

Abstract
Two natural saponins 1 and 2, isolated from Solanum indicum L., and containing 2,3-branched sugar moieties, have been efficiently synthesized. Partially protected monosaccharide and disaccharide donors were used to facilitate target synthesis. Stereo factors were critical in incorporating 2,3-branched sugars on steroid aglycones. Saponin 1 was synthesized in five steps and 30% overall yield, while saponin 2 was obtained using six straightforward sequential reactions in 31% overall yield. Saponin 2 shows promising cytotoxic activity toward human hepatocellular carcinoma BEL-7402 with an IC50 of <6 μg/mL.
Saponins constitute a structurally and biologically diverse class of molecules that are widely distributed in terrestrial plants and in certain marine organisms.1 Most traditional Chinese medicines contain saponins as major components and, thus, represent glycoconjugate templates in drug design and development.2 Dioscin (diosgenyl 2,4-di-O-α-L-rhamnopyranosyl-β-D-glucopyranoside), Pa (diosgenyl α-L-rhamnopyranosyl-(1→2)-[α-L-arabinofuranosyl-(1→4)]-β-D-glucopyranoside), methyl protodioscin (3-O-(2,4-di-O-α-L-rhamnopyranosyl-β-D-glucopyranosyl)-26-O-(β-D-glucopyranosyl)-22-methoxy-25(R)-furost-5-ene-3β,26-diol), and balanitin 7 (diosgenyl α-L-rhamnopyranosyl-(1→2)-[β-D-xylopyranosyl-(1→3)-β-D-glucopyranosyl-(1→4]-β-D-glucopyranoside) are all clinically recognized for their cardiovascular, antifungal, and antitumor activities.3 Both the carbohydrate and steroid components of saponins are structurally essential for their bioactivities.4 For example, removal of ester-linked tetrasaccharide from julibroside, a bioactive saponin isolated from Albizia julibrissin, dramatically decreases its cytotoxicity against KB cells,5 and the rhamnose moiety of solamargine (solasodinyl 2,4-di-O-α-L-rhamnopyranosyl-β-D-glucopyranoside) plays a crucial role in triggering cell death by apoptosis.6 The great difficulty associated with purification of closely related saponins from natural sources makes chemical synthesis the most realistic approach for obtaining homogeneous saponins. Significant effort has been directed toward the development of novel, powerful glycosylation reactions or strategies to access defined glycoconjugates.7 Natural saponins containing 2,3-branched carbohydrate moieties are typically assembled in two ways. The first strategy is to couple a 4,6-benzylidenated reducing end sugar unit to C-3 of the steroid or triterpene aglycone. Protection group manipulation (2-OH/3-OH), followed by glycosylation (3-OH/2-OH), deprotection (2-OH/3-OH), and a second glycosylation (2-OH/3-OH), furnishes the natural saponin having a 2,3-dibranched sugar structure.8 The disadvantages of this strategy are that it is a lengthy and low-efficiency synthesis. More importantly, it is sometimes problematic to remove the 2-acyl protecting group prior to a second glycosylation step.9 In the second strategy, a 2,3-branched oligosaccharide is first prepared and used to glycosylate the aglycone in the final step. Unfortunately, the final glycosylation step often affords an α,β mixture due to the absence of C-2 neighboring group participation.10,5b We recently described a new strategy for preparing natural saponins having 2,4-branched oligosaccharides that relies on partially protected glycosyl donors.11 We report here the extension of this strategy to the facile synthesis of natural saponins containing 2,3-branched oligosaccharides, diosgenyl 3-O-{α-L-rhamnopyranosyl-(1→2)-[β-D-glucopyranosyl-(1→3)]-β-D-galactopyranoside } (1) and diosgenyl 3-O-{α-L-rhamnopyranosyl-(1→2)-[β-D-xylopyranosyl-(1→3)]-β-D-galactopyranoside } (2). These saponins had previously been extracted from Solanum indicum L.,12 a traditional Chinese medicinal plant that has long been used for its antiinflammatory activity. In our previous study,11 we showed that direct coupling of isopropyl 4,6-O-benzylidene-1-thio-β-D-galactopyranoside (3) with diosgenin gave complicated mixtures. Thus, in our first attempt to synthesize 1, the 3-OH of 3 was regioselectively protected by 9-fluorenylmethyloxycarbonyl (Fmoc) using FmocCl in pyridine, yielding partially protected donor 4,13 which was used to glycosylate diosgenin affording saponin derivative 5 (Scheme 1) in 75% yield. Glycosylation of 5 with rhamnosyl imidate 9 gave disaccharide glycoside 6, and Et3N-promoted removal of Fmoc furnished acceptor 7 in one-pot (85% for two steps). In this straightforward strategy, the final glycosylation of saponin derivative 7 with glucosyl imidate 10 in CH2Cl2 with trimethylsilyl trifluoromethanesulfonate (TMSOTf) promotion was problematic. The crude protected saponin 8 was obtained in a low yield (~27%). MS analysis showed the presence of 8, but a sufficiently pure sample for NMR analysis could not be obtained. A detailed evaluation of this reaction suggested that the 3-OH of the galactosyl moiety was encircled by the 4,6-benzylidene group and the fully protected rhamnosyl residue, making the 3-OH group sterically inaccessible for glycosylation. To circumvent this problem, fully benzoylated donor 10 was replaced with a smaller acetylated glucosyl imidate 11; however, this reaction was sluggish, indicating that Scheme 1 was an impractical route to target saponin.
Scheme 1. Attempted Synthesis of Saponin 1a.

aReagents and conditions: (a) FmocCl, Pyr, DMAP, 0 °C to room temperature, 87%. (b) Diosgenin, NIS, TMSOTf, CH2Cl2, −42 °C, 75%. (c) TMSOTf, CH2Cl2, 0 °C, 88% for 6; 27% for 8. (d) Et3N, 85% from 5.
We next turned our attention to an alternative strategy for the efficient synthesis of natural saponin 1 utilizing a partially protected disaccharide donor 12 (Scheme 2). To these ends, sugar diol 3 was glycosylated with imidate donor 10 in anhydrous CH2Cl2 at 0 °C using TMSOTf catalyst, regioselectively affording the 1→3-linked disaccharide 12 in 86% yield.14 To confirm its structure, compound 12 was acetylated with Ac2O in pyridine to generate 12a. The 1H NMR spectra of 12a showed an H-2 signal (δ 5.31 ppm) shifted downfield from the corresponding H-2 signal of 12 (δ 3.90 ppm). Glycosylation of diosgenin with partially protected disaccharide donor 12 in dry CH2Cl2 at 0 °C, under the promotion of N-iodosuccinimide (NIS) and TMSOTf, afforded 13 in 63% yield. Introduction of the rhamnosyl moiety 9 into 13 under standard glycosylation conditions afforded the protected saponin derivative 8. Treatment of 8 in aqueous 80% acetic acid and 1 N NaOH gave the natural saponin 1 in five steps and a 30% overall yield.
Scheme 2. Successful Synthesis of Saponin 1a.

aReagents and conditions: (a) TMSOTf, CH2Cl2, 0 °C, 86% for 12; 84% for 8. (b) Ac2O, Pyr. (c) Diosgenin, NIS, TMSOTf, CH2Cl2, −42 °C, 63%. (d) Aqueous 80% AcOH, then aqueous 1 N NaOH, MeOH/CH2Cl2 2:1, 81%.
With the successful completion of the synthesis of saponin 1, our attention was turned to the synthesis of natural product 2, which we thought could be prepared in a similar way (Scheme 3). Surprisingly, when galactose diol 3 was glycosylated with acetylated xylopyranosyl imidate 14 (Scheme 4), the 1→2-linked disaccharide 15 was obtained as the major product (54%), together with the 2,3-dixylosylated trisaccharide (35%) and recovered starting material 3. To establish the linkage position, 15 was acetylated with acetic anhydride in pyridine to give 16. The 1H NMR spectrum of 16 showed the H-2 at δ 4.16 ppm and the H-3 at δ 4.93 ppm, confirming the presence of a 1→2 glycosidic bond in 15. The desired 1→3-linked disaccharide could not be obtained as a major product by changing solvent, catalyst, or reaction temperature. Curious about this regioselectivity, we undertook a detailed investigation of the scope of this reaction (Scheme 4). Glycosylation of galactosyl diol 3 with benzoylated xylopyranosyl imidate 17 afforded isopropyl 2,3,4-tri-O-benzoyl-β-D-(1→2)-4,6-O-benzylidene-1-thio-β-D-galactopyranoside 18 in 65% yield. The fully assigned 1H NMR spectrum of the acetylated derivative 19 confirmed its 1→2 linkage. Interestingly, the xylosyl residues in 18 and 19 adopt a 1C4 conformation, instead of the typical 4C1 form. This conformational assignment was supported by the small coupling constants observed for 19 (J1,2 = 3.9, J2,3 = J3,4 = 5.7, J4,5a = 4.2, J4,5b = 3.4 Hz). When glucopyranosyl 2,3-diol 20 was glycosylated with xylosyl donor 14, a 1→2-linked disaccharide 21 (48%) was again isolated. In contrast, glucosyl donor 10 predominantly afforded the β-(1→3)-linked disaccharide 23 (83%), as supported by 1H NMR spectra analysis of its chloro-acetylated derivative 24. Xylosylation of the diosgenyl galactopyranoside 25 also afforded the undesired β-(1→2) product 26, confirming that this was an unsuccessful strategy for synthesizing target 2. Realizing that regio- and stereochemical control is critical in synthesis of natural saponins and inspired by the observation that glucosyl and xylosyl donors show different reactivity in glycosylation (as observed in Scheme 4, 20 + 14 and 20 + 10), we attempted to xylosylate the 3-position of 7, despite our previous observation that incorporation of 10 into 7 had failed (Scheme 1). Thus, the acetyl-protected xylosyl imidate 14 was initially used to glycosylate the disaccharide glycosyl acceptor 7. Only trace amounts of product were obtained, and the rapid consumption of 14 was observed by TLC. In contrast, glycosylation of acceptor 7 with benzoylated donor 17, carried out in dry CH2Cl2 with catalytic TMSOTf, afforded the protected saponin derivative 28 in 78% isolated yield. The small proton coupling constants of the xylosyl residue (J1,2 < 1.0, J2,3 = J3,4 = 3.2, J4,5a < 1.0, J4,5b = 2.1 Hz) of saponin derivative 28 shows that benzoylated xylose residue takes the 1C4 conformation instead of typical 4C1 form. The xylosyl C-1 signal appears at δ 100.03 ppm in the 13C NMR spectra, confirming its β-linkage to the galactosyl residue. Deprotection of 28 using aqueous 80% acetic acid, followed by deacylation with 1 N NaOH furnished natural saponin 2 in six steps from commercially available isopropyl β-D-1-thiogalactopyranoside (IPTG) and in a 31% overall yield. A doublet, corresponding to xylosyl H-1, at δ 5.05 ppm (J = 7.5 Hz) in 1H NMR spectra indicates the expected xylosyl 4C1 conformation in natural product 2. The cytotoxicity of compounds 1 and 2 was examined using the procedure of Connolly et al.15 Saponin 2 showed good activity (IC50 < 6 μg/mL) toward human hepatocellular carcinoma BEL-7402 cells. The combinatorial synthesis of analogues of 2 is currently under investigation by our group.
Scheme 3.
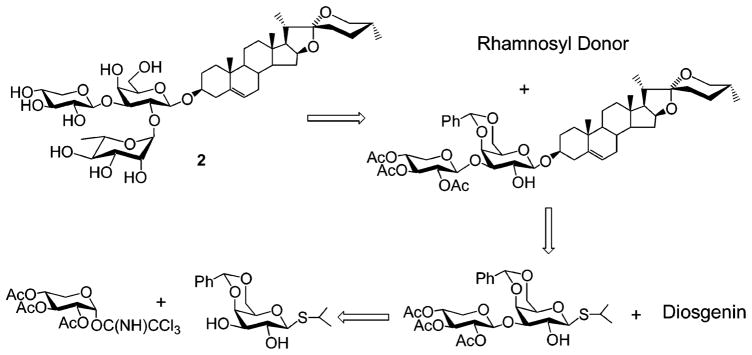
Retrosynthetic Analysis of Saponin 2
Scheme 4. Studies on Regioselective Glycosylationa.

aReagents and conditions: (a) TMSOTf, CH2Cl2, 0 °C, 54% for 15; 65% for 18; 48% for 21; 83% for 23; 51% for 26. (b) Ac2O, Pyr. (c) ClAcCl, Pyr.
In conclusion, an efficient and practical method has been developed for the synthesis of saponins with 2,3-branched oligosaccharide moieties. The key to this approach is the use of partially unprotected thioglycoside donors, resulting in significantly simplified protecting group manipulation and oligosaccharide assembly. The results of the present investigation should be of value in the synthesis of 2,3-branched natural steroidal glycosides. 16 Importantly, the application of this method in combinatorial chemistry may generate an efficient entry into libraries of more complex glycoconjugates.17
Experimental Section
Diosgenyl 3-O-Fluorenylmethoxycarbonyl-4,6-di-O-benzylidene-β-D-galactopyranoside (5)
To a solution of compound 4 (1.1 g, 2.01 mmol) and diosgenin (833 mg, 2.01 mmol) in anhydrous CH2Cl2 (10 mL) were added NIS (497 mg, 2.21 mmol) and a catalytic amount of TMSOTf (36 μL, 0.2 mmol) at −42 °C under N2 protection. The mixture was stirred under these conditions for 45 min, at the end of which time TLC indicated that the reaction was complete. The reaction mixture was neutralized with Et3N and concentrated to dryness. The residue was subjected to column chromatography on a silica gel column with 5:1 petroleum ether/EtOAc as the eluent to give 5 as a white solid (1.34 g, 75%); [α]20D −34° (c 1.3, CHCl3). Selected 1H NMR (CDCl3): δ 0.79 (d, 6 H, J = 6.2 Hz), 0.89–0.96 (m, 2 H), 0.97 (d, 3 H, J = 6.9 Hz), 1.03 (s, 3 H), 1.04–2.38 (m, 22 H), 3.37 (t, 1 H, J = 10.9 Hz), 3.46–3.50 (m, 2 H), 3.55–3.66 (m, 1 H), 4.04–4.09 (m, 2 H), 4.27–4.34 (m, 2 H), 4.35–4.50 (m, 5 H), 4.75 (dd, 1 H, J = 10.2, 3.7 Hz), 5.34 (br d, 1 H, J = 5.6 Hz), 5.51 (s, 1 H), 7.42–8.11 (13 H, Ph). Anal. Calcd for C55H66O10: C, 74.47; H, 7.50. Found: C, 74.69; H, 7.41.
Diosgenyl 2,3,4,6-Tetra-O-benzoyl-β-D-glucopyranosyl-(1→3)-4,6-di-O-benzylidene-β-D-galactopyranoside (13)
To a solution of compound 12 (452 mg, 0.50 mmol) and diosgenin (207 mg, 0.50 mmol) in anhydrous dichloromethane (8 mL) were added NIS (124 mg, 0.55 mmol) and a catalytic amount of TMSOTf (11 μL, 0.06 mmol) at −42 °C under N2 protection. The reaction mixture was stirred under these conditions for 45 min, at the end of which time TLC indicated the completion of the reaction. The mixture was then neutralized with Et3N and concentrated. The residue was subjected to column chromatography on a silica gel column with 3:1 petroleum ether/EtOAc as the eluent to give 13 as a white solid (391 mg, 63%); [α]20D −24° (c 1, CHCl3). Selected 1H NMR (CDCl3): δ 0.79 (d, 6 H, J = 6.3 Hz), 0.97 (d, 3 H, J = 6.9 Hz), 1.00 (s, 3 H), 3.23 (br s, 1 H), 3.37 (t, 1 H, J = 10.9 Hz), 3.45–3.57 (m, 2 H), 3.74 (dd, 1 H, J = 9.6, 3.3 Hz), 3.81–3.88 (m, 2 H), 4.15–4.22 (m, 3 H), 4.33 (d, 1 H, J = 7.6 Hz), 4.39 (q, 1 H, J = 7.5 Hz), 4.53 (dd, 1 H, J = 12.2, 5.3 Hz), 4.68 (dd, 1 H, J = 12.2, 3.0 Hz), 5.32 (d, 1 H, J = 5.0 Hz), 5.38 (d, 1 H, J = 7.9 Hz), 5.40 (s, 1 H), 5.57 (dd, 1 H, J = 9.6, 7.9 Hz), 5.69 (t, 1 H, J = 9.6 Hz), 5.91 (t, 1 H, J = 9.6 Hz), 7.25–8.05 (m, 25 H). Anal. Calcd for C74H82O17: C, 71.48; H, 6.65. Found: C, 71.69; H, 6.48.
Supplementary Material
Figure 1.

Structure of saponin target compounds 1 and 2.
Scheme 5. Total Synthesis of Natural Saponin 2a.

aReagents and conditions: (a) TMSOTf, CH2Cl2, 0 °C, 78%. (b) Aqueous 80% AcOH, then aqueous 1 N NaOH, MeOH/CH2Cl2 2:1, 85%.
Acknowledgments
This work was supported by National Basic Research Program of China (2003CB-415001), NNSF of China (20372081, 30330690), and the NIH (HL62244).
Footnotes
Supporting Information Available: Preparation and physical data for compounds 3–8, 12, 12a, 13, 1, 16, 19, 24, 25, 27, 28, and 2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Hostettmann K, Marston A. Saponins. Cambridge University Press; 1995. [Google Scholar]; (b) Iorizzi M, De Marino S, Zollo F. Curr Org Chem. 2001;5:951. [Google Scholar]
- 2.(a) Guo C, Fuchs PL. Tetrahedron Lett. 1998;39:1099. [Google Scholar]; (b) Deng S, Yu B, Lou Y, Hui Y. J Org Chem. 1999;64:202. doi: 10.1021/jo981685c. [DOI] [PubMed] [Google Scholar]; (c) Deng S, Yu B, Hui Y. Tetrahedron Lett. 1998;39:6511. [Google Scholar]; (d) Yu B, Xie J, Deng S, Hui Y. J Am Chem Soc. 1999;121:12196. [Google Scholar]; (e) Yu W, Jin Z. J Am Chem Soc. 2001;123:3369. doi: 10.1021/ja004098t. [DOI] [PubMed] [Google Scholar]; (f) Yu W, Jin Z. J Am Chem Soc. 2002;124:6576. doi: 10.1021/ja012119t. [DOI] [PubMed] [Google Scholar]; (g) Cheng MS, Wang QL, Tian Q, Song HY, Liu YX, Li Q, Xu X, Miao HD, Yao XS, Yang Z. J Org Chem. 2003;68:3658. doi: 10.1021/jo020683w. [DOI] [PubMed] [Google Scholar]
- 3.(a) Nakano K, Murakami K, Takaishi Y, Tomimatsu T, Nohara T. Chem Pharm Bull. 1989;37:116. [Google Scholar]; (b) Hufford CD, Liu S, Clark AM. J Nat Prod. 1988;51:94. doi: 10.1021/np50055a013. [DOI] [PubMed] [Google Scholar]; (c) Liu C, Chen Y. Acta Pharm Sinica. 1984;19:799. [PubMed] [Google Scholar]; (d) Namba T, Huang X, Shu Y, Huang S, Hattoti M, Kakiuchi N, Wang Q, Xu G. Planta Med. 1989;55:501. doi: 10.1055/s-2006-962080. [DOI] [PubMed] [Google Scholar]; (e) Zhou J. Pure Appl Chem. 1989;61:457. [Google Scholar]; (f) Hu K, Dong AJ, Yao XS, Kobayashi H, Iwasaki S. Planta Med. 1997;63:161. doi: 10.1055/s-2006-957636. [DOI] [PubMed] [Google Scholar]; (g) Nohara T, Yabuta H, Suenobu M, Hida R, Miyahara K, Kawasaki T. Chem Pharm Bull. 1973;21:1240. [Google Scholar]
- 4.Matsuda H, Pongpiriyadacha Y, Morikawa T, Kishi A, Kataoka K, Yoshikawa M. Bioorg Med Chem Lett. 2003;13:1101. doi: 10.1016/s0960-894x(03)00052-0. [DOI] [PubMed] [Google Scholar]
- 5.(a) Ikeda T, Fujiwara S, Araki K, Kinjo J, Nohara T, Miyoshi T. J Nat Prod. 1997;60:102. doi: 10.1021/np960556t. [DOI] [PubMed] [Google Scholar]; (b) Ikeda T, Kajimoto T, Kinjo J, Nakayama K, Nohara T. Tetrahedron Lett. 1998;39:3513. [Google Scholar]; (c) Du Y, Zhang M, Kong F. J Chem Soc, Perkin Trans. 2001;1:2289. [Google Scholar]
- 6.Chang LC, Tsai TR, Wang JJ, Lin CN, Kuo KW. Biochem Biophys Res Commun. 1998;242:21. doi: 10.1006/bbrc.1997.7903. [DOI] [PubMed] [Google Scholar]
- 7.For reviews see: Toshima K, Tasuta K. Chem Rev. 1993;93:1503.Schmidt RR, Kinzy W. Adv Carbohydr Chem Biochem. 1994;50:21. doi: 10.1016/s0065-2318(08)60150-x.Garegg PJ. Adv Carbohydr Chem Biochem. 1997;52:179. doi: 10.1016/s0065-2318(08)60091-8.
- 8.Li C, Yu B, Liu M, Hui Y. Carbohydr Res. 1998;306:189. doi: 10.1016/s0008-6215(97)10050-7. [DOI] [PubMed] [Google Scholar]
- 9.(a) Deng S, Yu B, Hui Y, Yu B, Han X. Carbohydr Res. 1999;317:53. doi: 10.1016/s0008-6215(99)00066-x. [DOI] [PubMed] [Google Scholar]; (b) Suhr R, Pfefferkorn P, Weingarten S, Thiem J. Org Biomol Chem. 2003;1:4373. doi: 10.1039/b309066c. [DOI] [PubMed] [Google Scholar]
- 10.(a) Liu M, Yu B, Hui Y. Tetrahedron Lett. 1998;39:415. [Google Scholar]; (b) Ikeda T, Miyashita H, Kajimoto T, Nohara T. Tetrahedron Lett. 2001;42:2353. [Google Scholar]
- 11.Du Y, Gu G, Wei G, Hua Y, Linhardt RJ. Org Lett. 2003;5:3627. doi: 10.1021/ol035353s. [DOI] [PubMed] [Google Scholar]
- 12.(a) Yahara S, Nakamura T, Someya Y, Matsumoto T, Yamashita T, Nohara T. Phytochemistry. 1996;43:1319. [Google Scholar]; (b) Chen P, Zheng G. Chinese Herbs and Compatibility. Science Press–Ohmsha, Ltd; Tokyo: 1997. [Google Scholar]
- 13.Roussel F, Knerr L, Grathwohl M, Schmidt RR. Org Lett. 2000;2:3043. doi: 10.1021/ol006081l. [DOI] [PubMed] [Google Scholar]
- 14.He H, Yang F, Du Y. Carbohydr Res. 2002;337:1673. doi: 10.1016/s0008-6215(02)00276-8. [DOI] [PubMed] [Google Scholar]
- 15.Connolly DT, Knight MB, Harakas NK, Wittwer AJ, Feder J. Anal Biochem. 1986;152:136. doi: 10.1016/0003-2697(86)90131-4. [DOI] [PubMed] [Google Scholar]
- 16.(a) Friedman M, Roitman JN, Kozukue N. J Agric Food Chem. 2003;51:2964. doi: 10.1021/jf021146f. [DOI] [PubMed] [Google Scholar]; (b) Putalun W, Xuan LJ, Tanaka H, Shoyama Y. J Nat Prod. 1999;62:181. doi: 10.1021/np980301a. [DOI] [PubMed] [Google Scholar]; (c) Wanyonyi AW, Chhabra SC, Mkoji G, Eilert U, Njue WM. Phytochemistry. 2002;59:79. doi: 10.1016/s0031-9422(01)00424-1. [DOI] [PubMed] [Google Scholar]; (d) Nakamura T, Komori C, Lee YY, Hashimoto F, Yahara S, Nohara T, Ejima A. Biol Pharm Bull. 1996;19:564. doi: 10.1248/bpb.19.564. [DOI] [PubMed] [Google Scholar]; (e) Mimaki Y, Ishibashi N, Ori K, Sashida Y. Phytochemistry. 1992;31:1753. doi: 10.1016/0031-9422(92)83141-k. [DOI] [PubMed] [Google Scholar]; (f) Dong M, Feng X, Wang B, Wu L, Ikejima T. Tetrahedron. 2001;57:501. [Google Scholar]; (g) Yokosuka A, Mimaki Y, Sashida Y. Phytochemistry. 2002;61:73. doi: 10.1016/s0031-9422(02)00168-1. [DOI] [PubMed] [Google Scholar]
- 17.Marcaurelle LA, Seeberger PH. Curr Opin Chem Biol. 2002;6:289. doi: 10.1016/s1367-5931(02)00322-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.