. capsulatus (Bath) and M. album BG8 also have the same local nitrogen coordination environments.
Figure 6.
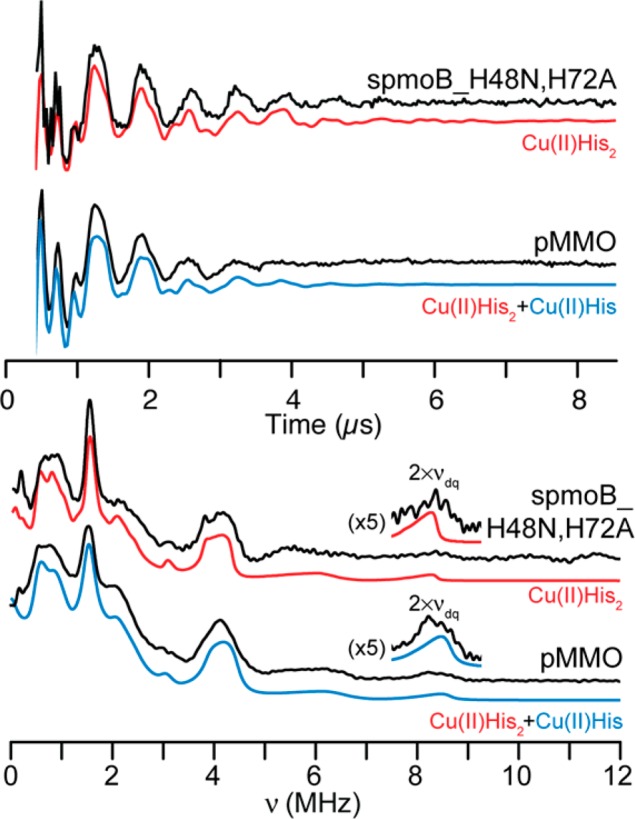
Three-pulse ESEEM time-domain waveforms (top) and Fourier transformed frequency-domain spectra for spmoB_H48N,H72A and M. capsulatus (Bath) pMMO in black. The Cu(II)His2 simulation for spmoB_H48N,H72A is shwon in red and the averaged sum of Cu(II)His2 and Cu(II)His for pMMO is shown in blue. The 2υdq combination features of the frequency-domain spectra are expanded five times as insets. Simulation parameters are given in the Experimental Section and Table 1. Spectral conditions for spmoB_H48N,H72A: νmw = 9.72 GHz, B0 = 3390 G, τ = 348 ns, T0 = 32 ns, ΔT = 32 ns, 256 points. Spectral parameters for pMMO: same as for spmoB_H48N,H72A except B0 = 3375 G.
The spectra shown in Figure 6 have been analyzed in detail. The responses reflect exact cancellation conditions, in which the nuclear Zeeman and hf terms are approximately equal in one electron-spin manifold. Thus, in this manifold, energy level separations are dominated by the nqi terms. The three features at 0.59, 0.94, and 1.53 MHz in the FT spectra of spmoB_H48N,H72A and pMMO (Figure 6, bottom) correspond to the nqi frequencies v–, v0, and v+, respectively. The broad feature, centered at 4.1 MHz observed in both samples corresponds to the ΔmI = ± 2 transition and is caused by the dipolar hf coupling and nqi within the other electron spin manifold, where the hf and nuclear Zeeman terms are noncanceling. In addition, each sample exhibits broad features at 2–3 MHz that can be assigned as combination lines and are positioned at the sums of the fundamental nqi frequencies (v0 + v+, v–+ v+, and 2 × v+). These combination lines are caused by relatively low probability nuclear spin transitions among the remote 14N nuclear spin states.
Of key importance, the weak feature observed for each sample at approximately 8 MHz can be assigned to the double quantum combination (2 × vdq) resulting from the addition of the ΔmI = ±2 splitting of two distinct remote histidyl 14N nuclei.50 Considering the ESEEM of spmoB_H48N,H72A, the presence of double-quantum features establishes that the ESEEM response arises from two essentially identical, remote histidyl 14N. Correspondingly, the ESEEM responses are well fit with parameters that are comparable to previously reported values for the remote 14N nuclei of Cu(II)-coordinated imidazoles42,47−51 (Figure 6). The EPR, ENDOR, and ESEEM results thus establish that the Cu(II) ion of spmoB corresponds to the single location within the crystallographic dicopper site of pMMO,6 ligated by the histidyl imidazoles of His137 and His139, which it binds in an approximately tetrahedral geometry.51
The EPR results presented above show that the Cu(II) of pMMO is incorporated in the valence-localized but tentatively assigned scrambled dicopper site, meaning that (roughly) half the intensity would derive from a Cu(II)His2 ion, as with spmoB, and the other half would come from Cu(II) in the other location associated with the dicopper site, with potential ligands His33 and the N-terminal amino group, with only His33 contributing to the ESEEM response. The pMMO ESEEM traces are fit well with such a summation (Figures 6 and S4).
The hyperfine and quadrupole parameters reported here for spmoB_H48N,H72A and the Cu(II)His2 of pMMO are in rough agreement with those reported by Lemos et al.,52 but the previous study was unable to detect the 14N double-quantum features in the ESEEM and, thus, did not identify the signal as coming from two histidines.
1H ENDOR Characterization
1H ENDOR measurements were performed to test for any 1HxO species coordinated to the Cu(II) ions at the dicopper site. Comparison of the CW 1H ENDOR spectra of spmoB_H48N,H72A and pMMO in H2O and D2O buffers reveals only the presence of an exchangeable proton signal (Figure 7) whose hyperfine coupling ranges from A⊥ = 2.3 MHz at g⊥ to A∥ = 6.3 MHz at g∥. An exchangeable proton with an analogous coupling was seen previously for pMMO within membrane fractions.46 These couplings are too small to be assigned to a terminal 1HxO ligand to Cu(II) or to a 1HxO bridge of a valence-localized mixed-valence dimer.53−55 Although other scenarios are possible, the couplings are compatible with the presence of a nearby water. These studies were performed on as-isolated samples, in which no hydroxyl group is observed. The 1H ENDOR spectroscopy of course cannot address the possible presence of an oxo bridge, as suggested for the active intermediate.56
Figure 7.
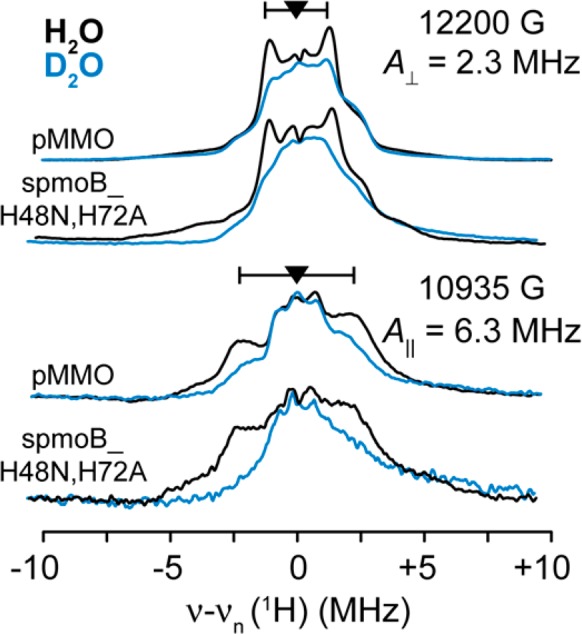
Proton exchange ENDOR of spmoB_H48N,H72A and pMMO. The Q-Band (∼35 GHz) CW ENDOR of spmoB_H48N,H72A and pMMO in H2O (black) and D2O (blue) are presented on a 1H Larmor centered axis collected at 2 K employing 1 G modulation amplitude, 100 kHz RF noise broadening, and 1 MHz s–1 scan rate. At g⊥, a sharp proton doublet feature of A⊥ = 2.3 MHz in both spmoB_H48N,H72A and pMMO is exchanged as observed in the ENDOR intensity difference between the H2O and D2O spectra. At g∥, the exchanged proton coupling is larger, A∥ = 6.3 MHz.
Discussion
Copper Oxidation States
EPR analysis indicates that the Cu(II) species in spmoB and pMMO are located at the proposed dicopper active site (Figures 3 and 4). The previous identification of the dicopper center of pMMO and spmoB as the active site is based on measured copper stoichiometries, activity measurements, EXAFS data, and oxygen reactivity studies.5,16,17 The EPR measurements reported here indicate that the dicopper site in pMMO contains one Cu(I) ion and one Cu(II) ion, proposed as a valence-localized mixed-valence Cu(I)Cu(II) pair, and that the monocopper site is present as Cu(I). The 1H ENDOR measurements show that the Cu(II) is not coordinated by a HxO ligand, so the two ions of the Cu(I)Cu(II) pair cannot be bridged by a hydroxo group in the as-isolated samples. The measurements do not rule out an oxo bridge, but we surmise that such a bridge would lead to valence delocalization.
The mixed-valence pair of pMMO is valence-localized, but EPR simulations suggest that the dicopper site is equally valence-scrambled between the two nonidentical positions within the dicopper site observed in the crystal structure. The inequivalence of the available ligands in the two positions (2His vs 1His) is consistent with differing EPR spectra for the two positions. In support of the possibility of a valence-localized, mixed-valence center, a recently characterized mixed-valence dicopper model compound showed valence localization, even though it exhibits two identical sites.57 The presence of a Cu(I)Cu(II) pair in the dicopper site of as-isolated purified pMMO is consistent with the observation that the putative oxygen adduct of this site, characterized by a 345 nm optical feature, is only formed upon reduction of the active site, presumably to Cu(I)Cu(I), followed by a reaction with O2 or H2O2.17
The spmoB protein contains a valence-localized Cu(II) in a single location within the dicopper site, assigned as occupying the location coordinated by His137 and His139, rather than a valence-scrambled dicopper pair as in pMMO. It may be that this spmoB Cu(II) is part of an ordered Cu(II)Cu(I) pair, but it is also possible that a disordered amino terminus, which contains the third histidine ligand (His33), results in incomplete assembly of the dicopper site of spmoB and that it contains only the Cu(II), and not its Cu(I) partner. In a recent crystal structure of a homologous amoB subunit, the five N-terminal residues are disordered.58 In the pMMO crystal structures, the N-terminal pmoB domain extensively contacts the transmembrane region, and it is likely that instability and disorder at the spmoB dicopper site results from its absence. Incomplete loading of the dicopper site of spmoB would also explain the enzyme’s reduced activity compared to pMMO,16 as only a small percentage of the active sites may be functional.
The mixed-valence dicopper center of pMMO is different from any dicopper center studied previously. Other dicopper centers with histidine ligation, such as those in hemocyanin and tyrosinase, are isolated in EPR-silent Cu(II)–OH––Cu(II) “met” forms59,60 and are converted to EPR-active delocalized Cu(1.5)–Cu(1.5) mixed valence states only upon addition of exogenous ligands.61,62 Those sites, however, contain a total of six, rather than three, histidine ligands and exhibit longer Cu–Cu distances. The 2.5–2.6 Å Cu–Cu distance observed for the dicopper center of pMMO7,8,10,11 is comparable to those in the mixed-valence CuA sites of cytochrome c oxidase and nitrous oxide reductase. However, the copper ions in those sites are bridged by two cysteine residues and also are in a delocalized Cu(1.5)–Cu(1.5) state.63 pH variations can convert the signal of a delocalized CuA site in engineered azurin to that of an apparent type 1 or type 2 Cu(II).64,65 However, the instability of spmoB precludes extensive pH variation and pMMO does not exhibit activity at the pH extremes used in those studies.66,67
It is noteworthy that both native pMMO and spmoB refolded in the presence of Cu(II) contain Cu(I). pMMO is typically purified aerobically, suggesting that the Cu(I) sites are not particularly oxygen sensitive. The spmoB proteins are loaded with Cu(II) during refolding, and active protein cannot be obtained by addition of copper in any oxidation state to the already refolded material. It is well documented that the metalloprotein folding state can affect reduction potential.68−70 For example, the average reduction potential for unfolded azurin is ∼130 mV higher than that of folded azurin.68 In addition, the copper loading of azurin occurs on a millisecond time scale for unfolded protein versus a minute to hour time scale for folded protein.70−72 In this case, Cu(I) binds favorably in the unfolded state because linear or trigonal coordination is readily accessible when the protein is unfolded. By analogy, Cu(I) might bind preferably to unfolded spmoB.
Copper Coordination Environment
The combined ENDOR and ESEEM data indicate that two histidine residues with equivalent hyperfine couplings are coordinated to the valence-localized Cu(II) ion of spmoB_H48N,H72A (Figures 5, 6). The hyperfine tensors of the remote nitrogens of the coordinated histidyl imidazoles are rotated by 120° relative to each other, consistent with tetrahedral symmetry for the Cu(II) coordination sphere. These results indicate that the EPR-active Cu(II) ion occupies the dicopper crystallographic site coordinated by residues His137 and His139 (Figure 1).16,45
The ESEEM of the Cu(II) ion in the valence-scrambled dicopper site of pMMO is correspondingly describable as a sum of responses from the two contributing valence isomers, one with the Cu(II) ion coordinated to His137 and His139, the other with Cu(II) coordinated to His33 and to the N-terminal amino group. Neither form exhibits the 1H signal associated with a coordinated water or hydroxide, either terminal or as a bridge to Cu(I) (Figure 7).
As detailed earlier, the ENDOR signal from 14N directly coordinated to Cu(II), denoted 14N1, exhibits a hyperfine coupling in good agreement with previously published CW and pulsed ENDOR results for pMMO. The hyperfine and quadrupole coupling parameters derived here from ESEEM responses for the remote nitrogens of the histidine ligands to Cu(II) also are in agreement with those reported previously. However, the EPR and ENDOR data presented here clearly demonstrate that the Cu(II) is located within the pmoB subunit in the dicopper active site,16 and not in the pmoA subunit.45 Furthermore, the presence of combination lines in the 14N ESEEM response show that the localized Cu(II) of pmoB is coordinated to two histidines, not one, and that the same is true for one of the two forms of the valence-scrambled dicopper site of pMMO.
Conclusions
Quantitative EPR analysis of spmoB and its variants has led to the assignment of the oxidation states of the M. capsulatus (Bath) pMMO copper centers: the crystallographic dicopper site contains a valence-localized Cu(I)Cu(II) pair, and the monocopper site, a Cu(I) ion. Although the valence-localized Cu(I)Cu(II) dicopper site contains one Cu(II) ion, it is proposed to be valence-scrambled between the two crystallographically identified locations. ENDOR/ESEEM measurements indicate that one location contains the Cu(II) ion coordinated by residues His137 and His139 and that the other has Cu(II) coordinated by His33 and the N-terminal amino group. The analysis was facilitated by the preparation of several new spmoB variants, and the overall similarity between spmoB and pMMO is promising for the use of spmoB as a model system. These results unambiguously resolve the origin of the type 2 Cu(II) EPR signal observed in pMMO samples for the past 20 years. Assignment of this signal to the active site provides an important new tool for future investigations of substrate and product binding.
Acknowledgments
The ICP-AES analysis was performed at the IMSERC facility funded by Northwestern University. The authors would like to thank Prof. M. Wasielewski (Northwestern University) for the use of their X-band pulse instrument.
Supporting Information Available
Table of previous quantitative EPR results from various methanotroph species; Table summary of EPR quantitation reported in this study; SDS-PAGE gel of expressed spmoB_wt, spmoB_H48N,H72A, spmoB_H33,137,139A, and spmoB_penta variants; methane oxidation activity of spmoB_wt, spmoB_H48N,H72A, and spmoB_H33,137,139A; PESTRE traces of spmoB_wt and spmoB_H48N,H72A; ESEEM simulation description of Figure 6, χ2 parameters for ESEEM simulations, and additional ESEEM simulation with two 14N nuclei with the same hyperfine parameters, but different quadrupole parameters for one remote 14N from an imidazole and one from an amino group. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† M.A.C. and G.E.C. contributed equally.
This work was supported by the National Institutes of Health (GM070473 to A.C.R., GM111097 to B.M.H., and F32GM097049 to M.A.C.) and the National Science Foundation (DGE-0824162 to G.E.C.). ESEEM results were obtained in the laboratory of Prof. M. R. Wasielewski as part of the ANSER Center EFRC (DOE, OSR-BES; DE-SC0001059).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hanson R. S.; Hanson T. E. Microbiol. Rev. 1996, 60, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periana R. A.; Bhalla G.; Tenn W. J. III; Young K. J. H.; Liu X. Y.; Mironev O.; Jones C. J.; Ziatdinov V. R. J. Mol. Catal. A 2004, 220, 7. [Google Scholar]
- Murrell J. C.; McDonald I. R.; Gilbert B. Trends Microbiol. 2000, 8, 221. [DOI] [PubMed] [Google Scholar]
- Tinberg C. E.; Lippard S. J. Acc. Chem. Res. 2011, 44, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culpepper M. A.; Rosenzweig A. C. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman R. L.; Rosenzweig A. C. Nature 2005, 434, 177. [DOI] [PubMed] [Google Scholar]
- Hakemian A. S.; Kondapalli K. C.; Telser J.; Hoffman B. M.; Stemmler T. L.; Rosenzweig A. C. Biochemistry 2008, 47, 6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S. M.; Rawat S.; Telser J.; Hoffman B. M.; Stemmler T. L.; Rosenzweig A. C. Biochemistry 2011, 50, 10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Op den Camp H. J.; Islam T.; Stott M. B.; Harhangi H. R.; Hynes A.; Schouten S.; Jetten M. S. M.; Birkeland N.-K.; Pol A.; Dunfield P. F. Environ. Microbiol. Rep. 2009, 1, 293. [DOI] [PubMed] [Google Scholar]
- Lieberman R. L.; Shrestha D. B.; Doan P. E.; Hoffman B. M.; Stemmler T. L.; Rosenzweig A. C. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman R. L.; Kondapalli K. C.; Shrestha D. B.; Hakemian A. S.; Smith S. M.; Telser J.; Kuzelka J.; Gupta R.; Borovik A. S.; Lippard S. J.; Hoffman B. M.; Rosenzweig A. C.; Stemmler T. L. Inorg. Chem. 2006, 45, 8372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S. I.; Yu S. S. F. Acc. Chem. Res. 2008, 41, 969. [DOI] [PubMed] [Google Scholar]
- Chan S. I.; Lu Y. J.; Nagababu P.; Maji S.; Hung M. C.; Lee M. M.; Hsu I. J.; Minh P. D.; Lai J. C. H.; Ng K. Y.; Ramalingam S.; Yu S. S. F.; Chan M. K. Angew. Chem., Int. Ed. 2013, 52, 3731. [DOI] [PubMed] [Google Scholar]
- Martinho M.; Choi D. W.; DiSpirito A. A.; Antholine W. E.; Semrau J. D.; Münck E. J. Am. Chem. Soc. 2007, 129, 15783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew E. F.; Tong D.; Coleman N. V.; Holmes A. J. Microbiology 2014, 160, 1267. [DOI] [PubMed] [Google Scholar]
- Balasubramanian R.; Smith S. M.; Rawat S.; Yatsunyk L. A.; Stemmler T. L.; Rosenzweig A. C. Nature 2010, 465, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culpepper M. A.; Cutsail G. E.; Hoffman B. M.; Rosenzweig A. C. J. Am. Chem. Soc. 2012, 134, 7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H.-H. T.; Shiemke A. K.; Jacobs S. J.; Hales B. J.; Lidstrom M. E.; Chan S. I. J. Biol. Chem. 1994, 269, 14995. [PubMed] [Google Scholar]
- Nguyen H. H.; Elliott S. J.; Yip J. H.; Chan S. I. J. Biol. Chem. 1998, 273, 7957. [DOI] [PubMed] [Google Scholar]
- Basu P.; Katterle B.; Andersson K. K.; Dalton H. Biochem. J. 2003, 369, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi D. W.; Kunz R. C.; Boyd E. S.; Semrau J. D.; Antholine W. E.; Han J. I.; Zahn J. A.; Boyd J. M.; de la Mora A. M.; DiSpirito A. A. J. Bacteriol. 2003, 185, 5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S. I.; Wang V. C. C.; Lai J. C. H.; Yu S. S. F.; Chen P. P. Y.; Chen K. H. C.; Chen C. L.; Chan M. K. Angew. Chem., Int. Ed. 2007, 46, 1992. [DOI] [PubMed] [Google Scholar]
- Besides the type 2 Cu(II) signal, Chan and co-workers report a broad isotropic signal at g ≈ 2.1, which they assigned to a ferromagnetically coupled trinuclear copper center.22,201,202 However, this signal has not been observed by other researchers.10,20,21,52,67 Notably, no EPR signal indicative of a delocalized dicopper site such as the CuA (Cu(1.5)-Cu(1.5)) site in cytochrome c oxidase.203,204
- Balasubramanian R.; Rosenzweig A. C. Acc. Chem. Res. 2007, 40, 573. [DOI] [PubMed] [Google Scholar]
- Smith S. M.; Balasubramanian R.; Rosenzweig A. C. Methods Enzymol. 2011, 495, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golombek A. P.; Hendrich M. P. J. Magn. Reson. 2003, 165, 33. [DOI] [PubMed] [Google Scholar]
- Stoll S.; Schweiger A. J. Magn. Reson. 2006, 178, 42. [DOI] [PubMed] [Google Scholar]
- Davoust C. E.; Doan P. E.; Hoffman B. M. J. Magn. Reson. 1996, 119, 38. [Google Scholar]
- Zipse H.; Artin E.; Wnuk S.; Lohman G. J. S.; Martino D.; Griffin R. G.; Kacprzak S.; Kaupp M.; Hoffman B.; Bennati M.; Stubbe J.; Lees N. J. Am. Chem. Soc. 2009, 131, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweiger A.; Jeschke G.. Principles of pulse electron paramagnetic resonance; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Epel B.; Gromov I.; Stoll S.; Schweiger A.; Goldfarb D. Concepts Magn. Reson. Part B 2005, 26B, 36. [Google Scholar]
- Epel B.; Arieli D.; Baute D.; Goldfarb D. J. Magn. Reson. 2003, 164, 78. [DOI] [PubMed] [Google Scholar]
- Mailer C.; Taylor C. P. S. Biochim. Biophys. Acta 1973, 322, 195. [DOI] [PubMed] [Google Scholar]
- Werst M. M.; Davoust C. E.; Hoffman B. M. J. Am. Chem. Soc. 1991, 113, 1533. [Google Scholar]
- Hoffman B. M.; Venters R. A.; Martinsen J. J. Magn. Reson. 1985, 62, 537. [Google Scholar]
- Doan P. E. J. Magn. Reson. 2011, 208, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan P. E.; Telser J.; Barney B. M.; Igarashi R. Y.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. J. Am. Chem. Soc. 2011, 133, 17329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutsail G. E. III; Doan P. E.; Hoffman B. M.; Meyer J.; Telser J. J. Biol. Inorg. Chem. 2012, 17, 1137. [DOI] [PubMed] [Google Scholar]
- Sun L.; Hernandez-Guzman J.; Warncke K. J. Magn. Reson. 2009, 200, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Interestingly, the small amount of copper that is quantified by ICP-AES in the spmoB_penta variant exhibits a very weak Cu(II) EPR signal with similar Cu(II) EPR parameters as spmoB_H33,137,139A, however, with a larger EPR line width, consistent with the above suggestion that the copper in spmoB_penta binds adventitiously at a different site.
- McDowell C. A.; Naito A.; Sastry D. L.; Cui Y. U.; Sha K.; Yu S. X. J. Mol. Struct. 1989, 195, 361. [Google Scholar]
- Colaneri M. J.; Peisach J. J. Am. Chem. Soc. 1995, 117, 6308. [Google Scholar]
- Mcdowell C. A.; Naito A. J. Magn. Reson. 1981, 45, 205. [Google Scholar]
- Calvo R.; Oseroff S. B.; Abache H. C. J. Chem. Phys. 1980, 72, 760. [Google Scholar]
- Elliott S. J.; Randall D. W.; Britt R. D.; Chan S. I. J. Am. Chem. Soc. 1998, 120, 3247. [Google Scholar]
- Katterle B.; Gvozdev R. I.; Abudu N.; Ljones T.; Andersson K. K. Biochem. J. 2002, 363, 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mims W. B.; Peisach J. J. Chem. Phys. 1978, 69, 4921. [Google Scholar]
- McCracken J.; Pember S.; Benkovic S. J.; Villafranca J. J.; Miller R. J.; Peisach J. J. Am. Chem. Soc. 1988, 110, 1069. [Google Scholar]
- Kofman V.; Farver O.; Pecht I.; Goldfarb D. J. Am. Chem. Soc. 1996, 118, 1201. [Google Scholar]
- Gunderson W. A.; Hernández-Guzmán J.; Karr J. W.; Sun L.; Szalai V. A.; Warncke K. J. Am. Chem. Soc. 2012, 134, 18330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal I.; Karlin K. D.; Strange R. W.; Blackburn N. J. J. Am. Chem. Soc. 1993, 115, 11259. [Google Scholar]
- Yuan H.; Collins M. L. P.; Antholine W. E. J. Inorg. Biochem. 1998, 72, 179–185. [DOI] [PubMed] [Google Scholar]
- Lemos S. S.; Collins M. L. P.; Eaton S. S.; Eaton G. R.; Antholine W. E. Biophys. J. 2000, 79, 1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton N. M.; Horsewill A. J. Mol. Phys. 1979, 37, 1349. [Google Scholar]
- Kim D.; Kim N. H.; Kim S. H. Angew. Chem., Int. Ed. 2012, 52, 1139. [Google Scholar]
- Burns C. S.; Aronoff-Spencer E.; Dunham C. M.; Lario P.; Avdievich N. I.; Antholine W. E.; Olmstead M. M.; Vrielink A.; Gerfen G. J.; Peisach J.; Scott W. G.; Millhauser G. L. Biochemistry 2002, 41, 3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon E. I.; Ginsbach J. W.; Heppner D. E.; Kieber-Emmons M. T.; Kjaergaard C. H.; Smeets P. J.; Tian L.; Woertink J. S. Faraday Discuss. 2011, 148, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halvagar M. R.; Solntsev P. V.; Lim H.; Hedman B.; Hodgson K. O.; Solomon E. I.; Cramer C. J.; Tolman W. B. J. Am. Chem. Soc. 2014, 136, 7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawton T. J.; Ham J.; Sun T.; Rosenzweig A. C.. Proteins2014, Feb 13. DOI: 10.1002/prot.24535 [Epub ahead of print]. [DOI]
- Solomon E. I.; Sundaram U. M.; Machonkin T. E. Chem. Rev. 1996, 96, 2563. [DOI] [PubMed] [Google Scholar]
- Jolley R. L. Jr.; Evans L. H.; Makino N.; Mason H. S. J. Biol. Chem. 1974, 249, 335. [PubMed] [Google Scholar]
- Himmelwright R. S.; Eickman N. C.; Lubien C. D.; Solomon E. I. J. Am. Chem. Soc. 1980, 102, 5378. [Google Scholar]
- Hepp A. F.; Himmelwright R. S.; Eickman N. C.; Solomon E. I. Biochem. Biophys. Res. Commun. 1979, 89, 1050. [DOI] [PubMed] [Google Scholar]
- Yoon J.; Solomon E. I. In High resolution EPR: applications to metalloenzymes and metals in medicine; Hanson G., Berliner L., Eds.; Springer: New York, NY, 2009; Vol. 28, p 471. [Google Scholar]
- Hwang H. J.; Lu Y. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 12842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X. J.; Gorelsky S. I.; Sarangi R.; Garner D. K.; Hwang H. J.; Hodgson K. O.; Hedman B.; Lu Y.; Solornon E. I. J. Am. Chem. Soc. 2008, 130, 5194. [DOI] [PubMed] [Google Scholar]
- Takeguchi M.; Miyakawa K.; Okura I. Biometals 1998, 11, 229. [DOI] [PubMed] [Google Scholar]
- Zahn J. A.; DiSpirito A. A. J. Bacteriol. 1996, 178, 1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J. R.; Wittung-Stafshede P.; Leckner J.; Malmström B. G.; Gray H. B. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monari S.; Millo D.; Ranieri A.; Di Rocco G.; van der Zwan G.; Gooijer C.; Peressini S.; Tavagnacco C.; Hildebrandt P.; Borsari M. J. Biol. Inorg. Chem. 2010, 15, 1233. [DOI] [PubMed] [Google Scholar]
- Palm-Espling M. E.; Niemiec M. S.; Wittung-Stafshede P. Biochim. Biophys. Acta 2012, 1823, 1594. [DOI] [PubMed] [Google Scholar]
- Pozdnyakova I.; Wittung-Stafshede P. J. Am. Chem. Soc. 2001, 123, 10135. [DOI] [PubMed] [Google Scholar]
- Pozdnyakova I.; Wittung-Stafshede P. Biochemistry 2001, 40, 13728. [DOI] [PubMed] [Google Scholar]
- Hung S.-C.; Chen C.-L.; Chen K. H.-C.; Yu S. S.-F.; Chan S. I. J. Chin. Chem. Soc. 2004, 51, 1229. [Google Scholar]
- Chen K. H.-C.; Chen C.-L.; Tseng C.-F.; Yu S. S.-F.; Ke S.-C.; Lee J.-F.; Nguyen H. T.; Elliott S. J.; Alben J. O.; Chan S. I. J. Chin. Chem. Soc. 2004, 51, 1081. [Google Scholar]
- Beinert H. Eur. J. Biochem. 1997, 245, 521. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Randall D. W.; Glaser T. Coord. Chem. Rev. 2000, 200, 595. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.