

Abstract
Tissue-engineered blood vessels (TEBVs) are promising in the replacement of diseased vascular tissues. However, it remains a great challenge to obtain a sufficient number of functional smooth muscle cells (SMCs) in a clinical setting to construct patient-specific TEBVs. In addition, it is critical to develop a scaffold to accommodate these cells and retain their functional phenotype for the regeneration of TEBVs. In this study, human induced pluripotent stem cells (iPSCs) were established from primary human aortic fibroblasts, and characterized with the pluripotency markers expression and cells’ capabilities to differentiate into all three germ layer cells. A highly efficient method was then developed to induce these human iPSCs into proliferative SMCs. After multiple times of expansion, the expanded SMCs retained the potential to be induced into the functional contractile phenotype of mature SMCs, which was characterized by the contractile response to carbachol treatment, up-regulation of specific collagen genes under transforming growth factor β1 treatment, and up-regulation of specific matrix metalloproteinase genes under cytokine stimulation. We also developed an advanced macroporous and nanofibrous (NF) poly(L-lactic acid) (PLLA) scaffold with suitable pore size and interpore connectivity to seed these human iPSC-derived SMCs and maintain their differentiated phenotype. Subcutaneous implantation of the SMC-scaffold construct in nude mice demonstrated vascular tissue formation, with robust collagenous matrix deposition inside the scaffold and the maintenance of differentiated SMC phenotype. Taken together, this study established an exciting approach towards the construction of patient-specific TEBVs. We established patient-specific human iPSCs, derived proliferative SMCs fore expansion, turned on their mature contractile SMC phenotype, and developed an advanced scaffold for these cells to regenerate vascular tissue in vivo.
Keywords: Human induced pluripotent stem cell, Smooth muscle cell, Nanofibrous scaffold, Tissue-engineered vascular tissue
1. Introduction
Cardiovascular disease remains the leading cause of mortality in the world, accounting for 17.3 million deaths per year [1]. Vascular bypass grafting or replacement is a major treatment for ischemic heart disease, aortic aneurysm and peripheral vascular disease [2]. In the US, there are about 1.4 million arterial bypass operations performed annually. However, many patients do not have suitable autologous vessels for arterial bypass or vascular replacement. The current synthetic vascular grafts are susceptible to thrombosis, infection, subsequent dehiscence of the anastomosis and pseudoaneurysm [3]. Particularly, in the pediatric population, the lack of growth potential of the synthetic vascular grafts requires multiple operations as a child grows [4]. Moreover, once infected (a mycotic aneurysm or infected graft), patients have to be treated with debridement and replacement with homografts from cadavers, creating a high risk of re-infection. Therefore current vascular grafts available for the treatment of ischemic heart disease, aortic aneurysms and peripheral vascular disease, and conduits designed for the pediatric population are far from ideal.
Limited autologous vessel source and the disadvantages of current synthetic vascular grafts drive the development of tissue-engineered (TE) blood vessels (TEBVs) that recapitulate the structure and function of native blood vessels. Three major approaches have been taken to construct TEBVs, including using de-cellularized matrices [5–7], engineered cell-sheets [8, 9] and biodegradable scaffolds [10]. While many efforts are being made in the development of biomaterials and optimization of the microenvironment for vascular tissue formation, cell source remains one of the bottleneck problems [11]. Mature vascular cells isolated from the donor vessels may not be sufficient in number and suffer from limited proliferation potentials and the rapidly declining cell functions during in vitro expansion. Alternatively, adult stem cells from various origins have been used to regenerate vascular tissue [12–15]. However, the proliferation and differentiation capabilities of adult stem cells decrease with donor aging, limiting their practical application in elderly patients who are in need of TEBVs the most. With the high proliferative capacity and the high differentiation potential to various types of cells, pluripotent stem cells have become a promising cell source in tissue engineering and regenerative medicine. However, their efficacy and potential advantages in constructing TEBVs require further investigation. Recently, we have successfully differentiated mouse embryonic stem cells (ESCs) and mouse induced pluripotent stem cells (iPSCs) into smooth muscle cells (SMCs), demonstrating the feasibility of harnessing pluripotent stem cells to serve as an advanced cell source for vascular engineering [16, 17]. However, many issues need to be addressed in translating animal studies towards the construction of the clinical relevant patient-specific TEBVs, including the establishment of facile protocol to generate and maintain feeder-free human iPSCs from clinically available patient donor cells, development of efficient methods to drive established human iPSCs into proliferative and safe SMCs, and refinement of the scaffold with beneficial microenvironment to promote and maintain the functional SMC phenotype of thus derived cells. In the present study, we aim at generating human iPSCs from patient primary aortic fibroblasts, developing a highly efficient differentiation procedure to drive the human iPSCs into functional SMCs, and finally testing the capability of the derived SMCs in constructing vascular tissues on pre-designed three-dimensional (3D) biodegradable scaffolds.
2. Materials and methods
2.1. Isolation and culture of primary aortic fibroblasts and SMCs
The aortic tissue from a heart transplant donor was obtained from The University of Michigan Cardiovascular Center. The use of the patient’s tissue was approved by the Institutional Review Board of The University of Michigan and written informed consent was obtained. The aortic tissue was sterile and the donor had no aortic or aortic valvular diseases. The specimens were washed twice with HBSS. The endothelial layer was removed, the tunica adventitia and tunica media was separated, and then the tissue was cut into pieces of about 1×1 mm. For isolation of primary fibroblasts, the tissue pieces were digested with a combination of 1 mg/ml of collagenase II (Washington Biochemical, Lakewood, NJ, USA) and 0.3 mg/ml of elastase (Sigma, St. Louis, MO, USA) in DMEM at 37 °C for 2–4 h, then the digestion was stopped by adding DMEM containing 10% FBS. The isolated cells were spun down, the supernatant was aspirated, and the cell pellets were resuspended with fresh culture medium. The cell suspension was passed through a 70 μm cell strainer. The collected cells were then plated into culture dishes. The cells were passaged after reaching 95% of confluence. For isolation of primary SMCs, the medial pieces of aorta were placed on a dish, and a small amount of medium was added for explant culture overnight following a previous report [15]. The medium was replenished the next day to cover the explants. The medium was changed every 3–4 d till the outgrown cell monolayer reached 95% of confluence (normally taking 2–3 wk). The cells were then passaged.
2.2. Generation of human iPSC lines
The iPSC 4-in-1 lentiviral vectors encoding OCT4, KLF4, SOX2 and c-MYC (OKSM) were constructed by engineering 2A-fusion of the four open reading frames behind human EF1a enhancer/promoter in pTYF backbone [18]. Lentivirus was generated as previously described [16]. Fibroblasts were seeded in a 6-well plate at 1×105 cells per well at one day before transduction, then incubated with the lentivirus supplemented with 4 μg/ml polybrene (Sigma) in DMEM overnight. The fresh medium was changed the next day. Six days post-infection, the cells were replated on irradiated mouse embryonic fibroblast (MEF) feeder layers. After 24 h of replating, the medium was switched to the human reprogramming medium (DMEM/F12, 20% knockout serum replacement, 1× non-essential amino acid, 2 mM L-glutamine, 0.1 mM 2-mercaptoethanol, 20 ng/ml FGF-2 and 1% penicillin-streptomycin) and the medium was changed every other day. Two weeks after transfection, the medium was changed daily. Most iPSC colonies appeared within 3 wk after transfection. The colonies resembling human ESCs morphology were mechanically picked into the single well of a 12-well plate. At least 12 colonies were picked for each transfection experiment.
2.3. Characterization of human iPSCs
For alkaline phosphatase (APS) staining, Alkaline Phosphatase Staining Kit II (Stemgent, Cambridge, MA, USA) was used following manufacturer’s instruction. Briefly, the culture medium was aspirated and the cells were washed with PBST. The cells were fixed at room temperature (RT) for 5 min. The fixed cells were washed with PBST and incubated with freshly prepared APS Substrate Solution for 5 to 15 min. The reaction was stopped and the cells were washed twice with PBS. For immunofluorescence staining, human iPSCs or their derivates were fixed in 4% paraformaldehyde for 20 min, permeabilized and blocked in 0.5% Triton X-100 with 5% normal goat serum in PBS, and incubated overnight with a panel of primary antibodies: rabbit anti-OCT4, rabbit anti-SOX2, rabbit anti-NANOG, mouse anti-TRA-1-60, mouse anti-TRA-1-81 (1:100, Stemgent); rat anti-SSEA3, mouse anti-SSEA4 (1:100, Developmental Studies Hybridoma Bank, Iowa City, IA, USA); mouse anti-TUJ1 (1:500, Covance, Princeton, NJ, USA); rabbit anti-α-1-fetoprotein (1:400, Dakocytomation, Glostrup, Denmark); mouse anti-α-SMA, mouse anti-CNN1 (1:500, Sigma); rabbit anti-SM22α (1:500, Abcam, Cambridge, MA, USA). Alexa 488 or Alexa 594 conjugated secondary goat anti-mouse, rat or rabbit IgG antibodies were added to the samples and incubated for 1 h. The antibodies used for immunofluorescence staining were listed in Supplemental Table S1. The cell nuclei were counter-stained with 4′,6-diamidino-2-phenylindole (DAPI; Life Technologies, Grand Island, NY, USA). The cells were rinsed, mounted and the fluorescence was observed under an Olympus BX53 fluorescence microscope. Images were captured using the Olympus DP controller software.
2.4. Differentiation of human iPSCs into SMCs
The differentiation of human iPSCs into SMCs was induced according to our previously reported human ESCs protocol with some modifications [19]. Briefly, human iPSCs were manually lifted, cut into pieces and transferred to an ultra-low-attachment dish (Corning, Corning, NY, USA) to generate floating embryoid bodies (EBs). EBs were cultured in human ESCs culture medium without bFGF supplement. Six-day old EBs were replated to 35-mm dishes pre-coated with 0.1% gelatin and cultured for an additional 6 d in DMEM plus 10% FBS. The outgrown cells from EBs were then dissociated with TrypLE Express (Life Technologies) digestion and cultured on Matrigel-coated dishes in complete smooth muscle growth medium 2 (SmGM2, Lonza, Walkersville, MD, USA). The derived cells were passaged when reaching 80–90% confluence. To further differentiate the cells into mature SMCs, the cells were cultured under a differentiation condition, in which the cells were grown on gelatin-coated dishes in SmGM2 basal medium plus 5% FBS for 5 d.
2.5. Contractility assay
After 5 d of differentiation, the cells were washed with PBS, stimulated with 1 mM carbachol and monitored for 30 min as previously described [12]. Images of the same field were captured every 1 minute for 30 min and compiled into movies using the DP controller software. The change of cell surface area from 0 min to 30 min of treatment was analyzed using Image J software.
2.6. RT-PCR and quantitative RT-PCR (qRT-PCR)
Total RNA was extracted using a RNeasy Mini kit (Qiagen, Valencia, CA, USA). The cDNA was synthesized from 0.5 μg of RNA with TaqMan Reverse Transcription Reagents (Life Technologies) in a 20 μl reaction volume. Primer sequences were listed in Supplemental Table S2. The qRT-PCR was performed on a StepOne Plus Real-Time System (Life Technologies) using iQ SYBR Green Supermix (Bio-Rad, Hercules, CA). Quantification of gene expression was assessed with the comparative cycle threshold (Ct) method. The relative amounts of mRNA for target genes were determined by subtracting the Ct values of these genes from the Ct value of the housekeeping gene GAPDH (ΔCt). Standard RT-PCR for some pluripotency genes expression was performed with primers described in Supplemental Table S3.
2.7. Flow cytometry
The cells were dissociated with 0.05% trypsin digestion into single cell and suspended in PBS, then fixed with the Fixation Solution from a Fixation/Permeabilization kit (eBiosciences, San Diego, CA, USA), and stained with primary and detection antibodies as described by the manufacturer. Briefly, 0.5 × 106 cells were resuspended in 400 μl of Fixation/Permeabilization Solution, kept at RT for 30 min, and washed twice with Perm/Wash Buffer. After being blocked with Blocking Solution for 30 min, the cells were double-stained against CNN1 and α-SMA. Briefly, the cells were incubated with the primary anti-CNN1 antibody in Blocking Solution for 1 h at RT, then the cells were washed twice and resuspended in 100 μl of Perm/Wash Buffer with APC-conjugated secondary antibody and FITC-conjugated anti-α-SMA antibody for 1 h at RT. Then the cells were washed twice and the resuspended cells in PBS were analyzed by flow cytometry. Isotype IgG1 and FITC-conjugated IgG2a were used as the negative control to CNN1 antibody and FITC-conjugated anti-α-SMA antibody, respectively. The antibodies used for flow cytometry were listed in Supplemental Table S1.
2.8. Fabrication of three-dimensional (3D) macroporous NF poly-L-lactide (PLLA) scaffolds
PLLA with an inherent viscosity of approximately 1.6 dl/g was purchased from Boehinger Ingelheim (Ingelheim, Germany). PLLA in tetrahydrofuran solution (10% wt/v) was cast into an assembled sugar template (formed from bound sugar spheres, 60–150 μm in diameter) under a mild vacuum. The polymer-sugar composite was phase separated at −20 °C overnight and then immersed into cyclohexane to exchange tetrahydrofuran for 2 d. The resulting composites were freeze-dried and the sugar spheres were leached out in distilled water and freeze-dried again to obtain high porous scaffolds. The scaffolds were cut into circular disks with dimensions of 5 mm in diameter and 1 mm in thickness.
2.9. Construction of tissue-engineered vascular tissues
The 3D NF PLLA scaffolds were pre-wetted in 70% ethanol for 30 min, washed three times with PBS for 30 min, and twice in the cell culture medium for 2 h each. 5×105 human iPSC-derived proliferative SMCs were seeded into each scaffold. After 24 h of incubation, the cell/scaffold constructs were subcutaneously implanted into nude mice. For implantation surgery, 6–8 wk old male nude mice (Charles River Laboratories, Wilmington, MA, USA) were used. Surgery was performed under general inhalation anesthesia with isofluorane. Two midsagittal incisions were made on the dorsa and one subcutaneous pocket was created on each side of each incision using blunt dissection. One cell-scaffold construct was implanted into each pocket. After placement of implants, the incisions were closed with staples. At the end of 2 wk of implantation period, the mice were euthanized and the implants were harvested. The animal procedures were performed according to the protocol approved by the University of Michigan Committee for Use and Care of laboratory Animals.
2.10. Scanning electron microscopy (SEM) observation
Samples were first rinsed in PBS, fixed in 2.5% glutaraldehyde and 2% paraformaldehyde overnight, and post-fixed in 1% osmium tetroxide for 1 h. Samples were dehydrated in increasing concentrations of ethanol and hexamethyldisilizane. The samples were then sputter-coated with gold and observed under a scanning electron microscope (Phillips XL30 FEG).
2.11. Histological observation of implants
The implants were washed in PBS, fixed with 3.7 % formaldehyde in PBS overnight, dehydrated through a graded series of ethanol, embedded in paraffin, and sectioned at a thickness of 5 μm. Sections were deparaffinized, rehydrated with a graded series of ethanol, and stained with H-E or Masson’s trichrome method. For immunohistochemistry (IHC) observation, following deparaffinization of sections, slides were treated with pepsin for antigen unmasking. After rinsing in deionized water, quenching of endogenous peroxidase activity was achieved by incubating sections in 3% hydrogen peroxide for 10 min. After washing with deionized water and blocking with serum, tissue sections were incubated with primary antibodies against α-SMA (Sigma), CNN1 (Sigma) or SM22α (Abcam) at 1:100 dilutions. Following PBS rinse, sections were incubated with biotinylated secondary antibodies, followed by avidin-biotin complex staining (R&D Systems, Minneapolis, MN, USA). The antibodies used for IHC staining were listed in Supplemental Table S1.
2.12. Statistics
Numerical data were reported as mean ± s.e.m. from triplicate cell culture (n = 3). The Student’s t-test was applied to test the significance between the groups. The value of p < 0.05 was considered to be statistically significant.
3. Results
3.1. Generation of human iPSCs from primary aortic fibroblasts
Using collagenase and elastase digestion method, we successfully isolated the primary fibroblasts from aortic tissue. The passage-2 cells of primary aortic fibroblasts were used for reprogramming. The protocol for reprogramming was illustrated in Fig. 1A. The human ESC-like colonies appeared 3 wk after transfection. After several rounds of passage and expansion, primary aortic fibroblast-derived iPSC lines were established. Each primary aortic fibroblast line was used to establish at least three iPSC lines. The established cell lines can be maintained and expanded both on MEF feeders and Matrigel-coated surfaces (Fig. 1B).
Fig. 1.

Establishment of human iPSCs from aortic fibroblasts. (A) Schematic diagram of the procedure. Aortic fibroblasts were isolated from the donor aorta, transfected with lentivirus containing OKSM and reprogrammed into iPSCs. (B) The morphology of primary aortic fibroblasts, putative iPSC colonies and established iPSC colonies cultured on mouse embryonic fibroblasts (MEF) feeders or Matrigel respectively. Scale bars = 200 μm.
3.2. Characterization of human iPSC colonies
At least twelve colonies were mechanically isolated from the culture of reprogrammed primary fibroblasts and passaged on fresh MEF feeder. At least three iPSC lines were successfully established from each reprogramming culture and the cells can be continuously passaged while maintaining human ESC-like morphology. The colonies of the established iPSC lines showed strong positive staining of APS activity. The pluipotency marker expression was assessed with immunofluorescence staining. Fig. 2A showed the representative iPSC colonies which strongly expressed transcription factors of OCT4, NANOG and SOX2 in nuclei, and other pluripotency markers of SSEA3, SSEA4, TRA-1-60 and TRA-1-81 on cell surface. The expression of OCT4, NANOG and SOX2 mRNA in the two human iPSC lines were also validated by RT-PCR (Fig. 2B). To conveniently generate feeder-free cells, the iPSCs were further maintained with standard procedure developed for feeder-free culture of human ESCs, and mechanically passaged on Matrigel-coated dishes with TeSR-E8 medium. Some of these cell lines have been maintained in continuous culture for over 6 months without noticeable loss of pluripotent properties (data not shown).
Fig. 2.

Pluripotency analysis of human iPSCs. (A) Human iPSC line 1 and line 2 generated from a normal donor were observed with alkaline phosphatase (APS) staining and immunofluorescence staining of pluripotency markers including OCT4, NANOG, SOX2, SSEA3, SSEA4, TRA-1-60 and TRA-1-81. Scale bars = 200 μm. (B) RT-PCR analysis of NANOG, OCT4 and SOX2 mRNA expression in MEFs, the two iPSC lines and H7 ESC line.
3.3. Pluripotency of human iPSCs
To further validate the pluripotency of established human iPSC lines, EB formation assay was performed in vitro and teratoma induction assay was performed in vivo. All the established iPSC lines we tested can differentiate into the three germ layers of endoderm, mesoderm and ectoderm in vitro, as Fig. 3 showed the positive staining of α-fetoprotein, α-SMA and TUJ1 respectively. Human iPSCs were further injected into SCID mice to develop teratomas. Histological observations revealed the presence of different types of tissues representing all three germ layers, including neural rosette (ectoderm), cartilage (mesoderm) and gut-like epithelia (endoderm) (data not shown).
Fig. 3.
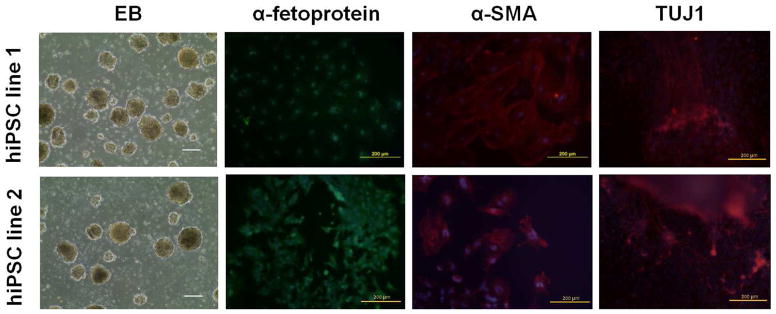
In vitro embryonic body (EB)-mediated differentiation of human iPSCs into three germ layers. EBs were cultured in ultra-low attachment dishes for 6 d then plated on gelatin-coated dishes for another 6 d. Differentiated cells were stained with antibodies against α-fetoprotein (endoderm marker), α-SMA (mesoderm marker) or TUJ1 (ectoderm marker) respectively. Nuclei were counter-stained with DAPI. Scale bars = 200 μm.
3.4. Differentiation of human iPSCs into SMCs
The differentiation procedure was illustrated in Fig. 4A and the results were shown in Fig. 4B. The human iPSCs were cultured in suspension to form EBs, then the EBs were placed on Matrigel in complete SmGM2 growth medium containing a growth factor cocktail of bFGF, EGF and insulin. After 30 d of culture, morphologically homogenous cell population dominated the culture, which showed very similar morphology to primary aortic SMCs cultured in the same growth medium (Fig. 4B, lower panel left). Importantly, these cells proliferated fast and can be continuously expanded in growth medium. To further obtain functional mature SMCs, these proliferative cells were then replated on gelatin-coated dishes in smooth muscle cell differentiation medium (SmGM2 basal medium plus 5% FBS). At the end of 5 d of differentiation culture, the cells elongated into a spindle-shaped morphology (Fig. 4B, lower panel right), with positive protein expression of SMC markers including α-SMA, CNN1 and SM22α shown by immunofluorescence staining (Fig. 4C). Compared to human primary aortic SMCs cultured in the same differentiation medium, human iPSC-derived SMCs had higher gene expression levels of α-SMA, CNN1 and SM22α (Fig. 4D). As expected, undifferentiated iPSCs had low expression levels of these SMC markers. In addition, flow cytometry assay revealed that 76.51% cells were co-stained with α-SMA and CNN1 (Fig. 4E). In addition, when the proliferative cells were injected into SCID mice, no teratoma tissues were developed (data not shown).
Fig. 4.

Differentiation of human iPSCs into vascular SMCs. (A) Schematic diagram of the procedure. EB, embryonic body; GM, growth medium; DM, differentiation medium. (B) Phase contrast observation of cells at different differentiation stages. SMGM, smooth muscle growth medium; SMDM, smooth muscle differentiation medium. (C) Immunofluorescence staining showed that primary human aortic SMCs and iPSC-derived SMCs highly expressed vascular SMC markers including α-SMA, CNN1 and SM22α. (D) Quantitative real-time PCR demonstrated that human iPSC-derived SMCs up-regulated the gene expression levels of SMC markers including α-SMA, CNN1 and SM22α, compared to undifferentiated hiPSCs. The primary SMCs served as a positive control. (E) Flow cytometry analysis of the vascular SMC marker expression of α-SMA and CNN1 in human iPSC-derived SMCs. Scale bars = 200 μm.
3.5. Functional assessment of human iPSC-derived SMCs
To examine whether the iPSC-derived SMCs were functional, their contractile potential under muscarinic agonist treatment were tested. The cells were treated with 1 mM carbachol for 30 min. Time-lapse microscopy showed that more than 50% of total tested SMCs contracted in response to carbachol treatment, in the same way as primary aortic SMCs responded (Fig. 5A and Supplementary Movies 1–3). Contracting cells exhibited a 20–30% change of cell surface area (n ≥ 8). To test collageneous matrices synthesis potentials, the expression levels of collagen 1A1 and 3A1 were measured in response to TGF-β1 treatment. Both collagen 1A1 and 3A1 were significantly increased in the two iPSC lines-derived SMCs (Fig. 6A). These response patterns were very similar to those observed in primary aortic SMCs. In addition, MMPs gene expression was detected after IL-1β stimulation. This response has been found to correlate with atherosclerosis and aneurysm formation. IL-1β treatment for 24 h significantly enhanced the expression levels of MMP 3, MMP 9 and MMP 12, while the expression level of MMP 2 was not altered. This expression profile was similar to the response of primary aortic SMCs (Fig. 6B). Collectively, these results suggested that the iPSC-derived SMCs were functional.
Fig. 5.

Contraction response of human iPSC-derived SMCs to muscarinic agonist. (A) Human iPSC-derived SMCs were cultured under differentiation conditions for 5 d, then the cells were treated with 1 mM carbachol, and the images were recorded every 1 min for consecutive 30 min. The images represented the cells before treatment and after treatment for 30 min. Arrows indicated the contracting cells. (B) The change of cell surface area was calculated and compared to that of primary SMCs. Scale bars = 100 μm (for iPSC-derived SMCs) or 200 μm (for primary SMCs).
Fig. 6.

Gene expression of collagens and matrix metalloproteinases (MMPs) in human iPSC-derived SMCs with growth factor or cytokine treatment. (A) Gene expression of collagen1A1 and collage3A1 under 5 ng/ml of TGF-β1 treatment for 24 h. (B) Gene expression of MMP 2, 3, 9 and 12 in response to 10 ng/ml IL-1β treatment for 24 h. *, P < 0.05 vs vehicle control; **, P < 0.01 vs vehicle control.
3.6. Construction of tissue-engineered vascular tissues with human iPSC-derived SMCs and 3D porous NF scaffolds
The NF scaffolds with macropore size of 60–150 μm (Fig. 7A, B) were developed to construct vascular tissues. After 24 h of seeding and culture, the iPSC-derived proliferative SMCs aggregated inside the pores of the scaffolds (Fig. 7C). After 2 wk of subcutaneous implantation in nude mice, the constructs were collected and subjected to histological observations. Vascular tissue formation was observed with H-E staining (Fig. 8A, B) or Masson’s trichrome staining (Fig. 8C, D). Particularly, trichrome staining results showed robust collagen deposition in the pores of the scaffolds. While there was no staining for immunohistochemical staining control (PBS instead of primary antibodies; Supplemental Fig. S1), positive staining of α-SMA (Fig. 8G, H), CNN1 (Fig. 8K, L) and SM22α (Fig. 8O, P) was observed in a vast majority of cells throughout the entire implants, which were similar to the human aorta section staining (Fig. 8E, F, I, J, M, N). These data indicated the maintenance of SMC differentiation phenotype of cells in the implants and the successful construction of vascular tissue.
Fig. 7.
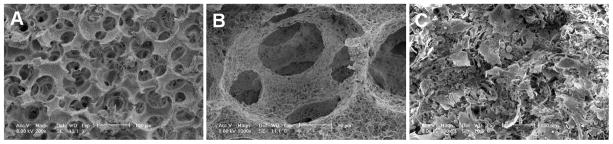
Seeding of human iPSC-derived proliferative SMCs into 3D scaffolds. SEM images of macroporous nanofibrous (NF) scaffolds (60–150 μm pore size) at low (A) and high (B) magnifications; and (C) human iPSC-derived proliferative SMCs seeded in these scaffolds for 24 h.
Fig. 8.

Construction of vascular tissues with human iPSC-derived SMCs and 3D scaffolds. Human iPSC-derived proliferative SMCs were seeded onto macroporous NF scaffolds and the cell-scaffold constructs were subcutaneously implanted into nude mice. After 2 wk of implantation, the constructs were retrieved for histological observation. H-E staining at low (A) and high (B) magnifications; Masson’s trichrome staining at low (C) and high (D) magnifications. IHC staining for α-SMA at low (G) and high (H) magnifications; CNN1 at low (K) and high (L) magnifications; SM22α at low (O) and high (P) magnifications. The human aorta tissue sections were stained correspondingly (E, F, I, J, M, N).
4. Discussion
Functional mature SMCs with a contractile phenotype are the major cell population in the vascular media, which play an important role in the homeostasis of normal vessels, such as contraction, relax and secretion of vasoactive regulators [20]. Our previous studies showed that primary human aortic SMCs grew well on tubular macroporous NF PLLA scaffolds, retained their differentiated phenotype, and could be used for vascular tissue engineering [21]. However, the difficulty in obtaining sufficient numbers of autologous cells for constructing TEBVs limits the practical application of such primary vascular SMCs. Moreover, primary vascular SMCs have low proliferation potential and suffer from culture senescence. SMCs have been demonstrated to be differentiated from alternative cell sources such as bone-marrow mesenchymal stem cells [12, 13], endothelial progenitor cells [14], adipose-derived stem cells [15], among many other adult stem cells. However, the proliferation and differentiation capabilities of adult stem cells significantly decrease with aging donors. Recently, the generation of patient-specific iPSCs, which could differentiate into various cell types, has brought substantial excitement to the field of tissue engineering and regenerative medicine [22]. In a previous report, we successfully established mouse iPSC lines and then differentiated them into SMCs with a short-term all-trans retinoid acid treatment [16]. However, obtaining patient-specific iPSC-derived SMCs to engineer human vascular grafts for clinic application remains a daunting challenge. In this study, we first established a facile procedure to generate human iPSCs from human aortic tissues and then efficiently differentiate these pluripotent cells into functional vascular SMCs, which could be easily scaled up to produce large numbers of functional cells with reproducible properties. A morphologically homogenous proliferative cell population was derived, which could be passaged and sequentially differentiated into functional SMCs. These proliferative SMCs intermediates allow us to rapidly expand a large number of cells before further inducing them into less-proliferative, contractile mature cells. In addition, when these proliferative cells were injected into SCID mice, no teratoma tissue was developed, minimizing the concern over the risk of teratoma formation in the future clinical application. The tissue engineered patient-specific blood vessels using the patient’s iPSCs have potential for the treatment of peripheral vascular disease, which is commonly seen in diabetic patients and renal failure patients. The generation of iPSCs from the fibroblasts of diabetic patients and renal failure patients need to be explored for such applications in the future.
Another challenge in the construction of TEBVs is to develop a suitable porous and biodegradable scaffold to support vascular cells growth and differentiation, and ultimately to form functional vascular tissue. Using a macroporous and NF PLLA scaffold and a control scaffold that does not have the NF feature, we found that the scaffold with the NF feature advantageously supported the growth and differentiation of mouse iPSC-derived SMCs [16]. In this study, we reduced pore size of the macroporous and NF PLLA scaffold (changing from 125–250 μm to 60–150 μm in pore diameter), and found that this NF scaffold with the smaller pore size better support human SMC growth and vascular tissue formation (unpublished data). This finding was corroborated by a recent report that a porous scaffold fabricated from ploy(glycerol sebacate) (PGS) with a smaller pore size resulted in a higher SMC density in the lumen and more ECM protein deposition compared to the scaffold with a larger pore size [23]. The reasonably smaller pore size may facilitate cell-cell interactions in the pores, possibly resulted in the increased ECM production. However, too small a pore size leads to inefficient cell seeding inside the scaffolds due to limited pore opening size and interconnection size between the pores. We chose a pore size of 60–150 μm because this size range allowed for efficient cell infiltration throughout the entire scaffolds while maintaining the SMC phenotype and promoting vascular tissue formation upon implantation.
Construction of a whole TEBV needs the incorporation of other cell types. The blood vessels are composed of three layers, namely the tunica intima, media and adventitia, which are mainly composed of endothelial cells, SMCs and fibroblasts, respectively. In this study, we primarily used human iPSC-derived SMCs to construct vascular tissues, due to the major role of tunica media in the maintenance of vessel tension and contraction-relaxation activities. In the literature, it has been suggested that co-culture with fibroblasts on the abluminal surface of the tubular constructs might increase collagen production and ECM organization, thus enhancing mechanical properties of the TE grafts [23]. In addition, lining up with a layer of endothelial cells inside the lumen of tubular constructs has been demonstrated to prevent thrombosis and protect SMCs from direct shear stress as well [24]. The fibroblasts and endothelial layers will be incorporated into our TE vascular tissues in future studies, and their mechanical properties will be evaluated for potential clinical application.
5. Conclusions
In summary, our study established an efficient approach towards generating patient-specific TEBVs. Large numbers of functional SMCs were efficiently obtained with our established reprogramming, expansion and differentiation protocols. Vascular tissue was constructed using these human iPSC-derived SMCs and an advantageous macroporous NF scaffold. Future studies are needed to regenerate the whole and functional patient-specific TEBVs.
Supplementary Material
Acknowledgments
The authors would like to acknowledge the financial support from the National Institute of Health (HL114038: PXM and YEC), University of Michigan Frankel Cardiovascular Center Inaugural Grant (BY), and University of Michigan MCubed grant (BY, YEC, and PXM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Laslett LJ, Alagona P, Jr, Clark BA, 3rd, Drozda JP, Jr, Saldivar F, Wilson SR, Poe C, Hart M. The worldwide environment of cardiovascular disease: prevalence, diagnosis, therapy, and policy issues: a report from the American College of Cardiology. J Am Coll Cardiol. 2012;60:S1–49. doi: 10.1016/j.jacc.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Isenberg BC, Williams C, Tranquillo RT. Small-diameter artificial arteries engineered in vitro. Circ Res. 2006;98:25–35. doi: 10.1161/01.RES.0000196867.12470.84. [DOI] [PubMed] [Google Scholar]
- 3.Kurobe H, Maxfield MW, Breuer CK, Shinoka T. Concise review: tissue-engineered vascular grafts for cardiac surgery: past, present, and future. Stem Cells Transl Med. 2012;1:566–71. doi: 10.5966/sctm.2012-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shinoka T, Breuer C. Tissue-engineered blood vessels in pediatric cardiac surgery. Yale J Biol Med. 2008;81:161–6. [PMC free article] [PubMed] [Google Scholar]
- 5.Schmidt CE, Baier JM. Acellular vascular tissues: natural biomaterials for tissue repair and tissue engineering. Biomaterials. 2000;21:2215–31. doi: 10.1016/s0142-9612(00)00148-4. [DOI] [PubMed] [Google Scholar]
- 6.Dahl SL, Kypson AP, Lawson JH, Blum JL, Strader JT, Li Y, et al. Readily available tissue-engineered vascular grafts. Sci Transl Med. 2011;3:68ra9. doi: 10.1126/scitranslmed.3001426. [DOI] [PubMed] [Google Scholar]
- 7.Hodde JP, Record RD, Tullius RS, Badylak SF. Retention of endothelial cell adherence to porcine-derived extracellular matrix after disinfection and sterilization. Tissue Eng. 2002;8:225–34. doi: 10.1089/107632702753724996. [DOI] [PubMed] [Google Scholar]
- 8.L’Heureux N, Germain L, Labbe R, Auger FA. In vitro construction of a human blood vessel from cultured vascular cells: a morphologic study. J Vasc Surg. 1993;17:499–509. doi: 10.1067/mva.1993.38251. [DOI] [PubMed] [Google Scholar]
- 9.Konig G, McAllister TN, Dusserre N, Garrido SA, Iyican C, Marini A, et al. Mechanical properties of completely autologous human tissue engineered blood vessels compared to human saphenous vein and mammary artery. Biomaterials. 2009;30:1542–50. doi: 10.1016/j.biomaterials.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Niklason LE, Gao J, Abbott WM, Hirschi KK, Houser S, Marini R, et al. Functional arteries grown in vitro. Science. 1999;284:489–93. doi: 10.1126/science.284.5413.489. [DOI] [PubMed] [Google Scholar]
- 11.Bajpai VK, Andreadis ST. Stem cell sources for vascular tissue engineering and regeneration. Tissue Eng Part B Rev. 2012;18:405–25. doi: 10.1089/ten.teb.2011.0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galmiche MC, Koteliansky VE, Briere J, Herve P, Charbord P. Stromal cells from human long-term marrow cultures are mesenchymal cells that differentiate following a vascular smooth muscle differentiation pathway. Blood. 1993;82:66–76. [PubMed] [Google Scholar]
- 13.Kashiwakura Y, Katoh Y, Tamayose K, Konishi H, Takaya N, Yuhara S, et al. Isolation of bone marrow stromal cell-derived smooth muscle cells by a human SM22alpha promoter: in vitro differentiation of putative smooth muscle progenitor cells of bone marrow. Circulation. 2003;107:2078–81. doi: 10.1161/01.CIR.0000070082.64414.B5. [DOI] [PubMed] [Google Scholar]
- 14.Li X, Tjwa M, Moons L, Fons P, Noel A, Ny A, et al. Revascularization of ischemic tissues by PDGF-CC via effects on endothelial cells and their progenitors. J Clin Invest. 2005;115:118–27. doi: 10.1172/JCI19189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriguez LV, Alfonso Z, Zhang R, Leung J, Wu B, Ignarro LJ. Clonogenic multipotent stem cells in human adipose tissue differentiate into functional smooth muscle cells. Proc Natl Acad Sci U S A. 2006;103:12167–72. doi: 10.1073/pnas.0604850103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xie C, Hu J, Ma H, Zhang J, Chang LJ, Chen YE, et al. Three-dimensional growth of iPS cell-derived smooth muscle cells on nanofibrous scaffolds. Biomaterials. 2011;32:4369–75. doi: 10.1016/j.biomaterials.2011.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu J, Xie C, Ma H, Yang B, Ma PX, Chen YE. Construction of vascular tissues with macro-porous nano-fibrous scaffolds and smooth muscle cells enriched from differentiated embryonic stem cells. PLoS One. 2012;7:e35580. doi: 10.1371/journal.pone.0035580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang LJ, Gay EE. The molecular genetics of lentiviral vectors--current and future perspectives. Curr Gene Ther. 2001;1:237–51. doi: 10.2174/1566523013348634. [DOI] [PubMed] [Google Scholar]
- 19.Xie CQ, Zhang J, Villacorta L, Cui T, Huang H, Chen YE. A highly efficient method to differentiate smooth muscle cells from human embryonic stem cells. Arterioscler Thromb Vasc Biol. 2007;27:e311–2. doi: 10.1161/ATVBAHA.107.154260. [DOI] [PubMed] [Google Scholar]
- 20.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–93. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 21.Hu J, Sun X, Ma H, Xie C, Chen YE, Ma PX. Porous nanofibrous PLLA scaffolds for vascular tissue engineering. Biomaterials. 2010;31:7971–7. doi: 10.1016/j.biomaterials.2010.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okano H, Nakamura M, Yoshida K, Okada Y, Tsuji O, Nori S, et al. Steps Toward Safe Cell Therapy Using Induced Pluripotent Stem Cells. Circulation Research. 2013;112:523–33. doi: 10.1161/CIRCRESAHA.111.256149. [DOI] [PubMed] [Google Scholar]
- 23.Lee KW, Stolz DB, Wang Y. Substantial expression of mature elastin in arterial constructs. Proc Natl Acad Sci U S A. 2011;108:2705–10. doi: 10.1073/pnas.1017834108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heyligers JM, Arts CH, Verhagen HJ, de Groot PG, Moll FL. Improving small-diameter vascular grafts: from the application of an endothelial cell lining to the construction of a tissue-engineered blood vessel. Ann Vasc Surg. 2005;19:448–56. doi: 10.1007/s10016-005-0026-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.